Upscaled CTAB-Based DNA Extraction and Real-Time PCR Assays for Fusarium culmorum and F. graminearum DNA in Plant Material with Reduced Sampling Error


Abstract
:1. Introduction
2. Materials and Methods
2.1. Fungal cultures
2.2. DNA isolation from pure fungal cultures grown in liquid media
2.3. Preparation of plant samples
2.4. Upscaled DNA extraction from plant material
2.5. Preparation of standards for quantitative real-time PCR
2.6. Real-time PCR
2.7. Fusarium DNA quantification in plant material
2.8. Box plot
3. Results and Discussion
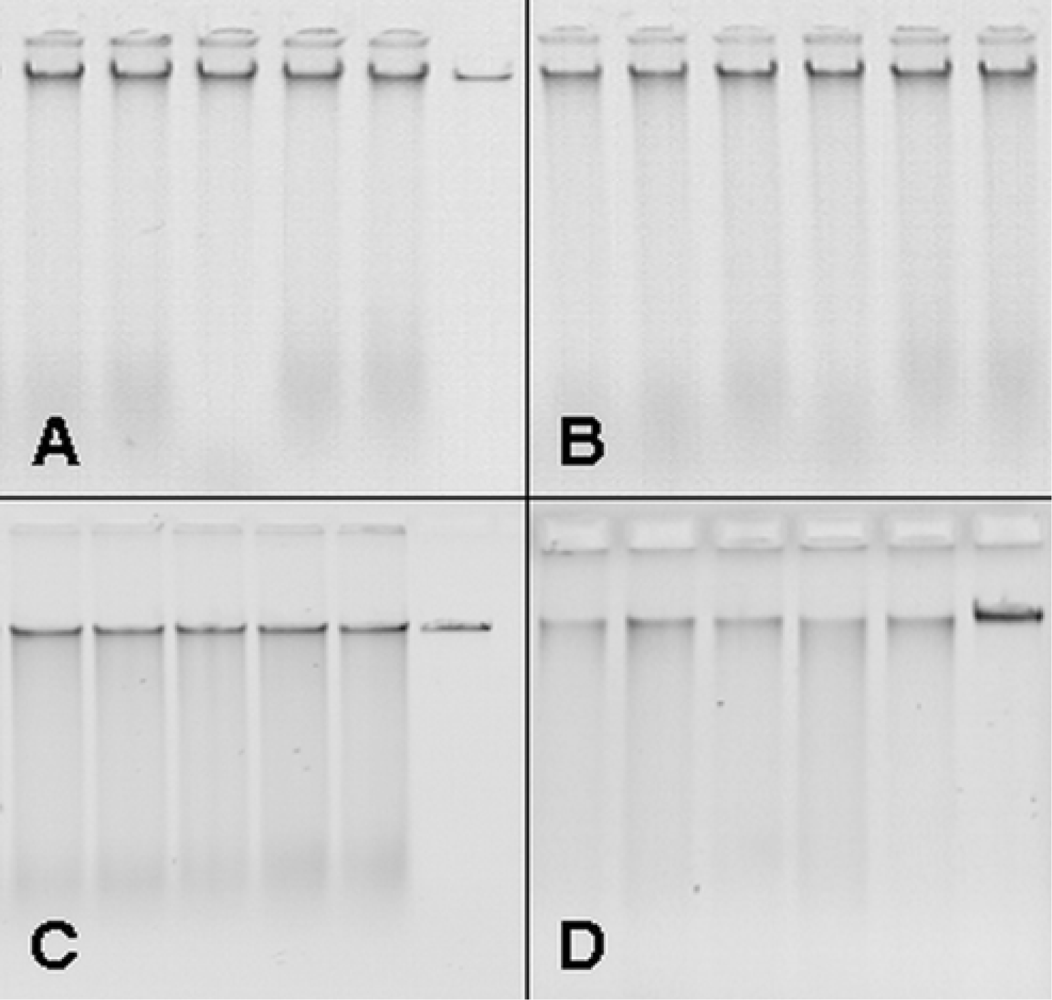
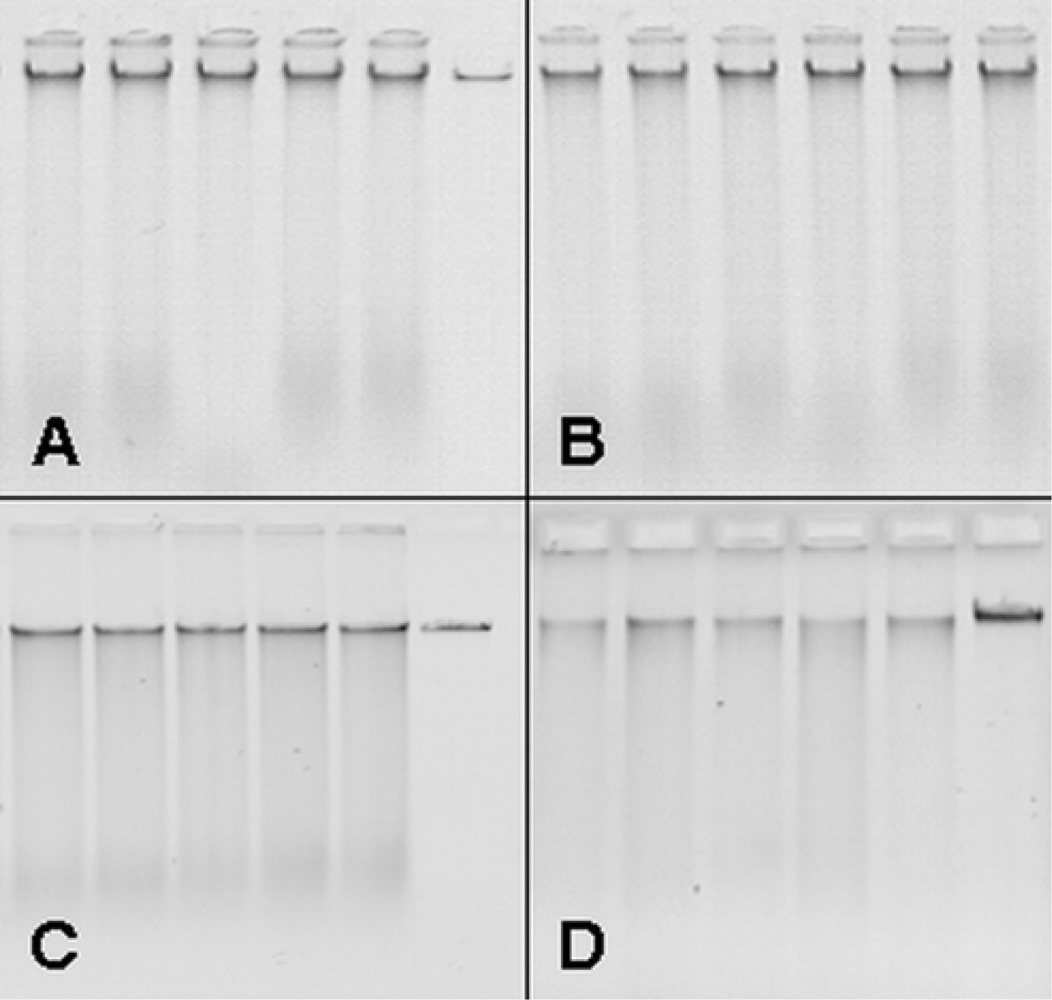
3.1. Upscaling and optimization of DNA extraction
3.2. Optimization of PCR conditions
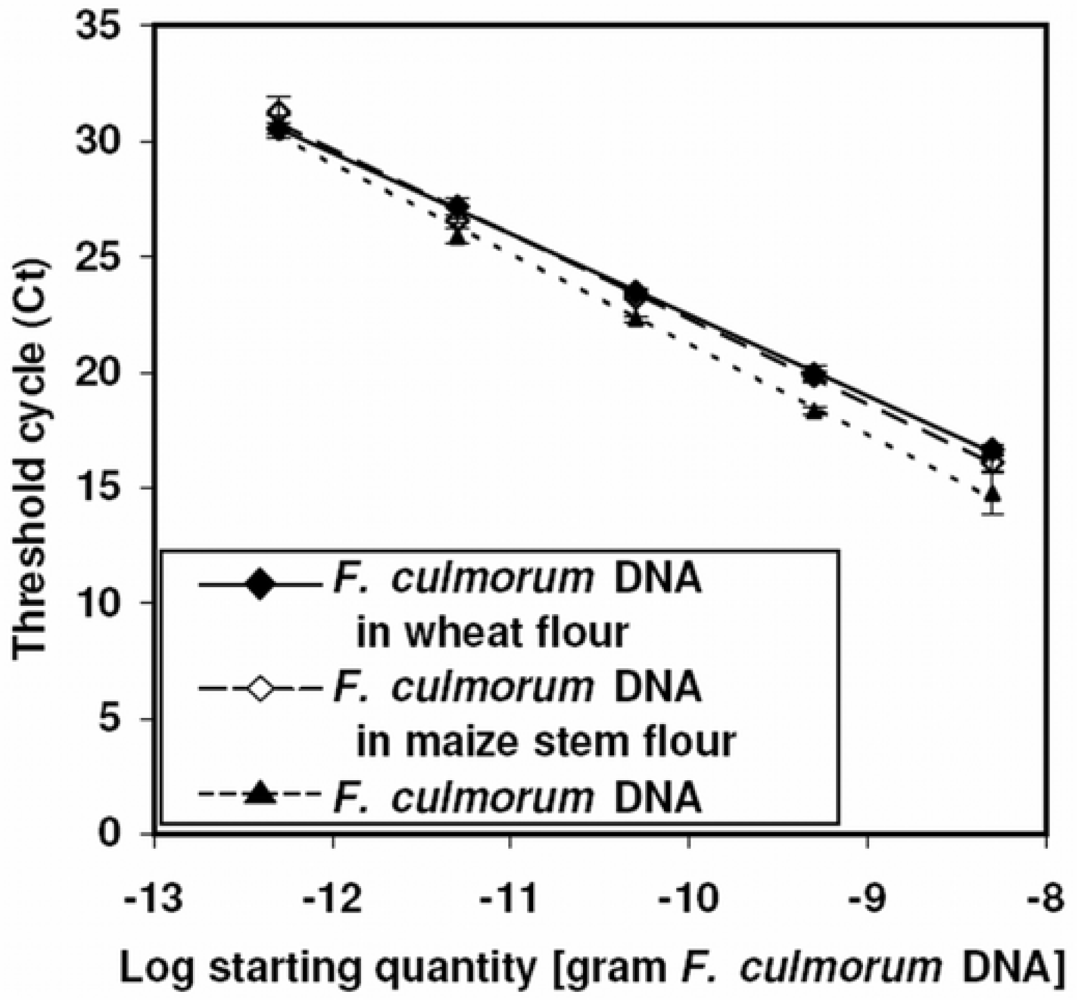
3.3. Matrix effects and calibration
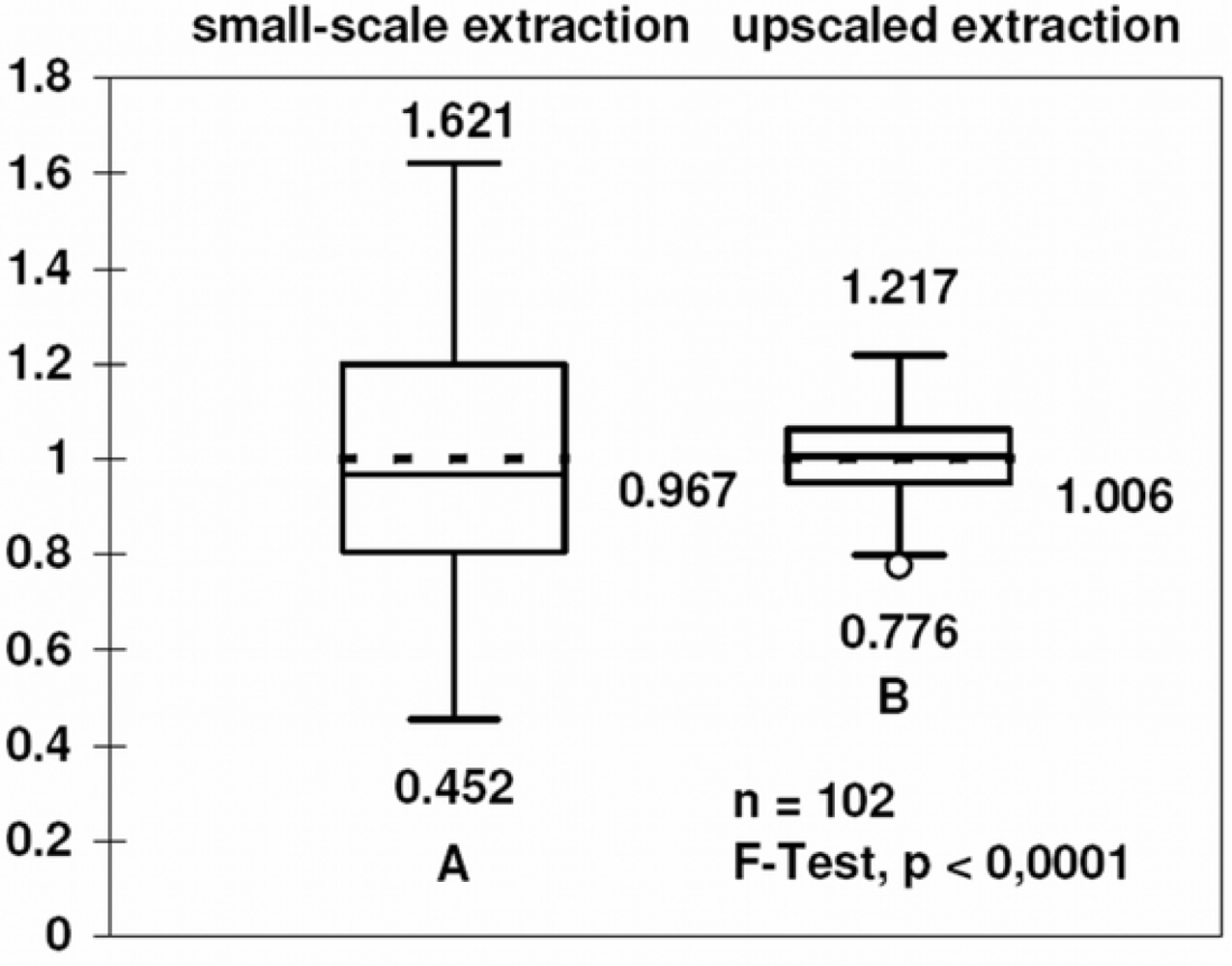
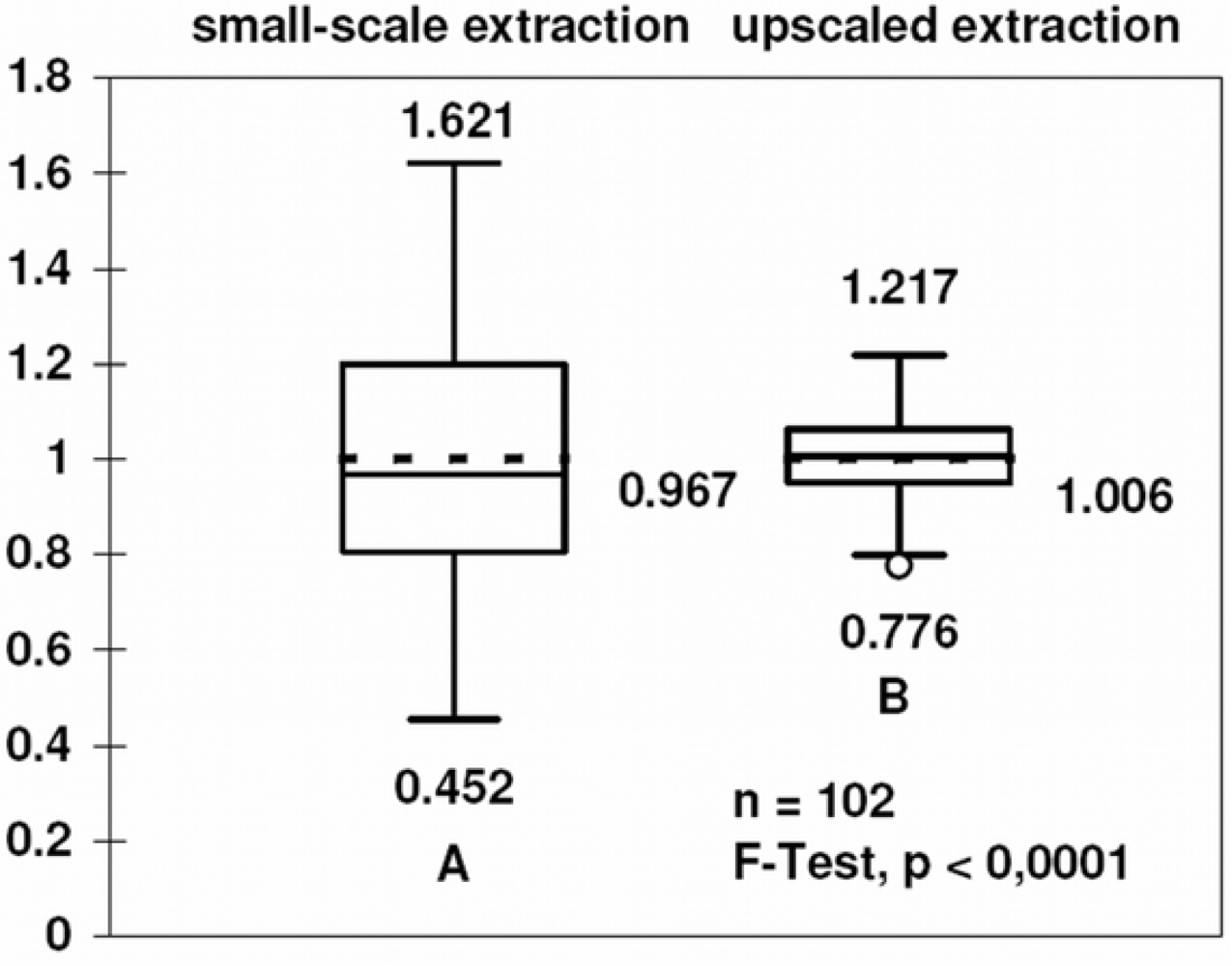
3.4. Comparison with other methods
4. Conclusions
Acknowledgments
References
- Leonard, KJ; Bushnell, WR (Eds.) Fusarium Head Blight of Wheat and Barley; The American Phytopathological Society: St. Paul, Minnesota, USA, 2004; pp. 1–530.
- Parry, DW; Jenkinson, P; McLeod, L. Fusarium ear blight (scab) in small grain cereals. Plant Pathol 1995, 44, 207–238. [Google Scholar]
- Peraica, M; Radic, B; Lucic, A; Pavlovic, M. Toxic effects of mycotoxins in humans. B. World Health Organ 1999, 77, 754–766. [Google Scholar]
- D'Mello, JPF; Placinta, CM; Macdonald, AMC. Fusarium mycotoxins: A review of global implications for animal health, welfare and productivity. Anim. Feed Sci. Tech 1999, 80, 183–205. [Google Scholar]
- Pestka, JJ; Smolinski, AT. Deoxynivalenol: Toxicology and potential effects on humans. J. Toxcol. Env. Health B 2005, 8, 39–69. [Google Scholar]
- Ecker, DJ; Sampath, R; Willett, P; Wyatt, JR; Samant, V; Massire, C; Hall, TA; Hari, K; McNeil, JA; Büchen-Osmond, C; Budowle, B. The Microbial Rosetta Stone Database: A compilation of global and emerging infectious microorganisms and bioterrorist threat agents. BMC Microbiol. 2005, 5, 19:1–19:17. [Google Scholar]
- Bai, GH; Shaner, G. Management and resistance in wheat and barley to Fusarium head blight. Annu. Rev. Phytopathol 2004, 42, 135–161. [Google Scholar]
- Miedaner, T; Wilde, F; Steiner, B; Buerstmayr, H; Korzun, V; Ebmeyer, E. Stacking quantitative trait loci (QTL) for Fusarium head blight resistance from non-adapted sources in an European elite spring wheat background and assessing their effects on deoxynivalenol (DON) content and disease severity. Theor. Appl. Genet 2006, 112, 562–569. [Google Scholar]
- Yuen, GY; Schoneweis, SD. Strategies for managing Fusarium head blight and deoxynivalenol accumulation in wheat. Int. J. Food Microbiol 2007, 119, 126–130. [Google Scholar]
- Waalwijk, C; Kastelein, P; de Vries, I; Kerényi, Z; van der Lee, T; Hesselink, T; Köhl, J; Kema, G. Major Changes in Fusarium spp. in Wheat in the Netherlands. Eur. J. Plant Pathol 2003, 109, 743–754. [Google Scholar]
- Xu, XM. Effects of environmental conditions on the development of Fusarium ear blight. Eur. J. Plant Pathol 2003, 109, 683–689. [Google Scholar]
- Schlüter, K; Kropf, U; Karlovsky, P. Untersuchungen zur systemischen Infektion von Fusarium culmorum an Winterweizen in Schleswig-Holstein. Gesunde Pflanzen 2006, 58, 107–116. [Google Scholar]
- Schlüter, K; Kropf, U; Karlovsky, P. Systemic development of Fusarium culmorum in winter wheat (German). Mitt. Biol. Bundesanst 2004, 396, 158–159. [Google Scholar]
- Nicholson, P; Chandler, E; Draeger, RC; Gosman, NE; Simpson, DR; Thomsett, M; Wilson, AH. Molecular tools to study epidemiology and toxicology of Fusarium head blight of cereals. Eur. J. Plant Pathol 2003, 109, 691–703. [Google Scholar]
- Waalwijk, C; van der Heide, R; de Vries, L; van der Lee, T; Schoen, C; Costrel-de Corainville, G; Hauser-Hahn, I; Kastelein, P; Kohl, J; Lonnet, P; Demarquet, T; Kema, GHJ. Quantitative detection of Fusarium species in wheat using TaqMan. Eur. J. Plant Pathol 2004, 110, 481–494. [Google Scholar]
- Schnerr, H; Vogel, RF; Niessen, L. Correlation between DNA of trichothecene-producing Fusarium species and deoxynivalenol concentrations in wheat-samples. Lett. Appl. Microbiol 2002, 35, 121–125. [Google Scholar]
- Yli-Mattila, T; Paavanen-Huhtala, S; Jestoi, M; Parikka, P; Hietaniemi, V; Gagkaeva, T; Sarlin, T; Haikara, A; Laaksonen, S; Rizzo, A. Real-time PCR detection and quantification of Fusarium poae, F. graminearum, F. sporotrichioides and F. langsethiae in cereal grains in Finland and Russia. Arch. Phytopath. Plant Protect 2008, 41, 243–260. [Google Scholar]
- Nicholson, P; Simpson, DR; Weston, G; Rezanoor, HN; Lees, AK; Parry, DW; Joyce, D. Detection and quantification of Fusarium culmorum and Fusarium graminearum in cereals using PCR assays. Physiol. Mol. Plant Pathol 1998, 53, 17–37. [Google Scholar]
- Eynck, C; Koopmann, B; Grunewaldt-Stoecker, G; Karlovsky, P; Tiedemann, A. Differential interactions of Verticillium longisporum and V. dahliae with Brassica napus detected with molecular and histological techniques. Eur. J. Plant Pathol 2007, 118, 259–274. [Google Scholar]
- Martin, KJ; Rygiewicz, PT. Fungal-specific PCR primers developed for analysis of the ITS region of environmental DNA extracts. BMC Microbiol. 2005, 5, 28:1–28:11. [Google Scholar]
- Edwards, SG; Pirgozliev, SR; Hare, MC; Jenkinson, P. Quantification of trichothecene-producing Fusarium species in harvested grain by competitive PCR to determine efficacies of fungicides against Fusarium head blight of winter wheat. Appl. Environ. Microbiol 2001, 67, 1575–1580. [Google Scholar]
- Pirgozliev, SR; Edwards, SG; Hare, MC; Jenkinson, P. Effect of dose rate of azoxystrobin and metconazole on the development of Fusarium head blight and the accumulation of deoxynivalenol (DON) in wheat grain. Eur. J. Plant Pathol 2002, 108, 469–478. [Google Scholar]
- Simpson, DR; Weston, GE; Turner, JA; Jennings, P; Nicholson, P. Differential control of head blight pathogens of wheat by fungicides and consequences for mycotoxin contamination of grain. Eur. J. Plant Pathol 2001, 107, 421–431. [Google Scholar]
- Edwards, SG; O'Callaghan, J; Dobson, ADW. PCR-based detection and quantification of mycotoxigenic fungi. Mycol. Res 2002, 106, 1005–1025. [Google Scholar]
- Reischer, GH; Lemmens, M; Farnleitner, A; Adler, A; Mach, RL. Quantification of Fusarium graminearum in infected wheat by species specific real-time PCR applying a TaqMan Probe. J. Microbiol. Methods 2004, 59, 141–146. [Google Scholar]
- Brandfass, C; Karlovsky, P. Simultaneous detection ofFusarium culmorum and F. graminearum in plant material by duplex PCR with melting curve analysis. BMC Microbiol. 2006, 6, 4:1–4:10. [Google Scholar]
- Murray, MG; Thompson, WF. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 1980, 8, 4321–4325. [Google Scholar]
- Schilling, AG; Moller, EM; Geiger, HH. Polymerase chain reaction-based assays for species-specific detection of Fusarium culmorum, F. graminearum, and F. avenaceum. Phytopathology 1996, 86, 515–522. [Google Scholar]
- Mulfinger, S; Niessen, L; Vogel, RF. PCR based quality control of toxigenic Fusarium spp. in brewing malt using ultrasonication for rapid sample preparation. Adv. Food Sci 2000, 22, 38–46. [Google Scholar]
- Wilson, IG. Inhibition and facilitation of nucleic acid amplification. Appl. Environ. Microbiol 1997, 63, 3741–3751. [Google Scholar]
- Manen, JF; Sinitsyna, O; Aeschbach, L; Markov, AV; Sinitsyn, A. A fully automatable enzymatic method for DNA extraction from plant tissues. BMC Plant Biol. 2005, 5, 23:1–23:9. [Google Scholar]
- Starkey, DE; Ward, TJ; Aoki, T; Gale, LR; Kistler, HC; Geiser, DM; Suga, H; Tóth, B; Varga, J; O'Donnell, K. Global molecular surveillance reveals novel Fusarium head blight species and trichothecene toxin diversity. Fungal Genet. Biol 2007, 44, 1191–1204. [Google Scholar]
- O'Donnell, K; Kistler, HC; Tacke, BK; Casper, HH. Gene genealogies reveal global phylogeographic structure and reproductive isolation among lineages of Fusarium graminearum, the fungus causing wheat scab. Proc. Natl. Acad. Sci. USA 2000, 97, 7905–7910. [Google Scholar]
- Ward, TJ; Bielawski, JP; Kistler, HC; Sullivan, E; O'Donnell, K. Ancestral polymorphism and adaptive evolution in the trichothecene mycotoxin gene cluster of phytopathogenic Fusarium. Proc. Natl. Acad. Sci. USA 2002, 99, 9278–9283. [Google Scholar]
- Winton, LM; Stone, JK; Watrud, LS; Hansen, EM. Simultaneous one-tube quantification of host and pathogen DNA with real-time polymerase chain reaction. Phytopathology 2002, 92, 112–116. [Google Scholar]
- Glasel, JA. Validity of Nucleic-Acid Purities Monitored by 260nm 280nm Absorbency Ratios. Biotechniques 1995, 18, 62–63. [Google Scholar]
- Huberman, JA. Importance of measuring nucleic-acid absorbency at 240 nm as well as at 260 and 280 nm. Biotechniques 1995, 18, 636. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
| Species | Number of Isolates |
|---|---|
| Alternaria alternata (Fries) Keissler | 1 |
| Cladosporium herbarum (Pers.) Link Ex SF Gray | 2 |
| Drechslera sorokiniana (Sacc.) Subram. & Jain | 1 |
| Fusarium acuminatum Ellis & Everh. | 5 |
| Fusarium avenaceum (Fr.) Sacc. | 2 |
| Fusarium compactum Wollenw. | 1 |
| Fusarium crookwellense Burgess, Nelson & Toussoun | 3 |
| Fusarium culmorum WG Smith | 12 |
| Fusarium equiseti (Corda) Sacc. | 5 |
| Fusarium graminearum Schwabe | 12 |
| Fusarium oxysporum (Schlech.) Snyder & Hansen | 1 |
| Fusarium poae (Peck) Wollenw. | 2 |
| Gibberella fujikuroi (Saw.) Wr. | 5 |
| Microdochium nivale (Fries) Samuels & Hallett var. majus | 1 |
| Microdochium nivale (Fries) Samuels & Hallett var. nivale | 3 |
| Pseudocercosporella herpotrichoides (Fron) Deighton | 3 |
| Rhizoctonia cerealis Van der Hoeven | 3 |
| Stagonospora nodorum (Berk.) Castellani & Germano | 1 |
| Sample type | Sample No. | Fusarium spp. DNA |
|---|---|---|
| Negative controls | 1 | - |
| 2 | 50 pg F. graminearum DNA in wheat flour matrix diluted 1:10 | |
| Standards | 3–4 | 0.5 pg F. culmorum DNA in wheat flour matrix diluted 1:10 |
| 5–6 | 5 pg F. culmorum DNA in wheat flour matrix diluted 1:10 | |
| 7–8 | 50 pg F. culmorum DNA in wheat flour matrix diluted 1:10 | |
| 9–10 | 0.5 ng F. culmorum DNA in wheat flour matrix diluted 1:10 |
| DNA extraction method | Sample size | Costs per sample |
|---|---|---|
| Commercial kit | 50–100 mg | 1.80–2.60 € |
| 1 g | 8.00–10.00 € | |
| CTAB based protocol described in this work | 1 g | 0.50 € |
| Taqman probes | SYBR Green-based detection | |
|---|---|---|
| TaqMan probe | 0.08–0.12 € | - |
| SYBR Green | - | 0.001 € |
| DNA polymerase, dNTPs, primers, PCR tubes and filter tips | 0.60–2.20€ | |
© 2008 by MDPI This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Brandfass, C.; Karlovsky, P. Upscaled CTAB-Based DNA Extraction and Real-Time PCR Assays for Fusarium culmorum and F. graminearum DNA in Plant Material with Reduced Sampling Error. Int. J. Mol. Sci. 2008, 9, 2306-2321. https://doi.org/10.3390/ijms9112306
Brandfass C, Karlovsky P. Upscaled CTAB-Based DNA Extraction and Real-Time PCR Assays for Fusarium culmorum and F. graminearum DNA in Plant Material with Reduced Sampling Error. International Journal of Molecular Sciences. 2008; 9(11):2306-2321. https://doi.org/10.3390/ijms9112306
Chicago/Turabian StyleBrandfass, Christoph, and Petr Karlovsky. 2008. "Upscaled CTAB-Based DNA Extraction and Real-Time PCR Assays for Fusarium culmorum and F. graminearum DNA in Plant Material with Reduced Sampling Error" International Journal of Molecular Sciences 9, no. 11: 2306-2321. https://doi.org/10.3390/ijms9112306
APA StyleBrandfass, C., & Karlovsky, P. (2008). Upscaled CTAB-Based DNA Extraction and Real-Time PCR Assays for Fusarium culmorum and F. graminearum DNA in Plant Material with Reduced Sampling Error. International Journal of Molecular Sciences, 9(11), 2306-2321. https://doi.org/10.3390/ijms9112306