N-(4-Bromophenyl)methoxycarbothioamide


Research Centre for Crystalline Materials, School of Science and Technology, Sunway University, No. 5 Jalan Universiti, Bandar Sunway 47500, Selangor Darul Ehsan, Malaysia
*
Author to whom correspondence should be addressed.
Molbank 2018, 2018(3), M1012; https://doi.org/10.3390/M1012
Submission received: 26 July 2018
/
Revised: 13 August 2018
/
Accepted: 13 August 2018
/
Published: 17 August 2018
(This article belongs to the Section Structure Determination)
{kind=link}
{kind=link}
{kind=link}
Abstract
:The synthesis, spectroscopic and crystallographic characterisation of the title compound, O-methyl-N-4-bromophenyl thiocarbamate, MeOC(=S)N(H)PhBr-4 (1), are described. Spectroscopy confirmed the formation of the compound and the molecular structure was determined crystallographically. Two independent but chemically similar molecules comprise the asymmetric unit of 1. The C‒S and C‒N bond lengths confirm the presence of the thioamide tautomer. The thione-S and amide-N‒H atoms are syn, enabling the formation of amide-N‒H…S(thione) hydrogen bonds between the two independent molecules that generates a two-molecule aggregate via an eight-membered {…HNCS}2 synthon. The aggregates are connected into a three-dimensional architecture via weak intermolecular interactions, including Br…π(4-bromophenyl), S…π(4-bromophenyl), and weak Br…S halogen bonding contacts. The overall molecular conformation, thioamide tautomer, and the presence of amide-N‒H…S(thione) hydrogen bonding in the crystal conform with expectation for this class of compound.
1. Introduction
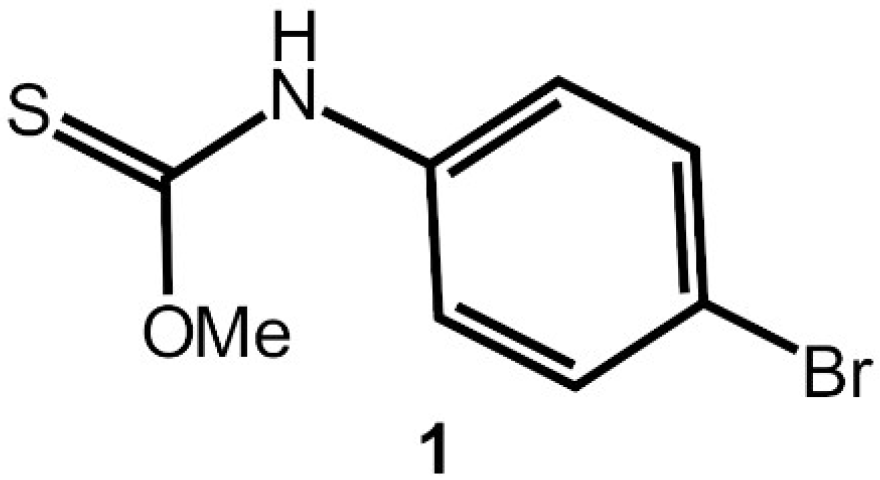
Interest in molecules that are related to the title thiocarbamate derivative, i.e., thione molecules of the general formula ROC(=S)N(H)R′ (for R, R′ = alkyl, aryl), is two-fold, relating to (i) the biological activity of their coinage metal derivatives and (ii) crystal engineering considerations. In the context of the former, anions that are derived from thiocarbamates complex phosphanegold(I) species to form molecules of the general formula R″3PAu[SC(OR)=NR′] (R, R′, R″ = alkyl, aryl), compounds that exhibit anti-cancer potential [1] and bactericidal properties [2]. Crystals of these species are often forthcoming enabling the evaluation of systematic variations of the R, R′, and R″ substituents upon the molecular packing and solid-state luminescence [3,4]. Indeed, the uncoordinated molecules are also of interest, as they, too exhibit varied supramolecular association depending on aryl-bound substituents [5,6], especially in circumstances when the nitrogen-bound groups carry pyridyl functionality [7]. The title compound, O-methyl (4-bromophenyl)carbamothioate (1) is a known compound, being first reported in 1935 [8,9,10] and a method for its synthesis has been patented [11]. Herein, the crystal and molecular structures of MeOC(=S)N(H)PhBr-4 (1), Scheme 1, along with spectroscopic (1H and 13C{1H} NMR, UV, and IR) characterisation, are described in continuation with the aforementioned systematic studies.
2. Results and Discussion
Compound 1 was prepared in an excellent yield (93%) by standard methods [5] and it was characterised by spectroscopy (see Supplementary Materials for original spectra). The NMR data showed the expected resonances and integration (1H). The most notable feature of the 13C{1H} spectrum was the resonance at δ 189.3, due to the quaternary carbon atom. Characteristic bands due to ν(N–H) [3229 (br)], ν(C–N) [1442 (s)], ν(C = S) [1205 (vs)], and ν(C–O) [1052 (s)] were noted in the IR spectrum. In the UV spectrum, a broad shoulder at 220 nm is assigned to a phenyl-bound n-π* transition, while the band at ca 280 nm is ascribed to a carbothioamide-centered π-π* transition [3]. The measured and calculated powder X-ray diffraction patterns for 1 (see Supplementary Materials). were in agreement, indicating that the results of the single crystal structure determination reported below are representative of the bulk material and that no phase change was apparent in the temperature range 100–298 K.
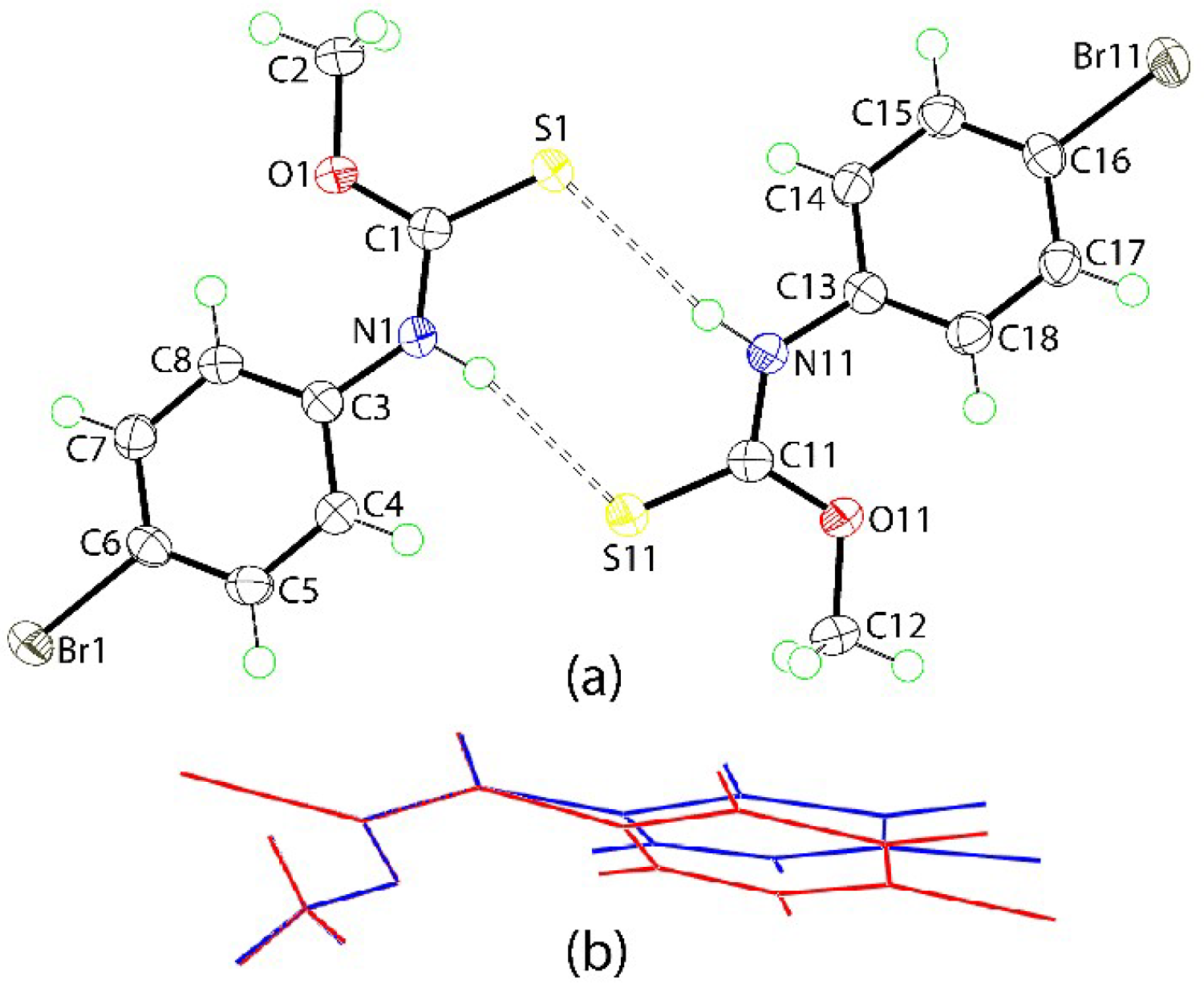
The molecular structures of the two independent molecules comprising the crystallographic asymmetric unit of 1 are shown in Figure 1a. The key geometric parameters for the two molecules are included in the figure caption and for the respective parameter, are equal to within experimental error. The magnitude of the C = S and C‒N bond lengths of ca 1.67 and 1.34–1.35 Å, respectively, as well as the presence of the thioamide-H atom confirm the thioamide assignment. The central CNOS chromophore is strictly planar with the r.m.s. deviation of the fitted atoms being 0.0008 Å and the maximum deviation from this plane is 0.0013(16) Å, for the C1 atom; the equivalent parameters for the S11-containing molecule are 0.0036 Å and 0.0022(6) Å (for the O11 atom). This planarity does not extend over the entire molecule with the dihedral angle between the CNOS and 4-bromo-C6 plane being 15.06(10)°; S11-molecule: 17.84(10)°. The angles about the C(quaternary) atom are significantly distorted from the ideal 120°, with the deviations being correlated with the presence of the C = S double-bond. Thus, for each molecule these angles span over 10° with the widest angle involving the S and O atoms, and the next widest involving the S and N atoms. The thione-S and amide-N‒H atoms are syn. The presence of a thione-S1 bond, the syn relationship between the thione-S and amide-N‒H atoms and trends in geometric parameters match those usually observed for molecules of this type [7]. An image showing the superimposition of the two independent molecules of 1 is shown in Figure 1b. From this, a close match is found for all but the 4-bromophenyl rings. The concordance between the molecules is exemplified in the values of the r.m.s. values of 0.0039 Å and 0.247° for bond lengths and angles, respectively [12].
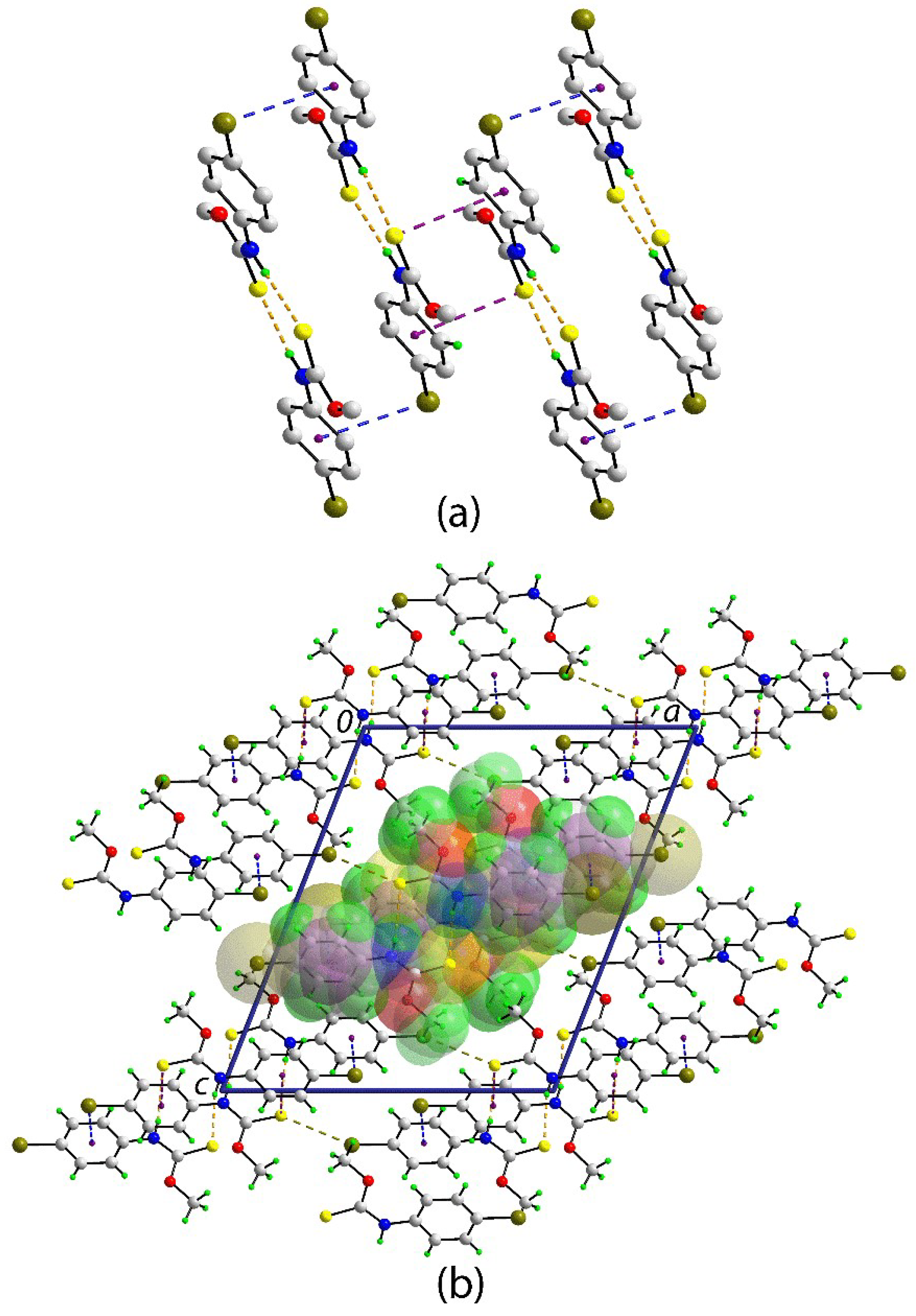
The molecular packing in the crystal of 1 features amide-N‒H…S(thione) hydrogen bonds between the two crystallographically independent molecules that generate an eight-membered {…HNCS}2 synthon and a two-molecule aggregate; the geometric parameters that characterise these hydrogen bonds are included in the caption to Figure 1. The aggregates are connected into a supramolecular chain along the b-axis via Br1…π(C13–C18) and S1…π(C3–C8) interactions, i.e., between unlike and like molecules, respectively, as illustrated in Figure 2a; geometric parameters for the specified intermolecular interactions are given in the figure caption. Globally, the chains pack into a layer parallel to (1 0 1), with the most prominent interactions between these being weak Br…S contacts. A view of the unit cell contents is shown in Figure 2b.
An interesting concept relating to the packing of molecules is the “isosteric principle”, which, stated simply, suggests that substituents with similar sizes will be isostructural/exhibit similar molecular packing, with the classic example being the chloro/methyl exchange [13]. Obviously, this principle will not hold if the substituent(s) concerned participate in significant, directional intermolecular interactions. There are several closely related crystal structures that are available for molecules of the general formula MeOC(=S)N(H)PhX-4, i.e., X = H [14], Me [15], and Cl [5]. Of these, the X = H compound adopts a distinctive molecular packing [15]. The X = Me compound crystallises in the triclinic space group P¯1 with Z′ = 2 [15], but is not isostructural with the X = Cl compound. The latter also crystallises with Z′ = 2 but in monoclinic space group P21/n and is isostructural with 1. There are differences in the molecular packing in the X = Br (1) and Cl crystals, however. While Br…π and S…π contacts are noted for 1, there are no such contacts within the van der Waals radii in the crystal of the X = Cl compound, intermolecular Cl…π and Cl…H interactions being formed instead [5].
In conclusion, the X-ray crystal structure determination reveals a structure that is consistent with spectroscopy and literature precedents, i.e., with a syn disposition of the thione-S and amide-H atoms in the molecular structure, and the presence of the amide-N‒H…S(thione) hydrogen bonds and an eight-membered {…HNCS}2 synthon in the molecular packing.
3. Materials and Methods
3.1. General Information
All of the chemicals and solvents were used as sourced without further purification. Except for 4-bromophenyl isothiocyanate, which was purchased from Sigma, all of the materials were purchased from Merck. Melting points were determined on a Biobase automatic melting point apparatus MP450. 1H and 13C{1H} NMR spectra were recorded in CDCl3 solution on a Bruker Ascend 400 MHz NMR spectrometer with chemical shifts relative to Me4Si; abbreviations for NMR assignments: s, singlet; d, doublet; br, broad. The UV spectrum was measured on a single-beam Shimadzu UV-3600 plus UV/VIS/NIR spectrophotometer in the range 200–800 nm for 1 taken up in an acetonitrile solution (2.5 × 10−6 M). IR spectra were measured on a Bruker Vertex 70v FTIR spectrophotometer from 4000 to 400 cm−1; abbreviations: br, broad; vs, very strong; s, strong. Elemental analyses were performed on a Leco TruSpec Micro CHN Elemental Analyser. Powder X-ray diffraction (XRD) was measured on a Rigaku Miniflex 600 X-ray diffractometer at 298 K while using Cu Kα (λ = 1.5418 Å) radiation in the 2θ range 5 to 40°. The comparison between experimental and calculated (from the CIF) PXRD patterns was performed with Rigaku′s PDXL2 software (https://www.rigaku.com/en/products/software/pdxl/overview).
3.2. Synthesis and Characterisation
In the present study, the title compound was prepared in the following manner. Sodium hydroxide (0.500 mmol, 0.0200 g) was dissolved in methanol (5 mL) and added to 4-bromophenyl isothiocyanate (0.500 mmol, 0.0107 g) in methanol (10 mL). The resulting mixture was stirred for 3 h at room temperature, followed by the addition of 5M hydrochloric acid dropwise, and then stirred for a further 1 h. The final product was extracted with chloroform (15 mL) and left for slow evaporation at room temperature, yielding colourless crystals suitable for X-ray crystallography after three weeks. Yield: 0.1144 g, 93%. M. pt: 97.2–98.5 °C cf. 99–100 °C [8], 101–102 °C [10] and 103–104 °C [11]. Anal. Calc′d for C8H8BrNOS: C, 39.04; H, 3.28; N, 5.69. Found: C, 39.14; H, 3.41; N, 5.47%. UV (acetonitrile, nm; ε in L cm−1 mol−1): 220 (8000), 279.5 (10000). FTIR (cm−1): 3229 (br) ν(N–H), 1442 (s) ν(C–N), 1205 (vs) ν(C = S), 1052 (s) ν(C–O). 1H NMR (CDCl3): 8.99 (s, br, 1H, NH), 7.44 (d, 2H, m-aryl-H, JHH = 8.6 Hz), 7.17 (s, br, 2H, o-aryl-H), 4.12 (s, 3H, CH3) ppm. 13C{1H} NMR (CDCl3): 189.3 (Cq), 136.1 (i-aryl-C), 132.1 (m-aryl-C), 123.4 (o-aryl-C), 118.6 (p-aryl-C), 58.9 (OCH3) ppm. FTIR (cm−1): 3229 (br) ν(N–H), 1442 (s) ν(C–N), 1205 (vs) ν(C = S), 1052 (s) ν(C–O).
3.3. Crystallography
Intensity data for 1 were measured at T = 100(2) K on a SuperNova Dual AtlasS2 diffractometer fitted with Cu Kα radiation so that θmax was 67.1°. Data reduction, including absorption correction, was accomplished with CrysAlis Pro [16]. Of the 20238 reflections measured, 3283 were unique (Rint = 0.024), and of these, 3195 data satisfied the I ≥ 2σ(I) criterion. The structure was solved by direct methods [17] and refined (anisotropic displacement parameters and C-bound H atoms in the riding model approximation) on F2 [18]. The positions of the nitrogen-bound hydrogen atoms were located from a difference map and refined with the distance constraint N‒H = 0.88±0.01 Å and with Uiso(H) = 1.2Ueq(N). A weighting scheme of the form w = 1/[σ2(Fo2) + (0.038P)2 + 1.155P] was introduced, where P = (Fo2 + 2Fc2)/3). Based on the refinement of 225 parameters, the final values of R and wR (all data) were 0.023 and 0.064, respectively. The molecular structure diagram was generated with ORTEP for Windows [19] and the packing diagram using DIAMOND [20].
Crystal data for C8H8BrNOS (1): M = 246.12, monoclinic, P21/n, a = 14.78070(10), b = 7.69460(10), c = 17.3719(2) Å, β = 111.4550(10)˚, V = 1838.82(4) Å3, Z = 4, Dx = 1.778 g cm-3, F(000) = 976, and μ = 7.819 mm−1. CCDC deposition number: 1857673.
Supplementary Materials
The following are available online. 1H and 13C{1H} NMR, UV and IR spectra, powder X-ray diffraction patterns (measured and simulated) and crystallographic data for 1 in Crystallographic Information File (CIF) format. CCDC 1857673 also contains the supplementary crystallographic data for this paper.
Author Contributions
C.I.Y. was the only experimentalist, who obtained and analyzed all data, apart from the X-ray crystallography, which was performed by E.R.T.T.
Funding
This research received no external funding. The APC was funded by Sunway University.
Acknowledgments
The X-ray crystallography laboratory at Sunway University is thanked for providing the X-ray intensity data.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ooi, K.K.; Yeo, C.I.; Mahandaran, T.; Ang, K.P.; Akim, A.M.; Cheah, Y.K.; Seng, H.L.; Tiekink, E.R.T. G2/M cell cycle arrest on HT-29 cancer cells and toxicity assessment of triphenylphosphanegold(I) carbonimidothioates, Ph3PAu[SC(OR)=NPh], R = Me, Et, and iPr, during zebrafish development. J. Inorg. Biochem. 2017, 166, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Yeo, C.I.; Sim, J.H.; Khoo, C.H.; Goh, Z.J.; Ang, K.P.; Cheah, Y.K.; Fairuz, Z.A.; Halim, S.N.B.A.; Ng, S.W.; Seng, H.L.; et al. Pathogenic Gram-positive bacteria are highly sensitive to triphenylphosphanegold(O-alkylthiocarbamates), Ph3PAu[SC(OR)=N(p-tolyl)] (R = Me, Et and iPr). Gold Bull. 2013, 46, 145–152. [Google Scholar] [CrossRef]
- Ho, S.Y.; Cheng, E.C.C.; Tiekink, E.R.T.; Yam, V.W.W. Luminescent phosphine gold(I) thiolates: Correlation between crystal structure and photoluminescent properties in [R3PAu{SC(OMe)=NC6H4NO2-4}] (R = Et, Cy, Ph) and [(Ph2P-R-PPh2){AuSC(OMe)=NC6H4NO2-4}2] (R = CH2, (CH2)2, (CH2)3, (CH2)4, Fc). Inorg. Chem. 2006, 45, 8165–8174. [Google Scholar] [CrossRef] [PubMed]
- Yeo, C.I.; Khoo, C.H.; Chu, W.C.; Chen, B.J.; Chu, P.L.; Sim, J.H.; Cheah, Y.K.; Ahmad, J.; Halim, S.N.A.; Seng, H.L.; et al. The importance of Au…π(aryl) interactions in the formation of spherical aggregates in binuclear phosphane gold(I) complexes of a bipodal thiocarbamate dianion: A combined crystallographic and computational study, and anti-microbial activity. RSC Adv. 2015, 52, 41401–41411. [Google Scholar] [CrossRef]
- Ho, S.Y.; Bettens, R.P.A.; Dakternieks, D.; Duthie, A.; Tiekink, E.R.T. Prevalence of the thioamide {...H‒N‒C=S}2 synthon—solid-state (X-ray crystallography), solution (NMR) and gas-phase (theoretical) structures of O-methyl-N-aryl-thiocarbamides. CrystEngComm 2005, 7, 682–689. [Google Scholar] [CrossRef]
- Kuan, F.S.; Mohr, F.; Tadbuppa, P.P.; Tiekink, E.R.T. Principles of crystal packing in O-isopropyl-N-aryl-thiocarbamides: iPrOC(=S)N(H)C6H4-4-Y: Y = H, Cl, and Me. CrystEngComm 2007, 9, 574–581. [Google Scholar] [CrossRef]
- Jotani, M.M.; Yeo, C.I.; Tiekink, E.R.T. A new monoclinic polymorph of N-(3-methylphenyl)ethoxycarbothioamide: Crystal structure and Hirshfeld surface analysis. Acta Crystallogr. E 2017, 73, 1889–1897. [Google Scholar] [CrossRef] [PubMed]
- Hunter, R.F.; Parken, E.R. 413. The unsaturation and tautomeric mobility of heterocyclic compounds. Part VI. The mobility of 5-substituted 1-hydroxybenzthiazoles, and the ultra-violet absorption of mobile and static derivatives of 1-hydroxybenzthiazole. J. Chem. Soc. 1935, 1755–1761. [Google Scholar] [CrossRef]
- Goeckeritz, D.; Pohloudek-Fabini, R. Halo and thiocyanato thiosemicarbazides and thiosemicarbazones and their behavior in paper chromatography. II. Pharmazie 1962, 17, 679–685. [Google Scholar]
- Goeckeritz, D.; Pohloudek-Fabini, R. Halo and thiocyanato thiourethans and their application to the determination of aliphatic alcohols by paper chromatography. Pharm. Zent. 1963, 102, 685–689. [Google Scholar]
- Bauman, R.A. Thioncarbamate esters. U.S. Patent US 3592835 A 19710713, 1971. [Google Scholar]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. D 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, M.R.; Jones, W.; Motherwell, W.D.S.; Shields, G.P. Crystal engineering and chloro-methyl interchange—A CSD analysis. Mol. Cryst. Liq. Cryst. 2001, 356, 337–353. [Google Scholar] [CrossRef]
- Ho, S.Y.; Lai, C.S.; Tiekink, E.R.T. O-Methyl N-phenylthiocarbamate. Acta Crystallogr. E 2003, 59, o1155–o1156. [Google Scholar] [CrossRef]
- Ho, S.Y.; Lai, C.S.; Tiekink, E.R.T. (E)-O-Methyl N-(4-methylphenyl)thiocarbamate. Acta Crystallogr. E 2007, 63, o1723–o1724. [Google Scholar] [CrossRef]
- Rigaku Oxford Diffraction. CrysAlis PRO; Agilent Technologies Inc.: Santa Clara, CA, USA, 2015. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Brandenburg, K. DIAMOND; Crystal Impact GbR: Bonn, Germany, 2006. [Google Scholar]
Sample Availability: Samples of the compound is not available from the authors. |
Scheme 1.
Chemical structure diagram for 1.

Figure 1.
(a) The molecular structures of the independent molecules of 1 showing atom labelling and displacement ellipsoids at the 70% probability level. Selected geometric parameters: C1 = S1 = 1.673(2) [1.665(2)], C1‒O1 = 1.327(2) [1.326(2)], C1‒N1 = 1.341(2) [1.346(3)] Å; S1‒C1‒O1 = 124.69(14) [124.52(15)], S1‒C1‒N1 = 122.22(15) [122.56(15)], and O1‒C1‒N1 = 113.09(17) [112.92(17)]°. The dashed lines indicate N–H...S hydrogen bonds between the independent molecules: N1–H1n…S11 = 2.528(18) Å, N1…S11 = 3.3891(17) Å with angle at H1n = 165(2)°; N11–H11n…S1 = 2.629(17) Å, N11…S1 = 3.4898(17) Å with angle at H11n = 167(2)°. (b) Superimposition of the S1- (red image) and inverted S11-containing (blue) molecules overlapped so the S, O, and N atoms are coincident.
Figure 1.
(a) The molecular structures of the independent molecules of 1 showing atom labelling and displacement ellipsoids at the 70% probability level. Selected geometric parameters: C1 = S1 = 1.673(2) [1.665(2)], C1‒O1 = 1.327(2) [1.326(2)], C1‒N1 = 1.341(2) [1.346(3)] Å; S1‒C1‒O1 = 124.69(14) [124.52(15)], S1‒C1‒N1 = 122.22(15) [122.56(15)], and O1‒C1‒N1 = 113.09(17) [112.92(17)]°. The dashed lines indicate N–H...S hydrogen bonds between the independent molecules: N1–H1n…S11 = 2.528(18) Å, N1…S11 = 3.3891(17) Å with angle at H1n = 165(2)°; N11–H11n…S1 = 2.629(17) Å, N11…S1 = 3.4898(17) Å with angle at H11n = 167(2)°. (b) Superimposition of the S1- (red image) and inverted S11-containing (blue) molecules overlapped so the S, O, and N atoms are coincident.

Figure 2.
The molecular packing in the crystal of 1: (a) a view of the supramolecular chain whereby two-molecule aggregates (sustained by N–H…S hydrogen bonds shown as orange dashed lines) are connected by Br…π(bromophenyl) and S…π(bromophenyl) interactions shown as blue and purple dashed lines, respectively. Non-participating hydrogen atoms have been omitted. (b) A view of the unit cell contents in projection down the b-axis. The Br…S contacts are indicated by dark-yellow dashed lines. One chain is indicated in space-filling mode. Selected geometric parameters of the specified intermolecular interactions: Br1…Cg(C13-C18)i = 3.6389(10) Å with angle at C6‒Br1 = 78.66(7)°. C1‒S1…Cg(C3-C8)ii = 3.6926(11) Å with angle at S1 = 79.70(8)°. Br11…S1iii = 3.6076(6) Å. Symmetry operations: (i) -x, 1-y, -z; (ii) -x, 2-y, -z; and, (iii) 1-x, 1-y, -z.
Figure 2.
The molecular packing in the crystal of 1: (a) a view of the supramolecular chain whereby two-molecule aggregates (sustained by N–H…S hydrogen bonds shown as orange dashed lines) are connected by Br…π(bromophenyl) and S…π(bromophenyl) interactions shown as blue and purple dashed lines, respectively. Non-participating hydrogen atoms have been omitted. (b) A view of the unit cell contents in projection down the b-axis. The Br…S contacts are indicated by dark-yellow dashed lines. One chain is indicated in space-filling mode. Selected geometric parameters of the specified intermolecular interactions: Br1…Cg(C13-C18)i = 3.6389(10) Å with angle at C6‒Br1 = 78.66(7)°. C1‒S1…Cg(C3-C8)ii = 3.6926(11) Å with angle at S1 = 79.70(8)°. Br11…S1iii = 3.6076(6) Å. Symmetry operations: (i) -x, 1-y, -z; (ii) -x, 2-y, -z; and, (iii) 1-x, 1-y, -z.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Yeo, C.I.; Tiekink, E.R.T. N-(4-Bromophenyl)methoxycarbothioamide. Molbank 2018, 2018, M1012. https://doi.org/10.3390/M1012
AMA Style
Yeo CI, Tiekink ERT. N-(4-Bromophenyl)methoxycarbothioamide. Molbank. 2018; 2018(3):M1012. https://doi.org/10.3390/M1012
Chicago/Turabian StyleYeo, Chien Ing, and Edward R.T. Tiekink. 2018. "N-(4-Bromophenyl)methoxycarbothioamide" Molbank 2018, no. 3: M1012. https://doi.org/10.3390/M1012
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.