Thick and Thin Filament Gene Mutations in Striated Muscle Diseases

Department of Pathology, Sahlgrenska University Hospital, S-413 45 Göteborg, Sweden
Int. J. Mol. Sci. 2008, 9(7), 1259-1275; https://doi.org/10.3390/ijms9071259
Submission received: 8 May 2008
/
Revised: 23 May 2008
/
Accepted: 12 June 2008
/
Published: 16 July 2008
(This article belongs to the Special Issue Muscle Contraction Mechanism, Motor Proteins Function and Molecular Aspects of Water)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The sarcomere is the fundamental unit of cardiac and skeletal muscle contraction. During the last ten years, there has been growing awareness of the etiology of skeletal and cardiac muscle diseases originating in the sarcomere, an important evolving field. Many sarcomeric diseases affect newborn children, i. e. are congenital myopathies. The discovery and characterization of several myopathies caused by mutations in myosin heavy chain genes, coding for the major component of skeletal muscle thick filaments, has led to the introduction of a new entity in the field of neuromuscular disorders: myosin myopathies. Recently, mutations in genes coding for skeletal muscle thin filaments, associated with various clinical features, have been identified. These mutations evoke distinct structural changes within the sarcomeric thin filament. Current knowledge regarding contractile protein dysfunction as it relates to disease pathogenesis has failed to decipher the mechanistic links between mutations identified in sarcomeric proteins and skeletal myopathies, which will no doubt require an integrated physiological approach. The discovery of additional genes associated with myopathies and the elucidation of the molecular mechanisms of pathogenesis will lead to improved and more accurate diagnosis, including prenatally, and to enhanced potential for prognosis, genetic counseling and developing possible treatments for these diseases. The goal of this review is to present recent progress in the identification of gene mutations from each of the major structural components of the sarcomere, the thick and thin filaments, related to skeletal muscle disease. The genetics and clinical manifestations of these disorders will be discussed.
1. Introduction
During the past decade, major advances have been made in defining the molecular basis of many genetically transmitted diseases. During this period, there has been growing awareness of the importance of sarcomeric protein mutations in the etiology of myopathy. Numerous mutations are currently associated with myopathy, with remarkable spectrum of phenotypic variation. In addition to a better understanding of the primary defect and basic molecular pathogenesis of disease, redefining the diagnostics of many disorders is another benefit of identifying the disease genes. These fundamental approaches will allow us to understand why a point mutation at one site in a sarcomeric protein (e.g. tropomyosin (TM)) leads to nemaline myopathy (NM), while a different point mutation causes distal arthrogryposis (DA). Improved understanding of the molecular basis of disease will probably allow targeting of pharmacological strategies as well as providing the cornerstone for gene therapy approaches.
In this review, disorders caused by mutation of sarcomeric thick and thin filament proteins will be discussed.
2. The Sarcomere
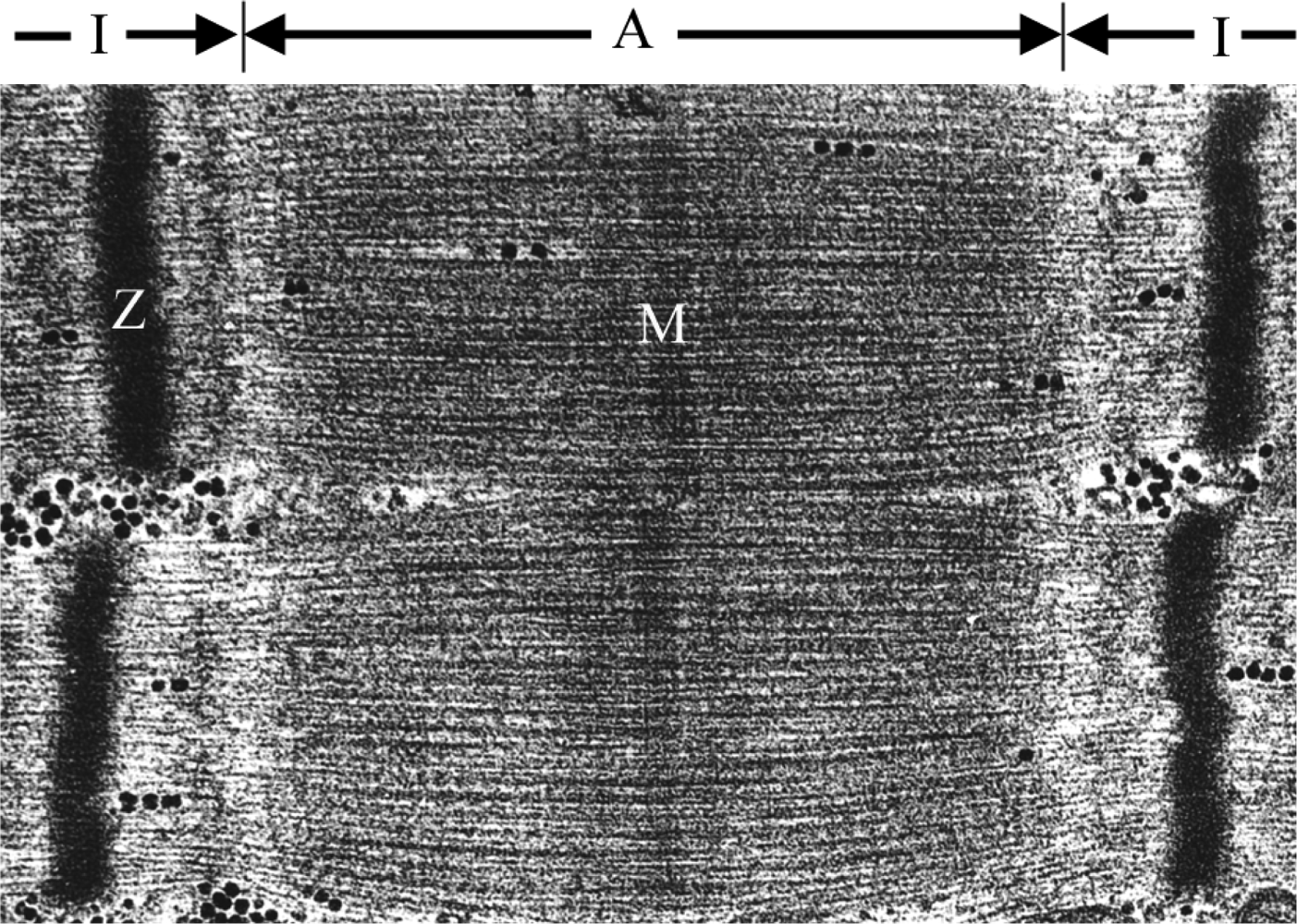
The sarcomere represents the basic contractile unit of both skeletal and cardiac muscle. It is a highly ordered structure composed of the thin and thick filaments, titin, and nebuline [1]. The characteristic striated appearance of muscle fibers is observable by electron microscopy as alternating light (I) and dark (A) bands (Figure 1). The principle components of striated muscle sarcomeres include parallel arrays of actin-containing thin filaments that span the I-band and overlap with myosin-containing thick filaments in the A-band. The thin filaments are anchored in the Z-disc and the thick filaments are similarly anchored in the M-band [1].
Skeletal muscle function depends on a precise alignment of actin and myosin filaments. This is achieved by accessory proteins, such as α-actinin, myomesin, M-protein, titin, desmin and myosin-binding proteins (MyBP)-C and -H, which link the different components and keep them aligned with each other [1] (Figure 2). The major component of the Z-line is α-actinin, which acts as an actin cross-linking protein and holds actin filaments in a lattice arrangement in the Z-disc [2, 3]. It has been proposed that myomesin and M-protein may connect titin and myosin filament systems and that myomesin plays a role in integrating thick filaments into assembling sarcomeres [4]. Titin, a huge protein which runs parallel to the filament array, forms a continuous filament system in myofibrils [1]. Desmin is the predominant intermediate filament protein of striated muscle [5] and contributes to maintaining the integrity and alignment of myofibrils [1]. MyBP-C is localized in seven stripes running parallel to the M-band (Figure 2). Because MyBP-C interacts with both the thick and titin filaments, its function may be to link them together and/or to align the thick filaments in the A-band [1]. In addition, MyBP-C reduces the critical concentration for myosin polymerization and the resulting filaments are longer and more uniform in length than those polymerized in its absence [6, 7]. Both myosin-binding proteins appear to aid in the assembly of vertebrate muscle thick filaments into their precise lengths [1]. It has also been suggested that MyBP-C and –H may be involved in regulating muscle contraction [8].
2.1. The Major Component of the Thick Filament: Myosin
Contraction of muscle is the result of cyclic interactions between the globular heads of the myosin molecules, also known as cross-bridges, and the actin filaments. The repetitive binding and release results in sliding of the thick filaments along the thin filaments powered by the hydrolysis of ATP. Myosin can be regarded as an ATPase that is activated by the binding of actin [9].
Myosin acts as a molecular motor that converts the chemical energy of ATP hydrolysis into mechanical force in eukaryotic cells [10]. Conventional myosin exists as a hexameric protein composed of two myosin heavy chain (MyHC) subunits and two pairs of non-identical light chain (MyLC) subunits (Figure 3). The MyHC has two functional domains. The globular, amino-terminal head domain, to which MyLCs bind, exhibits the motor function. The elongated alpha-helical coiled-coil carboxyl-terminal rod domain exhibits filament-forming properties [10]. The globular head that forms the cross-bridges contains the binding sites for actin and ATP [11].
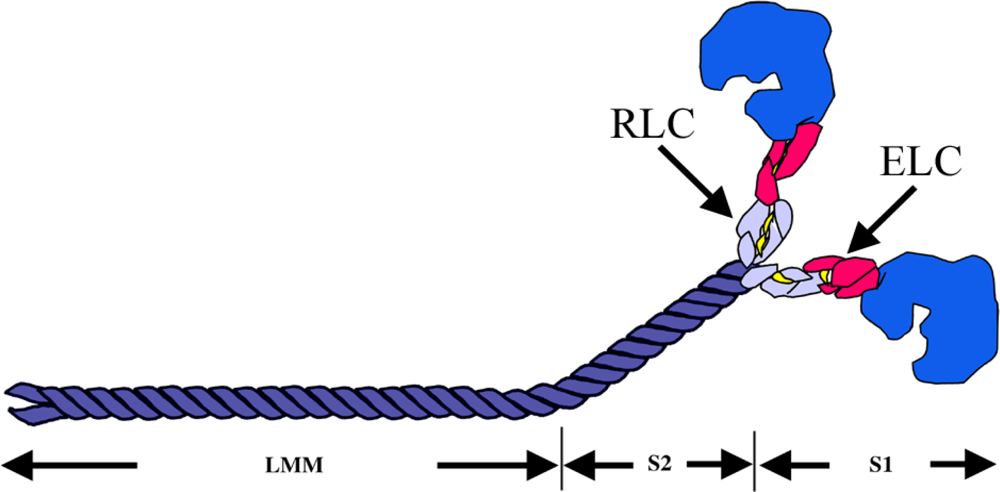
The MyHC can be cleaved by proteolytic enzymes into two subfragments, heavy meromyosin (HMM) and light meromyosin (LMM) [12, 13]. The HMM contains the head region, termed subfragment 1 (S1), and a portion of the coiled-coil-forming sequence referred to as subfragment 2 (S2) which connects the myosin heads to the thick filament. The LMM is the C-terminal proportion of the rod, which lies along the thick filAMent axis [14] (Figure 3).
The myosin motor domain is essentially constructed of three domains connected by flexible linkers. The 25-kDa amino-terminal nucleotide-binding domain is connected to the upper 50 kDa subdomain which is, in turn, connected to the lower 50-kDa subdomain. The third, 20-kDa domain is called the converter region (Figure 4). A long helix emerges from the converter domain and serves as the binding site for MyLCs. The essential light chains (ELC) occupy the binding site closest to the converter domain while the regulatory light chains (RLC) occupy the second site [15]. The binding sites are highly specific for their respective MyLCs [14]. The elongated neck region in S1 has been suggested to act as a lever arm to amplify small changes in the configuration of the motor domain into much larger displacements of actin [15].
Six striated muscle MyHCs are encoded by genes found in a tightly linked cluster on human chromosome 17 [16, 17]. The genes are arranged in the following order: MYH3, MYH2, MYH1, MYH8, MYH13 and MYH4 [18]. Cardiac MyHC isoforms encoded by MYH7 and MYH6 are located on chromosome 14 [19].
Three major MyHC isoforms are present in adult human limb muscle tissue: MyHC I, also called slow/beta MyHC (encoded by MYH7) is expressed in slow, type 1 muscle fibers and in heart ventricles; MyHC IIa (encoded by MYH2) is expressed in fast, type 2A muscle fibers and MyHC IIx (encoded by MYH1) is expressed in fast, type 2B muscle fibers [20]. In addition to the common MyHC isoforms found in fibers of adult human limb muscles, there are special MyHC isoforms expressed in some fibers in specific muscles. For example, very rapidly contracting fibers that express a specific MyHC isoform, extraocular MyHC, have been found in extraocular muscles [21–24]. Developing and regenerating muscle fibers express special MyHC isoforms, i.e. embryonic (encoded by MYH3) and perinatal (encoded by MYH8) MyHC. Embryonic MyHC is normally not expressed in postnatal human limb muscles unless there is ongoing muscle regeneration.
2.2. The Major Components of the Thin Filament: Actin, Tropmyosin, Troponin Complex and Nebulin
The thin filament is the main site of Ca+2 regulation and is composed of four components in striated muscle: actin, TM and troponin (Tn) with its three subunits [25] (Figure 5).
Actin is the principal protein component of the sarcomeric thin filaments. F-actin forms the backbone of the thin filament which can be viewed as a two-stranded helical structure [25].
TM is an actin-binding protein, composed of two α-helical chains forming a rod-shaped coiled-coil dimer. TM is one of the regulatory proteins in the thin filament and is localized head-to-tail along the length of the actin filament, providing stability; it is essential for myosin–actin interaction [25, 26]. Through Ca2+-dependent movement transmitted via the Tn complex, TM blocks or opens the myosin binding sites on actin [25]. So far, four different TM genes have been identified in the human genome, TPM1, TPM2, TPM3 and TPM4 [26].
There are three highly homologous major TM isoforms in human striated muscle: α-TM(α–TMfast), β-TM and γ-TM (α–TMslow) [26].
α-TM is a product of the TPM1 gene, β-TM is encoded by the TPM2 gene and γ-TM is encoded by the TPM3 gene. The muscle isoform encoded by TPM1 is predominantly expressed in cardiac muscle and fast, type 2 muscle fibers. TPM2 is mainly expressed in slow, type 1 and, to some extent, in fast muscle fibers and cardiac muscle. TPM3 is predominantly expressed in slow muscle fibers and is also expressed in the heart [27].
Tn exists as a complex of three component proteins: troponin C (TnC), troponin I (TnI) and troponin T (TnT). Each of the three proteins is a regulator of muscle contraction and plays distinct roles in the thin filament. TnC is the Ca2+ -binding subunit, TnI is the inhibitory subunit that binds to actin and inhibits actomyosin ATPase, and TnT is the TM-binding subunit that links the Tn complex to TM [25].
There are two TnC isoforms in human striated muscle, expressed by two separate genes, TNNC1 and TNNC2 [28, 29]. The product of the TNNC1 gene expressed in slow twitch skeletal muscle is also the isoform expressed in heart. The fast skeletal muscle TnC isoform is the product of TNNC2.
There are three TnI isoforms that are the products of separate genes in human striated muscle: fast skeletal TnI is the product of TNNI2, slow skeletal TnI is a product of TNNI1 and cardiac TnI is a product of TNNI3 [30].
3. Myopathies Involving Thick and Thin Filament Proteins
During the last decade, several protein components of the thick and thin filaments have been implicated in the pathogenesis of muscle diseases: slow/β-cardiac myosin heavy chain (MyHC I), fast type IIa myosin heavy chain (MyHC IIa), embryonic myosin heavy chain (MyHC-emb), perinatal myosin heavy chain (MyHC-peri), skeletal muscle alpha actin (α-actin), α-tropomyosin slow (γ-TM), β-tropomyosin (β-TM), slow TnT, fast TnT and fast TnI.
Furthermore, there are additional myopathies associated with mutations in the genes coding for other proteins of the sarcomere such as titin, desmin and components in the Z-disk, which is not included in this review.
3.1. Disorders-associated Mutations Found in Thick Filament Components
3.1.1. Myosin Heavy Chains
Mutation in the sarcomeric motor protein MyHC was first described in 1990 in association with a severe form of hypertrophic cardiomyopathy (HCM) [36].
During the past few years, MyHC mutations have been associated with different skeletal muscle diseases including autosomal dominant myopathy with congenital joint contractures, ophthalmoplegia and rimmed vacuoles (OMIM #605637), caused by a single point mutation in the fast IIa MyHC gene (MYH2) [37]; Laing early onset distal myopathy (OMIM #160500) [38–41] and myosin storage myopathy (OMIM #608358) [42–46], caused by different mutations in the slow/β-cardiac MyHC gene (MYH7); trismus-pseudocamptodatyly syndrome (TPS) (OMIM #158300), caused by mutation in perinatal MyHC (MYH8) [47, 48]; Freeman-Sheldon syndrome (FSS) (OMIM #193700) and Sheldon-Hall syndrome (SHS) (OMIM #601680), caused by mutations in embryonic MyHC (MYH3) [49].
3.1.1.1. MyHC IIa
Mutation of a MyHC gene was not described in association with pure skeletal muscle until 2000 [37]. This myopathy, also called “Autosomal dominant MyHC IIa myopathy” (OMIM #605637), is associated with a missense mutation in the MyHC IIa gene (MYH2). Clinical characteristics are congenital joint contractures, which normalize during early childhood, external ophthalmoplegia and predominantly proximal muscle weakness and atrophy. The course is frequently progressive in adulthood [50]. The mutation changes a highly conserved, negatively charged glutamate at position 706 into a positively charged lysine (E706K). The altered amino acid is located in the SH1 helix in the core of the myosin head.
Three additional mutations in the MYH2 gene have later been identified with myopathies in three families [51].
3.1.1.2. Slow/β-cardiac MyHC
Mutations in the cardiac/β-MyHC gene (MYH7) are a frequent cause of familial hypertrophic/dilated cardiomyopathy (HCM/DCM) [52, 53]. More than 190 MYH7 mutations with varying clinical penetrance have been described, some with relatively benign effects on life expectancy and others associated with a high incidence of sudden death ( http://www.hgmd.cf.ac.uk/ac/gene.php?gene=MYH7). Despite the knowledge that the slow/β-cardiac MyHC gene is also expressed in slow, type 1 skeletal muscle fibers, only a few studies have investigated the involvement of skeletal muscle in familial hypertrophic cardiomyopathy patients with identified missense mutations in the MYH7 gene [54, 55].
Laing early onset distal myopathy is identified and linked to the MYH7 gene on chromosome 14q11 [38]. Of the total number of identified mutations associated with this form of distal myopathy, the most prevalent are mutations located in the LMM region of the myosin tail of slow/β-cardiac MyHC [38–41]; only two cases of distal myopathy with cardiomyopathy have been associated with a mutation in the S1 region [56, 57].
Myosin storage myopathy is an additional myopathy associated with mutations in the MYH7 gene. It has been assigned various descriptive terms such as “myopathy with probable lysis of thick filaments” [58] and “hyaline body myopathy” [59, 60]. This myopathy is characterized by accumulation of slow/β cardiac myosin (MyHC I) in type I muscle fibers. Myosin storage myopathy has so far been associated with four mutations located in the distal rod region of slow/β-cardiac MyHC. The Arg1845Trp mutation has been reported in several unrelated cases [42, 44, 45]. This mutation has also been reported in cases with scapulo-peroneal myopathy [61].
3.1.1.3. Perinatal MyHC
To date, only one single mutation in perinatal MyHC (MYH8), R674Q, has been associated with TPS (OMIM #158300). The mutation has been identified in seven unrelated families with this disorder [47, 48]. TPS is a rare autosomal dominant distal arthrogryposis (DA7), characterized by camptodactyly of the fingers, apparent only upon dorsiflexion of the wrist (pseudocamptodactyly), and inability to completely open the mouth (trismus) [62].
3.1.1.4. Embryonic MyHC
DA syndromes are a group of sporadic and/or autosomal dominant disorders. The clinical features are multiple congenital contractures, where the hands and feet are tightly clenched and the fingers overlap [62, 63]. FSS, the most severe disorder with multiple congenital contractures and severe contractures of the orofacial muscles, and SHS, the most common DA, have recently been associated with mutations in the embryonic MyHC gene (MYH3) [49]. All of the MYH3 mutations associated with FSS except one are located in the myosin head. Interestingly, 6/20 investigated cases in a study of FSS carried an R672 substitution, which is paralogous to the R674 of perinatal MyHC (MYH8) [49]. However, the mutations that cause SHS are located throughout MYH3 [49].
A new study has recently reported novel MYH3 mutations associated with DA and demonstrated myopathic changes in muscle biopsy specimens from patients with DA and MYH3 mutations [64]. The results of this study add MYH3 to the list of MyHC genes involved in hereditary myosin myopathies.
3.1.2. Myosin Light Chains
3.1.2.1. Regulatory Light Chain RLC
Recent studies have shown that myosin RLC is one of the sarcomeric proteins associated with FHC [65–69]. Some of the RLC mutations were shown to be associated with a particular type of FHC, [67] whereas others presented with a more classic form of FHC, resulting in increased left ventricular wall thickness and abnormal ECG [65, 66, 68, 69].
Since the RLC gene (MYL2), like MYH7, is expressed in slow skeletal muscle, skeletal muscle biopsies from patients with the Glu22Lys MYL2 mutation exhibited abnormal skeletal muscle histology, similar to patients with ragged red fiber (RRF) myopathy [67].
3.1.2.2. Essential Light Chain ELC
Mutations in the ELC have been associated with FHC. The pathological phenotypes vary in severity but almost all ELC mutations result in sudden cardiac death at a young age [70].
3.2. Disorder-associated Mutations Found in Thin Filament Components
3.2.1. Actin
Mutations in the skeletal muscle alpha actin gene (ACTA1) have been associated with various skeletal muscle diseases, including actin myopathy (accumulation of actin), NM, intranuclear rod myopathy and congenital fiber type disproportion (CFTD) [72–74]. To date, more than 80 different ACTA1 mutations have been identified, distributed throughout the entire alpha actin gene. Most of these mutations are associated with NM. NM is defined by the presence of rod-shaped structures in muscle fibers, so-called nemaline rods, largely composed of α-actinin and actin. Other common pathological features are abnormal muscle fiber differentiation and fiber atrophy and/or hypotrophy. NM is genetically heterogeneous, with disease-causing mutations identified in different genes coding for various components of sarcomeric thin filament.
3.2.2.α-TM
Mutations in the α-TM gene (TPM1), mainly expressed in cardiac muscle, have been associated with HCM/DCM ( http://cardiogenomics.med.harvard.edu/home). There are currently 13 reported mutations in TPM1, of which two are associated with DCM and 11 are associated with HCM ( http://www.hgmd.cf.ac.uk/ac/gene.php?gene=TPM1).
3.2.3. β-TM
The TPM2 isoform of TM encodes for beta-TM, which is mainly expressed in slow, type 1 muscle fibers. Mutations in TPM2 have recently been identified as an important cause of neuromuscular disorders. These mutations have been associated with various clinical and muscle morphological phenotypes, including congenital NM (E117A), congenital myopathy without rods (Q147P) [75], early onset myopathy with cap structures (E41K and E139del) [76, 77], and congenital joint contractures with distal involvement DA type 1 (R91G) [78], and DA type 2B (R133W) [79]. Different regions of TM are in direct interaction with various thin filament proteins, which may explain why these various mutations lead to different phenotypic expressions.
3.2.4. γ-TM
γ-TM is the product of TPM3, which is mainly expressed in slow, type 1 muscle fibers. Mutations in TPM3 have been associated with both dominant and recessive NM [80–84]. Recently, mutations in TPM3 have been identified as the common cause of CFTD, a rare form of congenital myopathy with marked type 1 fiber hypotrophy as the main pathological finding on muscle biopsy85.
3.2.5. Tn Complex
Mutations in the fast skeletal TnI gene (TNNI2) have been associated with DA syndromes. The one missense mutation, Arg174Gln, and the nonsense mutation, Arg156X, have been reported in patients with DA type 2B [78]. In addition, a three-base pair deletion of a glutamate at position 167, has been reported in a three-generation family with DA type 2B [86] and one heterozygous three-base in-frame deletion, Lys176del, has been reported in a three-generation family with DA type 1 [87]. Later a novel three-base in-frame deletion, Lys175del, has been reported in a large Chinese family with DA type 2B [88]. All the reported mutations in the fast skeletal TnI gene associated with DA syndromes have been located in the exon 8 in the carboxy-terminal domain.
Mutations in cardiac TnI (TNNI3) have been associated with HCM, DCM and restrictive cardiomyopathy ( http://cardiogenomics.med.harvard.edu/home).
So far, no disease has been associated with mutations in the slow skeletal muscle TnI gene (TNNI1).
Mutations in the slow skeletal muscle TnT gene (TNNT1) are associated with Amish nemaline myopathy (ANM), a form of NM common among the Old Order Amish [89]. Recently, mutations in fast skeletal muscle TnT (TNNT3) have been associated with DA type 2B [90]. More than 30 mutations associated with HCM/DCM have been described in the cardiac TnT gene (TNNT2) ( http://cardiogenomics.med.harvard.edu/home).
3.2.6. Nebulin
Mutations in the nebulin gene (NEB) are a frequent cause of autosomal recessive NM (OMIM#256030). More than 60 NEB mutations have been associated with this form of the rare muscle disorder [93–96]. Recently, homozygous missense mutations in the nebulin gene (NEB) have been reported to cause a novel distal myopathy, “Distal nebulin myopathy” [97].
4. Conclusions
Despite the significant advances in knowledge of the etiology of skeletal muscle diseases originating in the thick and thin filaments, there is a relative lack of understanding regarding the precise mechanistic links between the primary structural mutations at the skeletal muscle sarcomere level and the defining characteristics of the disorders. Current knowledge will be improved by elucidating novel pathways of disease pathogenesis, required in order to apply biochemical and physiological experimental approaches, anchored in animal models, to clarifying the molecular mechanisms underlying skeletal myopathy pathogenesis.
In order to link genotype to phenotype, a broad range of both in vivo and in vitro studies focusing on the effects of independent mutations on sarcomeric protein function will be needed; the results may lead to better understanding of the disease process in general and hopefully identify targets for therapeutic intervention.
References
- Clark, KA; McElhinny, AS; Beckerle, MC; Gregorio, CC. Striated Muscle Cytoarchitecture: an Intricate Web of Form and Function. Annu Rev Cell Dev Biol 2002, 18, 637–706. [Google Scholar]
- Maruyama, K; Ebashi, S. Alpha-actinin, a New Structural Protein from Striated Muscle. II. Action on Actin. J Biochem 1965, 5, 13–19. [Google Scholar]
- Blanchard, A; Ohanian, V; Critchley, D. The Structure and Function of Alpha-actinin. J Muscle Res Cell Motil 1989, 10, 280–289. [Google Scholar]
- Ehler, E; Rothen, BM; Hammerle, SP; et al. Myofibrillogenesis in the Developing Chicken Heart: Assembly of Z-Disk, M-Line and the Thick Filaments. J Cell Sci 1999, 112, 1529–1539. [Google Scholar]
- Lazarides, E. Desmin and Intermediate Filaments in Muscle Cells. Results Probl Cell Differ 1980, 11, 124–131. [Google Scholar]
- Koretz, JF. Effects of C-protein on Synthetic Myosin Filament Structure. Biophys J 1979, 27, 433–446. [Google Scholar]
- Davis, JS. Interaction of C-protein with pH 8.0 Synthetic Thick Filaments Prepared from the Myosin of Vertebrate Skeletal Muscle. J Muscle Res Cell Motil 1988, 9, 174–183. [Google Scholar]
- Winegrad, S. Cardiac Myosin Binding Protein C. Circ Res 1999, 84, 1117–1126. [Google Scholar]
- Spudich, JA. How Molecular Motors Work. Nature 1994, 372, 515–518. [Google Scholar]
- Ruppel, KM; Spudich, JA. Structure-Function Analysis of the Motor Domain of Myosin. Annu Rev Cell Dev Biol 1996, 12, 543–573. [Google Scholar]
- Rayment, I; Holden, HM; Whittaker, M; et al. Structure of the Actin-Myosin Complex and Its Implications for Muscle Contraction. Science 1993, 261, 58–65. [Google Scholar]
- Chen, T; Reisler, E. Tryptic Digestion of Rabbit Skeletal Myofibrils: An Enzymatic Probe of Myosin Cross-Bridges. Biochemistry 1984, 23, 2400–2407. [Google Scholar]
- Assulin, O; Werber, MM; Muhlrad, A. Effect of the Integrity of the Myofibrillar Structure on the Tryptic Accessibility of a Hinge Region of the Myosin Rod. FEBS Lett 1986, 197, 328–334. [Google Scholar]
- Weiss, A; Schiaffino, S; Leinwand, LA. Comparative Sequence Analysis of the Complete Human Sarcomeric Myosin Heavy Chain Family: Implications for Functional Diversity. J Mol Biol 1999, 290, 61–75. [Google Scholar]
- Rayment, I; Rypniewski, WR; Schmidt-Base, K; et al. Three-Dimensional Structure of Myosin Subfragment-1: A Molecular Motor. Science 1993, 261, 50–58. [Google Scholar]
- Weiss, A; Leinwand, LA. The Mammalian Myosin Heavy Chain Gene Family. Annu Rev Cell Dev Biol 1996, 12, 417–439. [Google Scholar]
- Leinwand, LA; Saez, L; McNally, E; Nadal-Ginard, B. Isolation and Characterization of Human Myosin Heavy Chain Genes. Proc Natl Acad Sci USA 1983, 80, 3716–3720. [Google Scholar]
- Weiss, A; Mayer, DC; Leinwand, LA. Diversity of Myosin-based Motility: Multiple Genes and Functions. Soc Gen Physiol Ser 1994, 49, 159–171. [Google Scholar]
- Saez, LJ; Gianola, KM; McNally, EM; et al. Human Cardiac Myosin Heavy Chain Genes and Their Linkage in the Genome. Nucleic Acids Res 1987, 15, 5443–5459. [Google Scholar]
- Smerdu, V; Karsch-Mizrachi, I; Campione, M; et al. Type IIx Myosin Heavy Chain Transcripts are Expressed in Type IIb Fibers of Human Skeletal Muscle. Am J Physiol 1994, 267, C1723–1728. [Google Scholar]
- Pedrosa-Domellof, F; Holmgren, Y; Lucas, CA; et al. Human Extraocular Muscles: Unique Pattern of Myosin Heavy Chain Expression During Myotube Formation. Invest Ophthalmol Vis Sci 2000, 41, 1608–1616. [Google Scholar]
- Sartore, S; Mascarello, F; Rowlerson, A; et al. Fibre Types in Extraocular Muscles: A New Myosin Isoform in the Fast Fibres. J Muscle Res Cell Motil 1987, 8, 161–172. [Google Scholar]
- Asmussen, G; Traub, I; Pette, D. Electrophoretic Analysis of Myosin Heavy Chain Isoform Patterns in Extraocular Muscles of the Rat. FEBS Lett 1993, 335, 243–245. [Google Scholar]
- Wieczorek, DF; Periasamy, M; Butler-Browne, GS; et al. Co-expression of Multiple Myosin Heavy Chain Genes, in Addition to a Tissue-Specific One, in Extraocular Musculature. J Cell Biol 1985, 101, 618–629. [Google Scholar]
- Gordon, AM; Homsher, E; Regnier, M. Regulation of Contraction in Striated Muscle. Physiol Rev 2000, 80, 853–924. [Google Scholar]
- Perry, SV. Vertebrate Tropomyosin: Distribution, Properties and Function. J Muscle Res Cell Motil 2001, 22, 5–49. [Google Scholar]
- Pieples, K; Wieczorek, DF. Tropomyosin 3 Increases Striated Muscle Isoform Diversity. Biochemistry 2000, 39, 8291–8297. [Google Scholar]
- Gahlmann, R; Wade, R; Gunning, P; Kedes, L. Differential Expression of Slow and Fast Skeletal Muscle Troponin C. Slow Skeletal Muscle Troponin C is Expressed in Human Fibroblasts. J Mol Biol 1988, 201, 379–391. [Google Scholar]
- Schreier, T; Kedes, L; Gahlmann, R. Cloning, Structural Analysis, and Expression of the Human Slow Twitch Skeletal Muscle/Cardiac Troponin C Gene. J Biol Chem 1990, 265, 21247–21253. [Google Scholar]
- Syska, H; Perry, SV; Trayer, IP. A New Method of Preparation of Troponin I (Inhibitory Protein) Using Affinity Chromatography. Evidence for Three Different Forms of Troponin I in Striated Muscle. FEBS Lett 1974, 40, 253–257. [Google Scholar]
- Barton, PJ; Cullen, ME; Townsend, PJ; et al. Close Physical Linkage of Human Troponin Genes: Organization, Sequence, and Expression of the Locus Encoding Cardiac Troponin I and Slow Skeletal Troponin T. Genomics 1999, 57, 102–109. [Google Scholar]
- Huang, P; Jin, X; Chen, Y; et al. Use of a Mixed-Mode Packing and Voltage Tuning for Peptide Mixture Separation in Pressurized Capillary Electrochromatography with an Ion Trap Storage/Reflectron Time-of-Flight Mass Spectrometer Detector. Anal Chem 1999, 71, 1786–1791. [Google Scholar]
- Wang, K. Titin/Connectin and Nebulin: Giant Protein Rulers of Muscle Structure and Function. Adv Biophys 1996, 33, 123–134. [Google Scholar]
- Horowits, R; Luo, G; Zhang, JQ; Herrera, AH. Nebulin and Nebulin-Related Proteins in Striated Muscle. Adv Biophys 1996, 33, 143–150. [Google Scholar]
- McElhinny, AS; Schwach, C; Valichnac, M; et al. Nebulin Regulates the Assembly and Lengths of the Thin Filaments in Striated Muscle. J Cell Biol 2005, 170, 947–957. [Google Scholar]
- Geisterfer-Lowrance, AA; Kass, S; Tanigawa, G; et al. A Molecular Basis for Familial Hypertrophic Cardiomyopathy: A Beta Cardiac Myosin Heavy Chain Gene Missense Mutation. Cell 1990, 62, 999–1006. [Google Scholar]
- Martinsson, T; Oldfors, A; Darin, N; et al. Autosomal Dominant Myopathy: Missense Mutation (Glu-706 → Lys) in the Myosin Heavy Chain IIa Gene. Proc Natl Acad Sci USA 2000, 97, 14614–14619. [Google Scholar]
- Laing, NG; Laing, BA; Meredith, C; et al. Autosomal Dominant Distal Myopathy: Linkage to Chromosome 14. Am J Hum Genet 1995, 56, 422–427. [Google Scholar]
- Meredith, C; Herrmann, R; Parry, C; et al. Mutations in the Slow Skeletal Muscle Fiber Myosin Heavy Chain Gene (MYH7) Cause Laing Early-Onset Distal Myopathy (MPD1). Am J Hum Genet 2004, 75, 703–708. [Google Scholar]
- Lamont, PJ; Udd, B; Mastaglia, FL; et al. Laing Early Onset Distal Myopathy: Slow Myosin Defect with Variable Abnormalities on Muscle Biopsy. J Neurol Neurosurg Psychiatry 2006, 77, 208–215. [Google Scholar]
- Scoppetta, C; Casali, C; La Cesa, I; et al. Infantile Autosomal Dominant Distal Myopathy. Acta Neurol Scand 1995, 92, 122–126. [Google Scholar]
- Tajsharghi, H; Thornell, LE; Lindberg, C; et al. Myosin Storage Myopathy Associated with a Heterozygous Missense Mutation in MYH7. Ann Neurol 2003, 54, 494–500. [Google Scholar]
- Bohlega, S; Abu-Amero, SN; Wakil, SM; et al. Mutation of the Slow Myosin Heavy Chain Rod Domain Underlies Hyaline Body Myopathy. Neurology 2004, 62, 1518–1521. [Google Scholar]
- Laing, NG; Ceuterick-de Groote, C; Dye, DE; et al. Myosin Storage Myopathy: Slow Skeletal Myosin (MYH7) Mutation in Two Isolated Cases. Neurology 2005, 64, 527–529. [Google Scholar]
- Shingde, MV; Spring, PJ; Maxwell, A; et al. Myosin Storage (Hyaline Body) Myopathy: A Case Report. Neuromuscul Disord 2006, 16, 882–886. [Google Scholar]
- Dye, DE; Azzarelli, B; Goebel, HH; Laing, NG. Novel Slow-Skeletal Myosin (MYH7) Mutation in the Original Myosin Storage Myopathy Kindred. Neuromuscul Disord 2006, 16, 357–360. [Google Scholar]
- Veugelers, M; Bressan, M; McDermott, DA; et al. Mutation of Perinatal Myosin Heavy Chain Associated with a Carney Complex Variant. N Engl J Med 2004, 351, 460–469. [Google Scholar]
- Toydemir, RM; Chen, H; Proud, VK; et al. Trismus-pseudocamptodactyly Syndrome is Caused by Recurrent Mutation of MYH8. Am J Med Genet 2006, 140, 2387–2393. [Google Scholar]
- Toydemir, RM; Rutherford, A; Whitby, FG; et al. Mutations in Embryonic Myosin Heavy Chain (MYH3) Cause Freeman-Sheldon Syndrome and Sheldon-Hall Syndrome. Nat Genet 2006, 38, 561–565. [Google Scholar]
- Darin, N; Kyllerman, M; Wahlstrom, J; et al. Autosomal Dominant Myopathy with Congenital Joint Contractures, Ophthalmoplegia, and Rimmed Vacuoles. Ann Neurol 1998, 44, 242–248. [Google Scholar]
- Tajsharghi, H; Darin, N; Rekabdar, E; et al. Mutations and Sequence Variation in the Human Myosin Heavy Chain IIa Gene (MYH2). Eur J Hum Genet 2005, 13, 617–622. [Google Scholar]
- Watkins, H; Rosenzweig, A; Hwang, DS; et al. Characteristics and Prognostic Implications of Myosin Missense Mutations in Familial Hypertrophic Cardiomyopathy. N Engl J Med 1992, 326, 1108–1114. [Google Scholar]
- Seidman, JG; Seidman, C. The Genetic Basis for Cardiomyopathy: from Mutation Identification to Mechanistic Paradigms. Cell 2001, 104, 557–567. [Google Scholar]
- Fananapazir, L; Dalakas, MC; Cyran, F; et al. Missense Mutations in the Beta-Myosin Heavy-Chain Gene Cause Central Core Disease in Hypertrophic Cardiomyopathy. Proc Natl Acad Sci USA 1993, 90, 3993–3997. [Google Scholar]
- Cuda, G; Fananapazir, L; Epstein, ND; Sellers, JR. The in vitro Motility Activity of Beta-Cardiac Myosin Depends on the Nature of the Beta-Myosin Heavy Chain Gene Mutation in Hypertrophic Cardiomyopathy. J Muscle Res Cell Motil 1997, 18, 275–283. [Google Scholar]
- Darin, N; Tajsharghi, H; Ostman-Smith, I; et al. New Skeletal Myopathy and Cardiomyopathy Associated with a Missense Mutation in MYH7. Neurology 2007, 68, 2041–2042. [Google Scholar]
- Overeem, S; Schelhaas, HJ; Blijham, PJ; et al. Symptomatic Distal Myopathy with Cardiomyopathy due to a MYH7 Mutation. Neuromuscul Disord 2007, 17, 490–493. [Google Scholar]
- Cancilla, PA; Kalyanaraman, K; Verity, MA; et al. Familial Myopathy with Probable Lysis of Myofibrils in Type I Fibers. Neurology 1971, 21, 579–585. [Google Scholar]
- Barohn, RJ; Brumback, RA; Mendell, JR; et al. Hyaline Body Myopathy. Neuromuscul Disord 1994, 4, 257–262. [Google Scholar]
- Masuzugawa, S; Kuzuhara, S; Narita, Y; et al. Autosomal Dominant Hyaline Body Myopathy Presenting as Scapuloperoneal Syndrome: Clinical Features and Muscle Pathology. Neurology 1997, 48, 253–257. [Google Scholar]
- Pegoraro, E; Gavassini, BF; Borsato, C; et al. MYH7 Gene Mutation in Myosin Storage Myopathy and Scapulo-Peroneal Myopathy. Neuromuscul Disord 2007, 17, 321–329. [Google Scholar]
- Hall, JG; Reed, SD; Greene, G. The Distal Arthrogryposes: Delineation of New Entities-Review and Nosologic Discussion. Am J Med Genet 1982, 11, 185–239. [Google Scholar]
- Bamshad, M; Jorde, LB; Carey, JC. A Revised and Extended Classification of the Distal Arthrogryposes. Am J Med Genet 1996, 65, 277–281. [Google Scholar]
- Tajsharghi, H; Kimber, E; Kroksmark, AK; et al. Embryonic Myosin Heavy Chain Mutations Cause Distal Arthrogryposis and Developmental Myosin Myopathy That Persists Postnatally. Arch Neurol. in press.
- Andersen, PS; Havndrup, O; Bundgaard, H; et al. Myosin Light Chain Mutations in Familial Hypertrophic Cardiomyopathy: Phenotypic Presentation and Frequency in Danish and South African Populations. J Med Genet 2001, 38, E43. [Google Scholar]
- Flavigny, J; Richard, P; Isnard, R; et al. Identification of Two Novel Mutations in the Ventricular Regulatory Myosin Light Chain Gene (MYL2) Associated with Familial and Classical Forms of Hypertrophic Cardiomyopathy. J Mol Med 1998, 76, 208–214. [Google Scholar]
- Poetter, K; Jiang, H; Hassanzadeh, S; et al. Mutations in Either the Essential or Regulatory Light Chains of Myosin are Associated with a Rare Myopathy in Human Heart and Skeletal Muscle. Nat Genet 1996, 13, 63–69. [Google Scholar]
- Richard, P; Charron, P; Carrier, L; et al. Hypertrophic Cardiomyopathy: Distribution of Disease Genes, Spectrum of Mutations, and Implications for a Molecular Diagnosis Strategy. Circulation 2003, 107, 2227–2232. [Google Scholar]
- Kabaeva, ZT; Perrot, A; Wolter, B; et al. Systematic Analysis of the Regulatory and Essential Myosin Light Chain Genes: Genetic Variants and Mutations in Hypertrophic Cardiomyopathy. Eur J Hum Genet 2002, 10, 741–748. [Google Scholar]
- Hernandez, OM; Jones, M; Guzman, G; et al. Myosin Essential Light Chain in Health and Disease. Am J Physiol 2007, 292, H1643–1654. [Google Scholar]
- Morano, I. Tuning the Human Heart Molecular Motors by Myosin Light Chains. J Mol Med 1999, 77, 544–555. [Google Scholar]
- Nowak, KJ; Wattanasirichaigoon, D; Goebel, HH; et al. Mutations in the Skeletal Muscle Alpha-Actin Gene in Patients with Actin Myopathy and Nemaline Myopathy. Nat Genet 1999, 23, 208–212. [Google Scholar]
- Laing, NG; Clarke, NF; Dye, DE; et al. Actin Mutations are One Cause of Congenital Fibre Type Disproportion. Ann Neurol 2004, 56, 689–694. [Google Scholar]
- Kaindl, AM; Ruschendorf, F; Krause, S; et al. Missense Mutations of ACTA1 Cause Dominant Congenital Myopathy with Cores. J Med Genet 2004, 41, 842–848. [Google Scholar]
- Donner, K; Ollikainen, M; Ridanpaa, M; et al. Mutations in the Beta-Tropomyosin (TPM2) Gene-a Rare Cause of Nemaline Myopathy. Neuromuscul Disord 2002, 12, 151–158. [Google Scholar]
- Tajsharghi, H; Ohlsson, M; Lindberg, C; et al. Congenital Myopathy with Nemaline Rods and Cap Structures Caused by a Mutation in the Beta-Tropomyosin Gene (TPM2). Arch Neurol 2007, 64, 1334–1338. [Google Scholar]
- Lehtokari, VL; Ceuterick-de Groote, C; de Jonghe, P; et al. Cap Disease Caused by Heterozygous Deletion of the Beta-Tropomyosin Gene TPM2. Neuromuscul Disord 2007, 17, 433–442. [Google Scholar]
- Sung, SS; Brassington, AM; Grannatt, K; et al. Mutations in Genes Encoding Fast-Twitch Contractile Proteins Cause Distal Arthrogryposis Syndromes. Am J Hum Genet 2003, 72, 681–690. [Google Scholar]
- Tajsharghi, H; Kimber, E; Holmgren, D; et al. Distal Arthrogryposis and Muscle Weakness Associated with a Beta-Tropomyosin Mutation. Neurology 2007, 68, 772–775. [Google Scholar]
- Laing, NG; Wilton, SD; Akkari, PA; et al. A Mutation in the Alpha Tropomyosin Gene TPM3 Associated with Autosomal Dominant Nemaline Myopathy. Nat Genet 1995, 9, 75–79. [Google Scholar]
- Wattanasirichaigoon, D; Swoboda, KJ; Takada, F; et al. Mutations of the Slow Muscle Alpha-Tropomyosin Gene, TPM3, Are a Rare Cause of Nemaline Myopathy. Neurology 2002, 59, 613–617. [Google Scholar]
- Tan, P; Briner, J; Boltshauser, E; et al. Homozygosity for a Nonsense Mutation in the Alpha-Tropomyosin Slow Gene TPM3 in a Patient with Severe Infantile Nemaline Myopathy. Neuromuscul Disord 1999, 9, 573–579. [Google Scholar]
- Durling, HJ; Reilich, P; Muller-Hocker, J; et al. De Novo Missense Mutation in a Constitutively Expressed Exon of the Slow Alpha-Tropomyosin Gene TPM3 Associated with an Atypical, Sporadic Case of Nemaline Myopathy. Neuromuscul Disord 2002, 12, 947–951. [Google Scholar]
- Penisson-Besnier, I; Monnier, N; Toutain, A; et al. A Second Pedigree with Autosomal Dominant Nemaline Myopathy Caused by TPM3 Mutation: A Clinical and Pathological Study. Neuromuscul Disord 2007, 17, 330–337. [Google Scholar]
- Clarke, NF; Kolski, H; Dye, DE; et al. Mutations in TPM3 Are a Common Cause of Congenital Fiber Type Disproportion. Ann Neurol 2008, 63, 329–337. [Google Scholar]
- Shrimpton, AE; Hoo, JJ. A TNNI2 Mutation in a Family with Distal Arthrogryposis Type 2B. European journal of medical genetics 2006, 49, 201–206. [Google Scholar]
- Kimber, E; Tajsharghi, H; Kroksmark, AK; et al. A Mutation in the Fast Skeletal Muscle Troponin I Gene Causes Myopathy and Distal Arthrogryposis. Neurology 2006, 67, 597–601. [Google Scholar]
- Jiang, M; Zhao, X; Han, W; et al. A Novel Deletion in TNNI2 Causes Distal Arthrogryposis in a Large Chinese Family with Marked Variability of Expression. Hum Genet 2006, 120, 238–242. [Google Scholar]
- Johnston, JJ; Kelley, RI; Crawford, TO; et al. A Novel Nemaline Myopathy in the Amish Caused by a Mutation in Troponin T1. Am J Hum Genet 2000, 67, 814–821. [Google Scholar]
- Sung, SS; Brassington, AM; Krakowiak, PA; et al. Mutations in TNNT3 Cause Multiple Congenital Contractures: A Second Locus for Distal Arthrogryposis Type 2B. Am J Hum Genet 2003, 73, 212–214. [Google Scholar]
- Hoffmann, B; Schmidt-Traub, H; Perrot, A; et al. First Mutation in Cardiac Troponin C, L29Q, in a Patient with Hypertrophic Cardiomyopathy. Hum Mutat 2001, 17, 524. [Google Scholar]
- Mogensen, J; Murphy, RT; Shaw, T; et al. Severe Disease Expression of Cardiac Troponin C and T Mutations in Patients with Idiopathic Dilated Cardiomyopathy. J Am Coll Cardiol 2004, 44, 2033–2040. [Google Scholar]
- Pelin, K; Hilpela, P; Donner, K; et al. Mutations in the Nebulin Gene Associated with Autosomal Recessive Nemaline Myopathy. Proc Natl Acad Sci USA 1999, 96, 2305–2310. [Google Scholar]
- Pelin, K; Donner, K; Holmberg, M; et al. Nebulin Mutations in Autosomal Recessive Nemaline Myopathy: An Update. Neuromuscul Disord 2002, 12, 680–686. [Google Scholar]
- Wallgren-Pettersson, C; Donner, K; Sewry, C; et al. Mutations in the Nebulin Gene Can Cause Severe Congenital Nemaline Myopathy. Neuromuscul Disord 2002, 12, 674–679. [Google Scholar]
- Lehtokari, VL; Pelin, K; Sandbacka, M; et al. Identification of 45 Novel Mutations in the Nebulin Gene Associated with Autosomal Recessive Nemaline Myopathy. Hum Mutat 2006, 27, 946–956. [Google Scholar]
- Wallgren-Pettersson, C; Lehtokari, VL; Kalimo, H; et al. Distal Myopathy Caused by Homozygous Missense Mutations in the Nebulin Gene. Brain 2007, 130, 1465–1476. [Google Scholar]
Figure 1.
a) Ultrastructurally, the A-band corresponds to the thick filaments and includes a zone where the thin filaments overlap the thick filaments. The I-band is the zone in which the thin filaments do not overlap the thick filaments. The Z-line is a dark band in the centre of the I-band and the M-line runs down the center of the A-band.
Figure 1.
a) Ultrastructurally, the A-band corresponds to the thick filaments and includes a zone where the thin filaments overlap the thick filaments. The I-band is the zone in which the thin filaments do not overlap the thick filaments. The Z-line is a dark band in the centre of the I-band and the M-line runs down the center of the A-band.

Figure 2.
Illustration of the I-band, A-band, and M-line regions of the sarcomere. The thin filaments contain actin, tropomyosin, troponins C, I, and T and nebulin. The thick filaments are composed of myosin with the globular heads forming cross-bridges with thin filaments. Myosin-binding proteins, including MyBP-C, are associated with the thick filaments. The giant protein titin extend the length of an entire half sarcomere. The M-line contains different proteins, such as myomesin and M-protein.
Figure 2.
Illustration of the I-band, A-band, and M-line regions of the sarcomere. The thin filaments contain actin, tropomyosin, troponins C, I, and T and nebulin. The thick filaments are composed of myosin with the globular heads forming cross-bridges with thin filaments. Myosin-binding proteins, including MyBP-C, are associated with the thick filaments. The giant protein titin extend the length of an entire half sarcomere. The M-line contains different proteins, such as myomesin and M-protein.

Figure 3.
Schematic illustration of a myosin class II molecule, showing the essential light chains (ELC) and regulatory light chains (RLC) that wrap around the α-helical region of the S1. Two MyHC molecules intertwine via their α-helical regions to form a coiled-coil rod. Proteolytic fragments S1, S2 and LMM are indicated.
Figure 3.
Schematic illustration of a myosin class II molecule, showing the essential light chains (ELC) and regulatory light chains (RLC) that wrap around the α-helical region of the S1. Two MyHC molecules intertwine via their α-helical regions to form a coiled-coil rod. Proteolytic fragments S1, S2 and LMM are indicated.

Figure 4.
Ribbon representation of the chicken myosin S1 structure. The 25-, 50-, and 20-kDa segments of the heavy chain are shown as red, green, and purple ribbons, respectively. The upper and lower segments of the 50-kDa domain are indicated. The ATP- and actin-binding sites are shown. This figure was prepared using the program WebLab VIEWERLIFE 3.2.
Figure 4.
Ribbon representation of the chicken myosin S1 structure. The 25-, 50-, and 20-kDa segments of the heavy chain are shown as red, green, and purple ribbons, respectively. The upper and lower segments of the 50-kDa domain are indicated. The ATP- and actin-binding sites are shown. This figure was prepared using the program WebLab VIEWERLIFE 3.2.

Figure 5.
Schematic illustration of a part of the sarcomere, the contractile unit of muscle, composed of thick and thin filaments. Note that the illustration does not correspond to a notion of size.
Figure 5.
Schematic illustration of a part of the sarcomere, the contractile unit of muscle, composed of thick and thin filaments. Note that the illustration does not correspond to a notion of size.

Share and Cite
MDPI and ACS Style
Tajsharghi, H. Thick and Thin Filament Gene Mutations in Striated Muscle Diseases. Int. J. Mol. Sci. 2008, 9, 1259-1275. https://doi.org/10.3390/ijms9071259
AMA Style
Tajsharghi H. Thick and Thin Filament Gene Mutations in Striated Muscle Diseases. International Journal of Molecular Sciences. 2008; 9(7):1259-1275. https://doi.org/10.3390/ijms9071259
Chicago/Turabian StyleTajsharghi, Homa. 2008. "Thick and Thin Filament Gene Mutations in Striated Muscle Diseases" International Journal of Molecular Sciences 9, no. 7: 1259-1275. https://doi.org/10.3390/ijms9071259