Introduction
DNA microarray is the most attracting technology [
1,
2] in recent years, that will revolutionize the biological researches and clinical diagnosis. There are two general techniques for fabricating DNA microarray [
3]. Either pre-synthesized oligonucleotides or cDNA can be applied to the support, or in situ synthesize oligonucleotides on the support. Glass is chosen as a main support media for its low intrinsic fluorescence, chemical inertness and ability to prepare high density DNA array [
4].
A linker molecule, which attaches the nucleic acid probes on glass surface, should be introduced to increase the accurate and efficiency of hybridization between the tethered probe and target DNA sequences. The structural features of nucleic acid probes tethered to a solid support and the molecular basis of their interaction with targets in solution have direct relations to the properties of the linkers [
5,
6]. A suitable linker should meet three basic requirements: easy to synthesis, active end group (hydroxyl or amino group), and stable to harsh conditions used for synthesis, hybridization and denature environment for repeat uses. For a high quality DNA microarray, some properties of linker, like length, backbone flexibility and hydrophilic tendency should also be optimized [
7]. Unfortunately it is difficult to meet the requirements for conventional linkers. In the literatures, several useful linkers had been reported to bind oligonucleotides on glass surface by attaching pre-fabricated oligonucleotides or in situ synthesis of oligonucleotides. For spotting immobilization, the linkers were often short. To increase hybridization efficiency, DNA probes were prolonged by adding polyT to the tethered terminal, which increased the costs of probes [
8,
9]. For in situ synthesis, preparation of linkers was either time consuming or laborious [
4,
6,
7].
Terminally functionalized organosilyl compounds are wildly used to modify silica and glass to immobilize biomoleculars on the surface, such as enzyme, antibody and DNA etc. Trialkoxy silanes are most commonly used aminosilanes, like 3-aminopropyltrimethoxysilane (APTES). They have three reactive groups (trimethoxysilane) to form Si-O-Si linkages with glass hydroxysilane and between each other, which may result surface polymerization reactions and form multiple layers on the glass surface, so it leads to difficulties in the control, reproducibility, homogeneity, and stability of subsequent DNA attachment. Monoalkoxy silanes can produce monolayer on the surface but suffer from poor stability [
10]. To get consistent and stable modification, we chosen dialkoxy silane (N-(2-aminoethyl)-3-aminopropylmethyl dimethoxylsilane, AEAPS) to test its performance.
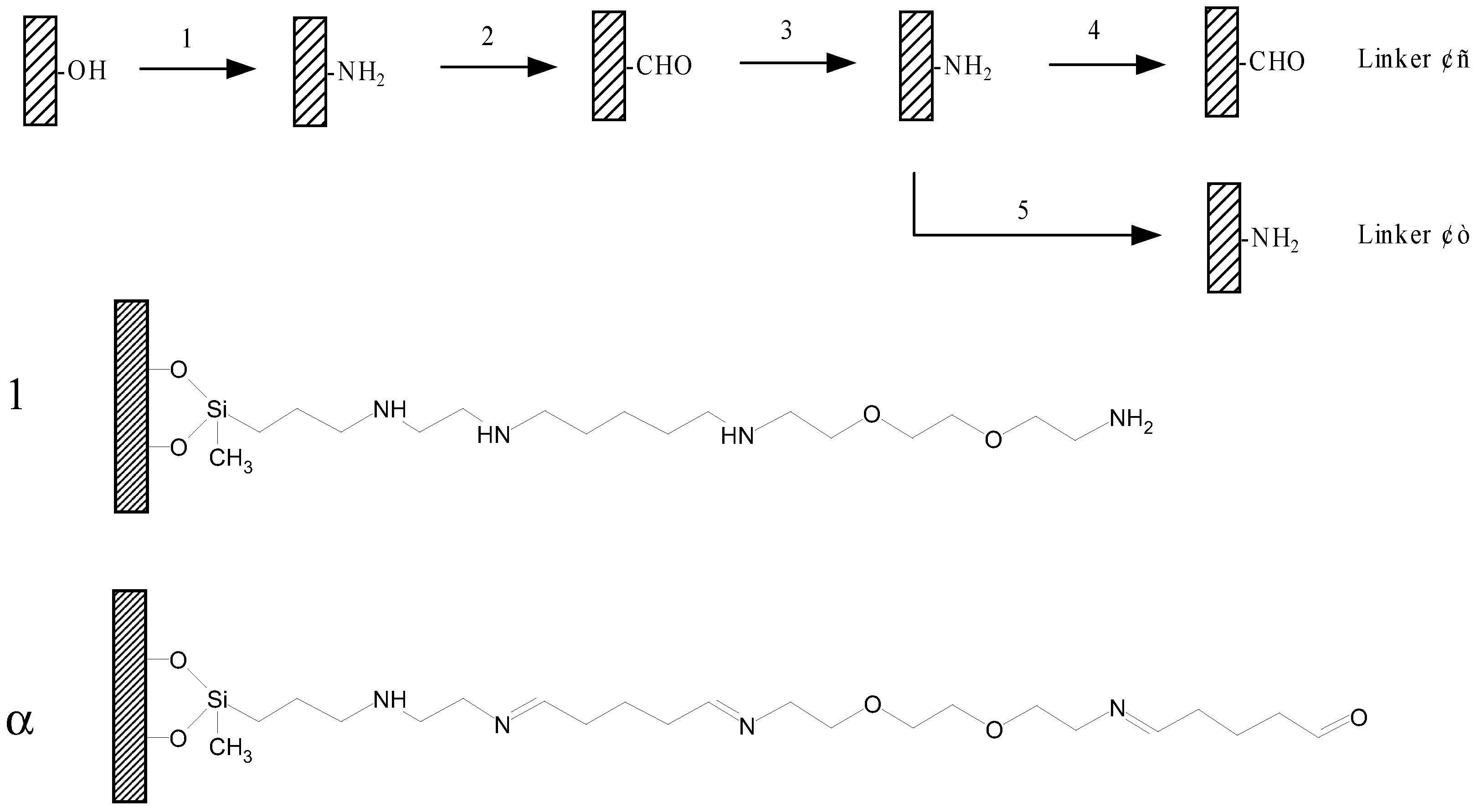
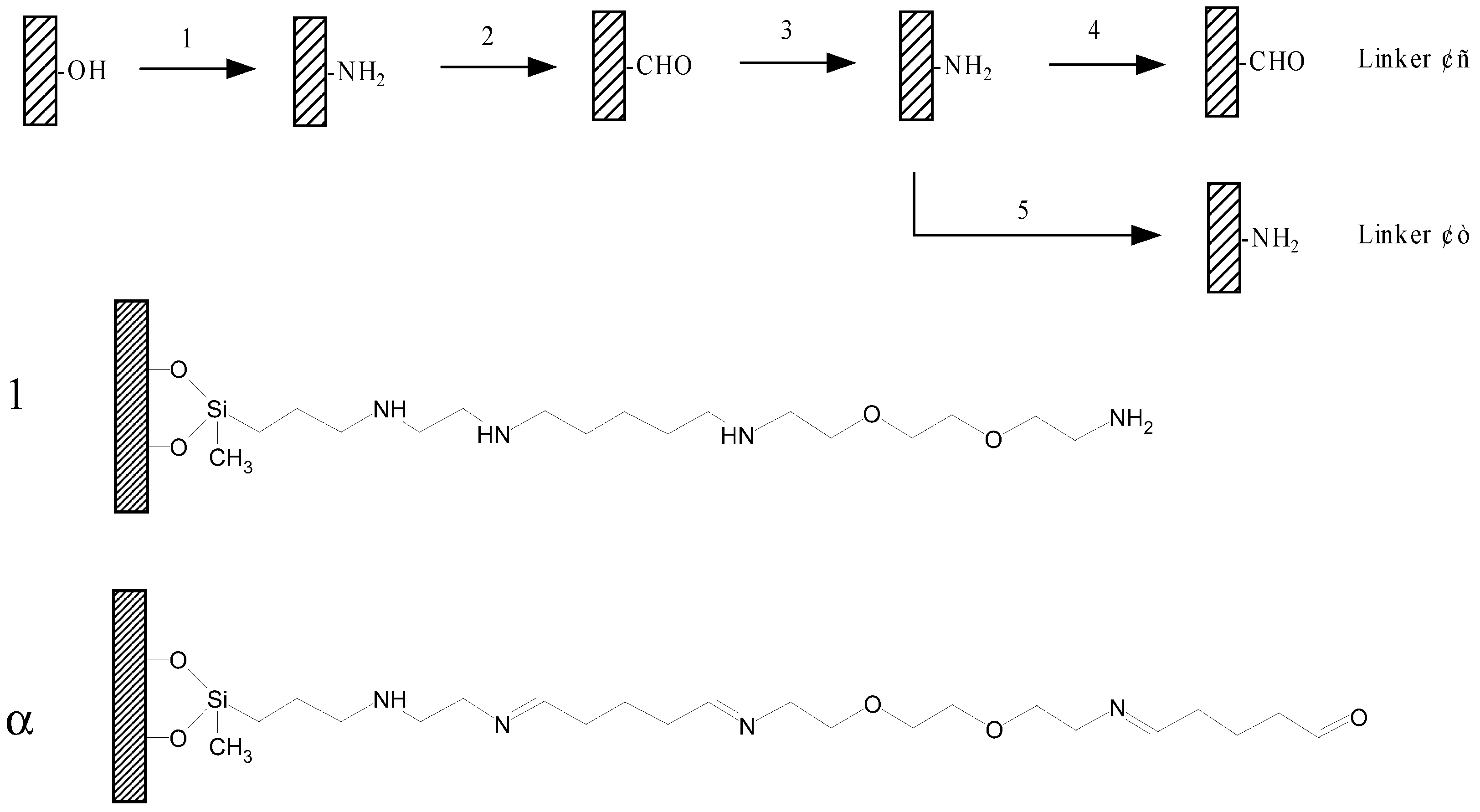
Figure 1.
Scheme of assembling of the linkers.
Figure 1.
Scheme of assembling of the linkers.
1. Silane; 2. Glutaraldehyde; 3. 1,2-bis (2-aminoethoxy) ethane; 4. Glutaraldehyde; 5. Reduction. Linker 1: The linker used for in situ synthesize oligonucleotides; Linker α: The linker used for binding pre-synthesized DNA.
This paper describes a method to form linkers by alternatively covalently assembling different molecular components on the glass surface. (outlined in
Figure 1). The method has been proved to be convenient, reliable and flexible. Two linkers with different terminal groups have been successfully prepared. The aldehyde linker is for attaching pre-prepared DNA, and amino linker is made for in situ synthesis oligonucleotide (see
Figure 1). Both of them are successfully used for oligonucleotides binding, and internal single base mismatch in 20mer oligonucleotides can be discriminated from complementary sequences.
Results and Discussion
Comparison of AEAPS and APTES were done for attaching oligonucleotides. Interestingly, AEAPS gave both better hybridization signals to noise ratio and reproducibility (see
Table 1). That is because AEAPS reacted with moisture much slower than APTES did. APTES tend to absorb moisture and form aggregations, which will greatly increase background signal.
Table 1.
Comparison of AEAPS and APTES.
Table 1.
Comparison of AEAPS and APTES.
| | Hybridization signal to noise ratio | Background signal |
| AEAPS | 25.6±6.9% | 231±2.1% |
| APTES | 21.3±7.5% | 352±3.7% |
The linkers can be prepared by simply dipping the slides in different solutions at room temperature. To change linker` length, we increased the reaction cycle or chose different regents, and by introducing rigid regents such as phenylendiisothiocyanate (PDC) we can easily modify the backbone rigidness. A consideration may be given for the homo-function reagents used, for some reactions may take place between two slide function groups and the homo-function reagents to form a bridge, thus reduce the number of function groups. There should have such side reactions, but the ratio must be very low because large excess homo-function regents used. An experiment was done to confirm that. A slide was modified with two different linkers (Linker 1.
![Sensors 01 00053 i006]()
-CH
2-CH
2-CH
2-NH-CH
2-CH
2-N=CH-CH
2-CH
2- CH
2-CHO; 2. Linker α). A 16mer oligonucleotide with 3` labeled amino group and 5` labeled FAM (5`FAM-CCT GAG AGA CTT AGC C-NH
2) was immobilized on these two regions. Fluorescence results showed no detectable difference between them. When immobilized with amino group labeled oligonucleotide and hybridized with its complementary FAM labeled oligonucleotide, about 20% higher hybridization signal was gotten for linker α than linker 1. This is due to the longer length of linker 2, which make targets easier to access tethered probes.
Efforts were paid to achieve uniform images when applied pre-fabricated oligonucleotides on slides. The unevenness of the hybridization images may cause quantification unreliable. Evaporation of immobilizing solution was avoided to prevent the change of concentration that may contribute to the unevenness of dots. Using ourselves developed protocol, uniform images were gotten with good reproducibility. Good discrimination was achieved between two 20mer oligonucleotides with one internal base mismatch (
Figure 2).
Figure 2.
Hybridization to tethered oligonucleotides.
Figure 2.
Hybridization to tethered oligonucleotides.
Diagonal spots were matched 20 mer oligonucleotides (5'NH2 AGG AGG CTA AGT CTC TCA GG 3'), which were separated by mismatched oligonucleotides (5'NH2 AGG AGG CTA AtT CTC TCA GG 3'). The slide were hybridized to FAM labeled complementary oligonucleotide (5'FAM CCT GAG AGA CTT AGC CTC CT 3') at 55.
For in situ synthesis of oligonucleotides on slide, an easy way was adopted to synthesis five different oligonucleotides on one slide in minimal steps. It is outlined in
Figure 3. After hybridization with FAM labeled oligonucleotide (5' FAM CCT GAG AGA CTT AGC CTC CT 3') at 50 , Position G gave much stronger signal than other places. The synthesis quality should be high enough for the hybridization, because large excess amount of monomer was used with prolonged coupling time. Another experiment was done to confirm that. On one slide two sequences were synthesized (T and T20), after that a FAM (6-carboxyfluorescein) monomer was coupled to both of the sequences. After deprotection these two areas were scanned on confocol microscope. By comparing the fluorescence intensity, as high as 98% step efficiency was achieved [
11]. Under standard final deprotection procedure (in concentrated ammonium hydroxide solution at 55 for 6-8 hours) at least 80% oligonucleotides were cleaved from glass [
4], which led to very weak fluorescence signal to be detected. Expedite monomers were used in this experiment to allow deprotection complete under mild conditions. Expedite phosphoramidite monomers except thymidine monomer are protected with tert- butylphenoxyacetyl (tBPA), which enable fast deprotection under milder conditions [
12].
Figure 3.
Schematic drawing of in situ synthesis of oligonucleotides.
Figure 3.
Schematic drawing of in situ synthesis of oligonucleotides.
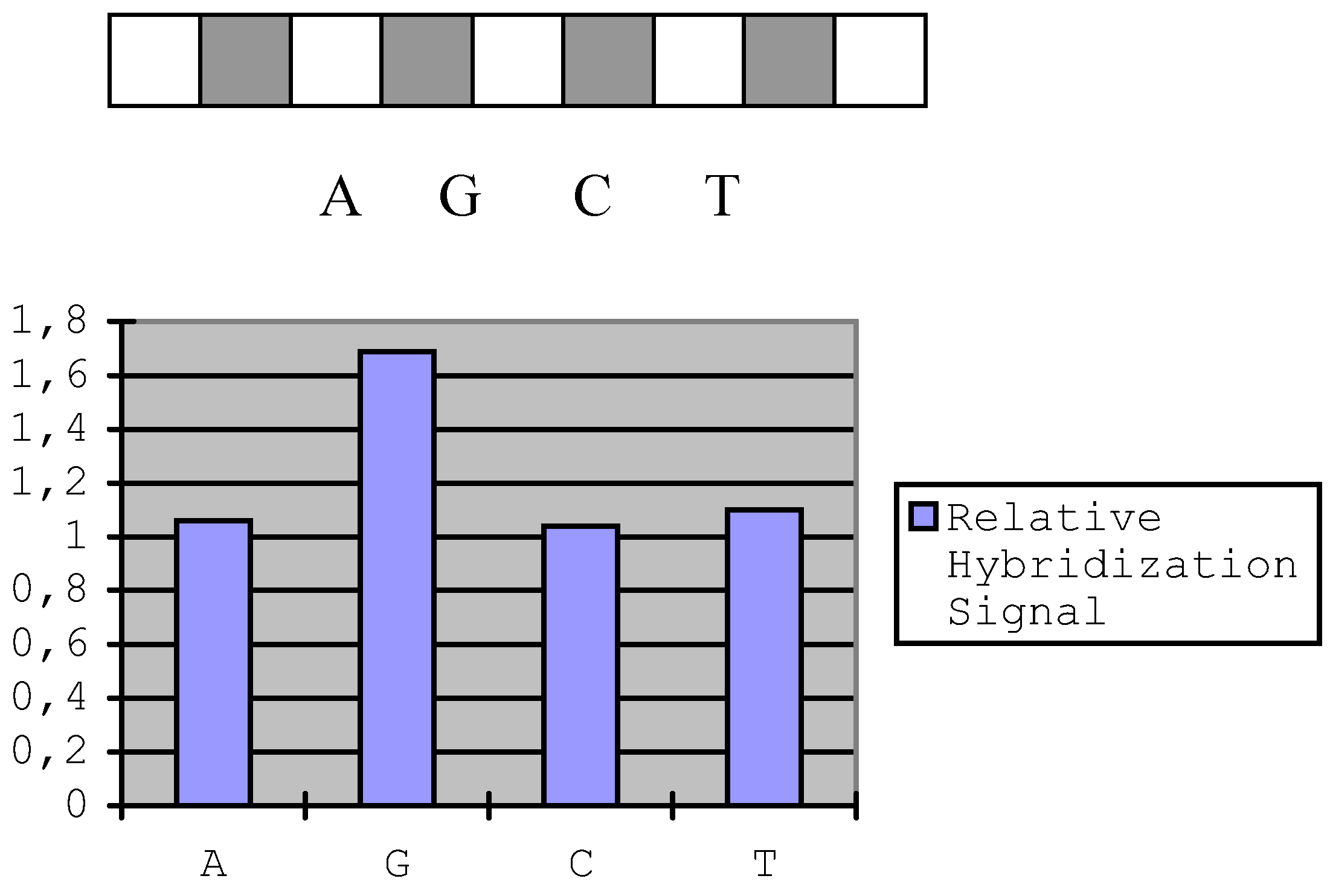
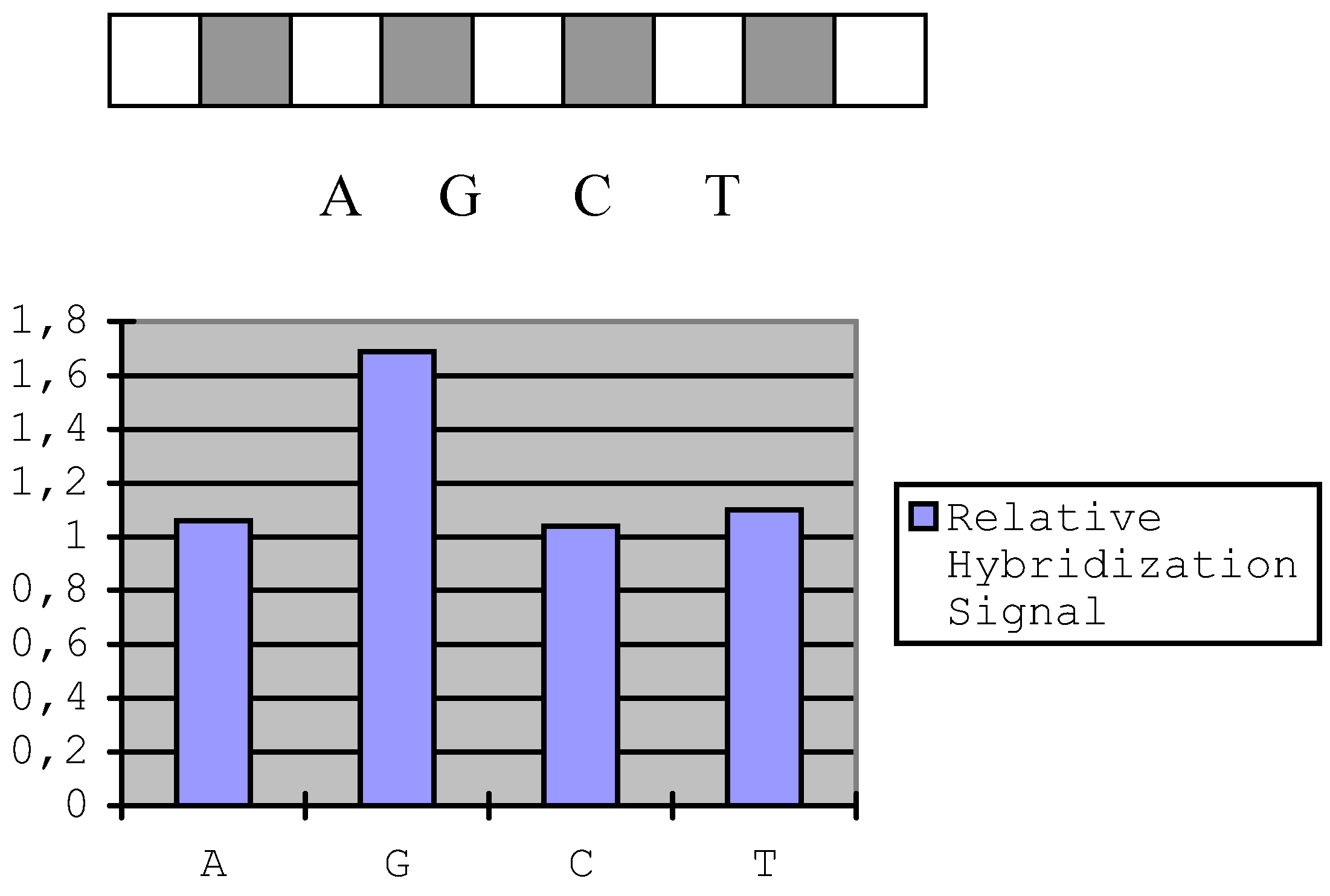
The silicone rubber rectangle cell (see Experimental section) was first put on the activated slide horizontally and synthesize a sequence of 9 mer (5' T CTC TCA GG 3’), then rotate the cell by 90° and put it on the A position. Monomer A was synthesized here, then move the cell to G, C and T positions to synthesize G, C, T on corresponding positions respectively. These syntheses were all conducted using Tritl On model. After that, the cell was put on the initially horizontal position and a sequence of 5' AGG AGG CTA A was synthesized using Tritl Off model. The sequences of different positions are:
![Sensors 01 00053 i001]()
: 5' AGG AGG CTA A∧T CTC TCA GG 3'
![Sensors 01 00053 i002]()
: 5' AGG AGG CTA AaT CTC TCA GG 3'
![Sensors 01 00053 i003]()
: 5' AGG AGG CTA AGT CTC TCA GG 3';
![Sensors 01 00053 i004]()
: 5' AGG AGG CTA AcT CTC TCA GG 3'; 5' AGG AGG CTA AtT CTC TCA GG 3'.
![Sensors 01 00053 i005]()
: 5' AGG AGG CTA AtT CTC TCA GG 3'.
Set the 19 mer oligonucleotide`s fluorescence intensity to 1, then calculated: A, 1.06; G, 1.69; C, 1.04 and T, 1.10. We compared our in situ synthesis linker with the linker used by MaskosError! Bookmark not defined.. The Maskos`s linker was prepared as their protocol. A 20-mer oligonucleotide sequence was synthesized on each slide, which was modified with corresponding linker. After hybridizing with complementary DNA, the hybridization fluorescence signal to noise ratio showed no much difference for them (our linker: 8.7; Maskos linker: 8.5).
Experimental
General
All chemical regents were obtained from TCI (Tokyo chemical industrial, Japan) and were used as received unless otherwise noted, and DNA synthesis regents were purchased from Applied Biosystems (USA). All references in the text to water refer to obtained from a Millipore™ purification system, >18 ΜΩ.
Linker synthesis
Typically, the procedure to prepare linker is described as follows: Glass slides were cleaned by immersion in 1:1 concentrated HCl:MeOH for 30 min, rinsed in water, immersed in concentrated H2SO4 for 30 min and rinsed in water before boiling in deionized water for several minutes prior to silanization.
Precleaned glass slides were immersed in 1% silane solution in 95% acetone/water for 5 min. The slides were washed five times with acetone and baked for 30 min at 110□.. Then put them in 5% glutaraldehyde in 1×PBS for 2 hours, and rinse slides for five times with water. They were immersed in 5% 1,2-bis (2-aminoethoxy) ethane aqueous solution for 2 hours, and rinsed with water. For in situ linkers, the slides were reduced by sodium borohydride solution (1.3g, 1×PBS 375ml, ethanol 125ml) for 5min. Thus a hydrophilic linker with 22 atoms was completed (
Figure 1), they were ready for in situ synthesis now. For binding pre-fabricated peptide or amino-labeled nucleic acids, another layer of glutaraldehyde was introduced on the slides in the same way.
Immobilization
Briefly, immobilization of pre-fabricated oligonucleotides was described as follows: Sodium carbonate buffer (0.1mol/L, PH9.0) was added to the oligonucleotides containing 5` amino group to produce a concentration of 40μM. Spot these ODNs on the slides under humid atmosphere (~70% humidity) by PixSys_5500 (Cartesian Corp) with spots size of 100μm and 300μm space. Put them in a wet box with face up for an hour at room temperature, then transfer the box to water bathe at 37°C for 2h. After that wash it with 0.1%SDS 2×5min, followed by completely wash with water. Then the slides were reduced by sodium borohydride solution (1.3g, 1×PBS 375ml, ethanol 125ml) for 5min, and washed by water.
In situ synthesis
In situ synthesis of oligonucleotides was carried out on the chip directed by a channel-system [Error! Bookmark not defined.]. Automated synthesizer (Expedite 8909 with Workstation software, Applied Biosystems, USA) was connected. Expedite monomers were used to allow fast deprotection of base protecting group under mild conditions, which did less harm to the sequence during the final deblock of exocyclic protection groups. The protocol of synthesis was modified with relative large amount of regents used (about four times of 1 μmol scale), and coupling time was prolonged to 3 min, then capping and oxidation steps were done. After the synthesis, putting the slide into a solution of ethylene diamine and ethanol (1:1) at room temperature for 10 hours to deprotect the oligonucleotides.
Hybridization
FAM (6-carboxyfluorescein) labeled oligonucleotide was diluted to 200nmol/L with hybridization solution which contain 6×SSC, 5×Denhardts, 0.5%SDS and 100μg/ml denatured salmon sperm DNA. Hybridization took place in a wet box for 2 hours. After hybridization, slides were washed with 2×SSC 0.1% SDS 5 min, 0.1×SSC 0.1% SDS 5 min, then wash briefly with water and blow to dry. Hybridization images were collected on a confocal microscope (Leica), and analyzed with the software.


 -CH2-CH2-CH2-NH-CH2-CH2-N=CH-CH2-CH2- CH2-CHO; 2. Linker α). A 16mer oligonucleotide with 3` labeled amino group and 5` labeled FAM (5`FAM-CCT GAG AGA CTT AGC C-NH2) was immobilized on these two regions. Fluorescence results showed no detectable difference between them. When immobilized with amino group labeled oligonucleotide and hybridized with its complementary FAM labeled oligonucleotide, about 20% higher hybridization signal was gotten for linker α than linker 1. This is due to the longer length of linker 2, which make targets easier to access tethered probes.
-CH2-CH2-CH2-NH-CH2-CH2-N=CH-CH2-CH2- CH2-CHO; 2. Linker α). A 16mer oligonucleotide with 3` labeled amino group and 5` labeled FAM (5`FAM-CCT GAG AGA CTT AGC C-NH2) was immobilized on these two regions. Fluorescence results showed no detectable difference between them. When immobilized with amino group labeled oligonucleotide and hybridized with its complementary FAM labeled oligonucleotide, about 20% higher hybridization signal was gotten for linker α than linker 1. This is due to the longer length of linker 2, which make targets easier to access tethered probes.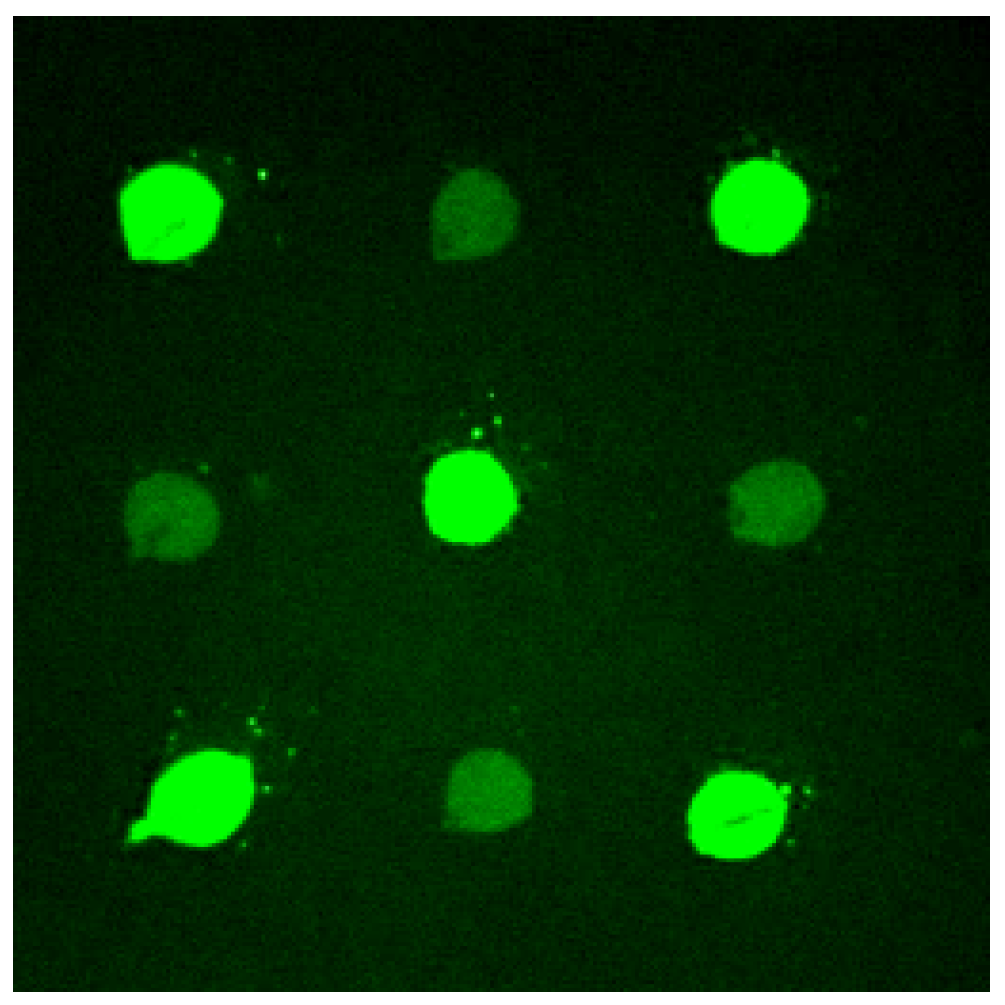





{kind=link}
{kind=link}
{kind=link}