
Synthesis of Norabietyl and Nordehydroabietyl Imidazolidine-2,4,5-Triones and Their Activity against Tyrosyl-DNA Phosphodiesterase 1
 , , ,
, , ,  and
and
Abstract
:1. Introduction
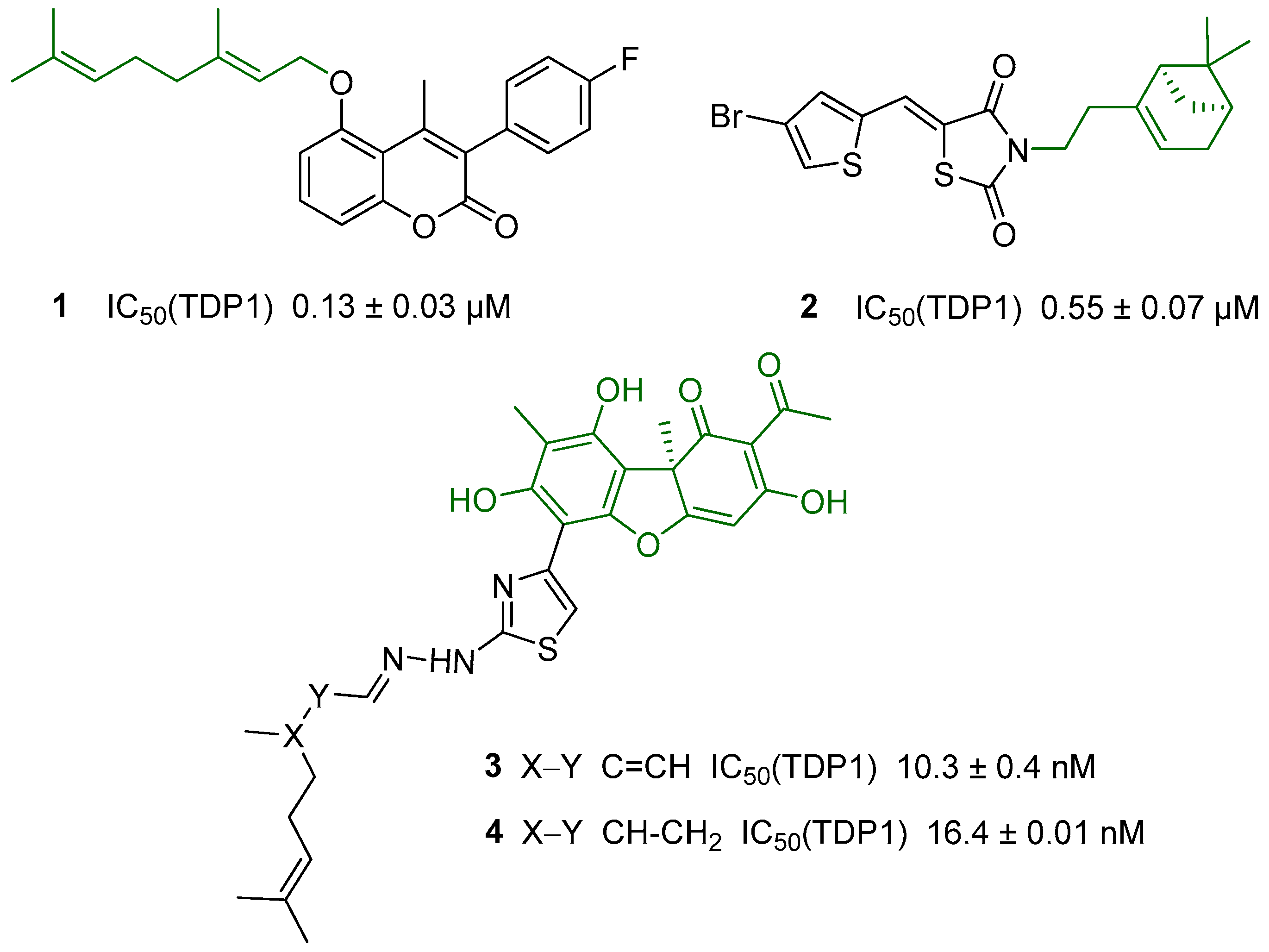
2. Results
3. Materials and Methods
- 1-(1-Adamantyl)-3-[(1R,4aR,10aR)-7-isopropyl-1,4a-dimethyl-1,2,3,4,4a,4b,5,6,10,10a-decahydrophenanthren-1-yl]imidazolidine-2,4,5-trione (8a) Yield 62%, white powder. M.p. 144.6 °C. 1H NMR (400 MHz, CDCl3, δ, ppm, J/Hz): 5.74 (1H, s, H-14), 5.35 (1H, s, H-7), 0.98 and 0.97 (3H both, d, J16, 15 = 6.8, Me-16 and Me-17), 0.81 (3H, s, Me-18), 1.75 (3H, s, Me-19), 2.76–3.01 (1H, m, H-15), 2.28–2.50 (8H, m, 2H-24, 2H-25, 2H-26, H-5, H-6), 1.91–2.27 (7H, m, H-27, H-28, H-29, 2H-12, H-6, H-9), 1.50–1.87 (12H, m, 2H-30, 2H-31, 2H-32, 2H-2, H-11, H-1, 2H-3), 1.11–1.36 (2H, m, H-1, H-11). 13C NMR (100 MHz, CDCl3, δ, ppm): 157.43, 156.23 and 154.60 (C-20, C-21 and C-22), 145.63 (C-13), 135.53 (C-8), 122.10 (C-14), 119.80 (C-7), 29.59 (C-27, C-28, C-29), 35.81 (C-30, C-31, C-32), 39.92 (C-24, C-25, C-26), 20.83 and 20.70 (Me-17 and Me-16), 21.26 (Me-18), 13.85 (Me-19), 34.75 (C-15), 50.62 (C-9), 45.44 (C-5), 18.80 (C-2), 22.54 (C-11), 24.44 (C-6), 27.28 (C-12), 35.29 (C-10), 37.26 (C-1), 35.96 (C-3), 68.20 (C-4), 62.19 (C-23). IR (KBr), ν, cm−1: 3100–3600, 2915, 1726, 1356. Found, m/z: 504.3350 [M]+. (C32H44O3N2)+. Calculated, m/z: 504.3347.
- 1-(2-Adamantyl)-3-[(1R,4aR,10aR)-7-isopropyl-1,4a-dimethyl-1,2,3,4,4a,4b,5,6,10,10a-decahydrophenanthren-1-yl]imidazolidine-2,4,5-trione (8b) Yield 64%, white powder. M.p. 111.4 °C. 1H NMR (400 MHz, CDCl3, δ, ppm, J/Hz): 5.74 (1H, s, H-14), 5.34 (1H, s, H-7), 4.14 (1H, s br, H-23), 2.82–3.00 (1H, m, H-15), 0.98 and 0.97 (3H both, d, J16, 15 = 6.8, Me-16 and Me-17), 0.81 (3H, s, Me-19), 1.77 (3H, s, Me-18), 2.39–2.55 (3H, m, H-5, H-24, H-25), 1.11–1.33 (2H, m, H-1a, H-11a), 2.01–2.28 (6H, m, 2H-26, 2H-28, H-1, H-6), 1.71–2.01 (12H, m, 2H-27, 2H-29, H-30, H-31, 2H-32, H-9, H-6, 2H-12), 1.58–1.71 (5H, m, H-11, 2H-2, 2H-3). 13C NMR (100 MHz, CDCl3, δ, ppm): 157.77, 155.73 and 155.61 (C-20, C-21 and C-22), 145.66 (C-13), 135.58 (C-8), 122.02 (C-14), 119.65 (C-7), 20.71 (Me-17 and Me-16), 21.28 (Me-18), 13.87 (Me-19), 62.54 (C-23), 50.63 (C-9), 45.48 (C-5), 34.74 (C-15), 30.35 and 30.51 (C-24 and C-25), 27.17 and 26.45 (C-30 and C-31), 18.77 (C-2), 22.54 (C-11), 35.95 (C-3), 24.39 (C-6), 27.28 (C-12), 35.31 (C-10), 32.58 and 32.46 (C-26 and C-28), 68.28 (C-4), 37.25 (C-1), 37.89 and 37.94 (C-27 and C-29), 37.20 (C-32). IR (KBr), ν, cm−1: 3100–3600, 2916, 1726, 1339. Found, m/z: 504.3353 [M]+. (C32H44O3N2)+. Calculated, m/z: 504.3347.
- 1-(1-Adamantyl)-3-[(1R,4aS,10aR)-7-isopropyl-1,4a-dimethyl-1,2,3,4,4a,9,10,10a-octahydrophenanthren-1-yl]imidazolidine-2,4,5-trione (9a) Yield 70%, white powder. M.p. 96.8 °C. 1H NMR (400 MHz, CDCl3, δ, ppm, J/Hz): 7.14 (1H, d, J = 8.1, H-11), 6.98 (1H, d, J = 8.1, H-12), 6.86 (1H, s, H-14), 1.74 (3H, s, Me-18), 1.20 (6H, d, J = 6.9, Me-16 and Me-17), 1.21 (3H, s, Me-19), 2.61–2.95 (5H, m, H-5, H-15, 2H-7, H-3), 2.37 (6H, br s, H-24, H-25, H-26), 2.19–2.30 (1H, m, H-1), 2.05–2.19 (3H, m, H-27, H-28, H-29), 1.62–1.71 (6H, m, 2H-30, 2H-31, 2H-32), 1.72–1.95 (4H, m, H-3, 2H-2, H-6), 1.46–1.59 (2H, m, H-1, H-6). 13C NMR (100 MHz, CDCl3, δ, ppm): 157.46, 156.24 and 154.62 (C-20, C-21 and C-22), 146.20 (C-9), 145.71 (C-13), 134.14 (C-8), 126.74 (C-14), 124.10 (C-11), 123.91 (C-12), 20.29 (Me-18), 23.83 and 23.86 (Me-17 and Me-16), 25.27 (Me-19), 33.31 (C-15), 44.60 (C-5), 29.54 (C-27, C-28, C-29), 19.13 (C-2), 19.93 (C-6), 29.66 (C-7), 35.77 (C-30, C-31, C-32), 39.89 (C-24, C-25, C-26), 69.24 (C-4), 38.44 (C-1), 36.80 and 35.10 (C-10 and C-3), 62.22 (C-23). IR (KBr), ν, cm−1: 2912, 1725, 1352. Found, m/z: 502.3187 [M]+. (C32H42O3N2)+. Calculated, m/z: 502.3190.
- 1-(2-Adamantyl)-3-[(1R,4aS,10aR)-7-isopropyl-1,4a-dimethyl-1,2,3,4,4a,9,10,10a-octahydrophenanthren-1-yl]imidazolidine-2,4,5-trione (9b) Yield 72%, white powder. M.p. 89.3 °C. 1H NMR (400 MHz, CDCl3, δ, ppm, J/Hz): 7.13 (1H, d, J = 8.2, H-11), 6.98 (1H, dd, J = 8.2, J = 1.7, H-12), 6.85 (1H, d, J = 1.7, H-14), 4.15 (1H, s br., H-23), 2.73–2.89 (4H, m, H-5, H-15, 2H-7), 2.65–2.73 (1H, m, H-3), 2.48–2.55 (2H, m, H-24, H-25), 2.21–2.29 (1H, m, H-1), 2.06–2.13 (2H, m, H-26, H-28), 1.71–1.98 (12H, m, 2H-27, 2H-29, H-30, H-31, 2H-32, H-3, 2H-2, H-6), 1.76 (3H, s, Me-18), 1.64–1.71 (2H, m, H-26, H-28), 1.48–1.60 (2H, m, H-1, H-6), 1.22 (3H, s, Me-19), 1.20 (6H, d, J = 6.9, Me-16 and Me-17). 13C NMR (100 MHz, CDCl3, δ, ppm): 157.68, 155.77 and 155.52 (C-20, C-21 and C-22), 146.08 (C-9), 145.67 (C-13), 134.03 (C-8), 126.69 (C-14), 124.00 (C-11), 123.86 (C-12), 69.25 (C-4), 62.55 (C-23), 44.61 (C-5), 38.38 (C-10), 37.77 (C-27 and C-29), 37.11 (C-32), 36.75 (C-1), 35.12 (C-3), 33.26 (C-15), 32.43 and 32.48 (C-26 and C-28), 30.25 and 30.27 (C-24 and C-25), 29.52 (C-7), 27.07 and 26.37 (C-30 and C-31), 25.19 (Me-19), 23.79 (Me-17 and Me-16), 20.14 (Me-18), 19.84 (C-6), 19.06 (C-2). IR (KBr), ν, cm−1: 2917, 1726, 1337. Found, m/z: 502.3188 [M]+. (C32H42O3N2)+. Calculated, m/z: 502.3190.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baglini, E.; Salerno, S.; Barresi, E.; Robello, M.; Da Settimo, F.; Taliani, S.; Marini, A.M. Multiple Topoisomerase I (TopoI), Topoisomerase II (TopoII) and Tyrosyl-DNA Phosphodiesterase (TDP) inhibitors in the development of anticancer drugs. Eur. J. Pharm. Sci. 2021, 156, 105594. [Google Scholar] [CrossRef]
- Yang, S.W.; Burgin, A.B.; Huizenga, B.N.; Robertson, C.A.; Yao, K.C.; Nash, H.A. A eukaryotic enzyme that can disjoin dead-end covalent complexes between DNA and type I topoisomerases. Proc. Natl. Acad. Sci. USA 1996, 93, 11534–11539. [Google Scholar] [CrossRef]
- Liang, X.; Wu, Q.; Luan, S.; Yin, Z.; He, C.; Yin, L.; Zou, Y.; Yuan, Z.; Li, L.; Song, X.; et al. A comprehensive review of topoisomerase inhibitors as anticancer agents in the past decade. Eur. J. Med. Chem. 2019, 171, 129–168. [Google Scholar] [CrossRef]
- Pommier, Y.; Huang, S.Y.N.; Gao, R.; Das, B.B.; Murai, J.; Marchand, C. Tyrosyl-DNA-phosphodiesterases (TDP1 and TDP2). DNA Repair 2014, 19, 114–129. [Google Scholar] [CrossRef]
- Huang, S.Y.N.; Murai, J.; Dalla Rosa, I.; Dexheimer, T.S.; Naumova, A.; Gmeiner, W.H.; Pommier, Y. TDP1 repairs nuclear and mitochondrial DNA damage induced by chain-terminating anticancer and antiviral nucleoside analogs. Nucleic Acids Res. 2013, 41, 7793–7803. [Google Scholar] [CrossRef]
- Inamdar, K.V.; Pouliot, J.J.; Zhou, T.; Lees-Miller, S.P.; Rasouli-Nia, A.; Povirk, L.F. Conversion of phosphoglycolate to phosphate termini on 3′ overhangs of DNA double strand breaks by the human tyrosyl-DNA phosphodiesterase hTdp1. J. Biol. Chem. 2002, 277, 27162–27168. [Google Scholar] [CrossRef]
- Zhou, T.; Lee, J.W.; Tatavarthi, H.; Lupski, J.R.; Valerie, K.; Povirk, L.F. Deficiency in 3′-phosphoglycolate processing in human cells with a hereditary mutation in tyrosyl-DNA phosphodiesterase (TDP1). Nucleic Acids Res. 2005, 33, 289–297. [Google Scholar] [CrossRef]
- Hirano, R.; Interthal, H.; Huang, C.; Nakamura, T.; Deguchi, K.; Choi, K.; Bhattacharjee, M.B.; Arimura, K.; Umehara, F.; Izumo, S.; et al. Spinocerebellar ataxia with axonal neuropathy: Consequence of a Tdp1 recessive neomorphic mutation? EMBO J. 2007, 26, 4732–4743. [Google Scholar] [CrossRef]
- Katyal, S.; El-Khamisy, S.F.; Russell, H.R.; Li, Y.; Ju, L.; Caldecott, K.W.; McKinnon, P.J. TDP1 facilitates chromosomal single-strand break repair in neurons and is neuroprotective in vivo. EMBO J. 2007, 26, 4720–4731. [Google Scholar] [CrossRef]
- Wei, X.; Wang, Z.; Hinson, C.; Yang, K. 3-DNA—Peptide/protein cross-links arising from abasic sites in vitro. Nucleic Acids Res. 2022, 50, 1–20. [Google Scholar] [CrossRef]
- Zakharenko, A.L.; Luzina, O.A.; Chepanova, A.A.; Dyrkheeva, N.S.; Salakhutdinov, N.F.; Lavrik, O.I. Natural Products and Their Derivatives as Inhibitors of the DNA Repair Enzyme Tyrosyl-DNA Phosphodiesterase 1. Int. J. Mol. Sci. 2023, 24, 5781. [Google Scholar] [CrossRef]
- Kugler, S.; Ossowicz, P.; Malarczyk-Matusiak, K.; Wierzbicka, E. Advances in Rosin-Based Chemicals: The Latest Recipes, Applications and Future Trends. Molecules 2019, 24, 1651. [Google Scholar] [CrossRef]
- Kovaleva, K.; Oleshko, O.; Mamontova, E.; Yarovaya, O.; Zakharova, O.; Zakharenko, A.; Kononova, A.; Dyrkheeva, N.; Cheresiz, S.; Pokrovsky, A.; et al. Dehydroabietylamine Ureas and Thioureas as Tyrosyl-DNA Phosphodiesterase 1 Inhibitors That Enhance the Antitumor Effect of Temozolomide on Glioblastoma Cells. J. Nat. Prod. 2019, 82, 2443–2450. [Google Scholar] [CrossRef]
- Kovaleva, K.; Yarovaya, O.; Ponomarev, K.; Cheresiz, S.; Azimirad, A.; Chernyshova, I.; Zakharenko, A.; Konev, V.; Khlebnikova, T.; Mozhaytsev, E.; et al. Design, Synthesis, and Molecular Docking Study of New Tyrosyl-DNA Phosphodiesterase 1 (TDP1) Inhibitors Combining Resin Acids and Adamantane Moieties. Pharmaceuticals 2021, 14, 422. [Google Scholar] [CrossRef]
- Burmistrov, V.; Morisseau, C.; Karlov, D.; Pitushkin, D.; Vernigora, A.; Rasskazova, E.; Butov, G.M.; Hammock, B.D. Bioisosteric substitution of adamantane with bicyclic lipophilic groups improves water solubility of human soluble epoxide hydrolase inhibitors. Bioorg. Med. Chem. Lett. 2020, 30, 127430. [Google Scholar] [CrossRef]
- Sato, T.; Komine, T.; Masahiro, N.; Renbu, M.; Naoki, K. Novel Parabanic Acid Derivative and Drug Having the Same as Active Ingredient. WO2011078370A1, 30 June 2011. [Google Scholar]
- Aboutabl, M.E.; Hassan, R.M.; El-Azzouny, A.A.S.; Aboul-Enein, M.N.; Abd-Allah, W.H. Design and synthesis of novel parabanic acid derivatives as anticonvulsants. Bioorg. Chem. 2019, 94, 103473. [Google Scholar] [CrossRef]
- Ienaga, K.; Nakamura, K.; Ishii, A. Hypolipidemic Agent Containing Imidazolidin-Trione Derivative. JPH07165581A, 27 June 1995. [Google Scholar]
- Ishii, A.; Kotani, T.; Nagaki, Y.; Shibayama, Y.; Toyomaki, Y.; Okukado, N.; Ienaga, K.; Okamoto, K. Highly selective aldose reductase inhibitors. 1. 3-(Arylalkyl)-2,4,5-trioxoimidazolidine-1-acetic acids. J. Med. Chem. 1996, 39, 1924–1927. [Google Scholar] [CrossRef]
- Burmistrov, V.; Morisseau, C.; D’yachenko, V.; Karlov, D.; Butov, G.M.; Hammock, B.D. Imidazolidine-2,4,5- and pirimidine-2,4,6-triones—New primary pharmacophore for soluble epoxide hydrolase inhibitors with enhanced water solubility. Bioorganic Med. Chem. Lett. 2019, 30, 126908. [Google Scholar] [CrossRef]
- Pejchal, V.; Stepankova, S.; Padelkova, Z.; Imramovsky, A.; Jampilek, J. 1,3-substituted imidazolidine-2,4,5-triones: Synthesis and inhibition of cholinergic enzymes. Molecules 2011, 16, 7565–7582. [Google Scholar] [CrossRef]
- Li, B.; Man, Y.; Bai, L.; Ji, H.; Shi, X.; Cui, D. Solution-Phase Parallel Syntheses of Herbicidal 1-Phenyl-2,4,5- Imidazolidinetriones and 2-Thioxo-4,5-Imidazolidinediones. High Throughput Screen 2013, 16, 78–82. [Google Scholar] [CrossRef]
- Rajabi, M.; Mansell, D.; Freeman, S.; Bryce, R.A. Structure-activity relationship of 2,4,5-trioxoimidazolidines as inhibitors of thymidine phosphorylase. Eur. J. Med. Chem. 2011, 46, 1165–1171. [Google Scholar] [CrossRef]
- Yao, H.; Liu, F.; Chen, J.; Li, Y.; Cui, J.; Qiao, C. Antischistosomal activity of N,N′-arylurea analogs against Schistosoma japonicum. Bioorg. Med. Chem. Lett. 2016, 26, 1386–1390. [Google Scholar] [CrossRef]
- Kunde, L.B.; Kalyani, V.S.; Gupte, S.P. Dibutyltin oxide catalyzed aminolysis of oxalate to carbamate, oxamate and derivatives of imidazolidine trione. Appl. Organomet. Chem. 2010, 24, 402–407. [Google Scholar] [CrossRef]
- Huang, W.G.; Wang, H.S.; Huang, G.B.; Wu, Y.M.; Pan, Y.M. Enantioselective friedel-crafts alkylation of N-methylindoles with nitroalkenes catalyzed by chiral bifunctional abietic-acid-derived thiourea-Zn II complexes. Eur. J. Org. Chem. 2012, 2012, 5839–5843. [Google Scholar] [CrossRef]
- Zakharenko, A.; Khomenko, T.; Zhukova, S.; Koval, O.; Zakharova, O.; Anarbaev, R.; Lebedeva, N.; Korchagina, D.; Komarova, N.; Vasiliev, V.; et al. Synthesis and biological evaluation of novel tyrosyl-DNA phosphodiesterase 1 inhibitors with a benzopentathiepine moiety. Bioorg. Med. Chem. 2015, 23, 2044–2052. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 * (TDP1), µM | CC50 ** (SNB19), µM |
|---|---|---|
| 8a | 0.52 ± 0.07 | >100 |
| 8b | 0.69 ± 0.07 | >100 |
| 9a | 0.78 ± 0.02 | 28.63 ± 1.16 |
| 9b | 0.96 ± 0.04 | >100 |
| Furamidine | 1.20 0.30 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovaleva, K.S.; Yarovaya, O.I.; Chernyshova, I.A.; Zakharenko, A.L.; Cheresiz, S.V.; Azimirad, A.; Pokrovsky, A.G.; Lavrik, O.I.; Salakhutdinov, N.F. Synthesis of Norabietyl and Nordehydroabietyl Imidazolidine-2,4,5-Triones and Their Activity against Tyrosyl-DNA Phosphodiesterase 1. Molbank 2023, 2023, M1743. https://doi.org/10.3390/M1743
Kovaleva KS, Yarovaya OI, Chernyshova IA, Zakharenko AL, Cheresiz SV, Azimirad A, Pokrovsky AG, Lavrik OI, Salakhutdinov NF. Synthesis of Norabietyl and Nordehydroabietyl Imidazolidine-2,4,5-Triones and Their Activity against Tyrosyl-DNA Phosphodiesterase 1. Molbank. 2023; 2023(4):M1743. https://doi.org/10.3390/M1743
Chicago/Turabian StyleKovaleva, Kseniya S., Olga I. Yarovaya, Irina A. Chernyshova, Alexandra L. Zakharenko, Sergey V. Cheresiz, Amirhossein Azimirad, Andrey G. Pokrovsky, Olga I. Lavrik, and Nariman F. Salakhutdinov. 2023. "Synthesis of Norabietyl and Nordehydroabietyl Imidazolidine-2,4,5-Triones and Their Activity against Tyrosyl-DNA Phosphodiesterase 1" Molbank 2023, no. 4: M1743. https://doi.org/10.3390/M1743