3. Materials and Methods
DIEA, DMF, EEDQ, ТЕА, cystamine dihydrochloride, 2,2′-dithiodipyridine, ethyl 4-bromobutyrate, spermine, thiocholesterol were obtained from Sigma-Aldrich (St. Louis, MO, USA); Boc2O, cesium carbonate were obtained from Fluka (Buchs, Switzerland); thiophenol was obtained from Merck (Darmstadt, Germany). Other solvents and reagents were purchased from Russian companies.
CH2Cl2, ТЕА, DIEA were refluxed with CaH2 and distilled prior to the reaction. EtOH was refluxed with magnesium turnings and iodine and distilled prior to the reaction. МеОН and DMF were kept over calcined molecular sieves 3 Å and 4 Å, respectively.
Column chromatography was carried out on silica gel Kieselgel 60 (0.040–0.063 mm, Merck). 1H-and 13C-NMR spectra were recorded on a Avance DPX-300 and Avance DRX-500 pulse Fourier transform spectrometers (Bruker, Karlsruhe, Germany) in CDCl3 unless otherwise stated. Chemical shifts were recorded in ppm on the δ scale relative to CHCl3 solvent residual peak (7.26 ppm for 1H and 77.0 ppm for 13C-NMR spectra). Coupling constants (J) are absolute values and recorded in Hz. Mass spectra were run on a Thermo Finnigan MAT 900XL-TRAP mass spectrometer and on a Orbitrap Fusion mass spectrometer (Thermo Scientific, Pittsburgh, PA, USA) with electrospray ionization (ESI). Melting points were determined on a IA9100 digital melting point apparatus (Electrothermal, Stone, Great Britain).
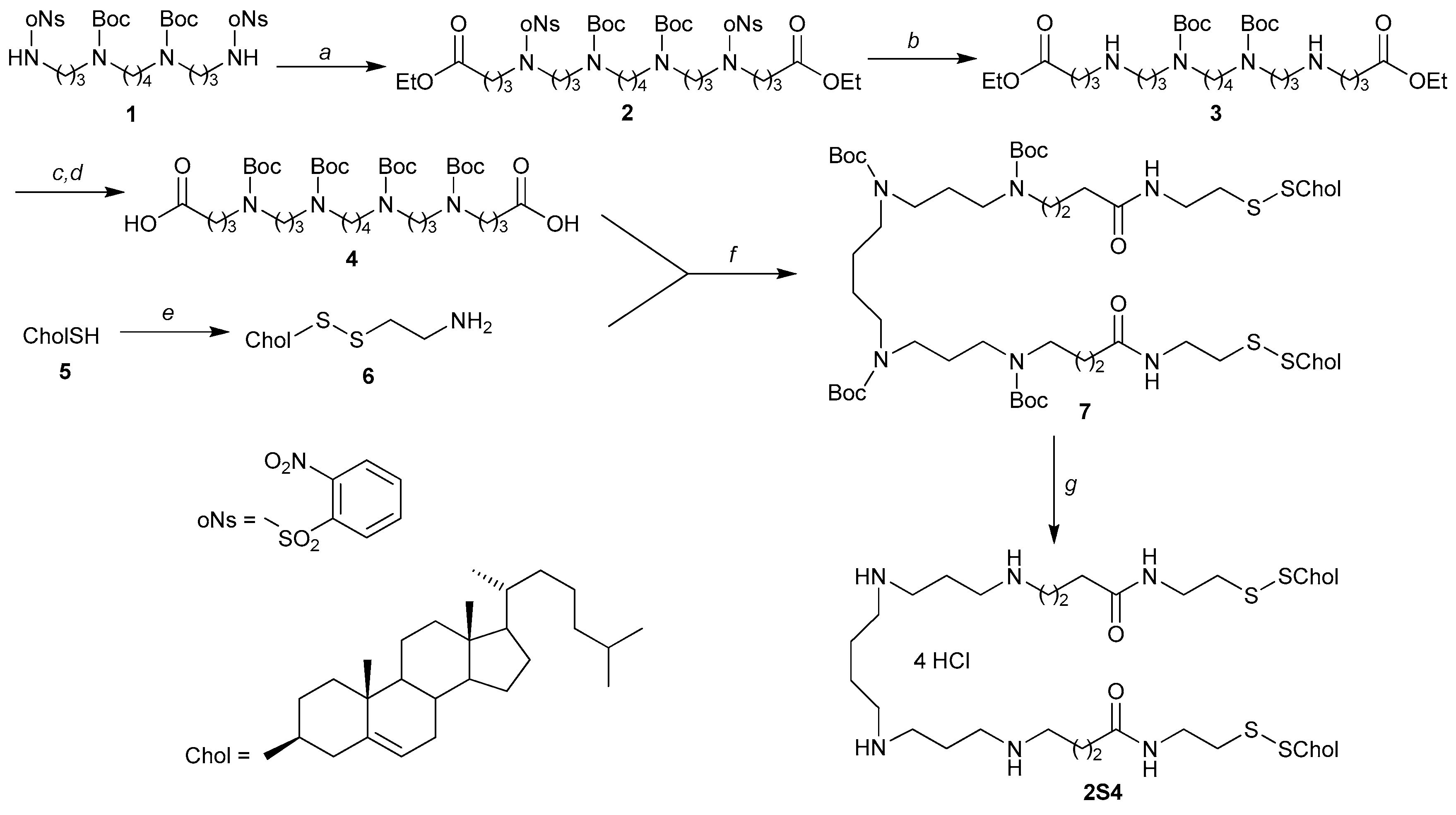
Diethyl N9,N14-Di(tert-butyloxycarbonyl)-N5,N18-bis(2-nitrobenzenesulfonyl)-5,8,14,18- tetraazadocosane-1,18-dioate (2). Cesium carbonate (0.84 g, 2.57 mmol) and ethyl 4-bromobutyrate (0.50 mL, 3.59 mmol) were added to a solution of compound 1 (0.80 g, 1.04 mmol) in anhydrous DMF (9 mL). The reaction mixture was stirred at 65 °С for 36 h, filtered on Celite® 545. The filtrate was evaporated to dryness, the residue was chromatographed on a silica gel column eluted with DCM-МеОН (120:1). The product 2 was obtained as a pale yellow oil (0.78 g, 76%). 1H-NMR (300 MHz): 1.18 (t, 6 Н, 2 CH2CH3, J = 7.1), 1.42 (br. s, 22 Н, 2 СМе3, NCH2(CH2)2CH2N), 1.76–1.81 (m, 4 Н, 2 NCH2CH2CH2N), 3.12–3.18 (m, 8 Н, 2 NCH2CH2CH2N, NCH2(CH2)2CH2N), 3.37–3.42 (m, 4 Н, 2 NCH2CH2CH2N), 4.09 (q, 4 Н, 2 CH2CH3, J = 7.1), 4.18 (s, 4 H, 2 CH2COO), 7.58–7.63 (m, 2 Н), 7.67–7.72 (m, 4 Н) and 8.04–8.07 (m, 2 Н, 2 С6Н4). 13С NMR (75 MHz): 14.33, 23.46, 27.48, 27.87, 28.58, 31.06, 31.25, 44.63, 45.48, 46.86, 60.66, 79.65, 124.31, 130.94, 131.81, 133.50, 133.65, 148.22, 155.54, 172.76. Anal. Calcd for C44H68N6O16S2: C, 52.79; H, 6.85; N, 8.39. Found: С, 52.55; Н, 6.68; N, 8.11.
Diethyl N9,N14-Di(tert-butyloxycarbonyl)-5,8,14,18-tetraazadocosane-1,22-dioate (3). Potassium carbonate K2CO3 (1.13 g, 8.14 mmol) was added to a solution of compound 2 (0.78 g, 0.79 mmol) in anhydrous DMF (10 mL), and the reaction mixture was stirred at 24 °С for 10 min. Then thiophenol (0.80 mL, 7.80 mmol) was added and the reaction mixture was additionally stirred at 24 °С for 2 h, filtered on Celite® 545. The filtrate was evaporated to dryness, the residue was chromatographed on a silica gel column eluted with DCM-МеОН-25% aq. ammonia (from 8:1:0.1 to 6:1:0.1). The product 3 was obtained as yellow oil (0.089 g, 18%). 1H-NMR (300 MHz): 1.24 (t, 6 H, J = 7.1, 2 CH3), 1.44 (br. s, 18 H, 2 Boc), 1.50–1.62 (m, 4 H, 2 CH2CH2CH2CH2), 2.07–2.26 (m, 8 H, 4 CH2CH2CH2), 2.46 (t, 4 H, J = 7.2, 2 CH2COOEt), 2.85–3.07 (m, 8 H, 4 CH2NH), 3.20–3.45 (m, 8 H, 4 CH2N), 4.10 (q, 4 H, J = 7.1, 2 CH2CH3). HRMS (ESI), m/z: 631.4641 [M + H]+. Calculated for C32H62N4O8: 631.4646 [M + H]+.
N5,N9,N14,N18-Tetra(tert-butyloxycarbonyl)-5,9,14,18-tetraazadocosane-1,22-dioic acid (4). A solution of compound 3 (0.089 g, 0.141 mmol) and anhydrous ТЕА (0.10 mL, 0.705 mmol) in anhydrous DCM (4 mL) was cooled to 0 °С. Вос2О (0.092 g, 0.423 mmol) in anhydrous DCM (0.50 mL) was added and the reaction mixture was stirred at 24 °С for 48 h, diluted with DCM (20 mL), washed with 3% aq. HCl (1 × 10 mL) and water (3 × 10 mL). The organic phase was dried over Na2SO4, filtered, and evaporated to dryness in vacuo. The residue was dissolved in МеОН (4 mL) and was added into a solution of NaOH (0.013 g, 0.33 mmol) in МеОН-Н2О (1.1 mL, 10:1 v/v). The reaction mixture was stirred at 24 °С for 56 h, then 0.5 M aq. HCl was added dropwise until рН 4, and the reaction mixture was evaporated to dryness in vacuo. The residue was chromatographed on a silica gel column eluted with DCM-МеОН-1% aq. АсОН (from 15:1:0.1 to 5:1:0.1). The product 4 was obtained as beige amorphous solid (0.025 g, 49%). 1H-NMR (CDCl3:CD3OD = 1:1, 300 MHz): 1.43 (br. s, 36 H, 4 Boc), 1.46–1.53 (m, 4, CH2CH2CH2CH2), 1.67–1.90 (m, 8 H, 4 CH2CH2CH2), 2.26 (t, 4 H, J = 7.2, 2 CH2COOH), 3.08–3.27 (m, 16 H, 8 CH2N). 13С NMR (75 MHz): 23.59, 25.66, 27.40, 27.93, 27.98, 29.36, 31.06, 44.88, 46.37, 46.82, 79.69, 79.83, 155.84, 175.34. MS (ESI), m/z: 774.08 [M]+, 797.49 [M + Na]+. Calculated for C38H70N4O12 774.50 [M]+, for C38H70N4NaO12 797.72 [M + Na]+.
2-[(Cholest-5-en-3β-yl)disulphanyl]ethanamine (6). Thiocholesterol (5) (0.18 g, 0.44 mmol) and anhydrous TEA (0.25 mL, 1.77 mmol) were added to a solution of cystamine dihydrochloride (0.050 g, 0.22 mmol) in DMF (10 mL) under argon atmosphere. The reaction mixture was further purged with argon for 5 min and stirred at 24 °С for 72 h, then evaporated to dryness in vacuo. The residue was chromatographed on a silica gel column eluted with DCM-МеОН-25% aq. ammonia (40:1:1). The product 8 was obtained as beige amorphous solid (0.092 g, 86%). 1H-NMR (300 MHz): 0.67 (s, 3 H, C(13)Me), 0.85 (d, 3 H, J = 6.6, C(25)Me), 0.87 (d, 3 H, J = 6.6, C(25)Me), 0.90 (d, 3 H, J = 6.2, C(20)Me), 1.00 (s, 3 H, C(10)Me), 1.03–1.67 (m, 21 H, Chol), 1.65–2.07 (m, 5 H, Chol), 2.17–2.40 (m, 2 H, H2C(4) Chol), 2.57–2.71 (m, 1 H, H(3) Chol), 2.75 (t, 2 H, J = 6.2, CH2S), 2.90–3.06 (m, 2 H, CH2NH2), 5.32–5.40 (m, 1 H, H(6) Chol). HRMS (ESI), m/z: 478.3535 [M + H]+. Calculated for C29H52NS2 478.3541 [M + H]+.
N8,N12,N17,N21-Tetra(tert-butyloxycarbonyl)-1,28-di[(cholest-5-en-3β-yl)disulphanyl]-4,25-dioxo-3,8,12,17,21,26-hexaazaoctacosane (7). Anhydrous DIEA (27 µL, 0.155 mmol) was added to a solution of compound 6 (0.037 g, 0.077 mmol) in anhydrous DCM (4 mL) and stirred for 10 min. Solutions of compound 4 (0.024 g, 0.031 mmol) in anhydrous DCM (3 mL) and EEDQ (0.026 g, 0.077 mmol) in anhydrous DCM (2 mL) were successively added to the stirring reaction mixture. After 48 h at 50 °С, the reaction mixture was cooled to 24 °С, diluted with DCM (30 mL), then washed with saturated aq. Na2СO3 (1 × 10 mL), water (1 × 10 mL), 0.2 M aq. HCl (1 × 10 mL), water (2 × 10 mL). The organic phase was dried over Na2SO4, filtered, and evaporated to dryness in vacuo. The residue was chromatographed on a silica gel column eluted with СHCl3-МеОН (from 80:1 to 60:1). The product 7 was obtained as pale yellow amorphous solid (0.037 g, 84%). 1H-NMR (300 MHz): 0.67 (s, 6 H, 2 C(13)Me), 0.84 (d, 6 H, J = 6.6, 2 C(25)Me), 0.86 (d, 3 H, J = 6.6, 2 C(25)Me), 0.89 (d, 6 H, J = 6.2, 2 C(20)Me), 1.00 (s, 6 H, 2 C(10)Me), 1.05–1.62 (m, 46 H, Chol, CH2CH2CH2CH2), 1.44 (br. s, 36 H, 4 Boc), 1.66–2.06 (m, 18 H, 4 CH2CH2CH2, Chol), 2.16 (t, 4 H, J = 7.2, 2 CH2CONH), 2.17–2.40 (m, 4 H, 2 H2C(4) Chol), 2.57–2.71 (m, 2 H, 2 H(3) Chol), 2.79 (t, 4 H, J = 6.2, 2 CH2S), 3.04–3.30 (m, 16 H, 8 NCH2), 3.46–3.62 (m, 4 H, 2 CH2NHCO), 5.31–5.39 (m, 2 H, 2 H(6) Chol). 13С NMR (125 MHz): 12.00, 18.86, 19.45, 21.09, 22.68, 22.93, 23.97, 24.41, 24.61, 26.02, 28.14, 28.34, 28.63, 29.16, 31.98, 32.02, 33.67, 35.91, 36.33, 36.90, 38.44, 39.08, 39.16, 39.66, 39.90, 42.46, 45.09, 46.18, 47.00, 50.23, 50.39, 53.53, 56.32, 56.90, 79.48, 79.80, 121.58, 141.50, 155.58, 172.69. HRMS (ESI), m/z: 1694.1792 [M + H]+, 847.5927 [M + 2H]2+. Calculated for C96H169N6O10S4: 1694.1783 [M + H]+, for C96H170N6O10S4: 847.5931 [M + 2H]2+.
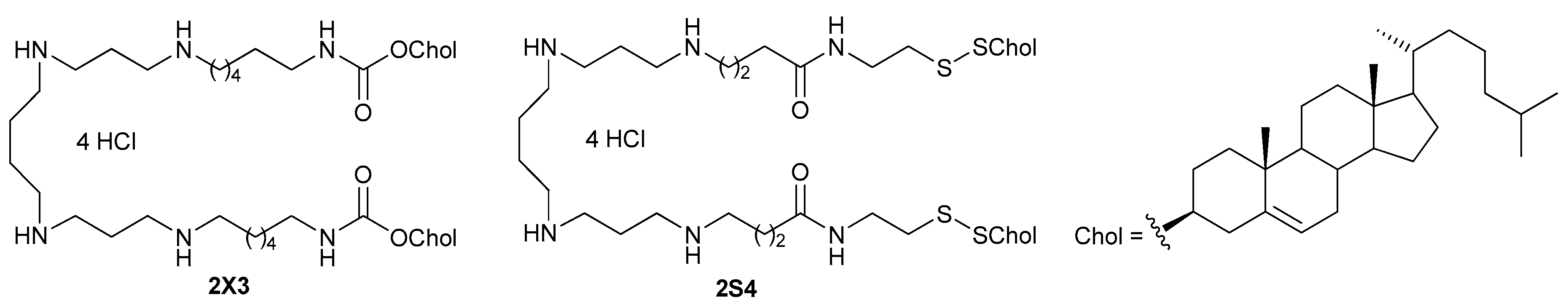
1,28-Di[(cholest-5-en-3β-yl)disulphanyl]-4,25-dioxo-3,8,12,17,21,26-hexaazaoctacosane tetrahydrochloride (2S4). A solution of 3 N HCl in anhydrous dioxane (6 mL) was added to a cooled (0 °C) solution of compound 7 (0.090 g, 0. 050 mmol) in anhydrous DCM (10 mL), and the reaction mixture was stirred at 24 °С for 24 h, then evaporated to dryness in vacuo. The residue was recrystallized successively from ethanol (5 mL) and diethyl ether (5 mL). The product 2S4 was obtained as white crystals (0.070 g, 91%), decompose without melting above 185 °С. 1H-NMR (CDCl3:CD3OD = 3:1, 500 MHz): 0.67 (s, 6 H, 2 C(13)Me), 0.84 (d, 6 H, J = 6.6, 2 C(25)Me), 0.85 (d, 3 H, J = 6.6, 2 C(25)Me), 0.90 (d, 6 H, J = 6.2, 2 C(20)Me), 0.99 (s, 6 H, 2 C(10)Me), 1.03–1.62 (m, 50 H, Chol, CH2CH2CH2CH2, 2 CH2CH2CH2), 1.66–2.06 (m, 14 H, 2 CH2CH2CH2CO, Chol), 2.12–2.17 (m, 4 H, 2 CH2CONH), 2.19–2.27 (m, 4 H, 2 CH2S), 2.30–2.38 (m, 4 H, 2 H2C(4) Chol), 2.55–2.63 (m, 2 H, 2 H(3) Chol), 3.30–3.38 (m, 16 H, 8 NCH2), 3.56–3.65 (m, 4 H, 2 CH2NHCO), 5.33–5.37 (m, 2 H, 2 H(6) Chol). HRMS (ESI), m/z: 1293.9682 [M + H]+, 647.4882 [M + 2H]2+. Calculated for C76H137N6O2S4: 1293.9686 [M + H]+, for C76H138N6O2S4: 647.4882 [M + 2H]2+.
Preparation of cationic liposomes (CLs). CLs were prepared by hydrating of thin lipid films. Briefly, polycationic amphiphile and lipid helper 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE, Avanti Polar Lipids, Alabaster, AL, USA) were dissolved at a molar ratio of 1:1 in a mixture of CHCl3 and CH3OH. Organic solvents were removed in vacuo. The lipid film obtained was dried for 4 h at 0.1 Torr to remove residual organic solvents. The dried lipid film was hydrated using deionized MilliQ water at 4 °C overnight, the resulting liposomal dispersion was sonicated for 15 min at 70–75 °C in a bath-type sonicator (Bandelin Sonorex Digitec DT 52H, Berlin, Germany), filtrated through a 0.45 μm pore polycarbonate membrane, flushed with argon and stored at 4 °C. The final polycationic amphiphile concentrations were 1 mM.
Preparation of CL/NA complexes. Prior to their use, the complexes of the CLs and NA were formed in a serum-free Opti-MEM medium (Invitrogen, Waltham, MA, USA) by vigorous mixing of nucleic acid (0.5 μg pEGFP-C2) and liposome suspensions taken at concentrations corresponding to the appropriate N/P (nitrogen to phosphate) ratio; the resulting mixtures were incubated for 20 min at 24 °С. 1 μg of DNA corresponds to 3.1 × 10−9 mol of phosphates.
Liposome and CL/NA complexes sizes and zeta potentials. The particle size and zeta potential were measured using a dynamic light scattering method by a Malvern Zetasizer Nano (Malvern Instruments Ltd., Malvern, UK) at 25 °C. For CL/NA complexes characterisation, 25 μL of nucleic acid solution prepared in MilliQ water was mixed with 25 μL of liposomes solution at N/P ratio 6/1. After incubation for 20 min at room temperature, 900 μL of water was added and the complexes were analysed using a 1-mL cuvette.
Cell lines and growth conditions. HEK 293 (human embryo kidney) cell lines were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) (Invitrogen, Waltham, MA, USA), 100 μg/mL penicillin, 100 μg/mL streptomycin, and 0.25 μg/mL amphotericin at 37 °C in a humidified atmosphere containing 5% CO2/95% air. The cells were plated in 24-well culture plates (at a density of 1.2 × 105 cells/well) and allowed to adhere overnight.
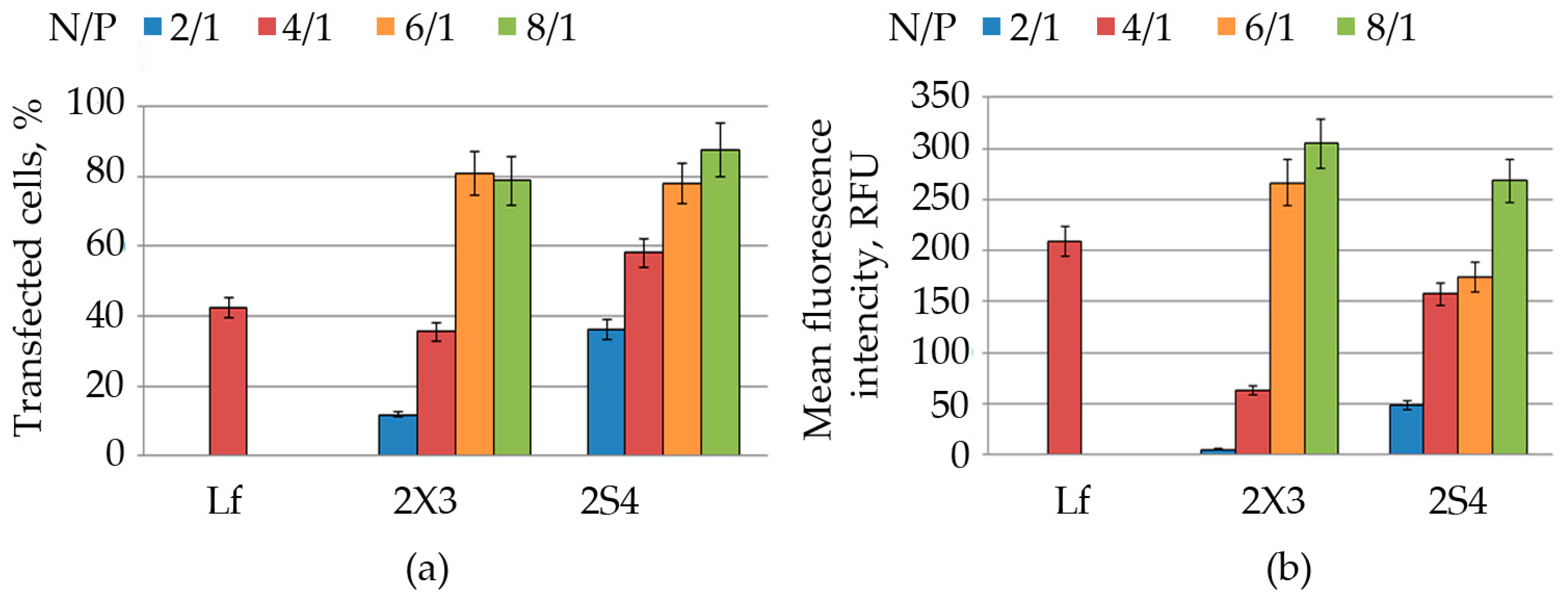
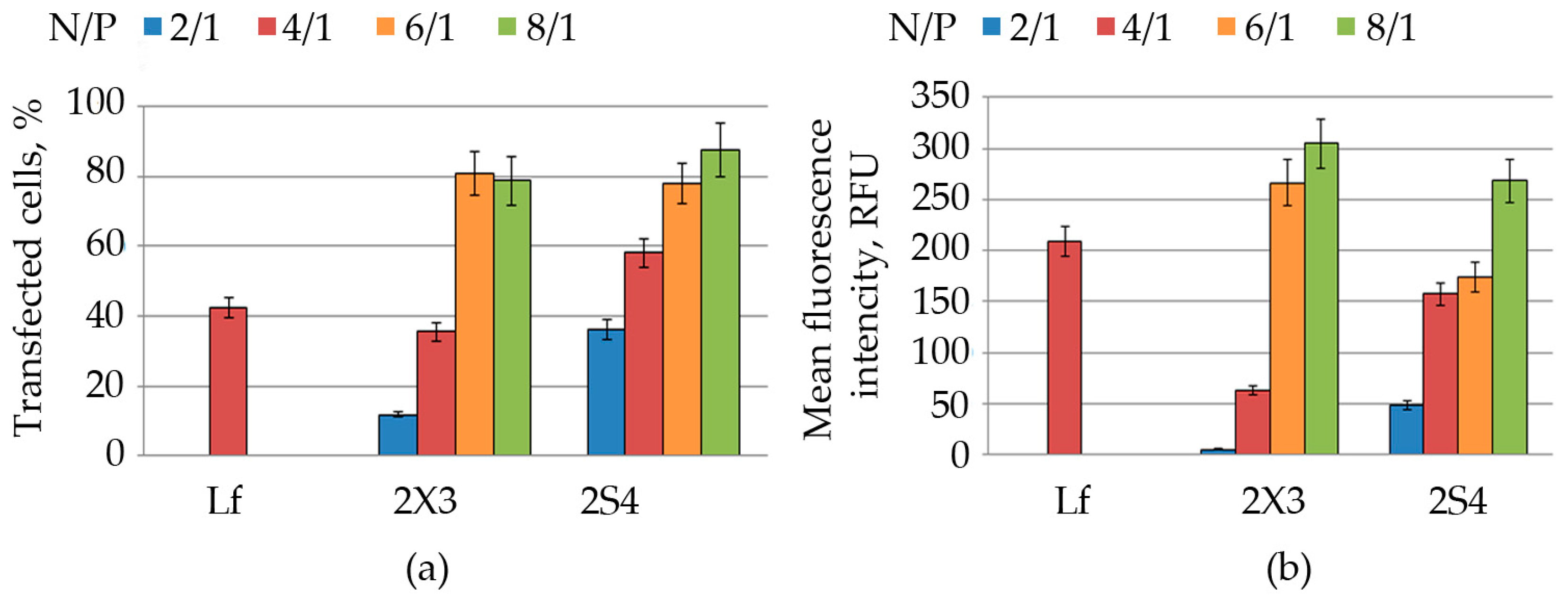
Cell transfection. HEK 293 (1.2 × 105 cells/well) cells were seeded in 24-well plates and grown as described above. On the day of the experiment, the culture medium of cells was replaced by 200 μL of fresh medium supplemented with 10% FBS. The CL/NA complexes at various N/P ratios (as described above) were added to the cells and incubated for 4 h. After the incubation, the cells were washed twice with PBS and then preserved in the DMEM medium (500 mL) with 10% FBS. The expression levels of EGFP were measured 48 h post transfection. All the experiments were performed in triplicate.
FACS analysis. Flow cytometry was used to characterize the transfection efficiency of cationic liposomes. Prior to analysis, cells were rinsed twice with PBS and detached from the plate by trypsin treatment (0.5 mg/mL in PBS) at 37 °C for 2 min. Trypsinized cells were then resuspended in the culture medium and collected by centrifugation (Contron T42K centrifuge, Centricon Instruments) at 1000 rpm for 10 min at 4 °C. The medium was removed, and the cells were washed with PBS and fixed with 4% formaldehyde in PBS. The resulting samples were assayed by flow cytometry using NovoCyte 3000 (Biosciences Inc., Allentown, PA, USA). A total of 2 × 104 cells were analyzed from each sample. All experimental points were prepared in triplicate for statistical analysis. The standard deviation did not exceed 7–9%.
Cell viability test. The relative amount of living cells after the incubation with liposomes or lipoplexes was determined by the MTT test. HEK 293 cells plated as described above in 96-well plates were incubated with cationic liposomes (final concentration in the well 1–80 μM) or lipoplexes (appropriate N/P ratio) for 4 h under serum-free conditions, then serum was added to each well and cells were additionally incubated for 20 h. After 24 h of incubation, a solution of MTT [3-(4,5-dimethylthiazolyl)-2,5-diphenyltetrasolium bromide] (Sigma, St. Louis, MO, USA) was added to a final concentration of 0.5 mg/mL, and cells were incubated for an additional 3 h. Then the culture medium was removed, formosan crystals were solubilized in DMSO, and the differences in absorbance at 570 and 620 nm were measured spectrophotometrically using Multiscan RC (Labsystems, Cergy-Pontoise, France). Results were expressed as mean values of measurements for three wells ± S.D.


{kind=link}
{kind=link}
{kind=link}