3-[2-(Oxiran-2-yl)ethyl]-1-{4-[(2-oxiran-2-yl)ethoxy]benzyl}imidazolium bis(Trifluoromethane)sulfonimide


1
Laboratory of Molecular and Thio-organic Chemistry, ENSICAEN, Normandy University, CNRS, 6 boulevard du Maréchal Juin, 14050 Caen, France
2
Lyon University, INSA Lyon, UMR CNRS 5223, IMP Polymer Materials Engineering, F-69621 Villeurbanne, France
*
Authors to whom correspondence should be addressed.
Molbank 2018, 2018(1), M974; https://doi.org/10.3390/M974
Submission received: 19 December 2017
/
Revised: 9 January 2018
/
Accepted: 10 January 2018
/
Published: 13 January 2018
(This article belongs to the Special Issue Heterocycles)
Abstract
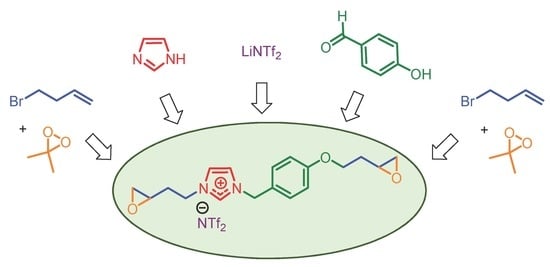
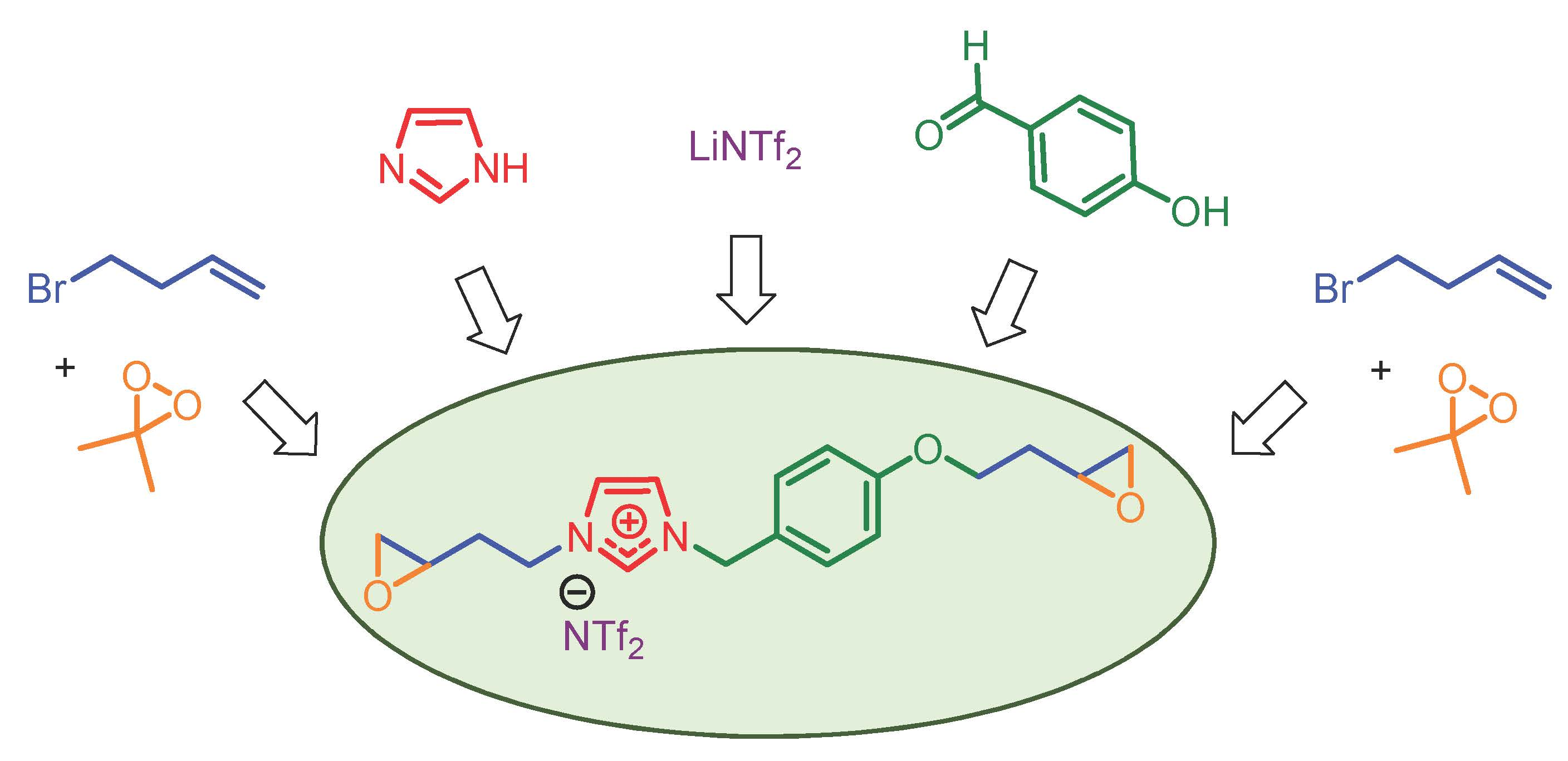
:The synthesis of 3-[2-(oxiran-2-yl)ethyl]-1-{4-[(2-oxiran-2-yl)ethoxy]benzyl}imidazolium bis(trifluoromethane)sulfonimide was achieved in seven steps with an overall yield of 63%. This ionic liquid monomer is based on an imidazolium core containing a phenoxy moiety and aliphatic chains with a reactive epoxide at both ends. This imidazolium was obtained by an efficient, reproducible, and scalable synthesis from inexpensive reagents. In the last step, a powerful oxidative agent was successfully used to generate two epoxy functions with only acetone as a byproduct. The monomer structure and intermediates were analyzed by NMR experiments, infrared, and mass spectra. In addition, the thermal properties of the diepoxide were evaluated by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC).
{kind=link}
{kind=link}
1. Introduction
Discovered as green solvents, ionic liquids (ILs) have become essential tools in many areas of research in chemistry and materials science. These salts are particularly attractive due to their unique properties such as a high thermal and chemical stability combined with a high conductivity, wide electrochemical window, low volatility, and nonflammability [1]. According to these additional characteristics, ionic liquids can be used for specific practical applications as surfactants [2], lubricants, plasticizers, reactive (or non-reactive) additives to polymer matrices, or simply as solvents in electrochemistry [3,4,5]. In the wide range of ionic liquids reported in the literature, it should be noted that those based on the imidazolium fragment have received increasing attention in recent years [6,7,8]. These ionic liquids with multifaceted roles can act as initiators in the polymerization of the epoxy prepolymer or directly as liquid monomers to design epoxy networks or polymerized ionic liquids [9,10]. In this field, the addition of terminal epoxides on the ionic liquid skeleton is a real challenge to facilitate access to new polymers with promising properties. To date, epichlorohydrin is the most valuable reagent to incorporate epoxides in these ionic monomers [11]. Unfortunately, this toxic and corrosive alkylating reagent limits the structural diversity of the monomers by adding only a methyloxirane moiety at the end of the sequence. In this context, we describe an easy and effective synthesis of an imidazolium monomer bearing epoxides for the future development of polymerized ionic liquids (PILs) [12].
2. Results
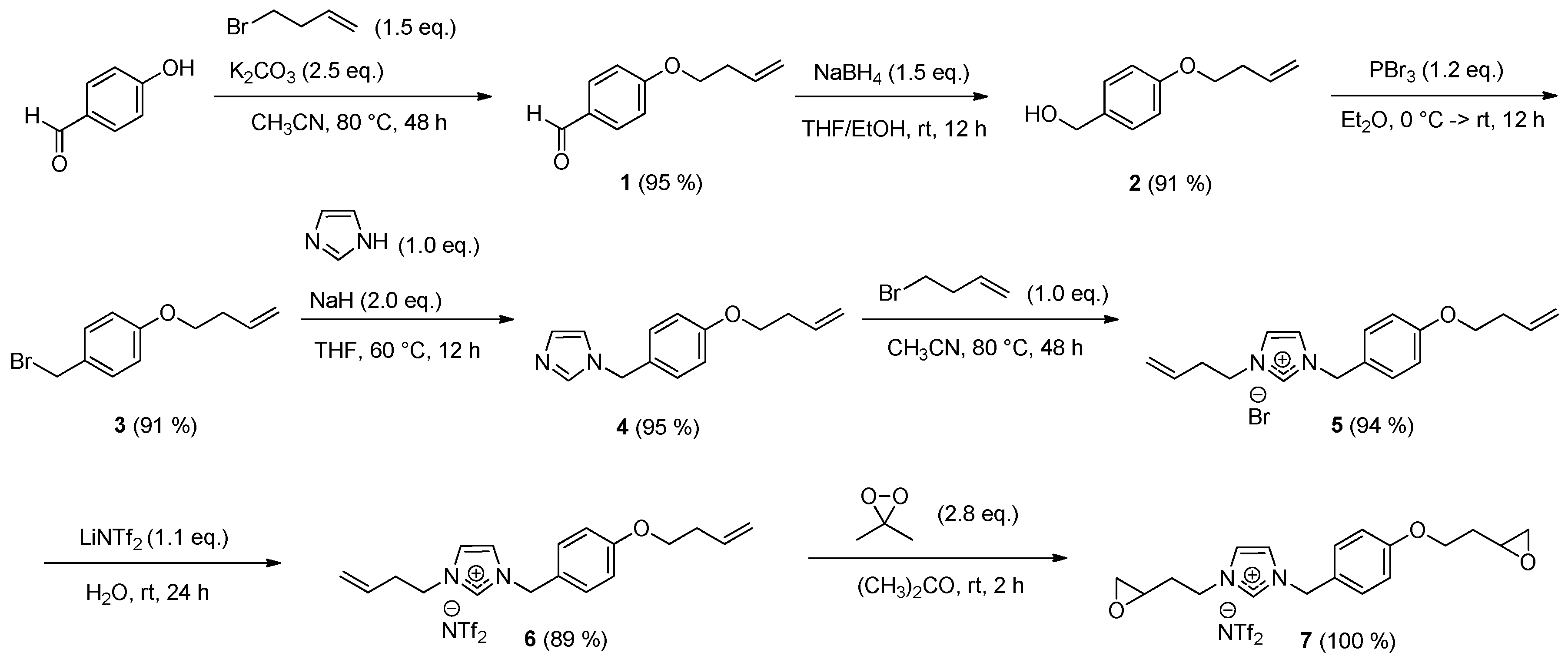
The imidazolium monomer 7 was synthesized in seven steps, as presented in Scheme 1.
In the first part, the commercial 4-hydroxybenzaldehyde was O-alkyled in acetonitrile with 4-bromo-1-butene in the presence of potassium carbonate. The aldehyde function of compound 1 was reduced to the alcohol using sodium borohydride in a mixture of tetrahydrofuran and methanol. Then, a bromation of compound 2 was realized with the addition of phosphorus tribromide in diethyl ether. This procedure gave compound 3 for the next step.
The imidazolium backbone was synthesized in three steps from imidazole. This heterocycle was deprotonated using sodium hydride in tetrahydrofuran and functionalized by alkylation using compound 3 obtained previously. In a second step, the quaternization of compound 4 with 4-bromo-1-butene gave imidazolium bromide 5. To exchange the bromine, the anion metathesis of this salt was performed in water for 24 h using the bis(trifluoromethane)sulfonimide lithium salt.
Finally, compound 6 was oxidized in the presence of freshly prepared dimethyldioxirane (2.8 eq.) in acetone [13,14] at room temperature. Compound 7 was obtained in quantitative yield after 2 h. Its purity was analyzed and confirmed by 1H-, 13C-, 19F-NMR, infrared, and mass spectra and its thermal behavior was also studied by TGA and DSC (see Supplementary Materials).
3. Discussion
The ionic monomer 7 was synthesized in 63% overall yield from 4-hydroxybenzaldehyde, 4-bromo-1-butene, bis(trifluoromethane)sulfonimide lithium salt, and imidazole. This procedure does not require any purification by silica gel column chromatography during the entire sequence, the final step being the preparation of the epoxides by a mild oxidation using dimethyldioxirane (DMDO). This oxidizing reagent generates diepoxide quantitatively with only inert acetone as the byproduct. This efficient pathway is reproducible and can be performed on a large scale in the laboratory. NMR experiments have confirmed that the monomer structure was obtained with excellent purity (no traces of byproducts have been detected). Thermal analyses were performed to investigate the purity and the thermal behavior of the diepoxide. The thermal stability was carried out by thermogravimetric analysis (TGA) using a Q500 (TA instruments) from 30 to 600 °C with a heating rate of 10 K min−1 under nitrogen atmosphere. The weight loss as a function of temperature was measured to determine the decomposition temperature and the percentage of degradation. Excellent thermal behavior of the epoxy monomer was noticed from the TGA curve (see Supplementary Materials) with a maximal degradation temperature of 453 °C and a weight loss of only 5 wt% at 300 °C and 20 wt% at 400 °C. Differential scanning calorimetry analysis was also performed to determine the glass transition temperature (Tg) on the DSC thermogram (see Supplementary Materials). Thus, we have observed a Tg of −31 °C during the first heating at a rate of 10 K min−1 under a nitrogen flow of 50 mL min−1.
4. Materials and Methods
4.1. General Information
All reagents were purchased from Sigma Aldrich, Alfa Aesar, or TCI and used as received without further purification. Solvents were used in RPE grade without further purification. Anhydrous solvents were obtained from a PURESOLV SPS400 apparatus developed by Innovative Technology Inc. (Hong Kong, China). 1H-, 13C-, and 19F-NMR spectra were recorded on a Bruker Avance III 400 MHz instrument (Wissembourg, France). Samples were dissolved in an appropriate deuterated solvent (CDCl3). The chemical shifts (δ) are expressed in ppm relative to internal tetramethylsilane for 1H and 13C nuclei, and coupling constants are indicated in Hz. Abbreviations for signal coupling are as follows: s = singlet; d = doublet; ddt = doublet of doublets of triplets; t = triplet; q = quartet; m = multiplet; br = broad signal. To assign the signals to the different proton and carbon atoms, as well as the relative stereochemistry of the cycloadducts, additional 2D NMR experiments (COSY, HSQC, HMBC) and NOESY experiments were performed. High-resolution mass spectra (HRMS) were performed on Xevo G2-XS Q-TOF WATERS (Milford, MA, USA) by electrospray ionization (ESI) or atmospheric solids analysis probe (ASAP). Infrared (IR) spectra were recorded with a Perkin Elmer 16 PC FTIR ATR spectrometer (Waltham, MA, USA), using the pure product (oil or solid). Thin layer chromatography (TLC) was run on pre-coated aluminum plates of silica gel 60 F-254 (Merck).
4.2. Synthesis of 1
To a solution of 4-hydroxybenzaldehyde (10.0 g, 81.9 mmol, 1.0 eq.) and K2CO3 (28.3 g, 204.7 mmol, 2.5 eq.) in CH3CN (300 mL), 4-bromo-1-butene (12.37 mL, 122.8 mmol, 1.5 eq.) was added. The mixture was refluxed at 80 °C for 48 h (the reaction advancement was monitored by TLC or NMR). After cooling to room temperature, the solvent was removed under reduced pressure. The residue was partitioned between CH2Cl2 and water and the aqueous layer was extracted with CH2Cl2 (2 × 10 mL). The combined organic extracts were washed with water, dried, and concentrated to afford the product 1 [15] as a pale yellow oil (13.77 g, 95%).
1H-NMR (400 MHz, CDCl3) δ 9.88 (s, 1H), 7.83 (d, J = 8.7 Hz, 2H), 7.00 (d, J = 8.7 Hz, 2H), 5.90 (ddt, J = 17.1, 10.3, 6.7 Hz, 1H), 5.12–5.21 (m, 2H), 4.10 (t, J = 6.7 Hz, 2H), 2.58 (q, J = 6.7 Hz, 2H). 13C-NMR (100 MHz, CDCl3) δ 191.0, 164.1, 134.0, 132.1, 130.1, 117.6, 114.9, 67.7, 33.5. IR (neat) cm−1 3078, 2938, 2739, 1686, 1598, 1576, 1508, 1250, 1157, 1024. HRMS m/z (ESI): calcd. for C11H13O2 [M + H]+: 177.0916, found: 177.0917.
4.3. Synthesis of
At 0 °C, compound 1 (5.0 g, 28.37 mmol, 1.0 eq.) in a THF/EtOH mixture (1:1 volume ratio, 40 mL) was added dropwise to a stirred solution of NaBH4 (1.61 g, 42.56 mmol, 1.5 eq.) in a THF/EtOH mixture (1:1 volume ratio, 40 mL). The mixture was stirred at room temperature for 12 h then cooled to 0 °C and quenched with aqueous NH4Cl solution. The volatiles were evaporated and the residue was extracted with EtOAc (2 × 20 mL). The combined organic extracts were washed with water (2 × 10 mL), dried over MgSO4, and concentrated under reduced pressure. The product 2 [15] was obtained as a pale yellow oil (4.62 g, 91%).
1H-NMR (400 MHz, CDCl3) δ 7.28 (d, J = 8.7 Hz, 2H,), 6.89 (d, J = 8.7 Hz, 2H), 5.91 (ddt, J = 17.1, 10.3, 6.7 Hz, 1H), 5.10–5.19 (m, 2H), 4.61 (s, 2H), 4.02 (t, J = 6.7 Hz, 2H), 2.55 (q, J = 6.7 Hz, 2H). 13C-NMR (100 MHz, CDCl3) δ 158.7, 134.5, 133.3, 128.8, 117.2, 114.8, 67.4, 65.2, 33.8. IR (neat) cm−1 3078, 2926, 2872, 1612, 1511, 1241, 1172, 1033, 994, 917. HRMS m/z (ASAP): calcd. for C11H14O2 [M]+: 178.0994, found: 178.0993.
4.4. Synthesis of 3
To a stirred solution of compound 2 (4.0 g, 22.44 mmol, 1.0 eq.) in Et2O (40 mL) at 0 °C was added slowly a solution of PBr3 (2.56 mL, 26.93 mmol, 1.2 eq.) in Et2O (20 mL). The mixture was stirred for 6 h at 0 °C and was stirred at room temperature for another 6 h. The reaction mixture was placed in an acetone ice bath, water was added, and the solution was extracted with diethyl ether. The organic extracts were washed with aqueous NaHCO3 solution, water then dried over MgSO4. The solvent was removed under reduced pressure. The product 3 [15] was obtained as a pale yellow liquid (4.94 g, 91%).
1H-NMR (400 MHz, CDCl3) δ 7.31 (d, J = 8.7 Hz, 2H), 6.88 (d, J = 8.7 Hz, 2H), 5.90 (ddt, J = 17.1, 10.3, 6.7 Hz, 1H), 5.10–5.20 (m, 2H), 4.50 (s, 2H), 4.01 (t, J = 6.7 Hz, 2H), 2.54 (q, J = 6.7 Hz, 2H). 13C-NMR (100 MHz, CDCl3) δ 159.1, 134.4, 130.6, 130.1, 117.3, 115.0, 67.4, 34.2, 33.7. IR (neat) cm−1 3077, 2924, 2870, 1610, 1509, 1246, 1174, 1033, 988, 916. HRMS m/z (ASAP): calcd. for C11H14OBr [M + H]+: 241.0228, found: 241.0229.
4.5. Synthesis of 4
To a solution of imidazole (564 mg, 8.29 mmol, 1.0 eq.) in THF (36 mL), NaH (60%) (664 mg, 16.59 mmol, 2.0 eq.) was added in small portions. The reaction mixture was stirred for 30 min. A solution of compound 3 (2.0 g, 8.29 mmol, 1.0 eq.) in THF (40 mL) was added and the reaction mixture was stirred at 60 °C for 12 h. The reaction was quenched with water (91 mL) and the solvent was removed under reduced pressure. Dichloromethane (92 mL) was added and the organic layer was thoroughly washed two times with water and then separated. The organic layer was dried over MgSO4 and the solvent was removed under vacuum to afford the product 4 as a yellow oil (1.79 g, 95%).
1H-NMR (400 MHz, CDCl3) δ 7.52 (s, 1H), 7.00–7.16 (m, 3H), 6.77–6.97 (m, 3H), 5.89 (ddt, J = 17.1, 10.3, 6.7 Hz, 1H), 4.98–5.22 (m, 4H), 4.00 (t, J = 6.7 Hz, 2H), 2.54 (q, J = 6.7 Hz, 2H). 13C-NMR (100 MHz, CDCl3) δ 159.0, 137.4, 134.4, 129.9, 129.0, 128.3, 119.2, 117.3, 115.1, 67.4, 50.5, 33.7. IR (neat) cm−1 3077, 2928, 2872, 1612, 1512, 1243, 1176, 1074, 1029, 905. HRMS m/z (ESI): calcd. for C14H17N2O [M + H]+: 229.1341, found: 229.1343.
4.6. Synthesis of 5
A solution of compound 4 (1.75 g, 7.65 mmol, 1.0 eq.) and 4-bromo-1-butene (0.77 mL, 7.65 mmol, 1.0 eq.) in CH3CN (50 mL) was stirred at 80 °C. After 48 h, the reaction mixture was cooled to room temperature and the solvent was removed under reduced pressure. The product 5 was obtained as a yellow oil (2.60 g, 94%).
1H-NMR (400 MHz, CDCl3) δ 10.65 (s, 1H), 7.39 (d, J = 8.7 Hz, 2H), 7.30–7.34 (m, 1H), 7.21–7.25 (m, 1H), 6.88 (d, J = 8.7 Hz, 2H), 5.72–5.94 (m, 2H), 5.48 (s, 2H), 4.99–5.22 (m, 4H), 4.42 (t, J = 6.7 Hz, 2H), 3.98 (t, J = 6.7 Hz, 2H), 2.66 (q, J = 6.7 Hz, 2H), 2.52 (q, J = 6.7 Hz, 2H). 13C-NMR (100 MHz, CDCl3) δ 159.9, 137.3, 134.2, 132.3, 130.7, 124.9, 122.1, 121.5, 119.9, 117.4, 115.5, 67.4, 53.1, 49.4, 34.5, 33.6. IR (neat) cm−1 3075, 2980, 1612, 1559, 1513, 1247, 1179, 1154, 1031, 919. HRMS m/z (ESI): calcd. for C18H23N2O [M]+: 283.1810, found: 283.1811.
4.7. Synthesis of 6
To a solution of compound 5 (2.52 g, 6.94 mmol, 1.0 eq.) in H2O (150 mL), bis(trifluoromethane)sulfonimide lithium salt (2.19 g, 7.64 mmol, 1.1 eq.) was added. The solution was stirred at room temperature for 24 h and then extracted with dichloromethane. The organic layer was washed several times with water, dried over MgSO4, and concentrated under reduced pressure. The product 6 was obtained as a yellow oil (3.46 g, 89%).
1H-NMR (400 MHz, CDCl3) δ 8.83 (s, 1H), 7.20–7.30 (m, 3H), 7.13–7.17 (m, 1H), 6.92 (d, J = 8.7 Hz, 2H), 5.89 (ddt, J = 17.1, 10.3, 6.7 Hz, 1H), 5.73 (ddt, J = 17.0, 10.2, 6.7 Hz, 1H), 4.98–5.28 (m, 6H), 4.26 (t, J = 6.7 Hz, 2H), 4.01 (t, J = 6.7 Hz, 2H), 2.50–2.63 (m, 4H). 13C-NMR (100 MHz, CDCl3) δ 160.2, 135.7, 134.3, 132.1, 130.6, 124.2, 122.4, 121.9, 120.1, 120.0 (q, JCF = 322.4 Hz), 117.4, 115.6, 67.5, 53.4, 49.6, 34.4, 33.6. 19F-NMR (376 MHz, CDCl3) δ −78.9. IR (neat) cm−1 3148, 3086, 2925, 1614, 1562, 1515, 1348, 1178, 1133, 1053. HRMS m/z (ESI): calcd. for C18H23N2O [M]+: 283.1810, found: 283.1811.
4.8. Synthesis of 7
To a solution of compound 6 (50 mg, 0.0887 mmol, 1.0 eq.) in acetone (1.00 mL), freshly prepared dimethyldioxirane (0.045 mol/L) (5.52 mL, 0.2484 mmol, 2.8 eq.) was added [15]. The reaction mixture was stirred at room temperature for 2 h (1H-NMR monitoring). Two drops of DMS was added to quench the reaction mixture and neutralized the excess of dimethyldioxirane. The crude was concentrated under reduced pressure. The product 7 was obtained as a yellow oil (53 mg, 100%).
1H-NMR (400 MHz, CDCl3) δ 8.98 (s, 1H), 7.28–7.33 (m, 3H), 7.11–7.13 (m, 1H), 6.95 (d, J = 8.7 Hz, 2H), 5.28 (s, 2H), 4.41 (t, J = 6.7 Hz, 2H), 4.09–4.17 (m, 2H), 3.12–3.18 (m, 1H), 2.98–3.04 (m, 1H), 2.78–2.86 (m, 2H), 2.42–2.60 (m, 3H), 2.10–2.20 (m, 1H), 1.75–1.96 (m, 2H). 13C-NMR (100 MHz, CDCl3) δ 160.1, 136.2, 130.7, 124.1, 122.8, 121.7, 120.0 (q, JCF = 322.4 Hz), 115.7, 65.0, 53.6, 49.7, 49.3, 47.9, 47.3, 46.7, 32.7, 32.5. 19F-NMR (376 MHz, CDCl3) δ −78.8. IR (neat) cm−1 3149, 2934, 1613, 1563, 1516, 1348, 1330, 1178, 1132, 1052. HRMS m/z (ESI): calcd. for C18H23N2O3 [M]+: 315.1709, found: 315.1712.
5. Conclusions
In conclusion, we have synthesized an imidazolium monomer incorporating an aromatic ring and terminal epoxides. This diepoxide salt was obtained in seven steps with an excellent overall yield of 63%. After optimization, the final compound was characterized by 1H-, 13C-, and 19F-NMR, DSC then TGA analyses to confirm its high purity and thermal stability. Further investigations of these imidazolium monomer bearing epoxides are in progress by our research team to explore their potential to create polymerized ionic liquids.
Supplementary Materials
Copies of the NMR spectra for all the compounds and TGA and DSC curves are available online.
Acknowledgments
The Ministery of Higher Education and Research, the Normandy region (fellowships to C.C.), CNRS and the European union (FEDER) are greatly acknowledged for funding this work. We thank the LABEX Synorg (ANR-11-LABX-0029) for financial support.
Author Contributions
J.B. and S.L. conceived and designed the project; C.C. performed the experiments and analyzed the IR, MS, and NMR spectral data; S.L. performed the TGA and DSC; all authors (C.C., J.R., S.L. and J.B.) contributed to literature research; C.C. and J.B. wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wasserscheid, P.; Welton, T. Ionic Liquids in Synthesis; Wiley-VCH: Weinheim, Germany, 2008; pp. 1–364. ISBN 3-527-30515-7. [Google Scholar]
- Livi, S.; Duchet-Rumeau, J.; Gérard, J.-F.; Pham, T.N. Polymers and Ionic Liquids: A Successful Wedding. Macromol. Chem. Phys. 2015, 216, 359–368. [Google Scholar] [CrossRef]
- Freemantle, M. An Introduction to Ionic Liquids; RSC Publishing: Cambridge, UK, 2010; pp. 1–281. ISBN 978-1-84755-161-0. [Google Scholar]
- Kubisa, P. Ionic liquids as solvents for polymerization processes-Progress and challenges. Prog. Polym. Sci. 2009, 34, 1333–1347. [Google Scholar] [CrossRef]
- Lu, J.; Yan, F.; Texter, J. Advanced applications of ionic liquids in polymer science. Prog. Polym. Sci. 2009, 34, 431–448. [Google Scholar] [CrossRef]
- Sanes, J.; Carrión-Vilches, F.-J.; Bermúdez, M.-D. New epoxy-ionic liquid dispersions. Room temperature ionic liquid as lubricant of epoxy resin-stainless steel contacts. e-Polymers 2017, 7. [Google Scholar] [CrossRef]
- Soares, B.G.; Livi, S.; Duchet-Rumeau, J.; Gérard, J.-F. Synthesis and Characterization of Epoxy/MCDEA Networks Modified with Imidazolium-Based Ionic Liquids. Macromol. Mater. Eng. 2011, 296, 826–834. [Google Scholar] [CrossRef]
- Maka, H.; Spychaj, T.; Pilawka, R. Epoxy Resin/Ionic Liquid Systems: The Influence of Imidazolium Cation Size and Anion Type on Reactivity and Thermomechanical Properties. Ind. Eng. Chem. Res. 2012, 51, 5197–5206. [Google Scholar] [CrossRef]
- Soares, B.G.; Silva, A.A.; Pereira, J.; Livi, S. Preparation of Epoxy/Jeffamine Networks Modified with Phosphonium Based Ionic Liquids. Macromol. Mater. Eng. 2015, 300, 312–319. [Google Scholar] [CrossRef]
- Leclère, M.; Livi, S.; Maréchal, M.; Picard, L.; Duchet-Rumeau, J. The properties of new epoxy networks swollen with ionic liquids. RSC Adv. 2016, 6, 56193–56204. [Google Scholar] [CrossRef]
- Matsumoto, K.; Endo, T. Design and synthesis of ionic-conductive epoxy-based networked polymers. React. Funct. Polym. 2013, 73, 278–282. [Google Scholar] [CrossRef]
- Yuan, J.; Antonietti, M. Poly(ionic liquid)s: Polymers expanding classical property profiles. Polymer 2011, 52, 1469–1482. [Google Scholar] [CrossRef]
- Chardin, C.; Rouden, J.; Livi, S.; Baudoux, J. Dimethyldioxirane (DMDO) as a valuable oxidant for the synthesis of polyfunctional aromatic imidazolium monomers bearing epoxides. Green Chem. 2017, 19, 5054–5059. [Google Scholar] [CrossRef]
- Taber, D.F.; DeMatteo, P.W.; Hassan, R.A. Simplified Preparation of Dimethyldioxirane (DMDO). Org. Synth. 2013, 90, 350–357. [Google Scholar] [CrossRef]
- Lipshutz, B.H.; Ghorai, S.; Leong, W.W.Y. Deprotection of Homoallyl (hallyl) Derivatives of Phenols, Alcohols, Acids, and Amines. J. Org. Chem. 2009, 74, 2854–2857. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Synthesis of 3-[2-(oxiran-2-yl)ethyl]-1-{4-[(2-oxiran-2-yl)ethoxy]benzyl}imidazolium bis(trifluoromethane)sulfonimide (7).
Scheme 1.
Synthesis of 3-[2-(oxiran-2-yl)ethyl]-1-{4-[(2-oxiran-2-yl)ethoxy]benzyl}imidazolium bis(trifluoromethane)sulfonimide (7).
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chardin, C.; Rouden, J.; Livi, S.; Baudoux, J. 3-[2-(Oxiran-2-yl)ethyl]-1-{4-[(2-oxiran-2-yl)ethoxy]benzyl}imidazolium bis(Trifluoromethane)sulfonimide. Molbank 2018, 2018, M974. https://doi.org/10.3390/M974
AMA Style
Chardin C, Rouden J, Livi S, Baudoux J. 3-[2-(Oxiran-2-yl)ethyl]-1-{4-[(2-oxiran-2-yl)ethoxy]benzyl}imidazolium bis(Trifluoromethane)sulfonimide. Molbank. 2018; 2018(1):M974. https://doi.org/10.3390/M974
Chicago/Turabian StyleChardin, Charline, Jacques Rouden, Sébastien Livi, and Jérôme Baudoux. 2018. "3-[2-(Oxiran-2-yl)ethyl]-1-{4-[(2-oxiran-2-yl)ethoxy]benzyl}imidazolium bis(Trifluoromethane)sulfonimide" Molbank 2018, no. 1: M974. https://doi.org/10.3390/M974
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.