Chemical Selectivity and Sensitivity of a 16-Channel Electronic Nose for Trace Vapour Detection

 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
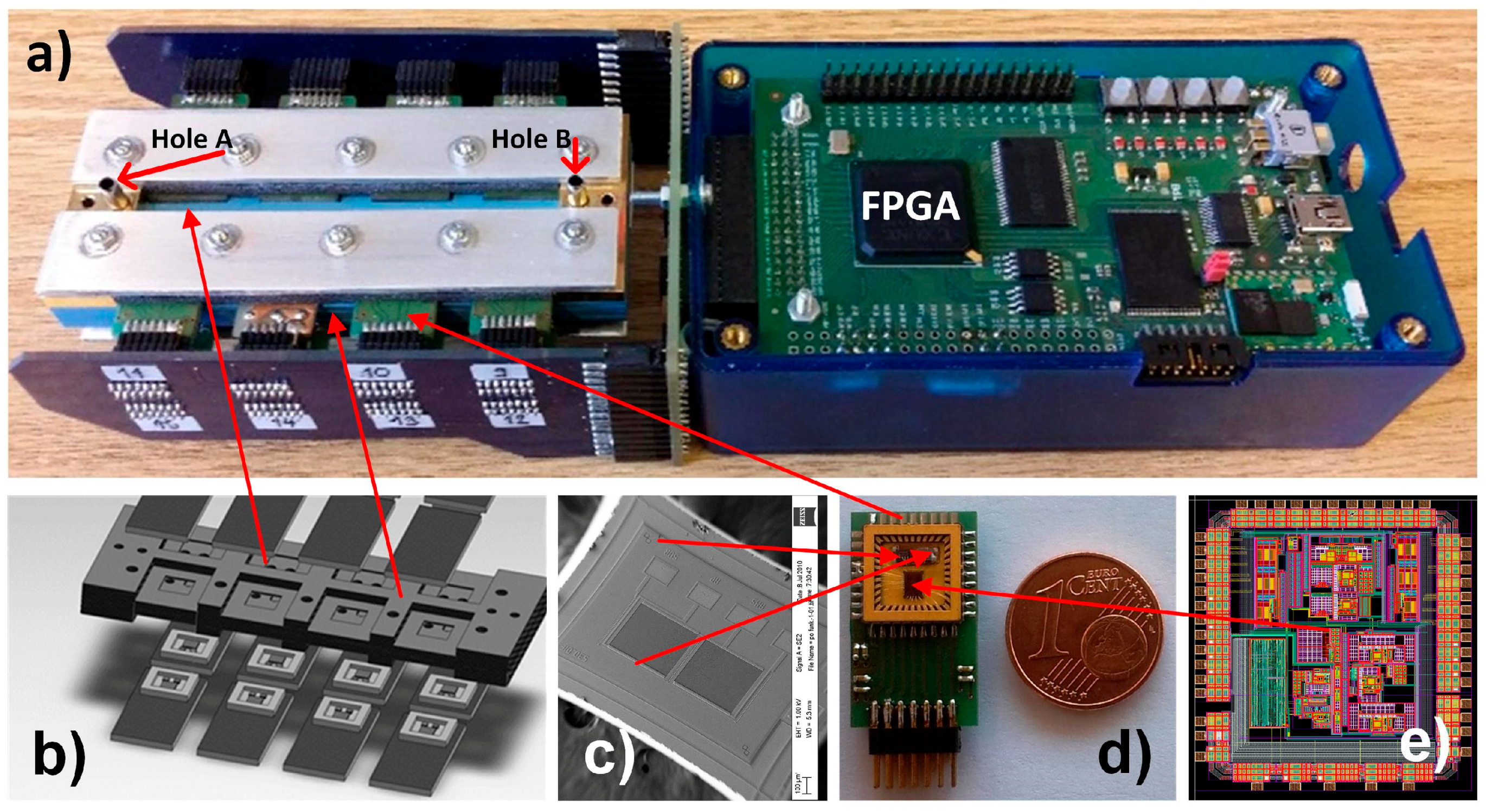
2.1. The Sensor
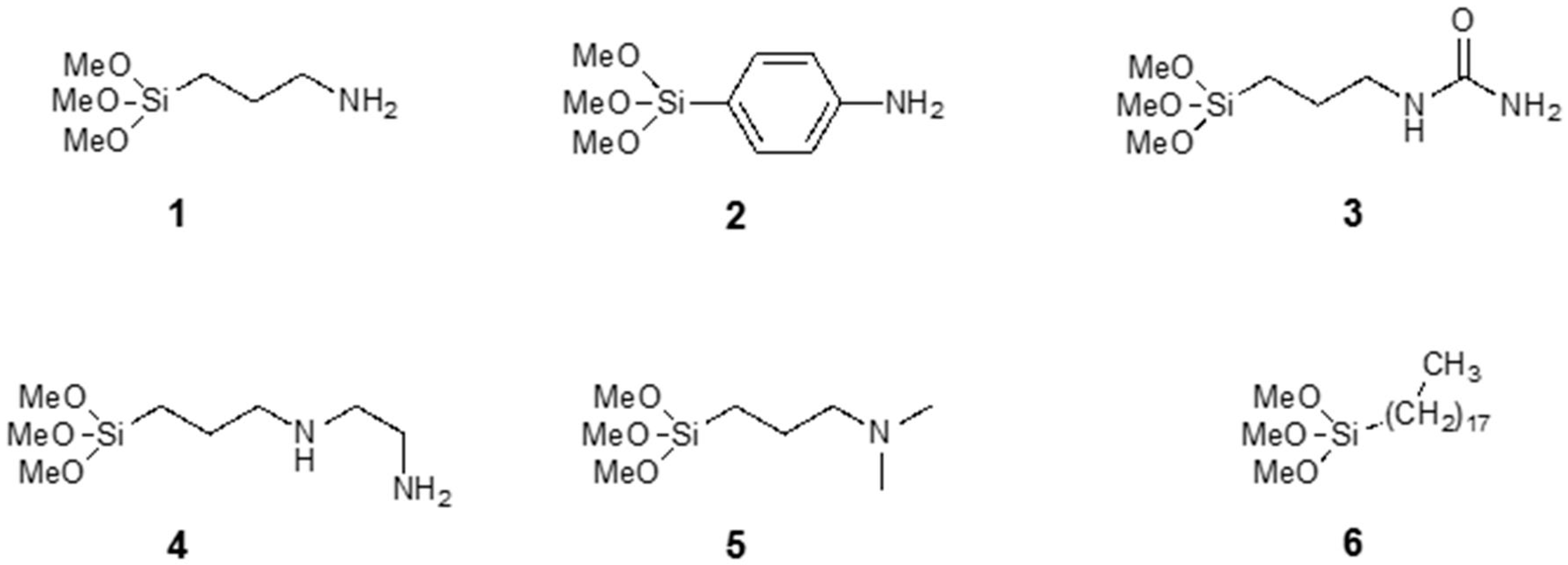
2.2. Chemical Functionalization
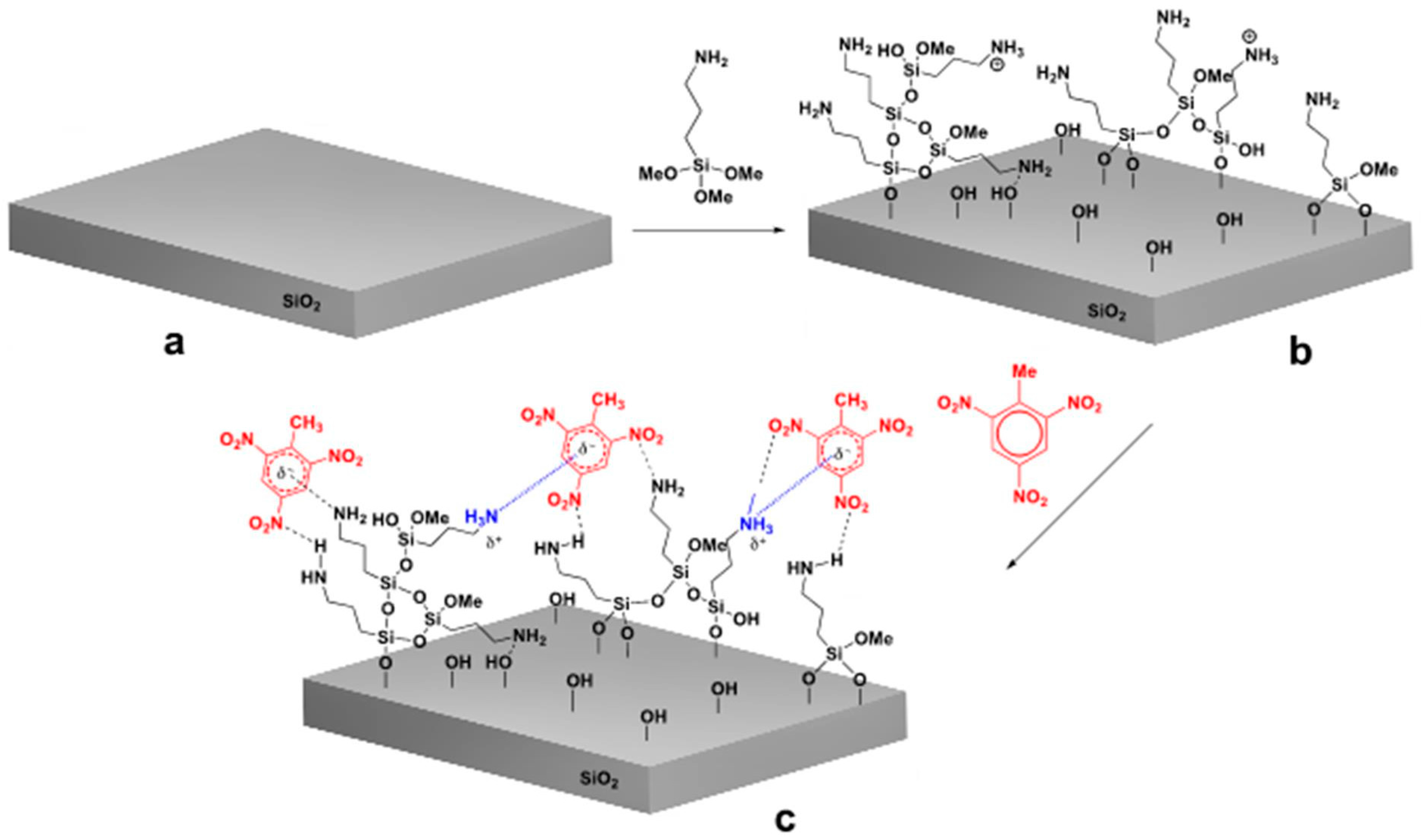
2.2.1. Surface Modifications of the Sensors
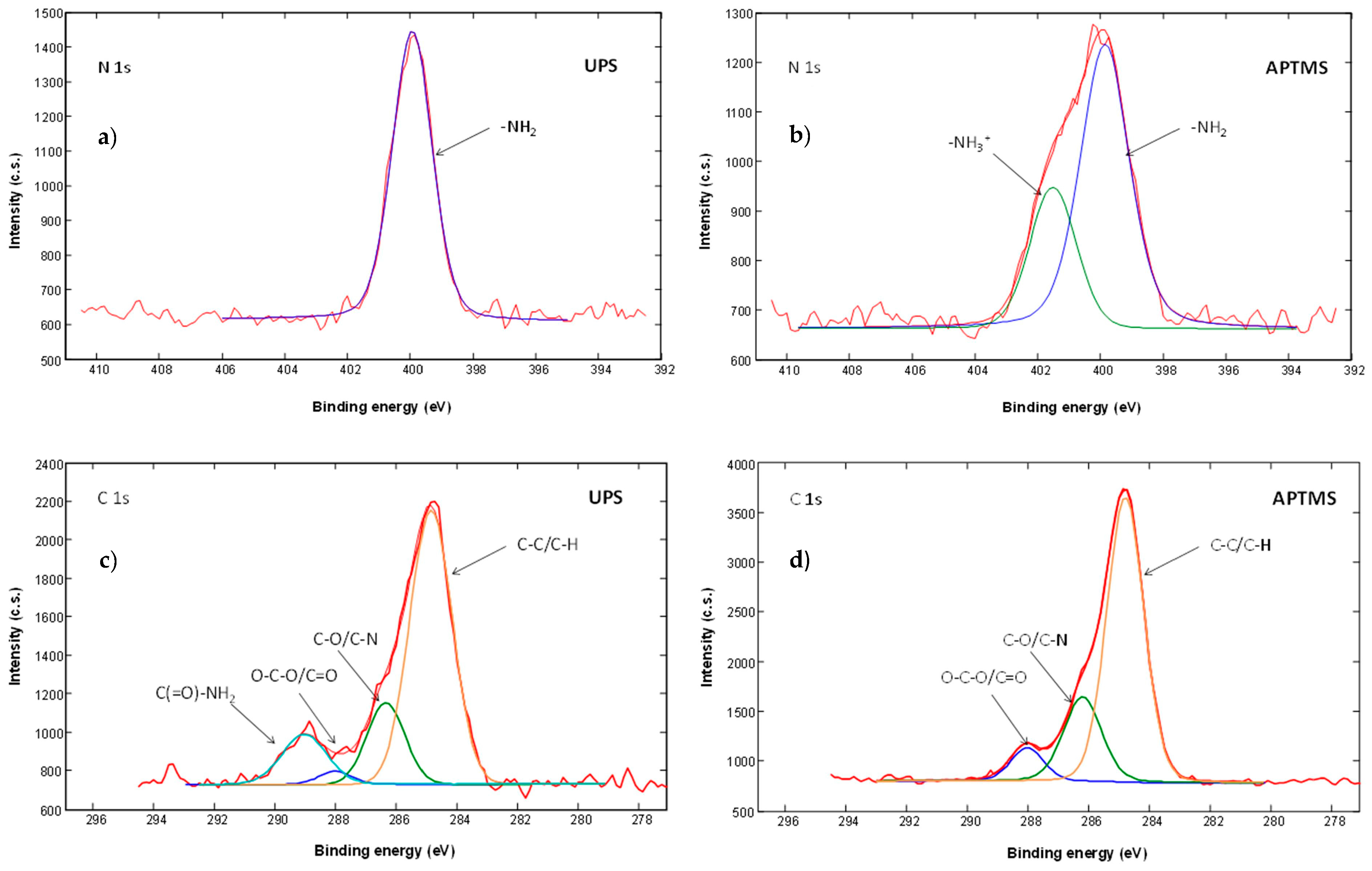
2.2.2. Surface Characterization
2.2.3. Results of Surface Characterization
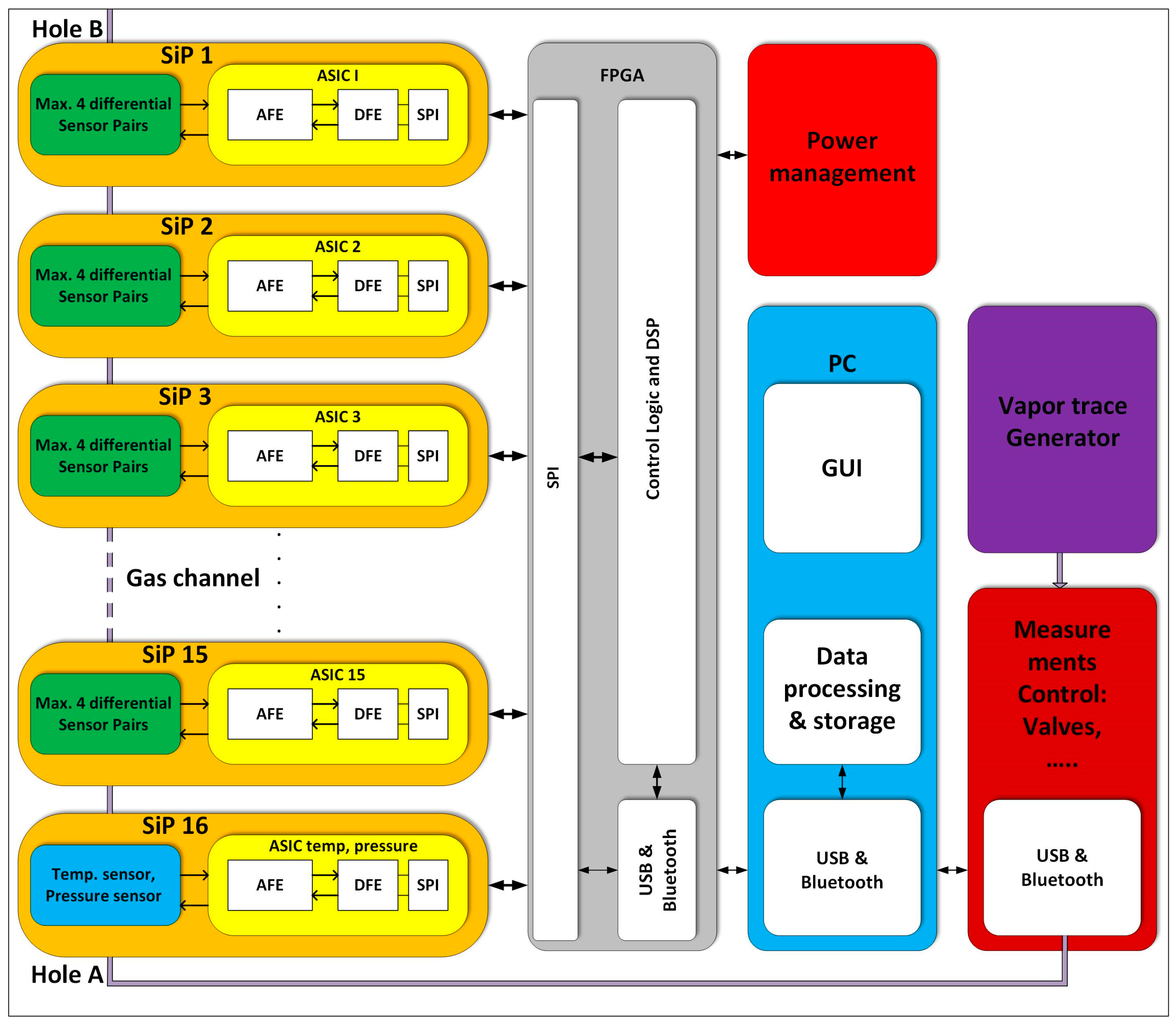
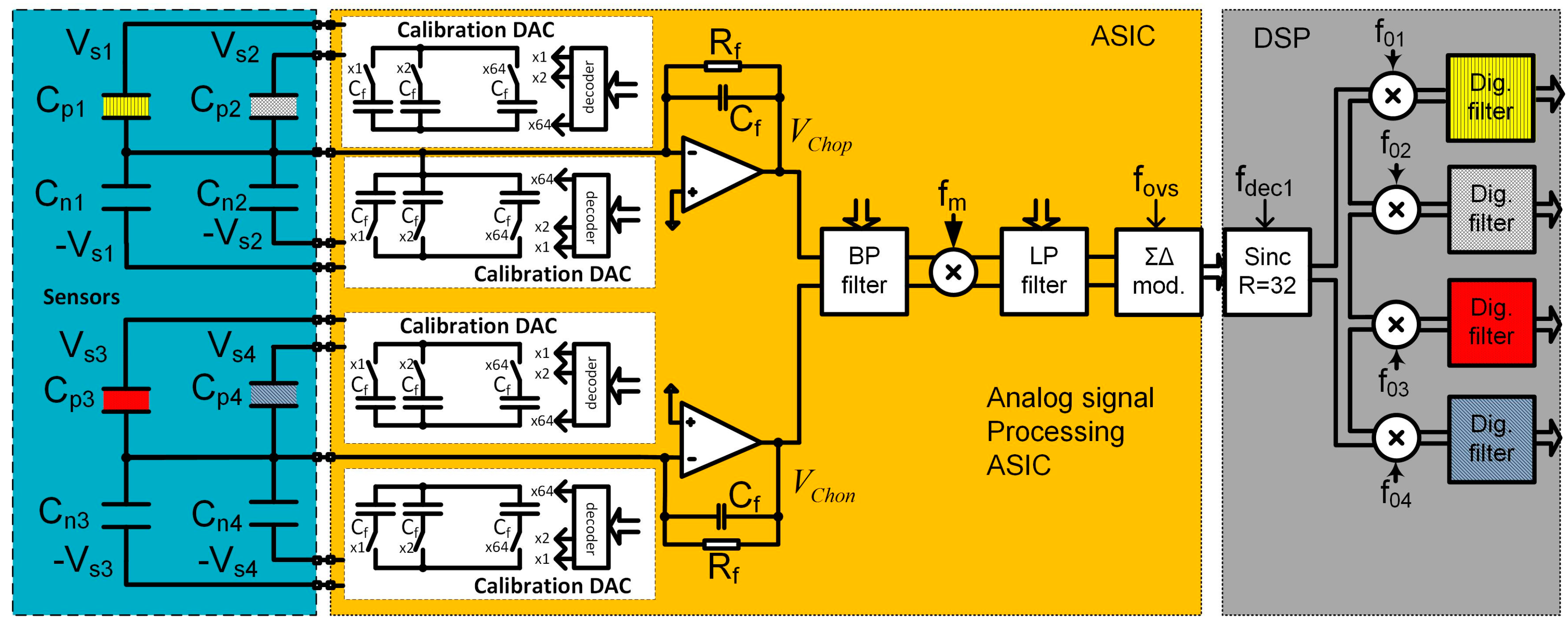
2.3. Electronic Detection System of the 16-Channel E-Nose
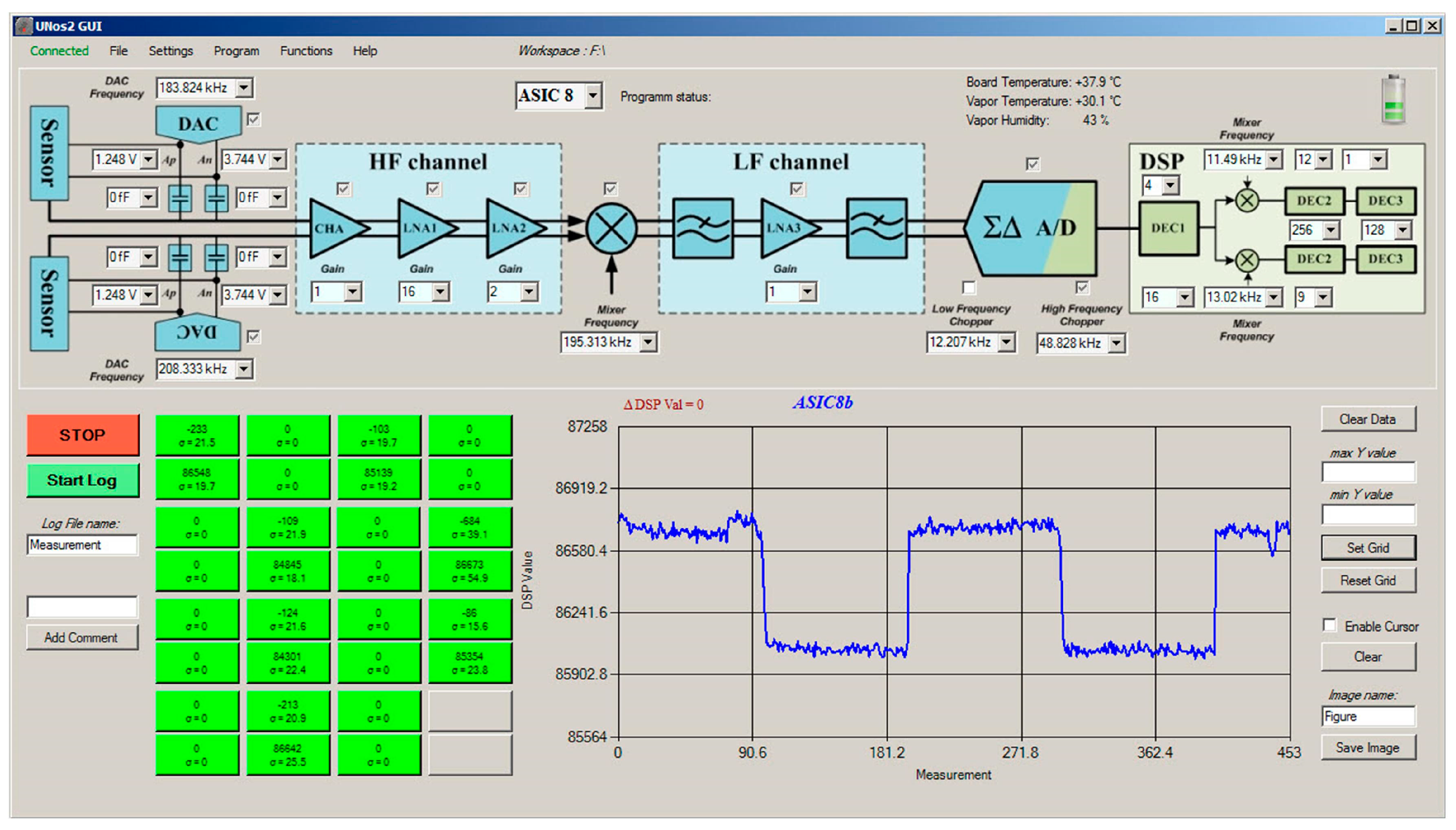
2.3.1. Sixteen-Channels Vapour Trace Detection System
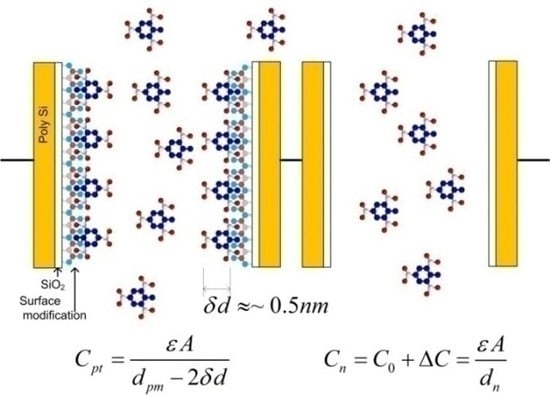
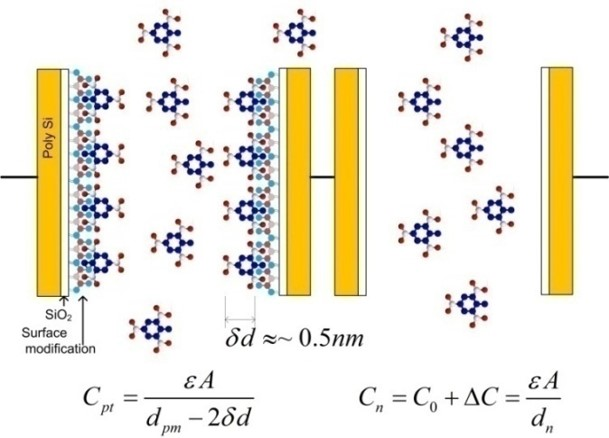
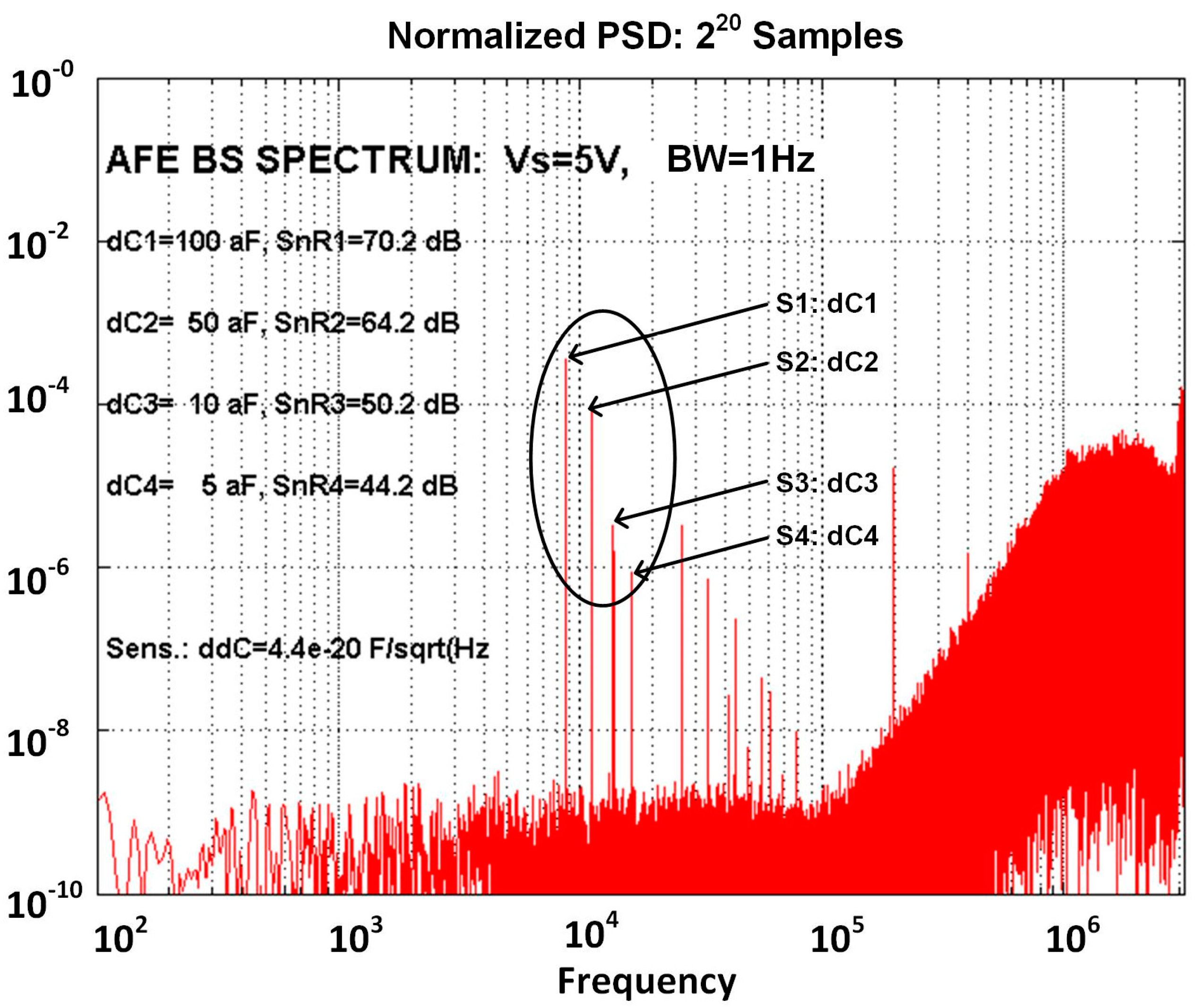
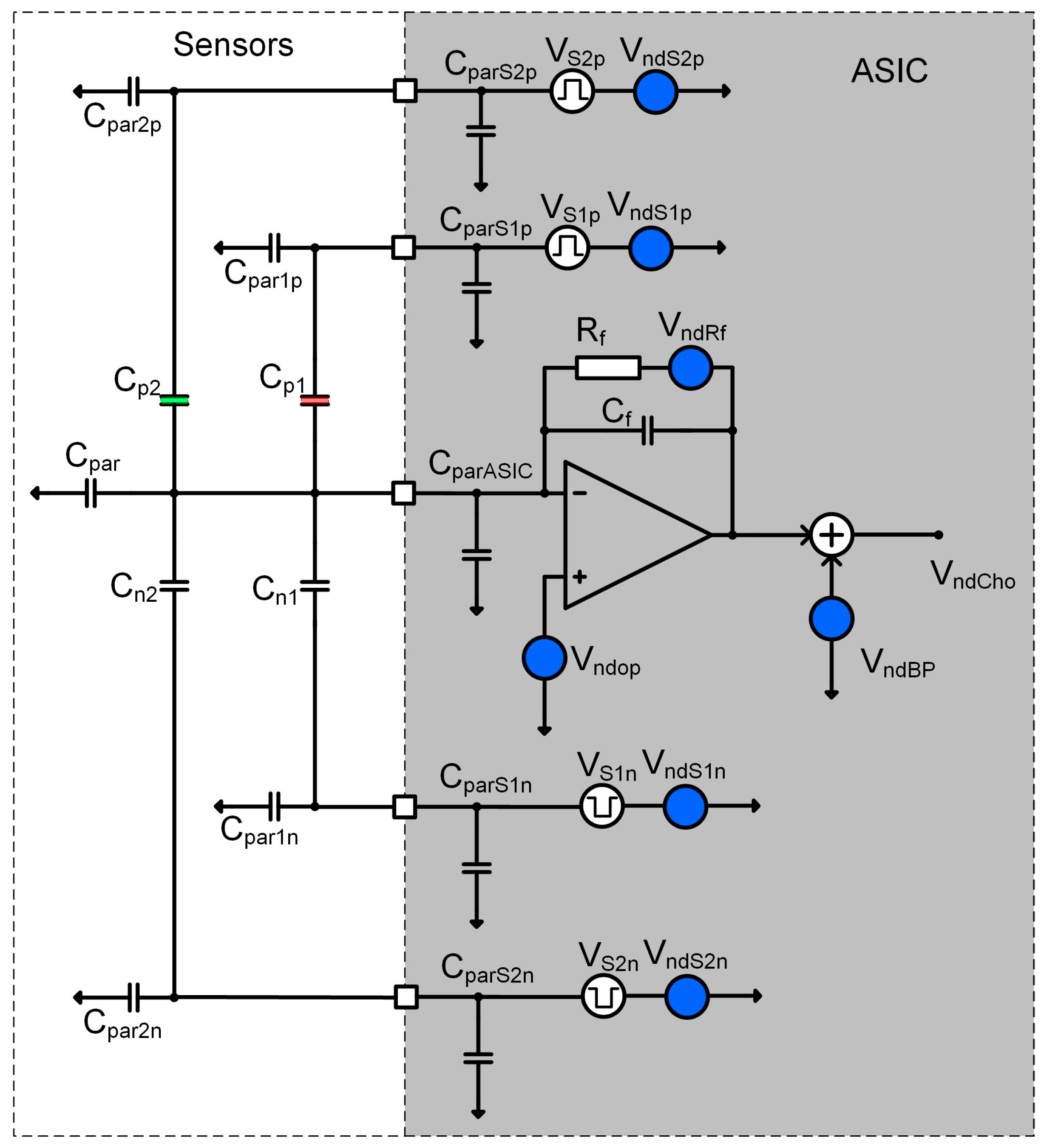
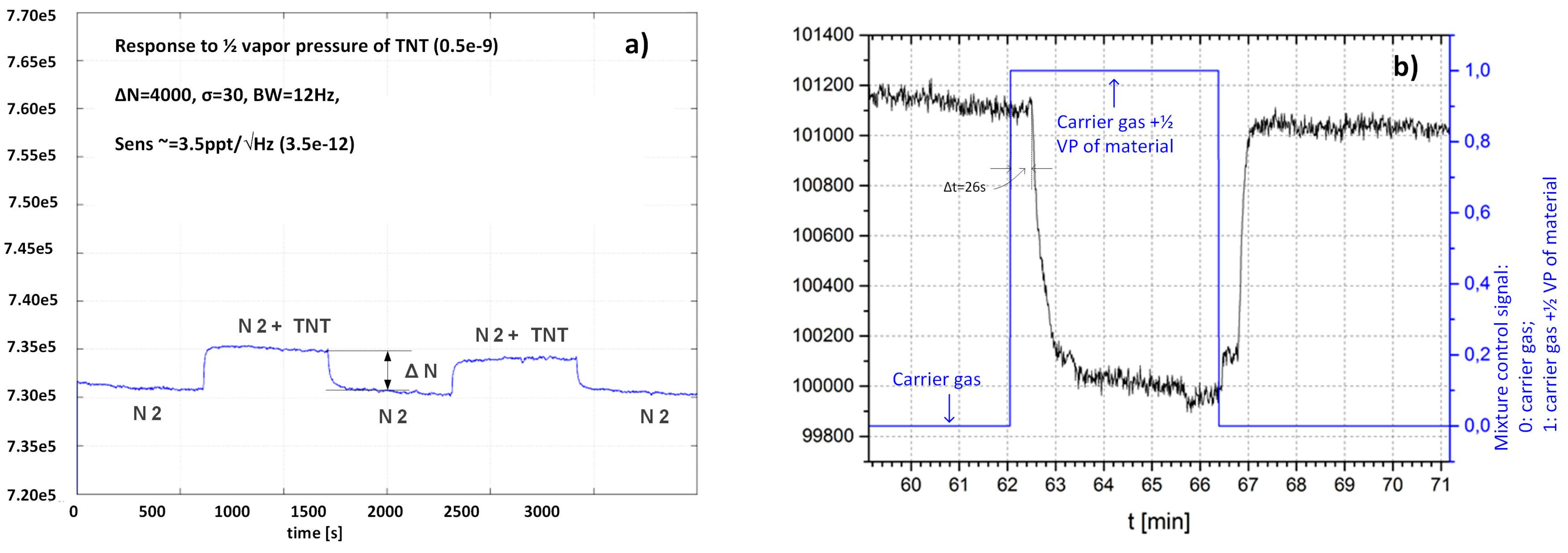
2.3.2. Sensitivity Considerations
- Differential COMB sensor capacitances: (Cpi, Cni), parasitic capacitances (Cparip, Cparin), the thickness and dielectric constant of the adsorption layer, the distance between the plates, etc.
- The noise level of corresponding charge amplifier around excitation signal frequency (Vndop, VndRf),
- The compatibility of the sensor and the charge amplifier, including parasitic capacitances (CparASIC),
- The excitation signal generator frequency (fsi), amplitude (Vsi) and the noise level (VndSpi),
- Input referred noise of all analogue blocks in the chain following charge amplifier (VndBP).
3. Measurements of the E-Nose Responses to Vapour Traces of Explosives and Solvents
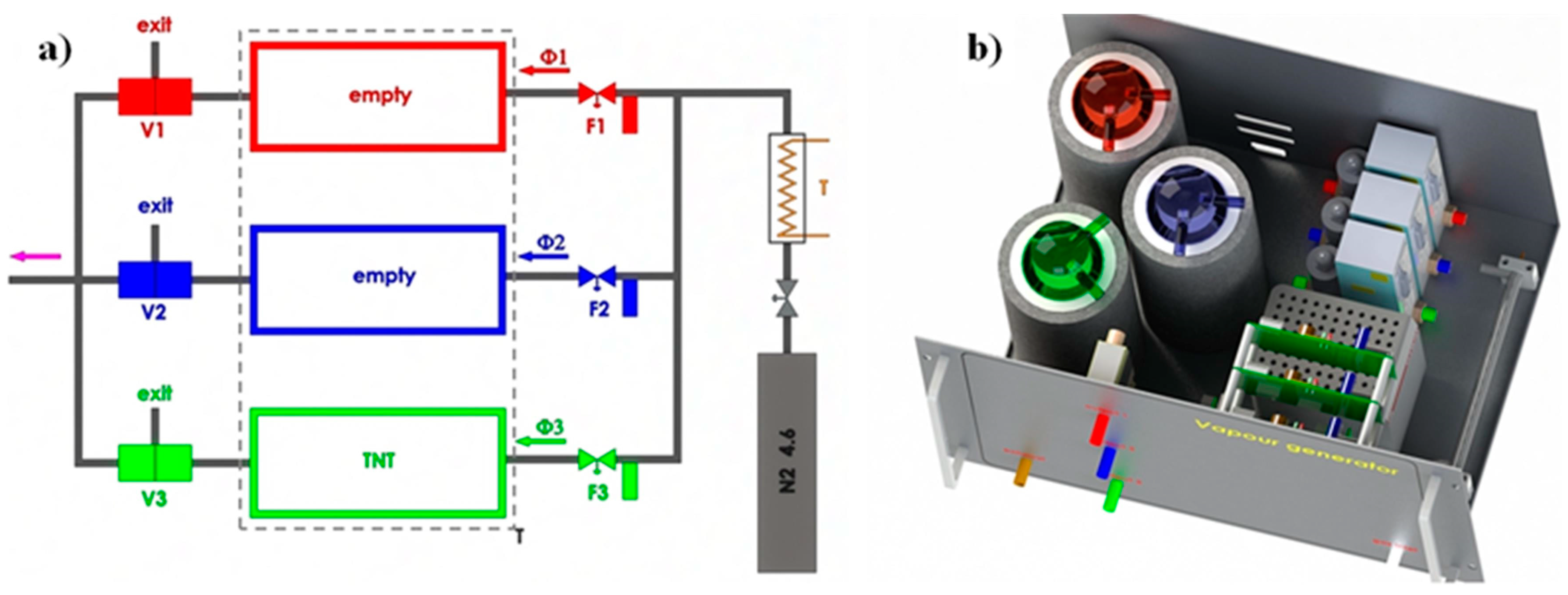
3.1. Experimental Set-Up and Protocols for Measurements Using Vapour Generator
3.2. Experimental Set-Up and Protocols for Sensing Solvents in Ambient Air
3.3. Signal Extraction and Background Correction
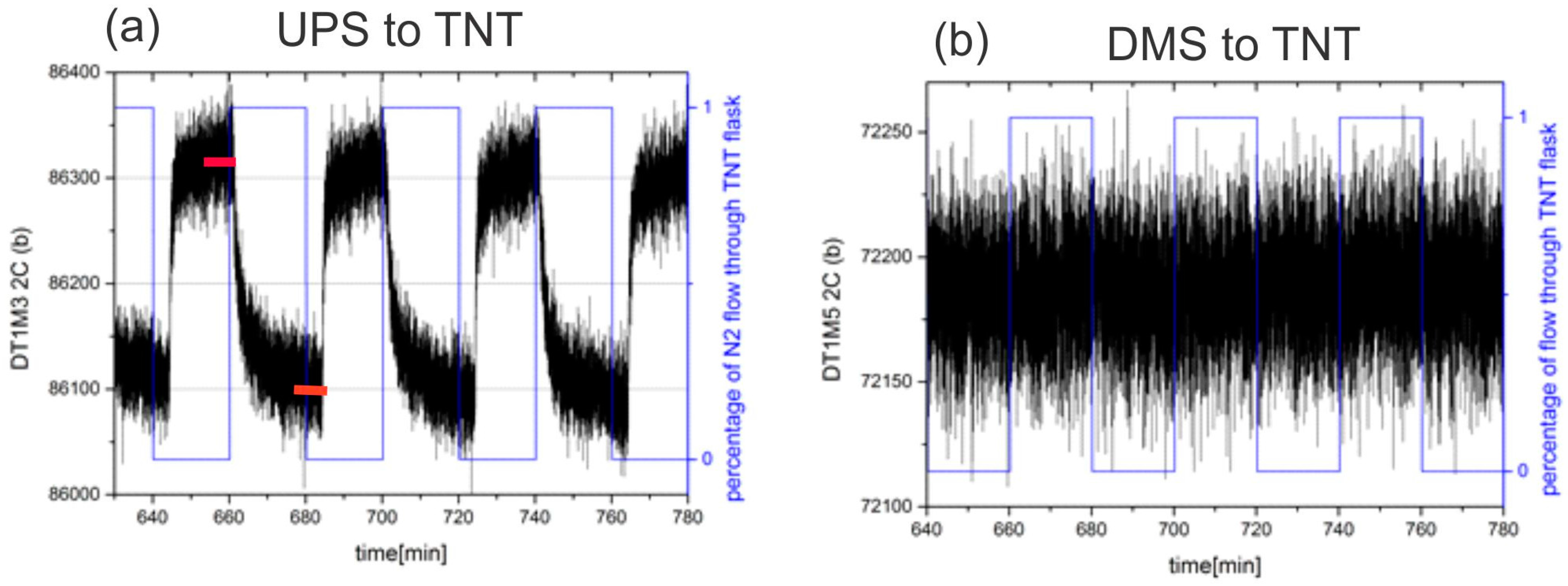
3.4. Measurement Results
3.5. Responses
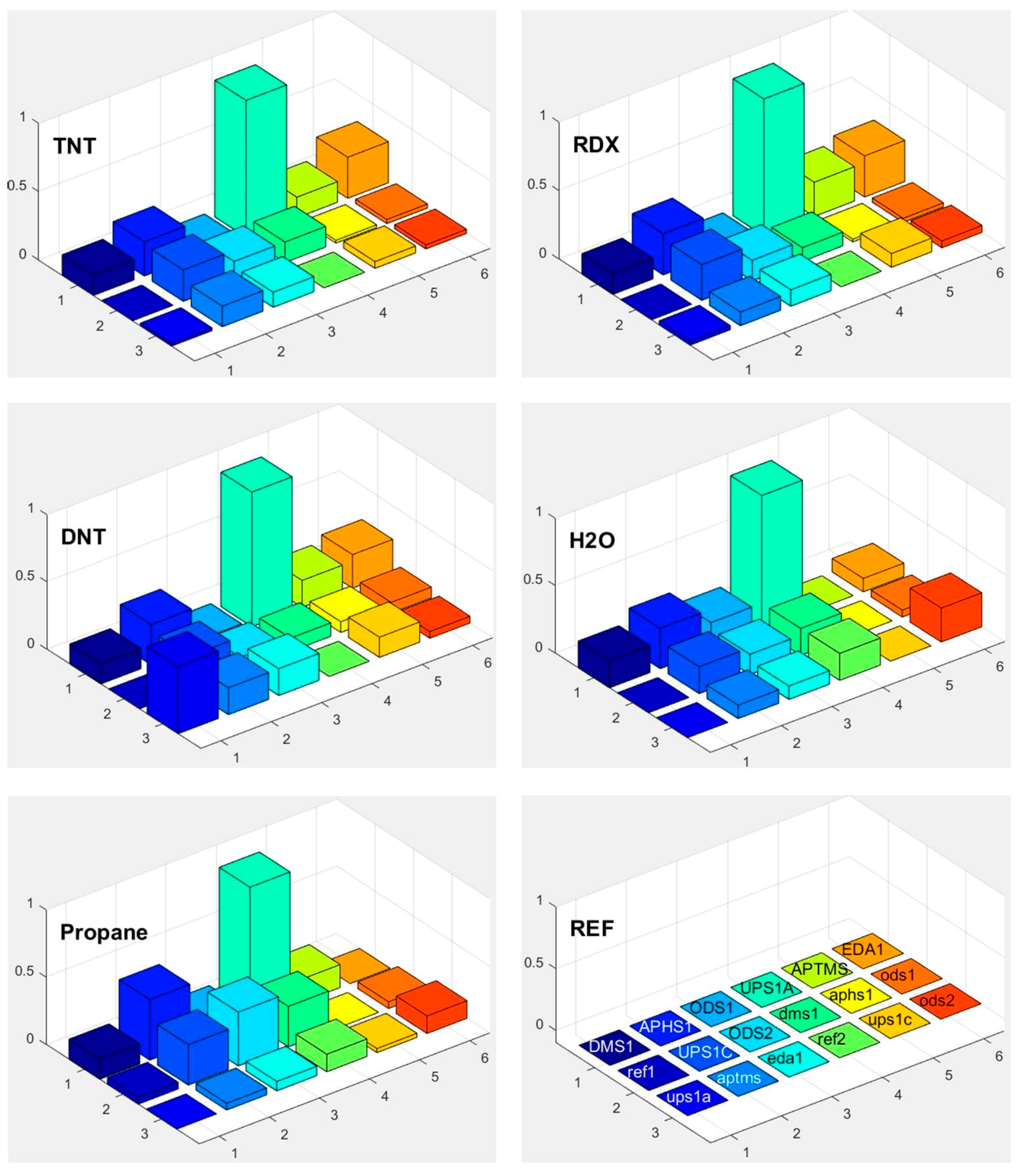
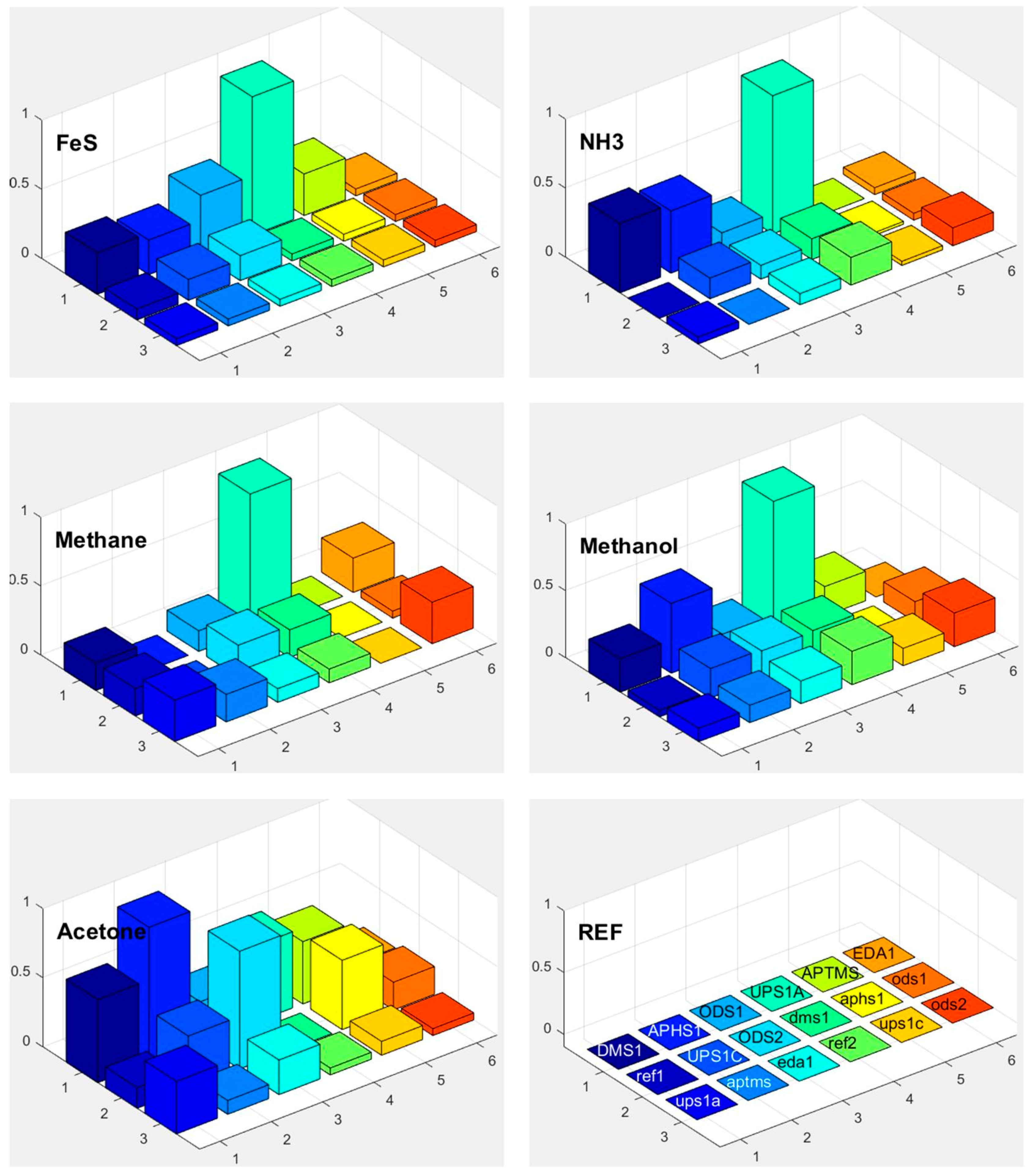
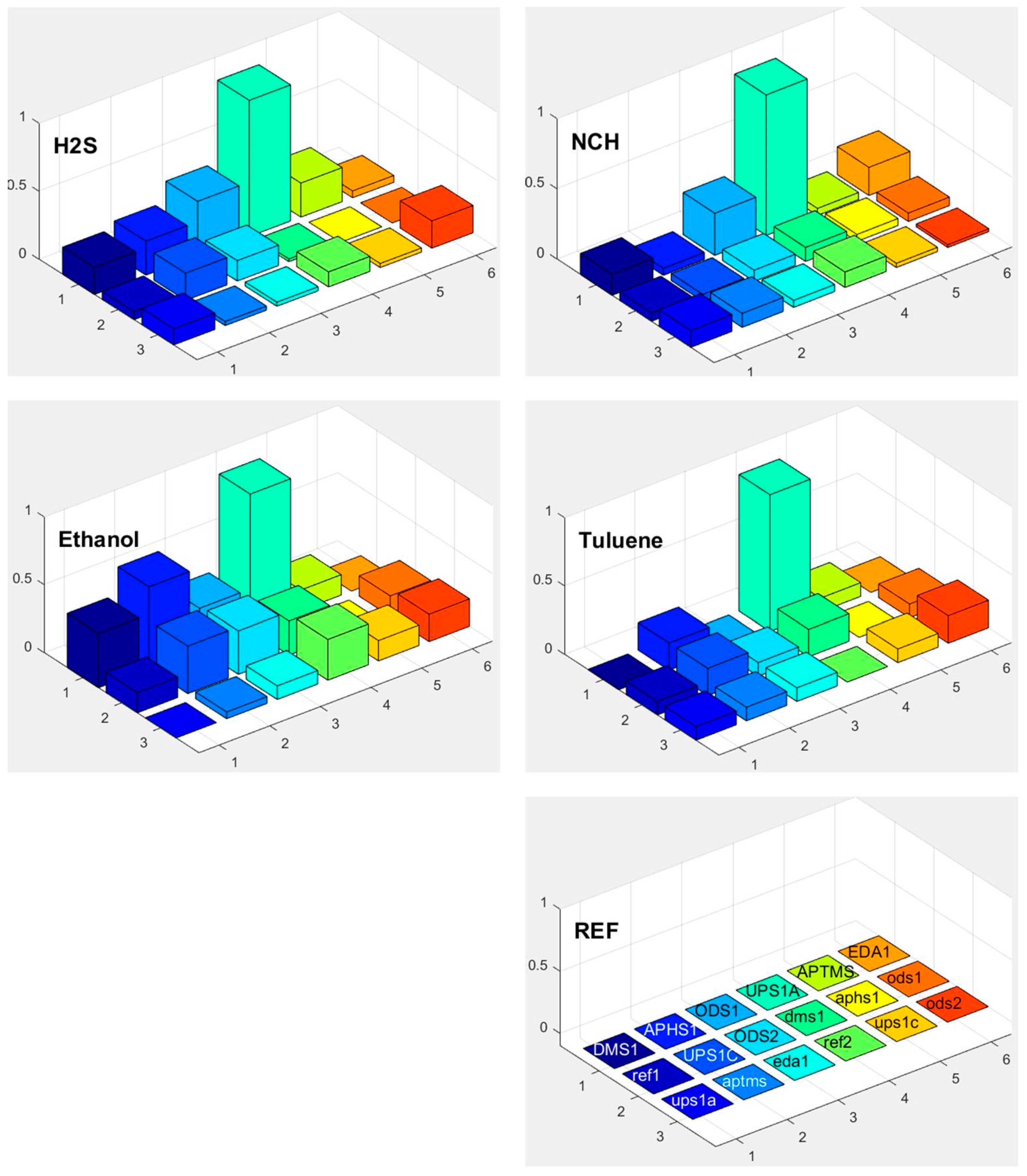
3.6. Selectivity in Surface Adsorption of Different Vapours
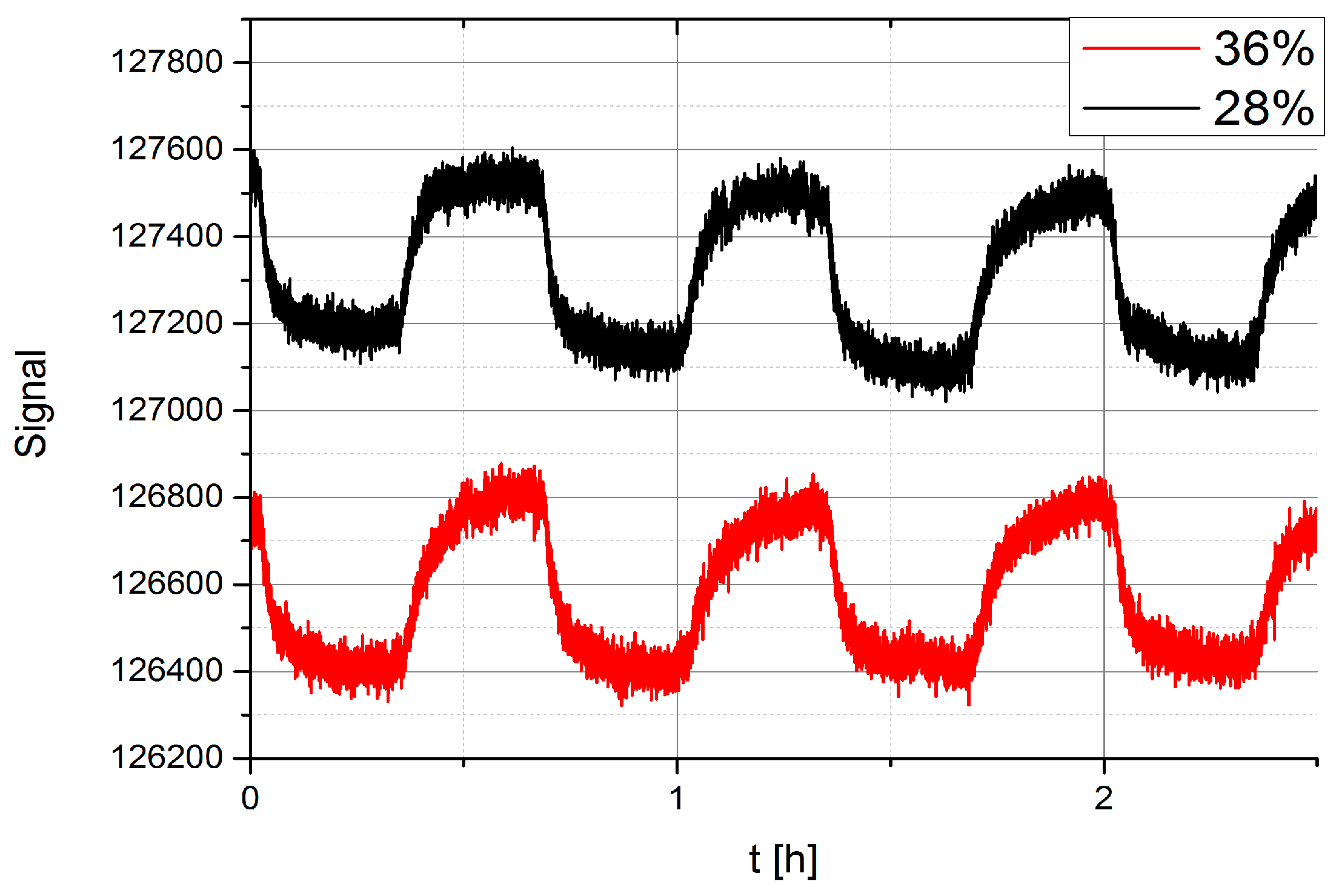
3.7. Sensor Response at Different Background Water Concentration
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Potyrailo, R.A. Multivariable Sensors for Ubiquitous Monitoring of Gases in the Era of Internet of Things and Industrial Internet. Chem. Rev. 2016, 116, 11877–11923. [Google Scholar] [CrossRef] [PubMed]
- Collin, W.R.; Serrano, G.; Wright, L.K.; Chang, H.; Nuñovero, N.; Zellers, E.T. Micro fabricated Gas Chromatograph for Rapid, Trace-Level Determination of Gas-Phase Explosive Marker Compounds. Anal. Chem. 2014, 86, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Gianchandani, Y.B. A Fully Micro-fabricated Gas Chromatograph with Complementary Capacitive Detectors for Indoor Pollutants. Microsyst. Nanoeng. 2016, 2, 15049–15100. [Google Scholar] [CrossRef]
- Pina, M.P.; Almazan, F.; Eguizabal, A.; Pellejero, I.; Urbiztondo, M.; Sese, J.; Santamaria, J.; Garcia-Romeo, D.; Calvo, B.; Medrano, N. Explosives Detection by Array of Si u-Cantilevers Coated with Titanosilicate-Type Nanoporous Materials. IEEE Sens. J. 2016, 16, 3435–3443. [Google Scholar] [CrossRef]
- Wang, J.; Yang, L.; Boriskina, S.; Yan, B.; Reinhard, B.M. Spectroscopic Ultra-Trace Detection of Nitroaromatic Gas Vapor on Rationally Designed Two-Dimensional Nanoparticle Cluster Arrays. Anal. Chem. 2011, 83, 2243–2249. [Google Scholar] [CrossRef] [PubMed]
- Potyrailo, R.A.; Larsen, M.; Riccobono, O. Detection of Individual Vapours and Their Mixtures Using a Selectivity Tenable Three-Dimensional Network of Plasmonic Nanoparticles. Angew. Chem. 2013, 125, 10550–10554. [Google Scholar] [CrossRef]
- Nagarkar, S.S.; Joarder, B.; Chaudhari, A.K.; Mukherjee, S.; Ghosh, S.K. Highly Selective Detection of Nitro Explosives by a Luminiscent Metal-Organic Framework. Angew. Chem. 2013, 125, 2953–2957. [Google Scholar] [CrossRef]
- Strle, D.; Stefane, B.; Nahtigal, U.; Zupanic, E.; Pozgan, F.; Kvasic, I.; Macek, M.; Trontelj, J.; Musevic, I. Surface-Functionalized COMB Capacitive Sensors and Electronics for Vapour Trace Detection of Explosives. IEEE Sens. J. 2012, 12, 1048–1057. [Google Scholar] [CrossRef]
- Strle, D.; Štefane, B.; Zupanič, E.; Trifkovič, M.; Maček, M.; Jakša, G.; Kvasič, I.; Muševič, I. Sensitivity Comparison of Vapor Trace Detection of Explosives Based on Chemo-Mechanical Sensing with Optical Detection and Capacitive Sensing with Electronic Detection. Sensors 2014, 14, 11467–11491. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Tillman, E.S.; Lewis, N.S. Detection and Classification of Volatile Organic Amines and Carboxylic Acids Using Arrays of Carbon Black-Dendrimer Composite Vapor Detectors. Chem. Mater. 2005, 17, 2904–2911. [Google Scholar] [CrossRef]
- Sysoev, V.V.; Kiselev, I.; Trouillet, V.; Bruns, M. Enhancing the gas selectivity of single-crystal SnO2:Pt thin-film chemiresistor microarray by SiO2 membrane coating. Sens. Actuators B Chem. 2013, 185, 59–69. [Google Scholar] [CrossRef]
- Mori, K.; Nagao, H.; Yoshihara, Y. The olfactory bulb: Coding and processing of odor molecule information. Science 1999, 286, 711–715. [Google Scholar] [CrossRef] [PubMed]
- Crooks, M.R.; Ricco, A.J. New Organic Materials Suitable for Use in Chemical Sensor Arrays. Acc. Chem. Res. 1998, 31, 219–227. [Google Scholar] [CrossRef]
- Crego-Calama, M.; Reinhoudt, D.N. New Materials for Metal Ion Sensing by Self Assembled Monolayers on Glass. Adv. Mater. 2001, 13, 1171–1174. [Google Scholar] [CrossRef]
- George, S.M.; Yoon, B.; Dameron, A.A. Surface Chemistry for Molecular Layer Deposition of Organic and Hybrid Organic-Inorganic Polymers. Acc. Chem. Res. 2009, 42, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Jiang, Y.; Zhao, H.; Zhu, N.; Lin, Y.; Yu, P. A Simple Assay of Direct Colorimetric Visualisation of Trinitrotoluene at Pico-molar Levels Using Gold Nanoparticles. Angew. Chem. Int. Ed. 2008, 120, 8729–8732. [Google Scholar]
- Engel, Y.; Elnathan, R.; Pevzner, A.; Davidi, G.; Flaxer, E.; Patolsky, F. Cover Picture: Supersensitive Detection of Explosives by Silicon Nanowire Arrays (Angew. Chem. Int. Ed. 38/2010). Angew. Chem. Int. Ed. 2010, 49, 6685. [Google Scholar] [CrossRef]
- Trogler, W.C.; Sohn, H.; Sailor, M.J.; Magde, D. Detection of Nitro-aromatic Explosives Based on Photoluminescence Polymers Containing Metalloles. J. Am. Chem. Soc. 2003, 125, 3821–3830. [Google Scholar]
- Jakša, G.; Štefane, B.; Kovač, J. Influence of Different Solvents on the Morphology of APTMS-Modified Silicon Surfaces. App. Surf. Sci. 2014, 315, 516–522. [Google Scholar] [CrossRef]
- Jakša, G.; Štefane, B.; Kovač, J. XPS and AFM characterization of aminosilanes with different numbers of bonding sites on a silicon wafer. Surf. Interface Anal. 2013, 45, 1709–1713. [Google Scholar] [CrossRef]
- Jakša, G. AFM and XPS Study of Aminosilanes on Si. Imaging Microsc. 2014, 2, 22–25. [Google Scholar]
- Jakša, G.; Kovač, J.; Štefane, B. XPS—In AFM Preiskava Silicijevih Površin, Modificiranih z Različnimi Aminosilani. Vakuumist 2014, 34, 4–8. [Google Scholar]
- Shircliff, R.A.; Stradins, P.; Moutinho, H.; Fennell, J.; Ghirardi, M.L.; Cowley, S.W.; Branz, H.M.; Martin, I.T. Angle-Resolved XPS Analysis and Characterization of Monolayer and Multilayer Silane Films for DNA Coupling to Silica. Langmuir 2013, 29, 4057–4067. [Google Scholar] [CrossRef] [PubMed]
- Keegan, N.; Suárez, G.; Spoors, J.A.; Ortiz, P.; Hedley, J.; McNeil, C.J. A microfabrication compatible approach to 3-Dimensonal patterning of bio-molecules at bio-MEMS and biosensor surfaces. In Proceedings of the Biomedical Circuits and Systems Conference, Beijing, China, 26–28 November 2009; pp. 17–20. [Google Scholar]
- Tran, T.H.; Lee, J.-W.; Lee, K.; Lee, Y.D.; Ju, B.-K. The gas sensing properties of single-walled carbon nanotubes deposited on an aminosilane monolayer. Sens. Actuators B Chem. 2007, 129, 67–71. [Google Scholar] [CrossRef]
- Moulder, J.F.; Stickle, W.F.; Sobol, P.E.; Bomben, K.D. Handbook of X-ray Photoelectron Spectroscopy; Physical Electronics Inc.: Eden Praire, MN, USA, 2008. [Google Scholar]
- Chen, J.; Xu, P.; Li, X. Self-assembling siloxabe bilayer directly on SiO2 surface of micro cantilevers for long term highly repeatable sensing to trace explosives. IOP Sci. Nanotechnol. 2010, 21, 1–10. [Google Scholar]
- Gradišek, A.; Slapničar, G.; Šorn, J.; Luštrek, M.; Gams, M.; Grad, J. Predicting Species Identity of Bumblebees through Analysis of Flight Buzzing Sounds. Bioacoustics 2017, 26, 63–76. [Google Scholar] [CrossRef]
- Esteva, A.; Kuprel, B.; Novoa, R.A.; Ko, J.; Swetter, S.M.; Blau, H.M.; Thrun, S. Dermatologist-level classification of skin cancer with deep neural networks. Nature 2017, 542, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Voosen, P. The AI detectives. Science 2017, 357, 22–27. [Google Scholar] [CrossRef] [PubMed]





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | C (at. %) | O (at. %) | Si (at. %) | N (at. %) |
|---|---|---|---|---|
| Reference (blank) sensor | 9.9 | 47.3 | 42.8 | |
| APTMS | 34.9 | 34.5 | 25.1 | 5.5 |
| APhS | 33.6 | 33.4 | 29.6 | 3.4 |
| UPS | 20.8 | 42.8 | 31.8 | 4.6 |
| EDA | 21.6 | 43.7 | 32 | 2.7 |
| DMS | 12.1 | 48.5 | 38 | 1.4 |
| ODS | 17.5 | 42.3 | 40.2 |
| Vapour | H2O | TNT | DNT |
|---|---|---|---|
| Density Ns(x)/N(N2) | 10−2 | 10−9 | 10−6 |
| Response ΔR | 1.2 × 10+3 | 4 × 10+3 | 1.7 × 10+3 |
| Scaled response Dscaled | 1.2 × 10+5 | 4 × 10+12 | 1.7 × 10+9 |
| Scaled response relative to H2O | 1 | 3.3 × 10+7 | 1.4 × 10+4 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strle, D.; Štefane, B.; Trifkovič, M.; Van Miden, M.; Kvasić, I.; Zupanič, E.; Muševič, I. Chemical Selectivity and Sensitivity of a 16-Channel Electronic Nose for Trace Vapour Detection. Sensors 2017, 17, 2845. https://doi.org/10.3390/s17122845
Strle D, Štefane B, Trifkovič M, Van Miden M, Kvasić I, Zupanič E, Muševič I. Chemical Selectivity and Sensitivity of a 16-Channel Electronic Nose for Trace Vapour Detection. Sensors. 2017; 17(12):2845. https://doi.org/10.3390/s17122845
Chicago/Turabian StyleStrle, Drago, Bogdan Štefane, Mario Trifkovič, Marion Van Miden, Ivan Kvasić, Erik Zupanič, and Igor Muševič. 2017. "Chemical Selectivity and Sensitivity of a 16-Channel Electronic Nose for Trace Vapour Detection" Sensors 17, no. 12: 2845. https://doi.org/10.3390/s17122845