

1-{(1S,2S,4R)-7,7-Dimethyl-1-[(pyrrolidin-1-yl)methyl]bicyclo [2.2.1]heptan-2-yl}-1H-benzo[d]imidazole


Faculty of Chemistry and Chemical Technology, University of Ljubljana, Večna pot 113, SI-1000 Ljubljana, Slovenia
*
Author to whom correspondence should be addressed.
Molbank 2023, 2023(1), M1538; https://doi.org/10.3390/M1538
Submission received: 8 December 2022
/
Revised: 19 December 2022
/
Accepted: 3 January 2023
/
Published: 6 January 2023
(This article belongs to the Topic Organocatalysis and Transition-Metal Catalysis: Key Trends in Synthetic Chemistry and Challenges)
{kind=link}
{kind=link}
Abstract
:A three-step synthesis of 1-{(1S,2S,4R)-7,7-dimethyl-1-[(pyrrolidin-1-yl)methyl]bicyclo[2.2.1]heptan-2-yl}-1H-benzo[d]imidazole, prepared from camphor derived diamine, is disclosed. The absolute configuration at the chiral center bearing benzo[d]imidazole moiety was confirmed by NOESY. The structure of a newly synthesized compound was confirmed by 1H- and 13C-NMR, 2D NMR, IR spectroscopy, and high resolution mass-spectrometry.
1. Introduction
In the last three decades, the field of asymmetric organocatalysis has experienced exponential growth, leading to numerous mechanistic studies and synthetic applications. The most attractive features of organocatalysis are easy-to-handle reaction conditions, environmentally friendly reagents without potentially toxic metal ions, and readily available tunable organocatalysts operating via different substrate activation modes. Nowadays, organocatalysis is an established methodology within asymmetric catalysis [1,2,3,4,5] and is increasingly used in the pharmaceutical and biotechnological industries [6,7].
Bifunctional hydrogen-bond donor organocatalysts are at the forefront of noncovalent organocatalysis. They allow simultaneous activation and coordination of both nucleophilic and electrophilic reactants. A typically used H-bond donor bifunctional organocatalyst is a derivative of a chiral 1,2-diamine scaffold such as cyclohexane-1,2-diamine or quinuclidine, which contains a tertiary amine functionality and a hydrogen-bond donor moiety. Thiourea and (thio)squaramide double hydrogen-bond donors are the most common and best [1,8,9,10,11]. Efficient bifunctional organocatalysts based on camphor-1,3-diamine were recently developed in our group [12,13]. Since the number of hydrogen bond donors used in organocatalysis is limited, the introduction of new hydrogen-bond donors would increase the number of available H-bond donor bifunctional organocatalysts and possibly broaden the substrate scope. In this context, we reported the synthesis of enaminone and benzenediamine hydrogen bond donors based on the chiral quinuclidine scaffold [14].
2. Results and Discussion
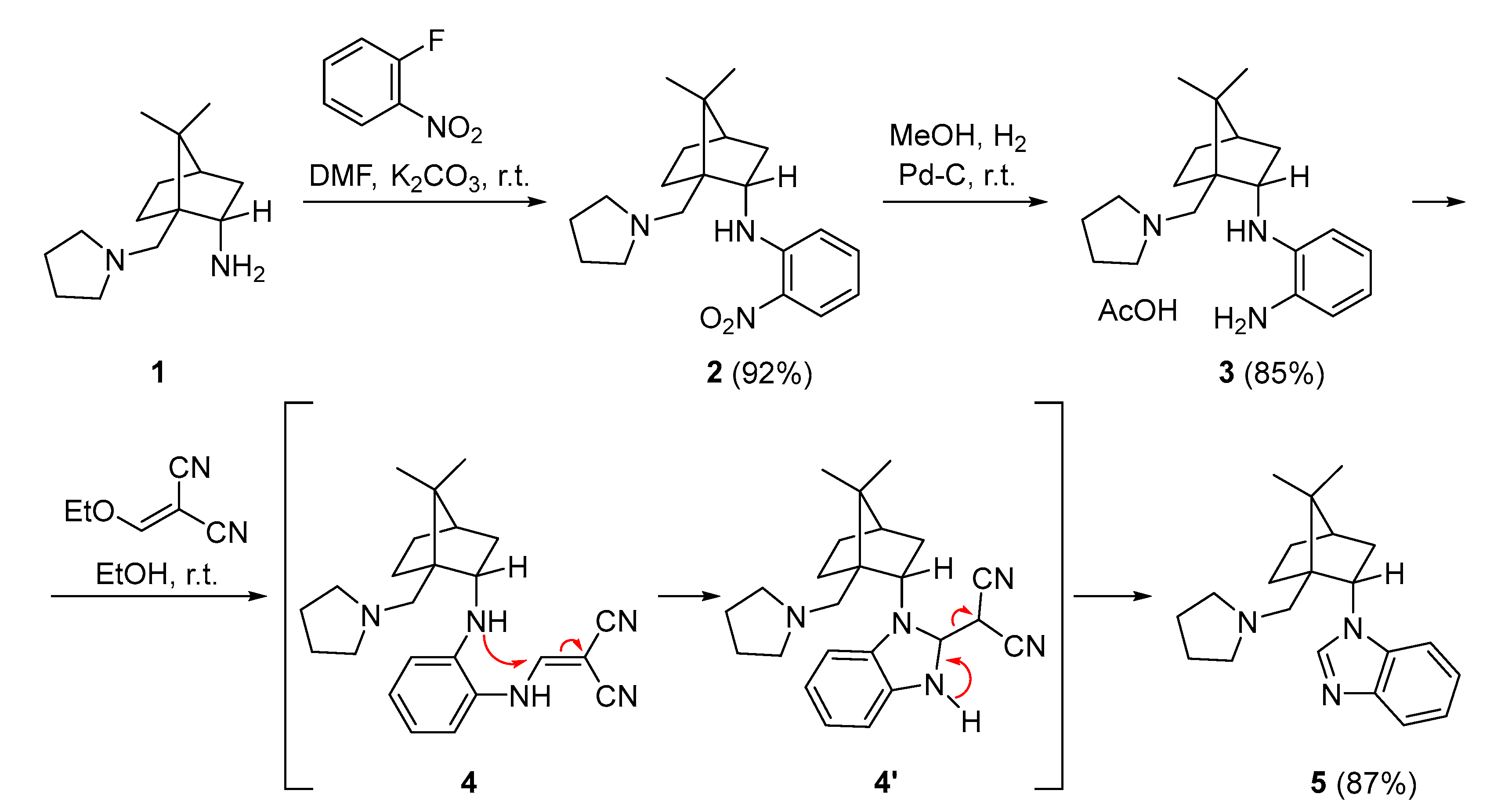
Recently, we reported the synthesis and catalytic activity of (S)-quininamine-based organocatalysts bearing enaminone and benzenediamine hydrogen bond donors [14]. In extension of this work, the developed three-step synthesis was used for the preparation of camphor-derived organocatalysts bearing benzenediamine hydrogen bond donor 4. Starting from diamine 1 [12], the nucleophilic aromatic substitution with 2-fluoronitrobenzene provided a nitroaniline derivative 2. The following catalytic hydrogenation of 2 furnished benzenediamine derivative 3, isolated as a salt solvates with acetic acid after column chromatography. Finally, treatment of the primary amino group of 3 with commercial 2-(ethoxymethylene)malononitrile was expected to provide the enaminone-benzenediamine derivative 4 with a Michael addition-elimination reaction. Instead, camphor-derived benzo[d]imidazole 5 was isolated in an 87% yield (Scheme 1). The formation of product 5 could be rationalized by the initial formation of the desired enamine 4, which in a 5-exo-trig cyclization gave imidazoline 4′. Elimination of the malononitrile provided the final aromatic benzo[d]imidazole 5. A closely similar reaction for the formation of benzo[d]imidazole systems, applying diethyl 2-(methoxymethylene)malonate, was reported by Pfizer in 2003 [16]. The benzimidazole core is usually prepared from ortho-phenylenediamine by cyclization with a carbonyl compound or its equivalent under various reaction conditions [17]. C1 reagents used include DMSO [18], aldehydes [19,20], orthoesters [21], formamides [22], carboxylic acids [23] and others [17].
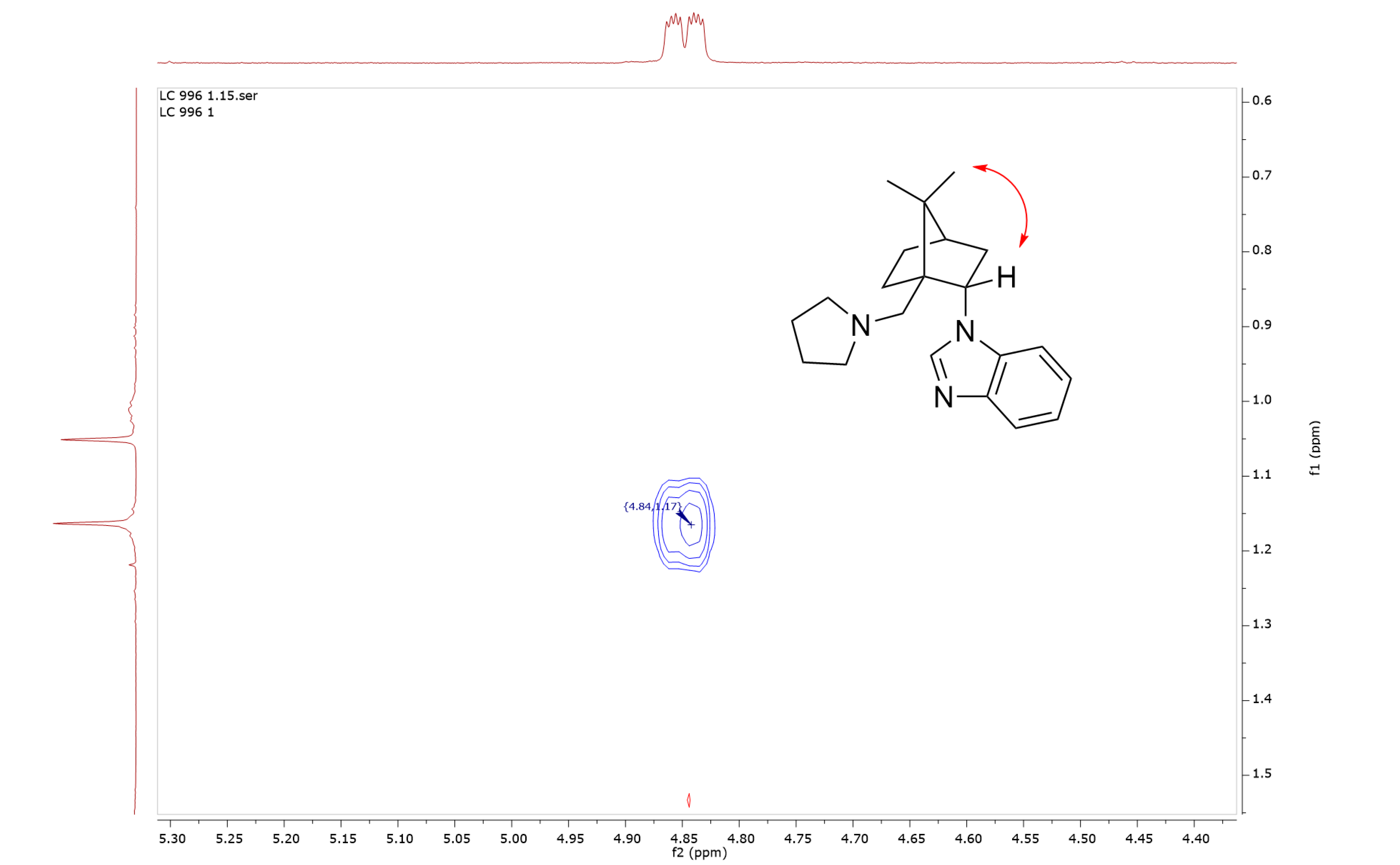
The structure of compound 5 was confirmed by spectroscopic methods (1H- and 13C-NMR, 2D-NMR, IR, and high-resolution mass spectrometry). The absolute configuration at the chiral center bearing benzo[d]imidazole moiety was confirmed by NOESY on the basis of the cross-peak between the methyl group and the exo-H(2) proton (Figure 1). The intermediate 2 was characterized by 1H- and 13C-NMR, 2D-NMR, IR, and melting point while the intermediate 3 was characterized by 1H- and 13C-NMR (In Supplementary Materials). Intermediates 4 and 4′ were not observed.
In conclusion, camphor-derived benzo[d]imidazole 5 was synthesized in three-steps from diamine 1. In view of the synthetic accessibility of benzo[d]imidazole 5, a small library can easily be prepared and studied further. In addition, alkylation of benzo[d]imidazole 5 could lead to camphor-based N-heterocyclic carbene precursors [15].
3. Materials and Methods
Solvents for extractions and chromatography were of technical grade and were distilled prior to use. Extracts were dried over technical grade anhydrous Na2SO4. Melting points were determined on a Kofler micro hot stage and on SRS OptiMelt MPA100—Automated Melting Point System (Stanford Research Systems, Sunnyvale, CA, USA). The NMR spectra were obtained on a Bruker UltraShield 500 plus (Bruker, Billerica, MA, USA) at 500 MHz for 1H and 126 MHz for 13C nucleus, using CDCl3 with TMS as the internal standard, as solvents. Mass spectra were recorded on an Agilent 6224 Accurate Mass TOF LC/MS (Agilent Technologies, Santa Clara, CA, USA), IR spectra on a Perkin-Elmer Spectrum BX FTIR spectrophotometer (PerkinElmer, Waltham, MA, USA). Column chromatography (CC) was performed on silica gel (Silica gel 60, particle size: 0.035–0.070 mm (Sigma-Aldrich, St. Louis, MO, USA)). All the commercially available chemicals used were purchased from Sigma-Aldrich (St. Louis, MO, USA). Catalytic hydrogenation was performed on a Parr Pressure Reaction Hydrogenation Apparatus (Moline, IL, USA). The optical rotation of optical active substances was measured on a Perkin Elmer 241 MC Polarimeter (PerkinElmer, Waltham, MA, USA) equipped with a Na lamp (sodium emission lines at 589.0 nm) at 20 °C.
(1S,2S,4R)-7,7-Dimethyl-1-[(pyrrolidin-1-yl)methyl]bicyclo[2.2.1]heptan-2-amine (1) [12] was prepared following the literature procedure.
3.1. Synthesis of (1S,2S,4R)-7,7-Dimethyl-N-(2-nitrophenyl)-1-[(pyrrolidin-1-yl)methyl]bicyclo[2.2.1]heptan-2-amine (2)
A mixture of (1S,2S,4R)-7,7-dimethyl-1-[(pyrrolidin-1-yl)methyl]bicyclo[2.2.1]heptan-2-amine (1) (1.75 mmol, 343 mg), 1-fluoro-2-nitrobenzene (1.75 mmol, 0.185 mL), K2CO3 (1.75 mmol, 0.242 g), and DMF (5 mL) was stirred at 25 °C for 24 h. Volatile components were evaporated in vacuo. The residue was purified by column chromatography (EtOAc). Fractions containing the product 2 were combined and volatile components evaporated in vacuo. Yield: 553 mg (1.61 mmol, 92%) of yellow solid; mp = 83.5–85.3 °C. 1H-NMR (500 MHz, CDCl3): δ 0.95 (s, 3H); 1.02 (s, 3H); 1.01–1.06 (m, 1H); 1.24 (ddd, J = 4.4, 9.5, 12.3 Hz, 1H); 1.58–1.68 (m, 3H); 1.68–1.84 (m, 4H); 2.23 (ddd, J = 4.1, 9.5, 13.5 Hz, 1H); 2.38–2.49 (m, 4H); 2.69 (q, J = 7.3 Hz, 2H); 2.82 (d, J = 13.4 Hz, 1H); 3.81 (d, J = 10.1 Hz, 1H); 6.57 (ddd, J = 1.3, 6.9, 8.4 Hz, 1H); 6.71 (dd, J = 1.3, 8.9 Hz, 1H); 7.35 (ddd, J = 1.7, 6.9, 8.7 Hz, 1H); 8.14 (dd, J = 1.7, 8.7 Hz, 1H); 9.01 (s, 1H). 13C-NMR (126 MHz, CDCl3): δ 19.40, 20.23, 24.08, 26.66, 28.14, 38.28, 45.28, 48.43, 52.25, 56.73, 58.33, 58.48, 114.60, 115.41, 126.94, 132.46, 135.61, 145.90.
3.2. Synthesis of N1-{(1S,2S,4R)-7,7-Dimethyl-1-[(pyrrolidin-1-yl)methyl]bicyclo[2.2.1]heptan-2-yl}benzene-1,2-diamine (3)
A mixture of (1S,2S,4R)-7,7-dimethyl-N-(2-nitrophenyl)-1-[(pyrrolidin-1-yl)methyl]bicyclo-[2.2.1]heptan-2-amine (2) (0.83 mmol, 285 mg), Pd-C (ω = 10%, 20 mg), and MeOH (5 mL) was shaken in a Paar shaker hydrogenation apparatus in H2 atmosphere (3 bar) at 25 °C for 6 h. The reaction mixture was filtrated to remove Pd-C, volatile components were evaporated in vacuo. The residue was purified by column chromatography (Silica Gel 60; EtOAc/MeOH/AcOH = 4:1:0.1). Fractions containing the product 3 were combined and volatile components evaporated in vacuo. Yield: 262 mg (0.70 mmol, 85%, acetic acid to amine 3 in a 1:1 ratio) of colorless oil. νmax 3378, 2954, 2877, 2791, 1616, 1571, 1507, 1441, 1416, 1389, 1354, 1323, 1254, 1231, 1156, 1069, 1037, 910, 868, 777, 740, 695, 670 cm−1. 1H-NMR (500 MHz, CDCl3): δ 0.95 (s, 3H); 0.98–1.03 (m, 1H); 1.02 (s, 3H); 1.33 (ddd, J = 4.6, 9.5, 12.4 Hz, 1H); 1.62–1.69 (m, 1H); 1.71 (t, J = 4.6 Hz, 1H); 1.79–1.88 (m, 5H); 1.99 (s, 3H); 2.41–2.57 (m, 2H); 2.88 (d, J = 2.5 Hz, 2H); 2.94 (br s, 2H); 3.12 (br s, 2H); 3.71 (ddd, J = 1.8, 3.9, 9.6 Hz, 1H); 6.18 (br s, 4H); 6.49 (dd, J = 1.3, 7.6 Hz, 1H); 6.62–6.67 (m, 2H); 6.71 (ddd, J = 2.7, 6.2, 7.7 Hz, 1H). 13C-NMR (126 MHz, CDCl3): δ 19.33, 20.16, 22.93, 23.76, 26.34, 28.27, 38.34, 45.03, 49.77, 51.13, 56.93, 57.01, 60.65, 112.16, 115.23, 118.59, 119.04, 136.11, 136.25, 176.69.
3.3. Synthesis of 1-{(1S,2S,4R)-7,7-Dimethyl-1-[(pyrrolidin-1-yl)methyl]bicyclo[2.2.1]heptan-2-yl}-1H-benzo[d]imidazole (5)
A mixture of N1-{(1S,2S,4R)-7,7-dimethyl-1-[(pyrrolidin-1-yl)methyl]bicyclo[2.2.1]heptan-2-yl}benzene-1,2-diamine (3) (157 mg, 0.42 mmol; acetic acid to amine 3 in a 1:1 ratio) and 2-(ethoxymethylene)malononitrile (61 mg, 0.50 mmol) in dichloromethane (2 mL) was stirred at 25 °C for 24 h. Volatile components were evaporated in vacuo. The residue was purified by column chromatography (Silica Gel 60; EtOAc/petroleum ether = 3:1). Fractions containing the product 5 were combined and volatile components evaporated in vacuo. Yield: 118 mg (0.365 mmol, 87%) of colorless semisolid. [α]Dr.t. = −55.9 (0.16, MeOH). EI-HRMS: m/z = 324.2429 (MH+); C21H29N3 requires: m/z = 324.2434 (MH+); νmax 2958, 2781, 2192, 1613, 1562, 1482, 1456, 1420, 1390, 1370, 1351, 1329, 1284, 1225, 1196, 1111, 1073, 1009, 907, 888, 797, 783, 766, 737, 643 cm−1. 1H-NMR (500 MHz, CDCl3): δ 0.98–1.10 (m, 2H); 1.06 (s, 3H); 1.17 (s, 3H); 1.17–1.26 (m, 2H); 1.56–1.66 (m, 2H); 1.73–1.83 (m, 1H); 1.86 (t, J = 4.5 Hz, 1H); 1.89–2.01 (m, 3H); 2.03–2.09 (m, 1H); 2.14–2.24 (m, 2H); 2.36 (d, J = 13.1 Hz, 1H); 2.59–2.69 (m, 1H); 2.78 (d, J = 13.2 Hz, 1H); 4.86 (ddd, J = 2.4, 5.0, 11.9 Hz, 1H); 7.19–7.25 (m, 2H); 7.39–7.44 (m, 1H); 7.71–7.77 (m, 1H); 8.17 (s, 1H). 13C-NMR (126 MHz, CDCl3): δ 19.41, 20.79, 23.50, 27.00, 28.52, 37.42, 45.03, 50.85, 54.00, 55.64, 57.07, 59.81, 111.64, 119.62, 121.82, 121.96, 135.87, 142.22, 142.72.
Supplementary Materials
Synthesis and characterization data; Copies of 1H- and 13C-NMR spectra; copies of 2D spectra; Copies of HRMS reports; IR spectra.
Author Contributions
Conceptualization, L.C., U.G., J.S. and B.Š.; methodology, L.C. and U.G.; software, L.C., U.G., J.S. and B.Š.; validation, L.C., U.G., J.S., F.P. and B.Š.; formal analysis, U.G. and L.C.; investigation, L.C. and U.G.; resources, L.C., U.G. and J.S.; data curation, L.C., U.G., J.S. and B.Š.; writing—original draft preparation, L.C., U.G., J.S. and B.Š.; writing—review and editing, L.C., U.G., J.S., F.P. and B.Š.; visualization, L.C., U.G., B.Š. and J.S.; supervision, U.G.; project administration, U.G. and J.S.; funding acquisition, U.G. and J.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Slovenian Research Agency through grant P1-0179.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We thank the EN-FIST Centre of Excellence, Dunajska 156, 1000 Ljubljana, Slovenia, for the use of their BX FTIR spectrophotometer and Agilent 1260 Infinity LC for the HPLC analyses.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Torres, R.R. (Ed.) Stereoselective Organocatalysis: Bond Formation Methodologies and Activation Modes, 1st ed.; JohnWiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- List, B. Asymmetric Organocatalysis 1. Lewis Base and Acid Catalysis. In Science of Synthesis; Georg Thieme Verlag KG: Stuttgart, Germany, 2012. [Google Scholar]
- Dalko, P.I. Comprehensive Enantioselective Organocatalysis: Catalysts, Reactions, and Applications; Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar]
- Pellissier, H. Asymmetric organocatalysis. Tetrahedron 2007, 63, 9267–9331. [Google Scholar] [CrossRef]
- List, B.; MacMillan, D.W.C. For the Development of Asymmetric Organocatalysis. Available online: https://www.nobelprize.org/prizes/chemistry/2021/advanced-information/ (accessed on 30 November 2022).
- De Figueiredo, R.M.; Christmann, M. Organocatalytic Synthesis of Drugs and Bioactive Natural Products. Eur. J. Org. Chem. 2007, 2007, 2575–2600. [Google Scholar] [CrossRef]
- Sun, B.-F. Total synthesis of natural and pharmaceutical products powered by organocatalytic reactions. Tetrahedron Lett. 2015, 56, 2133–2140. [Google Scholar] [CrossRef] [Green Version]
- Okino, T.; Hoashi, Y.; Takemoto, Y. Enantioselective Michael Reaction of Malonates to Nitroolefins Catalyzed by Bifunctional Organocatalysts. J. Am. Chem. Soc. 2003, 125, 12672–12673. [Google Scholar] [CrossRef] [PubMed]
- Malerich, J.P.; Hagihara, K.; Rawal, V.H. Chiral Squaramide Derivatives are Excellent Hydrogen Bond Donor Catalysts. J. Am. Chem. Soc. 2008, 130, 14416–14417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rombola, M.; Sumaria, C.S.; Montgomery, T.D.; Rawal, V.H. Development of Chiral, Bifunctional Thiosquaramides: Enantioselective Michael Additions of Barbituric Acids to Nitroalkenes. J. Am. Chem. Soc. 2017, 139, 5297–5300. [Google Scholar] [CrossRef]
- Alemán, J.; Parra, A.; Jiang, H.; Jørgensen, K.A. Squaramides: Bridging from Molecular Recognition to Bifunctional Organocatalysis. Chem. Eur. J. 2011, 17, 6890–6899. [Google Scholar] [CrossRef] [PubMed]
- Ričko, S.; Svete, J.; Štefane, B.; Perdih, A.; Golobič, A.; Meden, A.; Grošelj, U. 1,3-Diamine-Derived Bifunctional Organocatalyst Prepared from Camphor. Adv. Synth. Catal. 2016, 358, 3786–3796. [Google Scholar] [CrossRef]
- Ričko, S.; Meden, A.; Ivančič, A.; Perdih, S.; Štefane, B.; Svete, J.; Grošelj, U. Organocatalyzed Deracemization of Δ2-Pyrrolin-4-ones. Adv. Synth. Catal. 2017, 359, 2288–2296. [Google Scholar] [CrossRef]
- Ciber, L.; Požgan, F.; Brodnik, H.; Štefane, B.; Svete, J.; Grošelj, U. Synthesis and Catalytic Activity of Organocatalysts Based on Enaminone and Benzenediamine Hydrogen Bond Donors. Catalysts 2022, 12, 1132. [Google Scholar] [CrossRef]
- Grošelj, U. Camphor-Derivatives in Asymmetric Organocatalysis—Synthesis and Application. Curr. Org. Chem. 2015, 19, 2048–2074. [Google Scholar] [CrossRef]
- Singer, R.A.; Ginsburg, P.H. Process for The Preparation Of 1,3-Substituted Indenes and Aryl-Fusedazapolycyclic Componunds. U.S. Patent 2003/0060624 A1, 27 March 2003. [Google Scholar]
- Faheem, M.; Rathaur, A.; Pandey, A.; Kumar Singh, V.; Tiwari, A.K. A Review on the Modern Synthetic Approach of Benzimidazole Candidate. ChemistrySelect 2020, 5, 3981–3994. [Google Scholar] [CrossRef]
- Zhu, X.; Zhang, F.; Kuang, D.; Deng, G.; Yang, Y.; Yu, J.; Liang, Y. K2S as Sulfur Source and DMSO as Carbon Source for the Synthesis of 2-Unsubstituted Benzothiazoles. Org. Lett. 2020, 22, 3789–3793. [Google Scholar] [CrossRef] [PubMed]
- Ryabukhin, S.V.; Plaskon, A.S.; Volochnyuk, D.M.; Tolmachev, A.A. Synthesis of Fused Imidazoles and Benzothiazoles from (Hetero)Aromatic ortho-Diamines or ortho-Aminothiophenol and Aldehydes Promoted by Chlorotrimethylsilane. Synthesis 2006, 21, 3715–3726. [Google Scholar] [CrossRef]
- Bahrami, K.; Khodaei, M.M.; Naali, F. Mild and Highly Efficient Method for the Synthesis of 2-Arylbenzimidazoles and 2-Arylbenzothiazoles. J. Org. Chem. 2008, 73, 6835–6837. [Google Scholar] [CrossRef] [PubMed]
- Bastug, G.; Eviolitte, C.; Markó, I.E. Functionalized Orthoesters as Powerful Building Blocks for the Efficient Preparation of Heteroaromatic Bicycles. Org. Lett. 2012, 14, 3502–3505. [Google Scholar] [CrossRef] [PubMed]
- Nale, D.B.; Bhanage, B.M. N-Substituted Formamides as C1-Sources for the Synthesis of Benzimidazole and Benzothiazole Derivatives by Using Zinc Catalysts. Synlett 2015, 26, 2835–2842. [Google Scholar] [CrossRef]
- Wang, Y.; Sarris, K.; Sauer, D.R.; Djuric, S.W. A simple and efficient one step synthesis of benzoxazoles and benzimidazoles from carboxylic acids. Tetrahedron Lett. 2006, 47, 4823–4826. [Google Scholar] [CrossRef]
Scheme 1.
Synthesis of camphor derived benzo[d]imidazole 5.

Figure 1.
Section of the NOESY spectra of camphor derived benzo[d]imidazole 5 displaying the cross-peak between the methyl group and the exo-H(2) proton.
Figure 1.
Section of the NOESY spectra of camphor derived benzo[d]imidazole 5 displaying the cross-peak between the methyl group and the exo-H(2) proton.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ciber, L.; Požgan, F.; Svete, J.; Štefane, B.; Grošelj, U. 1-{(1S,2S,4R)-7,7-Dimethyl-1-[(pyrrolidin-1-yl)methyl]bicyclo [2.2.1]heptan-2-yl}-1H-benzo[d]imidazole. Molbank 2023, 2023, M1538. https://doi.org/10.3390/M1538
AMA Style
Ciber L, Požgan F, Svete J, Štefane B, Grošelj U. 1-{(1S,2S,4R)-7,7-Dimethyl-1-[(pyrrolidin-1-yl)methyl]bicyclo [2.2.1]heptan-2-yl}-1H-benzo[d]imidazole. Molbank. 2023; 2023(1):M1538. https://doi.org/10.3390/M1538
Chicago/Turabian StyleCiber, Luka, Franc Požgan, Jurij Svete, Bogdan Štefane, and Uroš Grošelj. 2023. "1-{(1S,2S,4R)-7,7-Dimethyl-1-[(pyrrolidin-1-yl)methyl]bicyclo [2.2.1]heptan-2-yl}-1H-benzo[d]imidazole" Molbank 2023, no. 1: M1538. https://doi.org/10.3390/M1538
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.