Three Dimensional Pharmacophore Modelling of Monoamine oxidase-A (MAO-A) inhibitors

Abstract
:1. Introduction
2. Computational Method
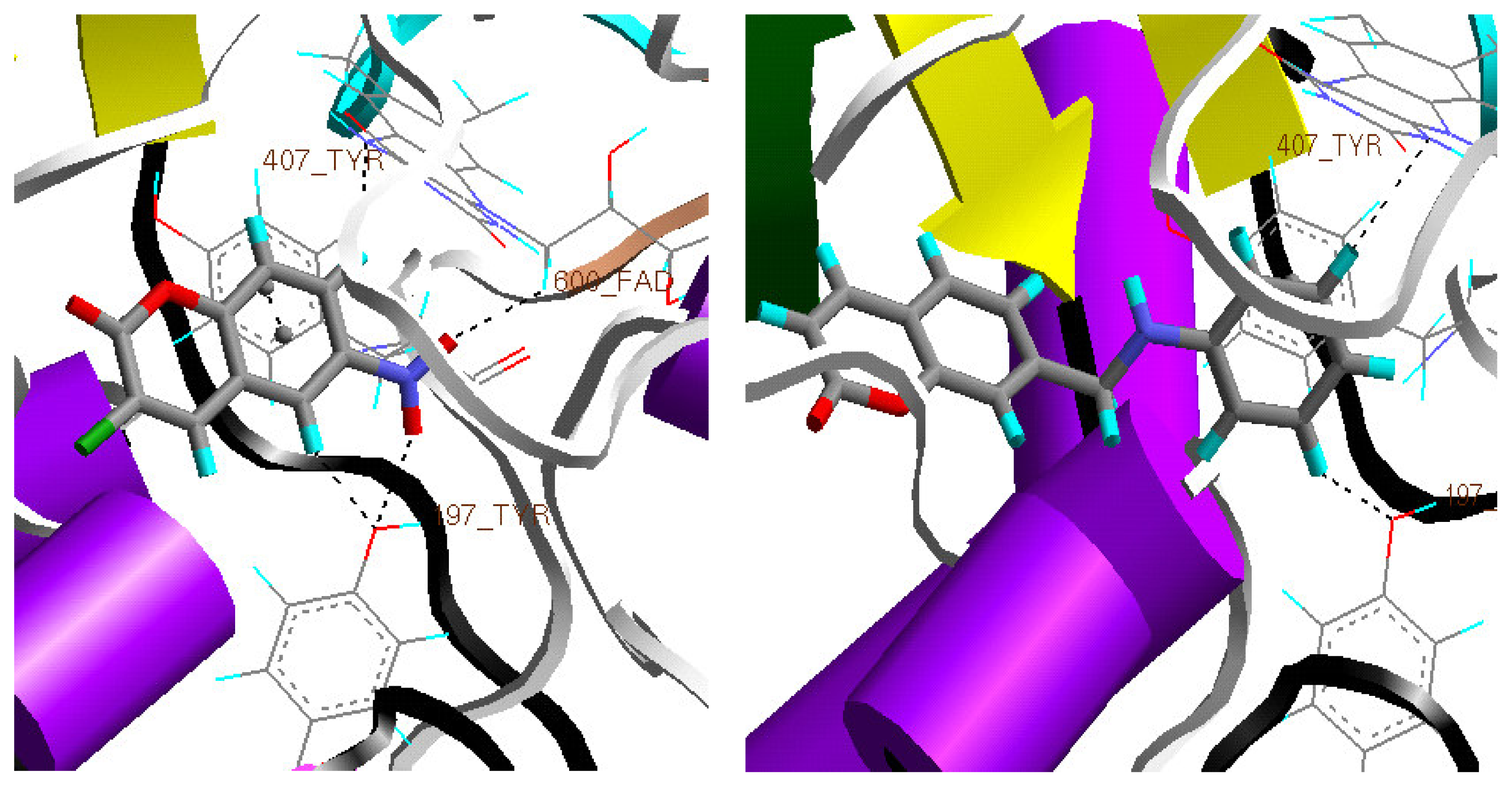
Docking
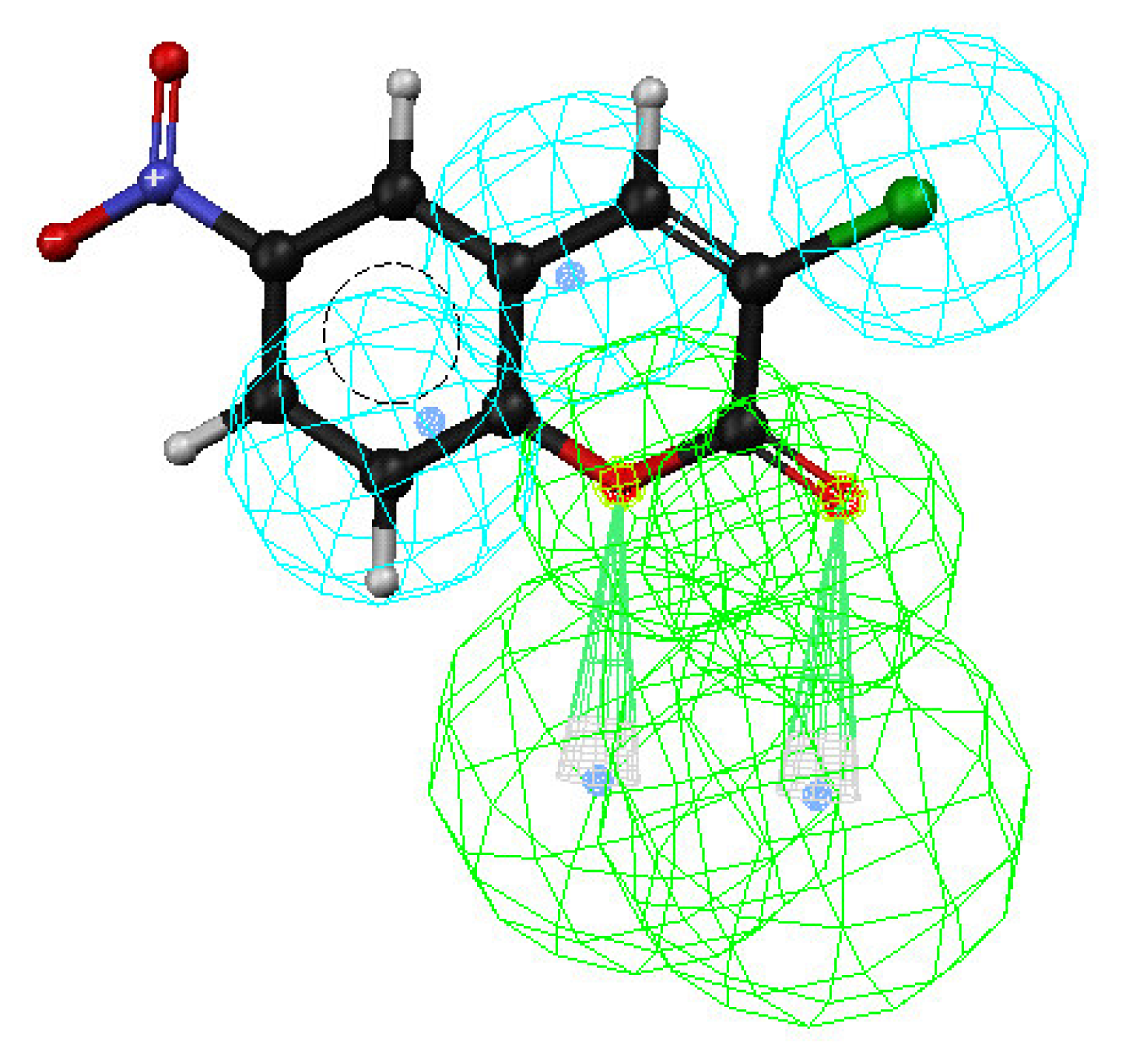
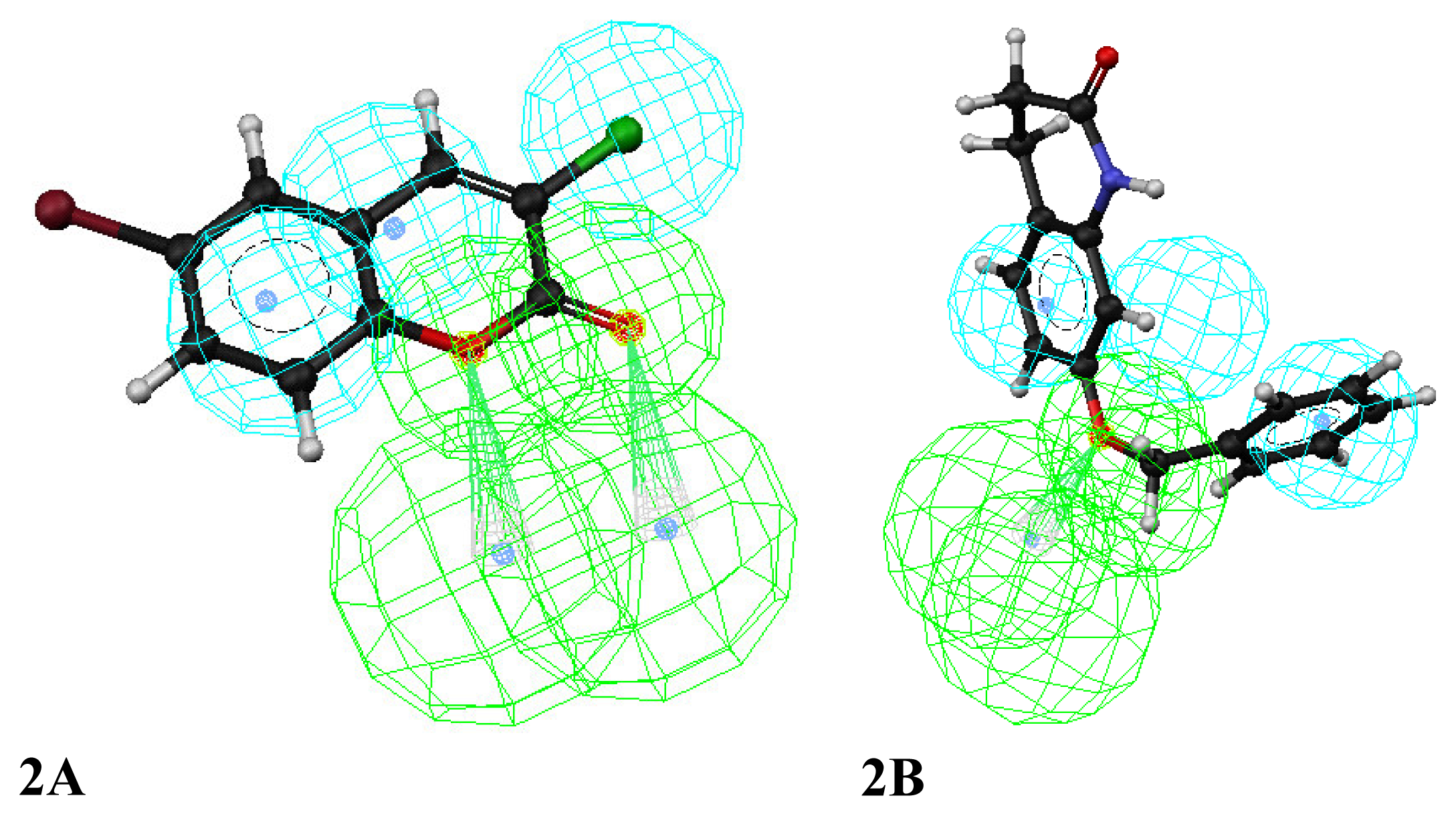
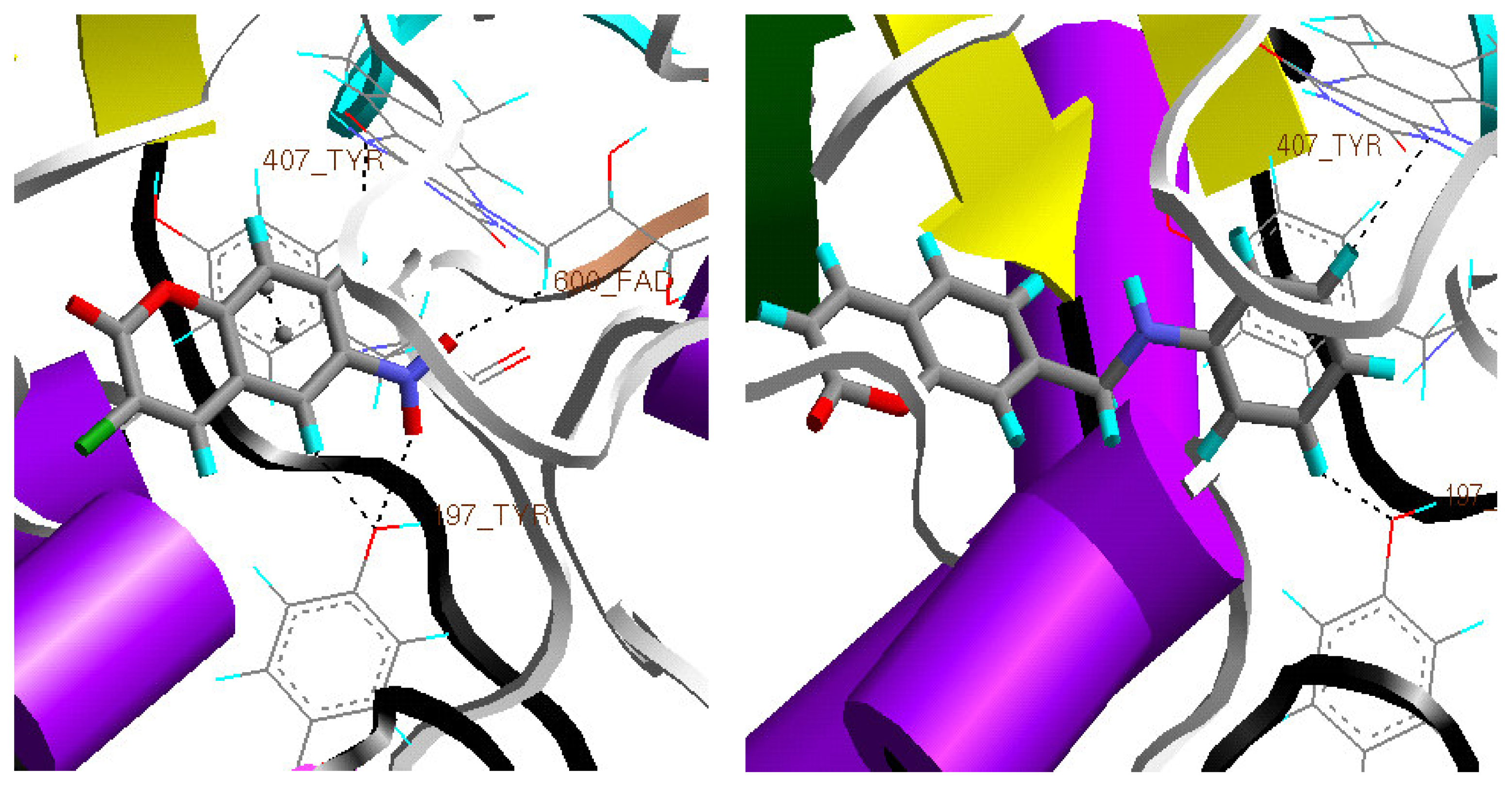
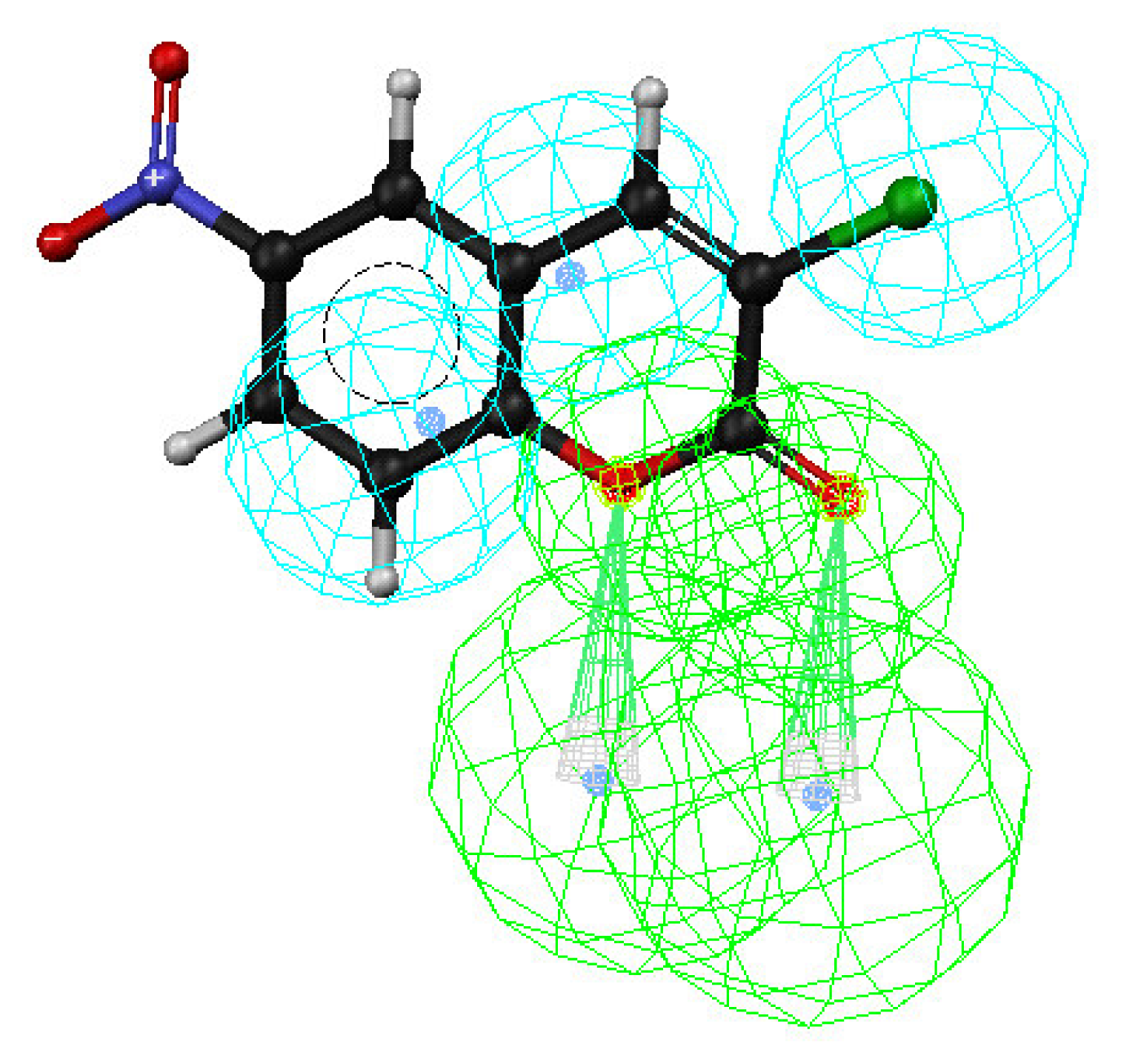
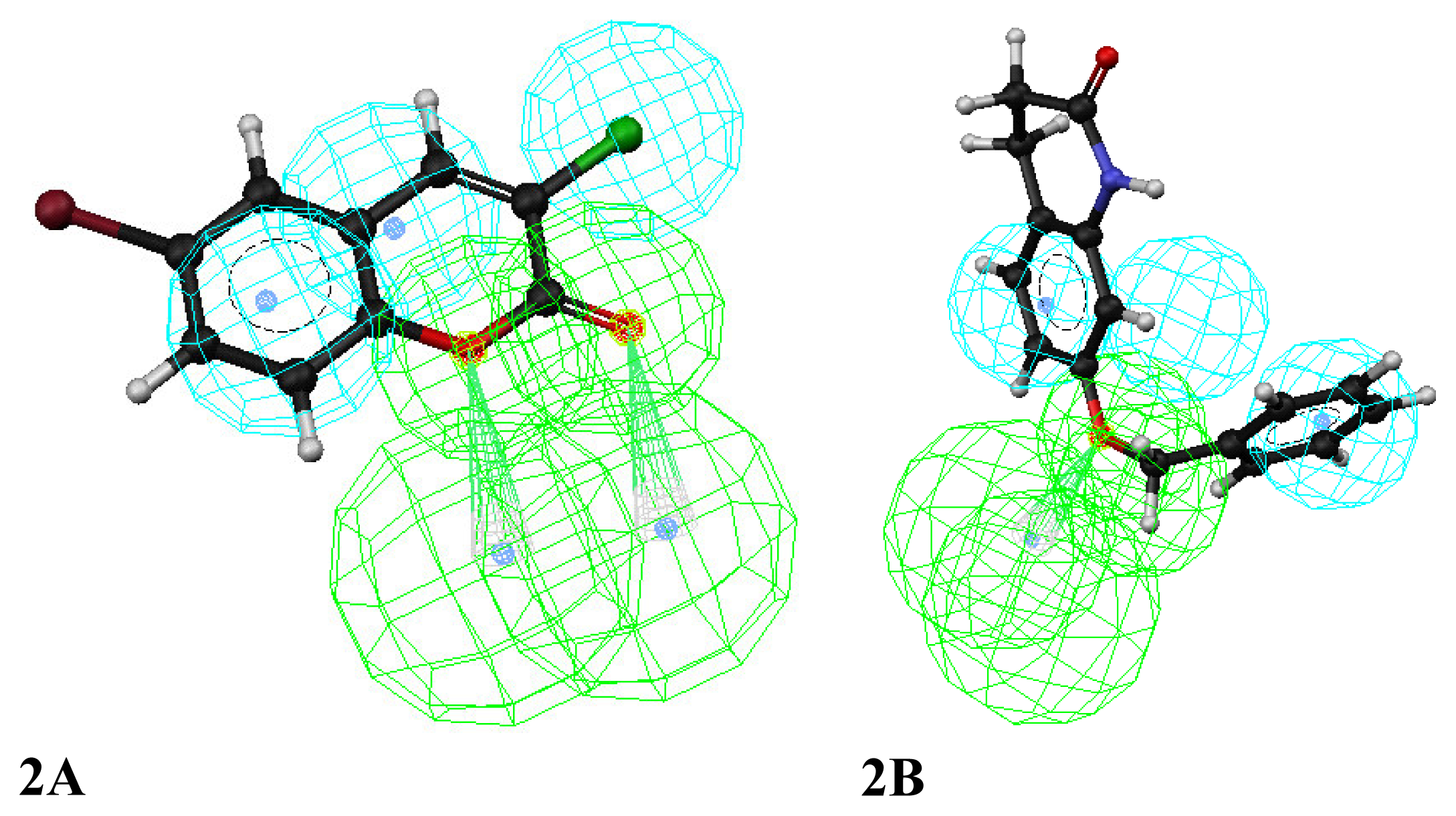
3. Results and Discussion
Enrichment factor analysis
Docking
4. Conclusion
Supplementary Information

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Reference | ||
|---|---|---|---|
| 1 | MAOB specific |  | Eur. J. Med. Chem.1995, 30, 471–482 |
| 2 |  | Eur. J. Med. Chem.1995, 30, 471–482 | |
| 3 |  | Eur. J. Med. Chem.1995, 30, 471–482 | |
| 4 | 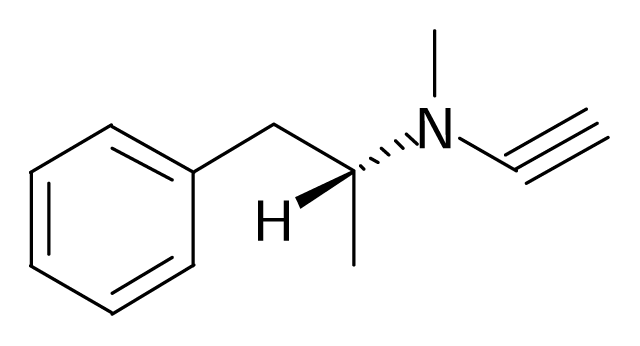 | J. Enzyme Inhib. Med. Chem.2003, 18, 339–347 | |
| 5 |  | J. Med. Chem.1992, 35, 3705–3713 | |
| 6 | 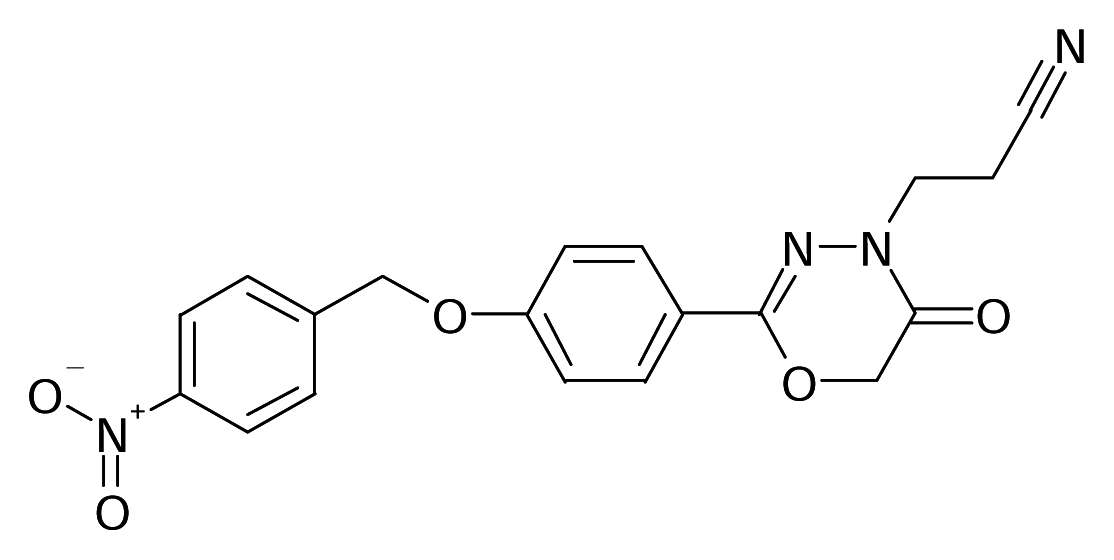 | J. Med. Chem.1993, 36, 1157–1167 | |
| 7 |  | J. Med. Chem.1993, 36, 1157–1167 | |
| 8 |  | J. Med. Chem.1993, 36, 1157–1167 | |
| 9 |  | J. Med. Chem.1993, 36, 1157–1167 | |
| 10 | 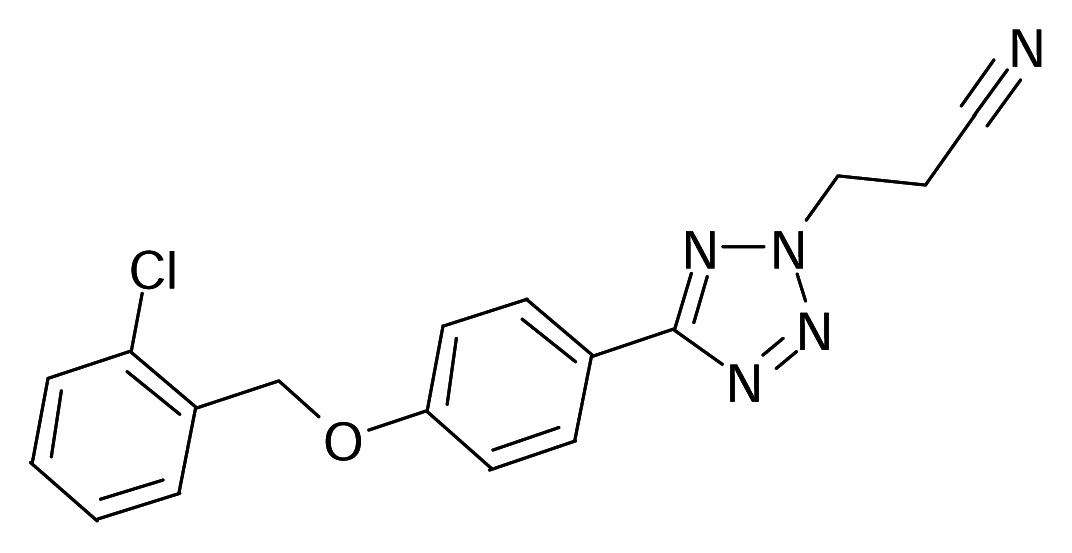 | J. Med. Chem.1995, 38, 4786–4792 | |
| 11 | MAOA Specific |  | J. Med. Chem.2004, 47, 3455–3461 |
| 12 |  | J. Med. Chem.1994, 37, 2085–2089 | |
| 13 |  | J. Med. Chem.1994, 37, 2085–2089 | |
| 14 |  | J. Med. Chem.1994, 37, 2085–2089 | |
| 15 |  | J. Med. Chem.1993, 36, 1157–1167 | |
| 16 |  | J. Med. Chem.1991, 34, 2931–2933 | |
| 17 |  | J. Med. Chem.1991, 34, 2931–2933 | |
| 18 |  | J. Med. Chem.2005, 48, 2407–2419 | |
| 19 | 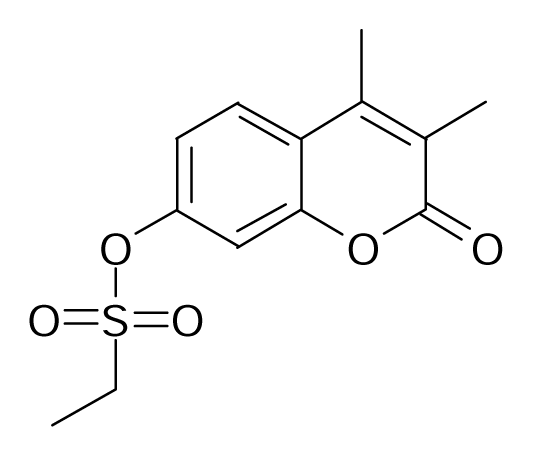 | Bioorg. Med. Chem. Lett.1994, 4, 1195–1198 | |
| 20 |  | J. Med. Chem.1998, 41, 2118–2125 | |
| 21 |  | J. Med. Chem.1998, 41, 2118–2125 | |
| 22 |  | Eur. J. Med. Chem.1999, 34, 137–151 | |
| 23 |  | J. Med. Chem.1997, 40, 2466–2473 | |
| 24 |  | J. Med. Chem.1996, 39, 1857–1863 | |
| 25 | Mixed |  | J. Med. Chem.1996, 39, 1857–1863 |
| 26 |  | J. Med. Chem.2002, 45, 5260–5279 | |
| 27 |  | J. Med. Chem.1994, 37, 2085–2089 | |
| 28 | 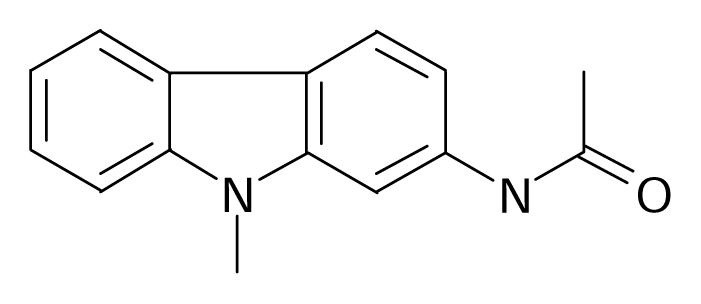 | J. Med. Chem.1994, 37, 2085–2089 | |
| 29 |  | J. Med. Chem.1994, 37, 2085–2089 | |
| 30 |  | J. Enzyme Inhib. Med. Chem.2003, 18, 339–347 | |
| 31 |  | J. Enzyme Inhib. Med. Chem.2003, 18, 339–347 | |
| 32 |  | J. Enzyme Inhib. Med. Chem.2003, 18, 339–347 | |
| 33 |  | J. Med. Chem.2004, 47, 3455–3461 | |
| 34 |  | J. Med. Chem.2004, 47, 3455–3461 | |
| 35 |  | J. Med. Chem.2005, 48, 664–670 | |
| 36 | 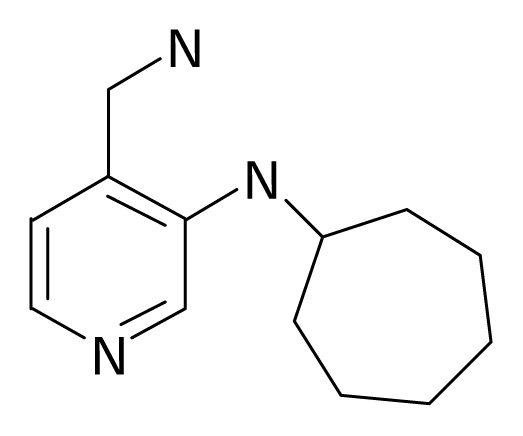 | J. Med. Chem.2005, 48, 664–670 | |
| 37 | 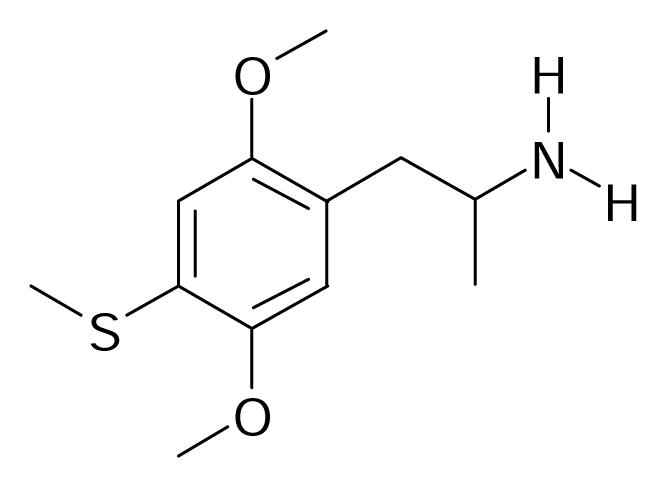 | J. Med. Chem.2005, 48, 2407–2419 | |
| 38 |  | J. Med. Chem.2005, 48, 2407–2419 | |
| 39 | 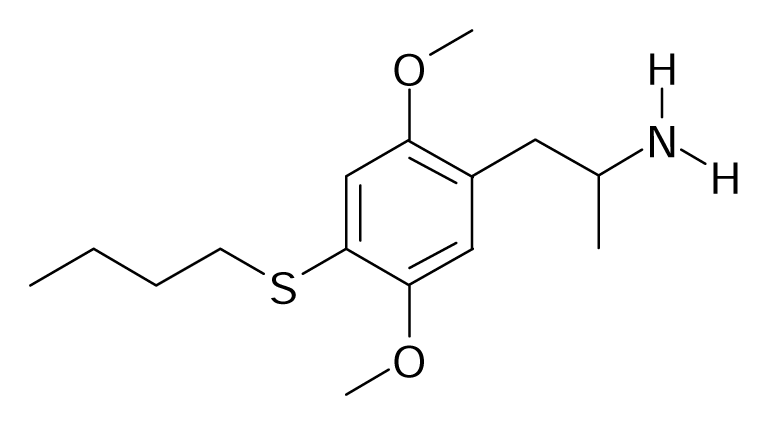 | J. Med. Chem.2005, 48, 2407–2419 | |
| 40 |  | J. Med. Chem.2005, 48, 2407–2419 | |
| 41 |  | J. Med. Chem.2005, 48, 2407–2419 | |
| 42 |  | J. Med. Chem.2005, 48, 2407–2419 | |
| 43 |  | J. Med. Chem.2005, 48, 2407–2419 | |
| 44 |  | J. Med. Chem.2005, 48, 2407–2419 | |
| 45 |  | J. Med. Chem.2005, 48, 2407–2419 | |
| 46 |  | J. Med. Chem.2005, 48, 2407–2419 | |
| 47 |  | J. Med. Chem.2005, 48, 2407–2419 | |
| 48 |  | Bioorg. Med. Chem. Lett.1994, 4, 1195–1198 | |
| 49 |  | Bioorg. Med. Chem. Lett.1994, 4, 1195–1198 | |
| 50 |  | Bioorg. Med. Chem. Lett.1994, 4, 1195–1198 | |
| 51 |  | Bioorg. Med. Chem. Lett.1994, 4, 1195–1198 | |
| 52 |  | J. Med. Chem.2002, 45, 5260–5279 | |
| 53 |  | J. Med. Chem.2002, 45, 5260–5279 | |
| 54 |  | J. Med. Chem.2002, 45, 5260–5279 | |
| 55 |  | J. Med. Chem.2002, 45, 5260–5279 | |
| 56 |  | J. Med. Chem.2002, 45, 5260–5279 | |
| 57 | 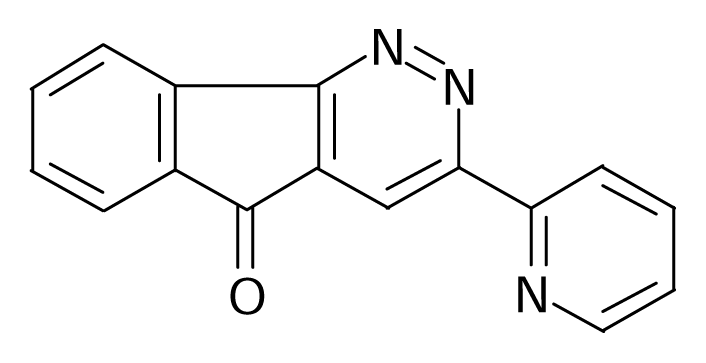 | J. Med. Chem.1995, 38, 3874–3883 | |
| 58 |  | J. Med. Chem.1995, 38, 3874–3883 | |
| 59 |  | Bazinaprine | |
| 60 |  | J. Med. Chem.1994, 37, 2085–2089 | |
| 61 |  | J. Med. Chem.1993, 36, 1157–1167 | |
| 62 | 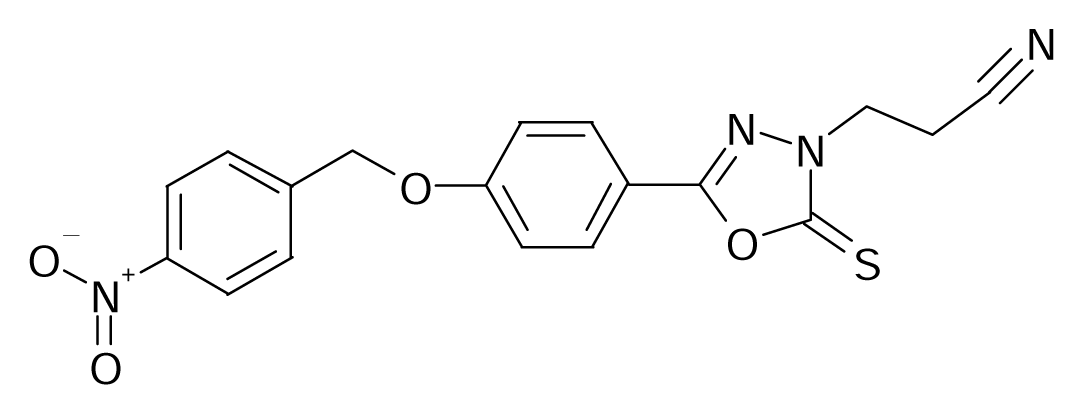 | J. Med. Chem.1993, 36, 1157–1167 | |
| 63 |  | J. Med. Chem.1993, 36, 1157–1167 | |
| 64 |  | J. Med. Chem.1993, 36, 1157–1167 | |
| 65 |  | J. Med. Chem.1993, 36, 1157–1167 | |
| 66 |  | J. Med. Chem.1992, 35, 3705–3713 | |
| 67 |  | J. Med. Chem.1992, 35, 3705–3713 | |
| 68 | 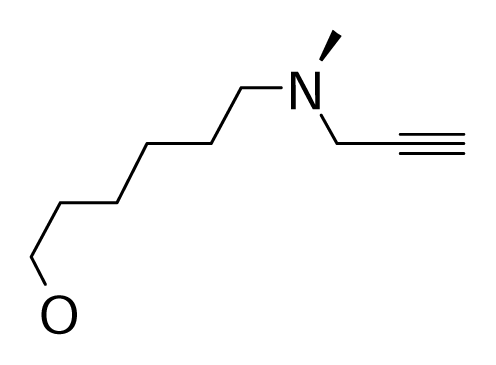 | J. Med. Chem.1992, 35, 3705–3713 | |
| 69 | 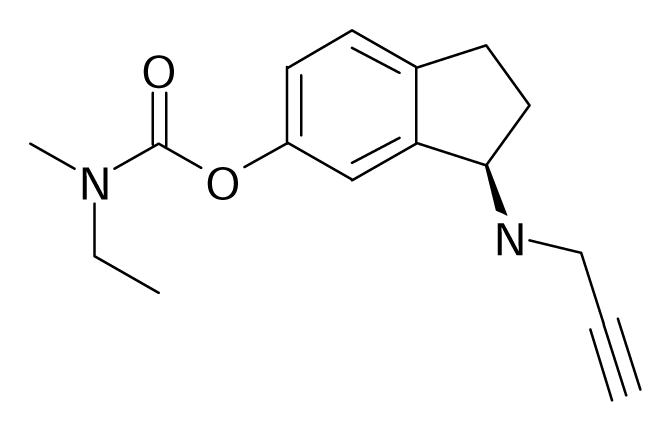 | J. Med. Chem.2004, 47, 1760–1766 | |
| 70 |  | J. Med. Chem.2004, 47, 3455–3461 | |
| 71 |  | J. Med. Chem.2004, 47, 3455–3461 | |
| 72 |  | J. Med. Chem.2004, 47, 3455–3461 | |
| 73 |  | J. Med. Chem.2004, 47, 3455–3461 | |
| 74 |  | J. Med. Chem.2004, 47, 3455–3461 | |
| 75 |  | J. Med. Chem.2004, 47, 3455–3461 | |
| 76 |  | J. Med. Chem.2004, 47, 3455–3461 | |
| 77 |  | J. Med. Chem.2004, 47, 3455–3461 | |
| 78 |  | J. Med. Chem.2004, 47, 3455–3461 | |
| 79 |  | J. Med. Chem.2005, 48, 664–670 | |
| 80 |  | J. Med. Chem.2005, 48, 664–670 | |
| 81 |  | J. Med. Chem.2006, 49, 3743–3747 | |
| 82 |  | Bioorg. Med. Chem. Lett.1996, 6, 115–120 | |
| 83 |  | Bioorg. Med. Chem. Lett.1996, 6, 115–120 | |
| 84 |  | Bioorg. Med. Chem. Lett.1996, 6, 115–120 | |
| 85 |  | Bioorg. Med. Chem. Lett.1996, 6, 115–120 | |
| 86 |  | Bioorg. Med. Chem. Lett.1996, 6, 115–120 | |
| 87 |  | Bioorg. Med. Chem. Lett.1996, 6, 115–120 | |
| 88 |  | Bioorg. Med. Chem. Lett.1996, 6, 115–120 | |
| 89 |  | Bioorg. Med. Chem. Lett.1996, 6, 115–120 | |
| 90 |  | Bioorg. Med. Chem. Lett.1996, 6, 115–120 | |
| 91 |  | Bioorg. Med. Chem. Lett.1996, 6, 115–120 | |
| 92 | 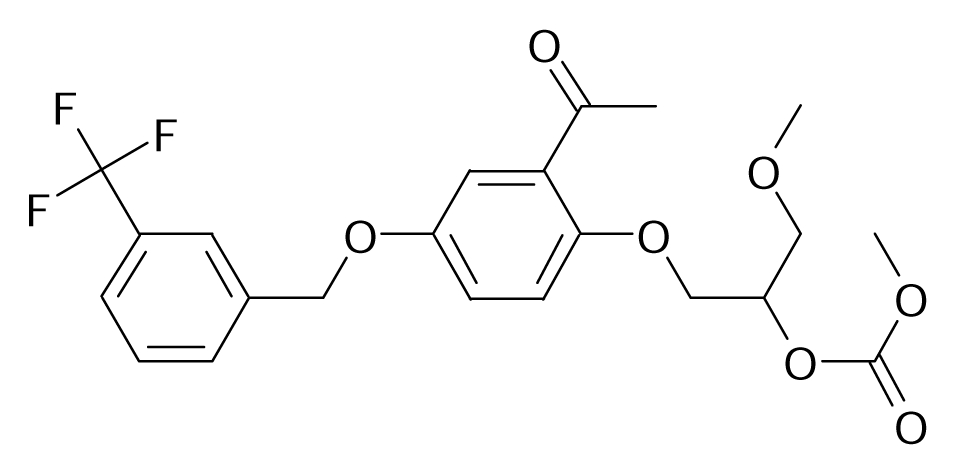 | Bioorg. Med. Chem. Lett.1996, 6, 115–120 | |
| 93 |  | Bioorg. Med. Chem. Lett.1996, 6, 115–120 | |
| 94 |  | Bioorg. Med. Chem. Lett.1994, 4, 1195–1198 | |
| 95 |  | Bioorg. Med. Chem. Lett.1994, 4, 1195–1198 | |
| 96 |  | Bioorg. Med. Chem. Lett.1994, 4, 1195–1198 | |
| 97 |  | Bioorg. Med. Chem. Lett.1994, 4, 1195–1198 | |
| 98 |  | Bioorg. Med. Chem. Lett.1994, 4, 1195–1198 | |
| 99 | 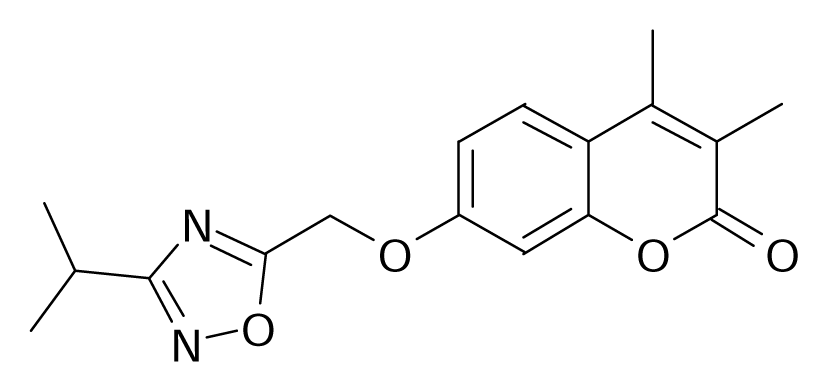 | Bioorg. Med. Chem. Lett.1994, 4, 1195–1198 | |
| 100 | 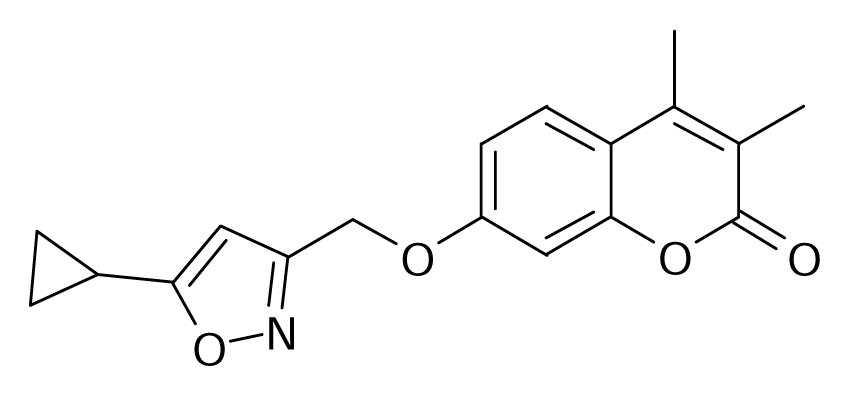 | Bioorg. Med. Chem. Lett.1994, 4, 1195–1198 | |
| 101 |  | Bioorg. Med. Chem. Lett.1994, 4, 1195–1198 | |
| 102 |  | Bioorg. Med. Chem. Lett.2001, 11, 2715–2717 | |
| 103 |  | Bioorg. Med. Chem. Lett.2001, 11, 2715–2717 | |
| 104 |  | Bioorg. Med. Chem. Lett.2001, 11, 2715–2717 | |
| 105 |  | Bioorg. Med. Chem. Lett.2001, 11, 2715–2717 | |
| 106 |  | Bioorg. Med. Chem. Lett.2001, 11, 2715–2717 | |
| 107 |  | Bioorg. Med. Chem. Lett.2001, 11, 2715–2717 | |
| 108 |  | Bioorg. Med. Chem. Lett.2002, 12, 2121–2123 | |
| 109 |  | Bioorg. Med. Chem. Lett.2002, 12, 2121–2123 | |
| 110 |  | Bioorg. Med. Chem. Lett.2002, 12, 2121–2123 | |
| 111 | 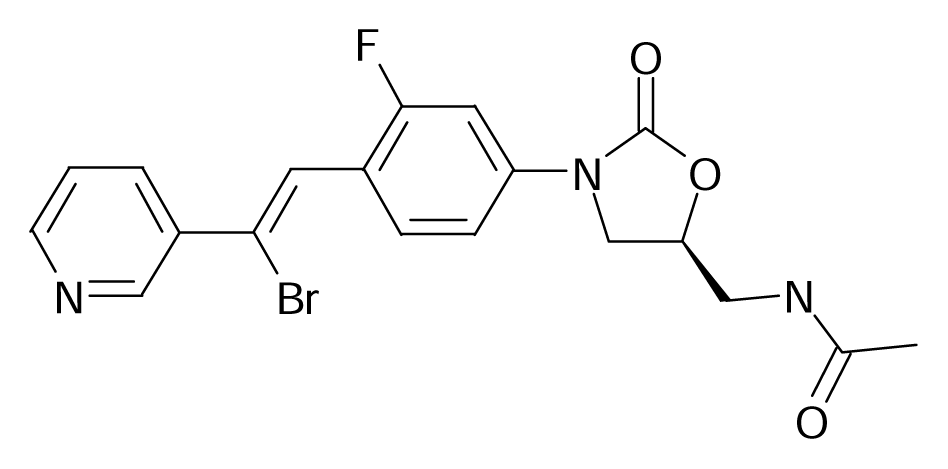 | Bioorg. Med. Chem. Lett.2002, 12, 2121–2123 | |
| 112 |  | Bioorg. Med. Chem. Lett.2002, 12, 2121–2123 | |
| 113 |  | J. Med. Chem.1998, 41, 2118–2125 | |
| 114 |  | J. Med. Chem.1998, 41, 3812–3820 | |
| 115 |  | J. Med. Chem.1995, 38, 3874–3883 | |
| 116 |  | J. Med. Chem.1998, 41, 3812–3820 | |
| 117 |  | J. Med. Chem.1998, 41, 3812–3820 | |
| 118 |  | Bioorg. Med. Chem.2004, 12, 273–279 | |
| 119 |  | Bioorg. Med. Chem.2005, 13, 773–783 | |
| 120 |  | Eur. J. Med. Chem.1992, 27, 45–52 | |
| 121 |  | Eur. J. Med. Chem.1992, 27, 45–52 | |
| 122 |  | Eur. J. Med. Chem.1992, 27, 939–948 | |
| 123 |  | Eur. J. Med. Chem.1995, 30, 471–482 | |
| 124 |  | Eur. J. Med. Chem.1995, 30, 471–482 | |
| 125 |  | Eur. J. Med. Chem.1995, 30, 471–482 |
 | |||
|---|---|---|---|
| No. | Structures | No. | Structures |
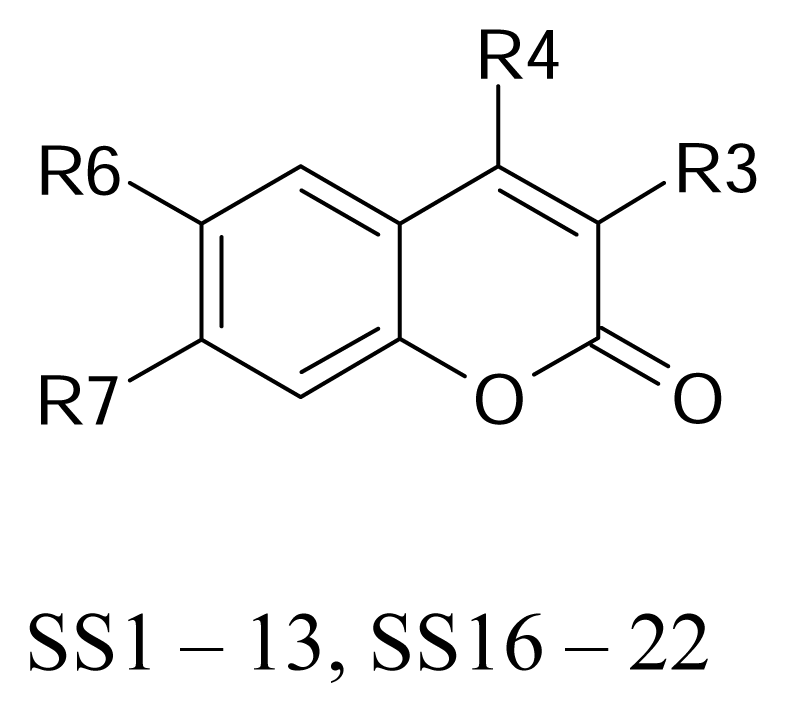
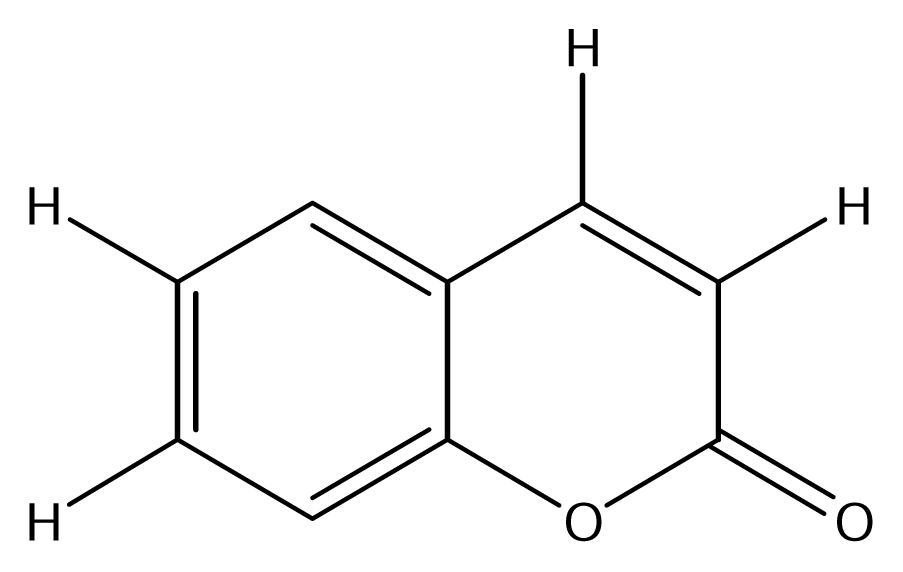
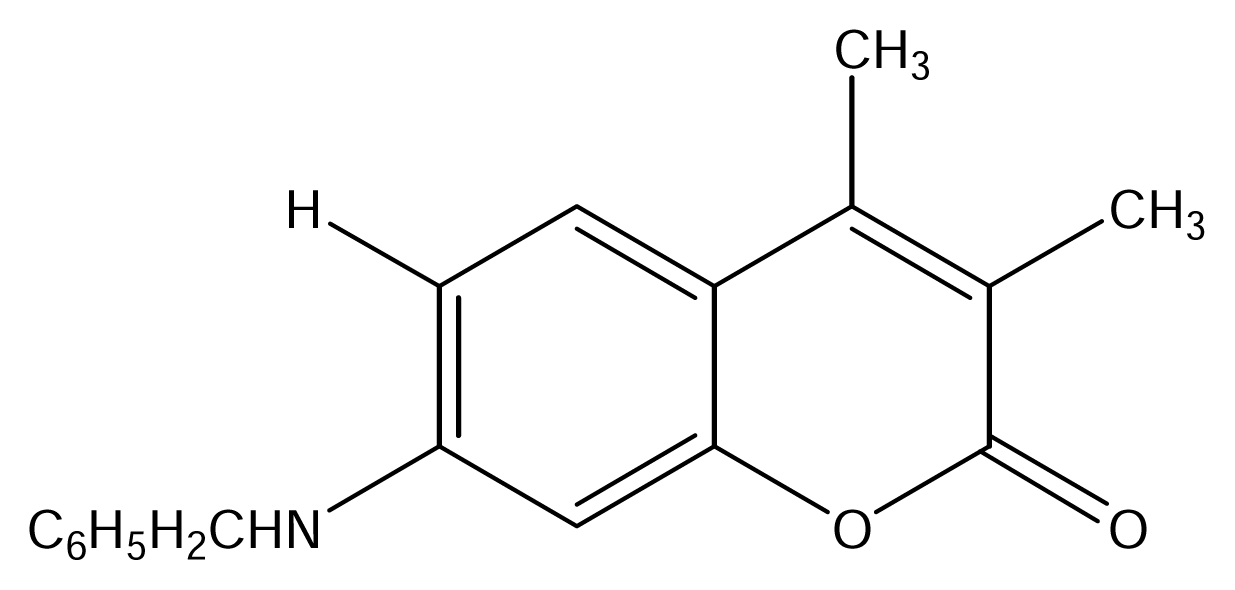
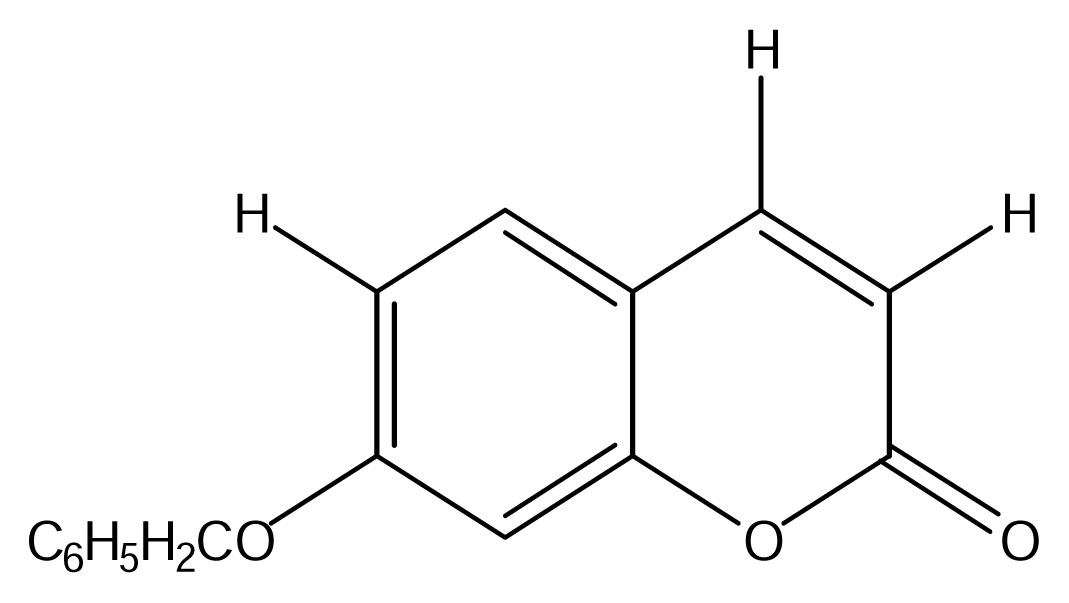
| SS01 |  | SS09 |  |
| SS02 |  | SS10 |  |
| SS03 |  | SS11 |  |
| SS04 |  | SS12 |  |
| SS05 |  | SS13 |  |
| SS06 |  | SS16 |  |
| SS07 | 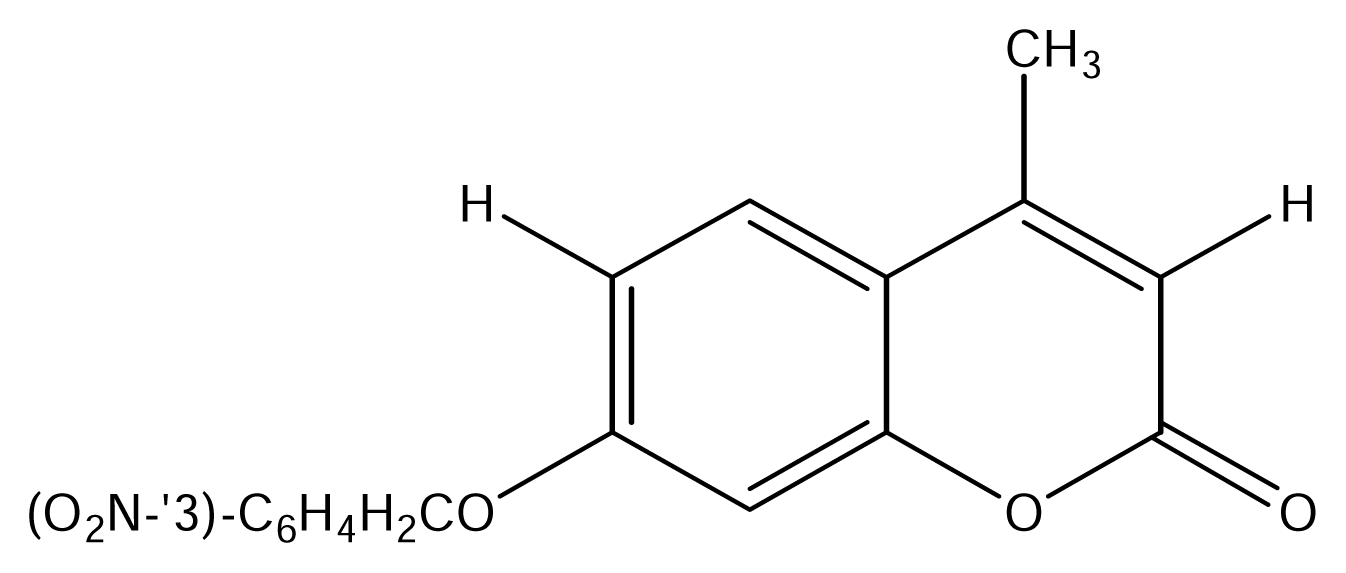 | SS17 |  |
| SS08 |  | SS18a | |
| SS19 |  | SS20 |  |
| SS21 | 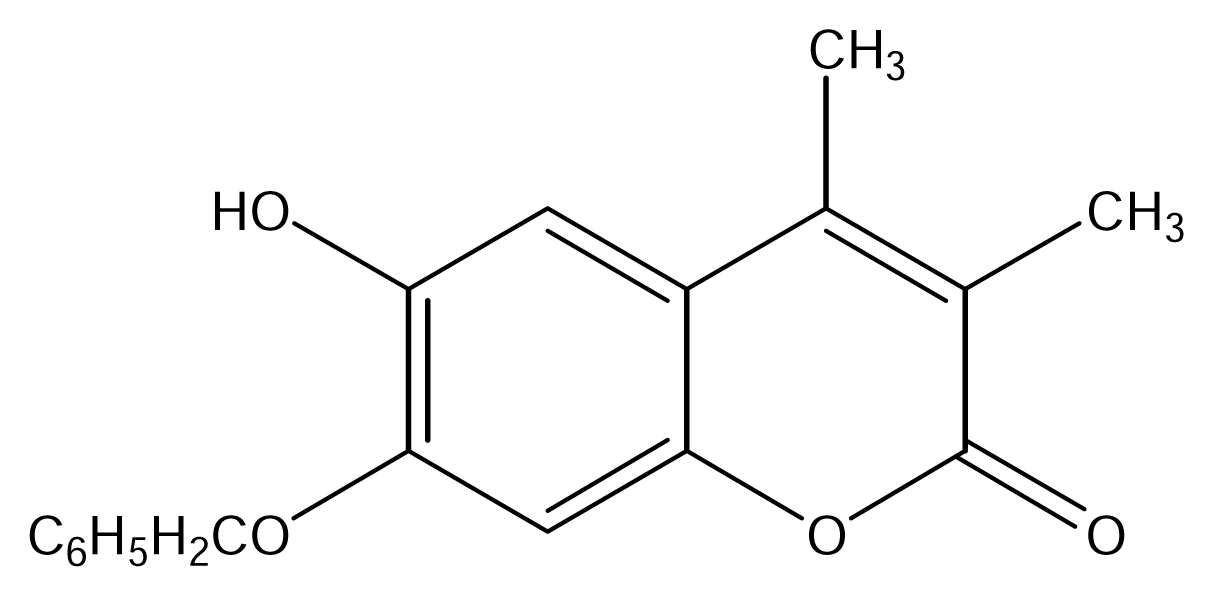 | SS22 |  |
 |  | ||
|---|---|---|---|
| No. | Structures | No. | Structures |
| SS14 |  | SS24 |  |
| SS15 |  | SS25 |  |
| SS23 |  | SS26 |  |
 | |||
|---|---|---|---|
| No. | Structures | No. | Structures |
| SS27 |  | SS33 |  |
| SS28 |  | SS34 |  |
| SS29 |  | SS35 |  |
| SS30 |  | SS36 |  |
| SS31 | 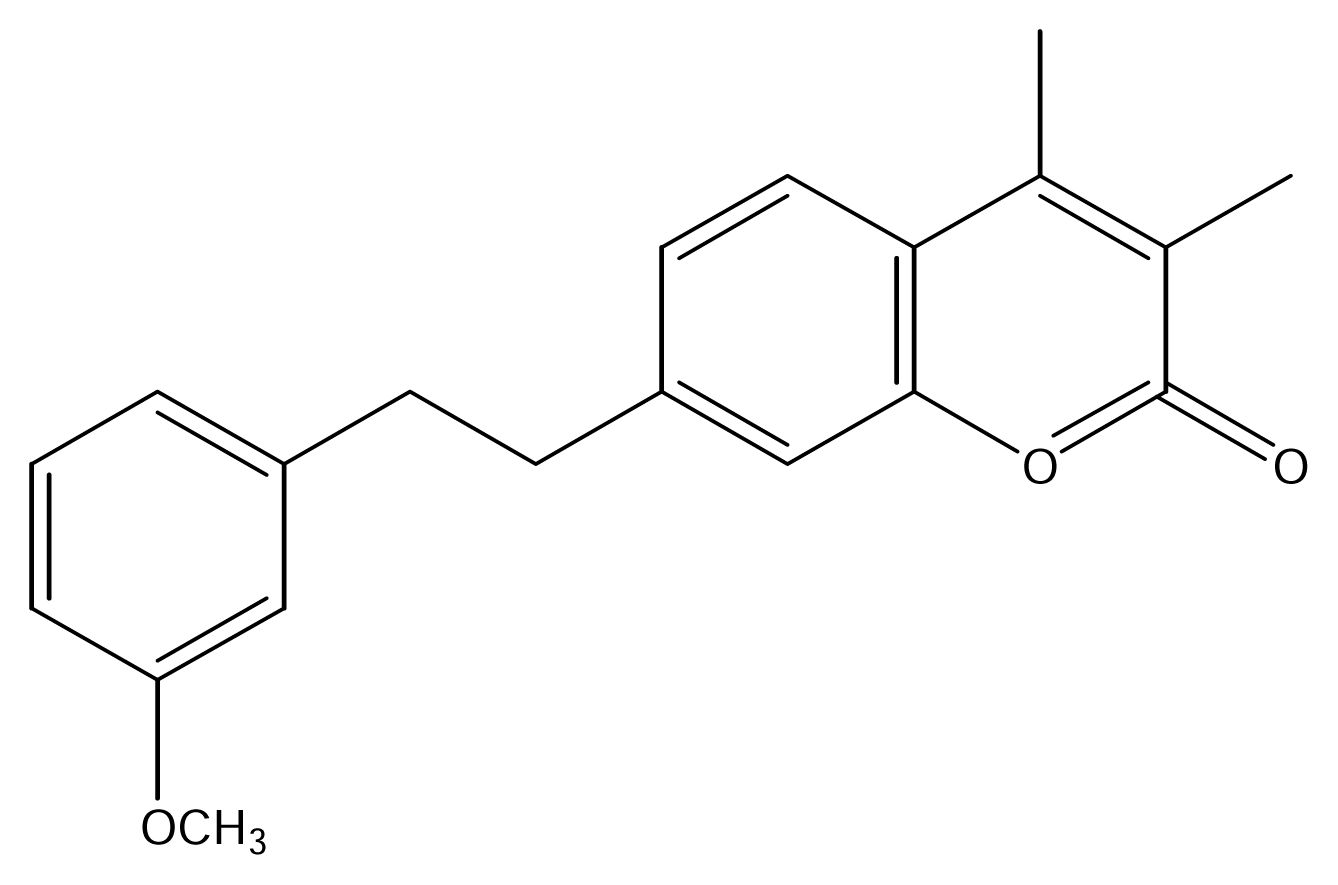 | SS37 |  |
| SS32 |  | SS38 |  |
| SS39 |  | SS40 |  |
| SS41 |  | SS42 |  |
| SS43 |  | SS44 |  |
| SS45 |  | SS47 |  |
| SS47 |  | SS48 |  |
| SS49 |  | ||
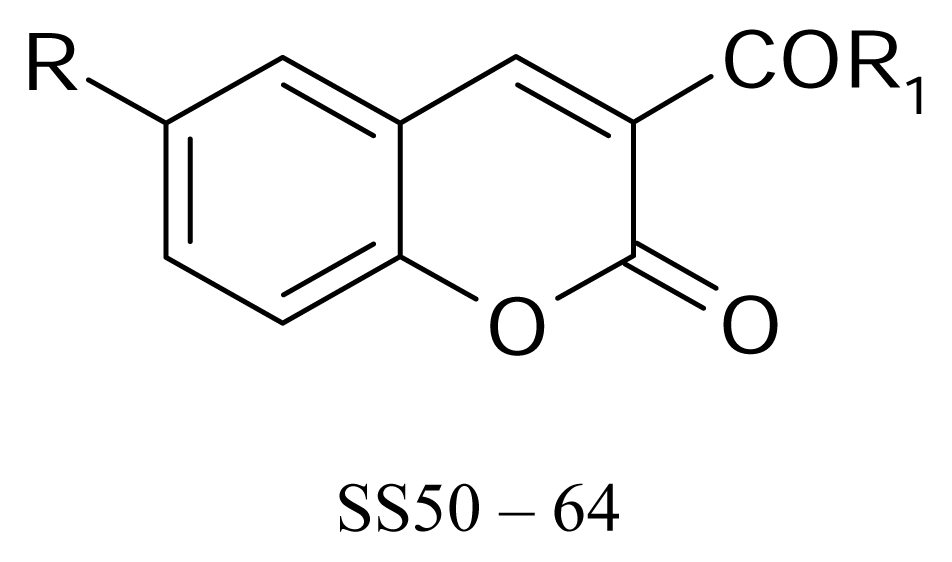 | |||
|---|---|---|---|
| No. | Structures | No. | Structures |
| SS50 |  | SS51 |  |
| SS52 |  | SS53 |  |
| SS54 |  | SS55 |  |
| SS56 |  | SS57 |  |
| SS60 |  | SS61 |  |
| SS62 |  | SS63 |  |
| SS64 |  | ||
| Sl. No. | Observed Activity | Estimated Activity |
|---|---|---|
| 1 | 0.0000407 | 0.000039 |
| 2 | 0.00000676 | 0.000021 |
| 3 | 0.000000389 | 0.000022 |
| 4 | 0.0000417 | 0.00002 |
| 5 | 0.00000195 | 0.0000022 |
| 6 | 0.00000195 | 0.000018 |
| 7 | 0.0000234 | 0.00000031 |
| 8 | 0.000000126 | 0.0000022 |
| 9 | 0.000000192 | 0.0000023 |
| 10 | 0.00000158 | 0.00000041 |
| 11 | 0.000001 | 0.000002 |
| 12 | 0.00000355 | 0.0000014 |
| 13 | 0.00000158 | 0.0000016 |
| 14 | 0.00000158 | 0.00003 |
| 15 | 0.0000162 | 0.000026 |
| 16 | 0.00000676 | 0.0000014 |
| 17 | 0.00000138 | 0.00000036 |
| 18 | 0.0000000758 | 0.0000026 |
| 19 | 0.000000562 | 0.00000018 |
| 20 | 0.0000000707 | 0.000000096 |
| 21 | 0.000112 | 0.0000022 |
| 22 | 0.00000933 | 0.0000023 |
| 23 | 0.000000407 | 0.000012 |
| 24 | 0.0000218 | 0.000027 |
| 25 | 0.0000398 | 0.000023 |
| 26 | 0.00000363 | 0.000027 |
| 27 | 0.000102 | 0.00000013 |
| 28 | 0.00000229 | 0.00000046 |
| 29 | 0.000000416 | 0.00000025 |
| 30 | 0.00000331 | 0.00000034 |
| 31 | 0.000000416 | 0.00000024 |
| 32 | 0.00000151 | 0.000000066 |
| 33 | 0.00000588 | 0.00000037 |
| 34 | 0.000001 | 0.000000064 |
| 35 | 0.00000707 | 0.00000045 |
| 36 | 0.000000575 | 0.00000044 |
| 37 | 0.00000122 | 0.00000029 |
| 38 | 0.00000191 | 0.00000011 |
| 39 | 0.000000218 | 0.00000026 |
| 40 | 0.0000000758 | 0.00000012 |
| 41 | 0.00000371 | 0.00000021 |
| 42 | 0.000000123 | 0.000000093 |
| 43 | 0.000000123 | 0.00000011 |
| 44 | 0.0000001 | 0.00000036 |
| 45 | 0.000000831 | 0.00000044 |
| 46 | 0.000000123 | 0.00000065 |
| 47 | 0.000000676 | 0.0000002 |
| 48 | 0.000000114 | 0.00000059 |
| 49 | 0.000000181 | 0.00000045 |
| 50 | 0.000000691 | 0.00000031 |
| 51 | 0.000000229 | 0.00000027 |
| 52 | 0.00000019 | 0.0000002 |
| 53 | 0.000000208 | 0.0000002 |
| 54 | 0.000000501 | 0.00000018 |
| 55 | 0.000000257 | 0.000039 |
| 56 | 0.0000151 | 0.00003 |
| 57 | 0.000012 | 0.00003 |
| 60 | 0.0000000676 | 0.00000007 |
| 61 | 0.0000000602 | 0.000000039 |
| 62 | 0.0000000288 | 0.000000037 |
| 63 | 0.0000000199 | 0.000000036 |
| 64 | 0.0000000301 | 0.000000016 |
Acknowledgement
Reference
- Geha, R.M.; Chen, K.; Wouters, J.; Ooms, F.; Shih, J.C. Analysis of conserved active site residues in monoamine oxidase A and B and their three-dimensional molecular modeling. J. Biol. Chem 2002, 277, 17209–17216. [Google Scholar]
- Gnerre, C.; Catto, M.; Leonetti, F.; Weber, P.; Carrupt, P.A.; Altomare, C.; Carotti, A.; Testa, B. Inhibition of monoamine oxidases by functionalized coumarin derivatives: biological activities, QSARs, and 3D-QSARs. J. Med. Chem 2000, 43, 4747–4758. [Google Scholar]
- Chimenti, F.; Secci, D.; Bolasco, A.; Chimenti, P.; Granese, A.M; Befani, O.; Turini, P.; Alcaro, S.; Ortuso, F. Inhibition of monoamine oxidases by coumarin-3-acyl derivatives: biological activity and computational study. Bioorg. Med. Chem. Lett 2004, 14, 3697–3703. [Google Scholar]
- CATALYST; Accelrys Inc: San Diego, CA.
- Cerius2; Accelrys Inc: San Diego, CA.
- Santana, L.; Uriarte, E.; Diaz, H. G.; Zagotto, G.; Otero, R.S.; Alvarez, E.M. A QSAR model for in silico screening of MAO-A inhibitors. Prediction, synthesis, and biological assay of novel coumarins. J. Med. Chem 2006, 49, 1149–1156. [Google Scholar]
- Parada, M.R.; Fierro, A.; Vasquez, P. I.; Cassels, B.K. A QSAR model for in silico screening of MAO-A inhibitors. Prediction, synthesis, and biological assay of novel coumarins. Curr. Enz. Inhib 2005, 1, 85–95. [Google Scholar]
© 2007 by MDPI Reproduction is permitted for noncommercial purposes.
Share and Cite
Sairam, K.V.V.M.; Khar, R.K.; Mukherjee, R.; Jain, S.K. Three Dimensional Pharmacophore Modelling of Monoamine oxidase-A (MAO-A) inhibitors. Int. J. Mol. Sci. 2007, 8, 894-919. https://doi.org/10.3390/i8090894
Sairam KVVM, Khar RK, Mukherjee R, Jain SK. Three Dimensional Pharmacophore Modelling of Monoamine oxidase-A (MAO-A) inhibitors. International Journal of Molecular Sciences. 2007; 8(9):894-919. https://doi.org/10.3390/i8090894
Chicago/Turabian StyleSairam, Kalapatapu V.V.M., Roop K. Khar, Rama Mukherjee, and Swatantra K. Jain. 2007. "Three Dimensional Pharmacophore Modelling of Monoamine oxidase-A (MAO-A) inhibitors" International Journal of Molecular Sciences 8, no. 9: 894-919. https://doi.org/10.3390/i8090894
APA StyleSairam, K. V. V. M., Khar, R. K., Mukherjee, R., & Jain, S. K. (2007). Three Dimensional Pharmacophore Modelling of Monoamine oxidase-A (MAO-A) inhibitors. International Journal of Molecular Sciences, 8(9), 894-919. https://doi.org/10.3390/i8090894