The Use of Flow-Injection Analysis with Chemiluminescence Detection of Aqueous Ferrous Iron in Waters Containing High Concentrations of Organic Compounds

Abstract
:
1. Introduction
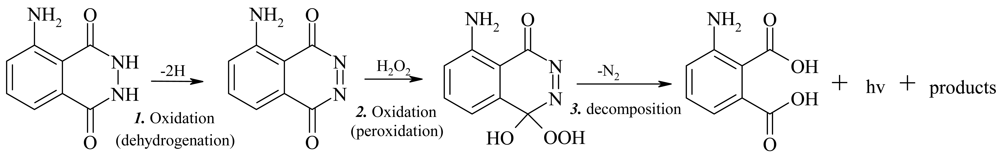
1.1. Chemiluminescence of Luminol
2. Experimental
2.1. Reagents and Samples
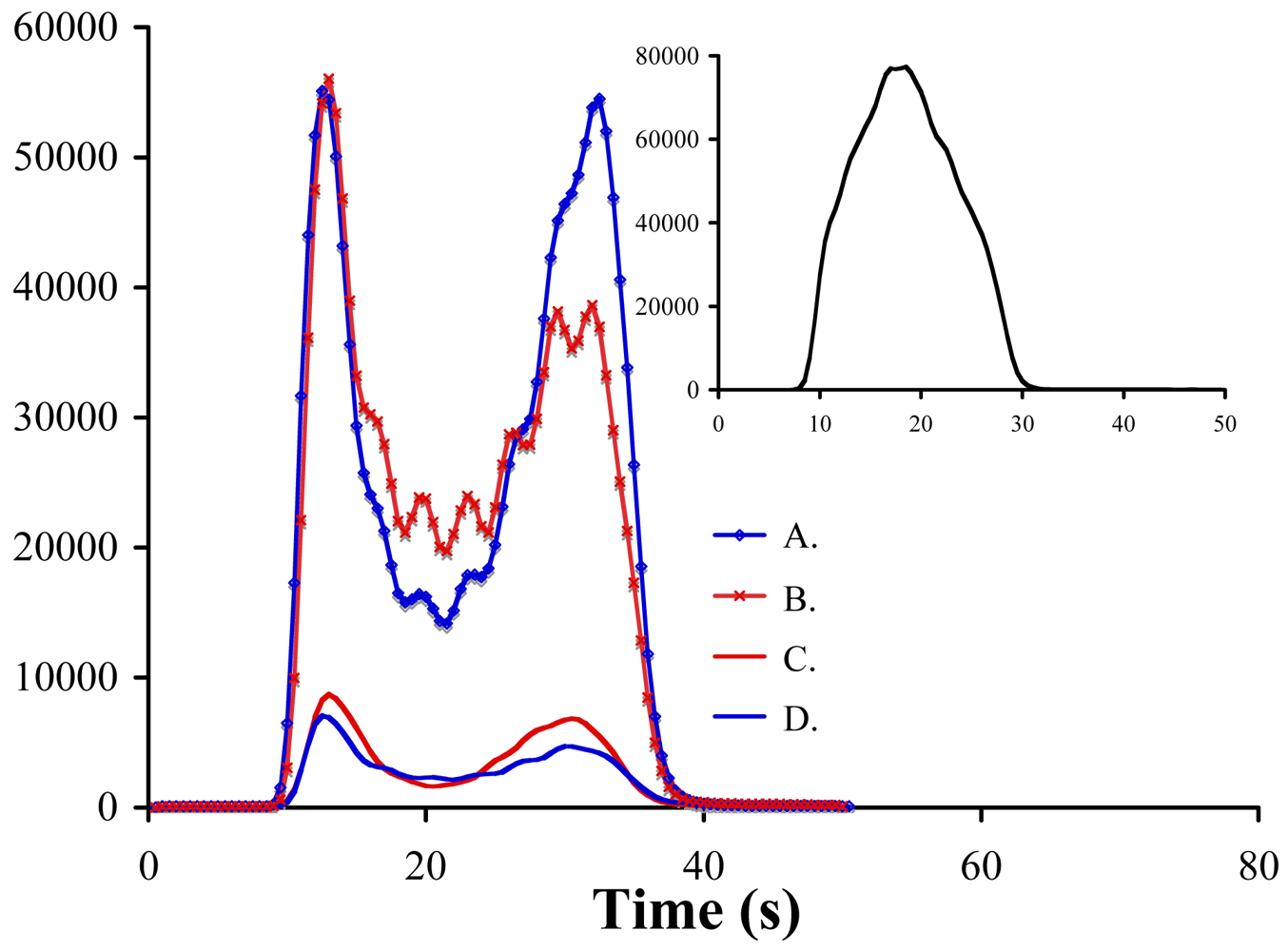
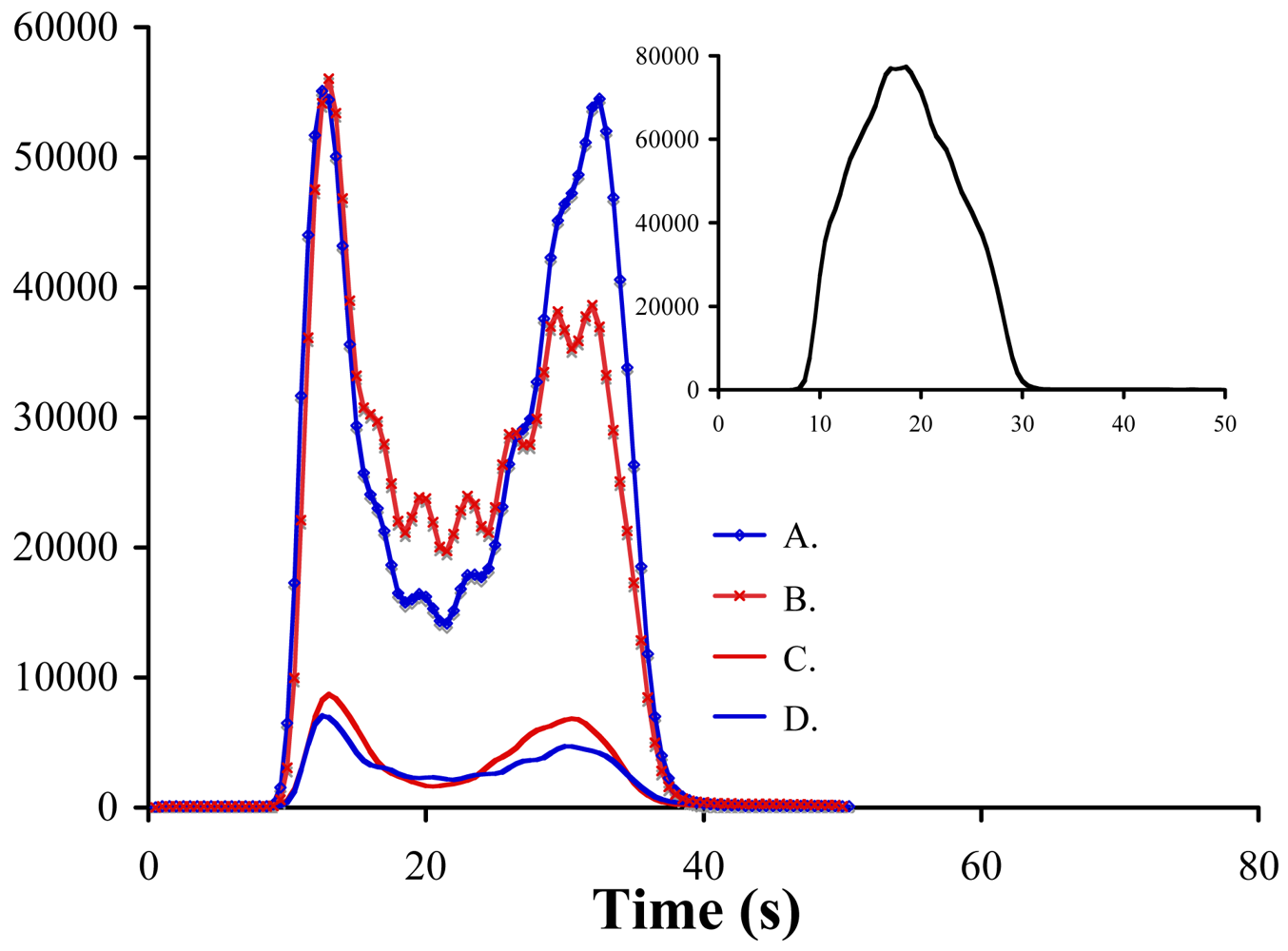
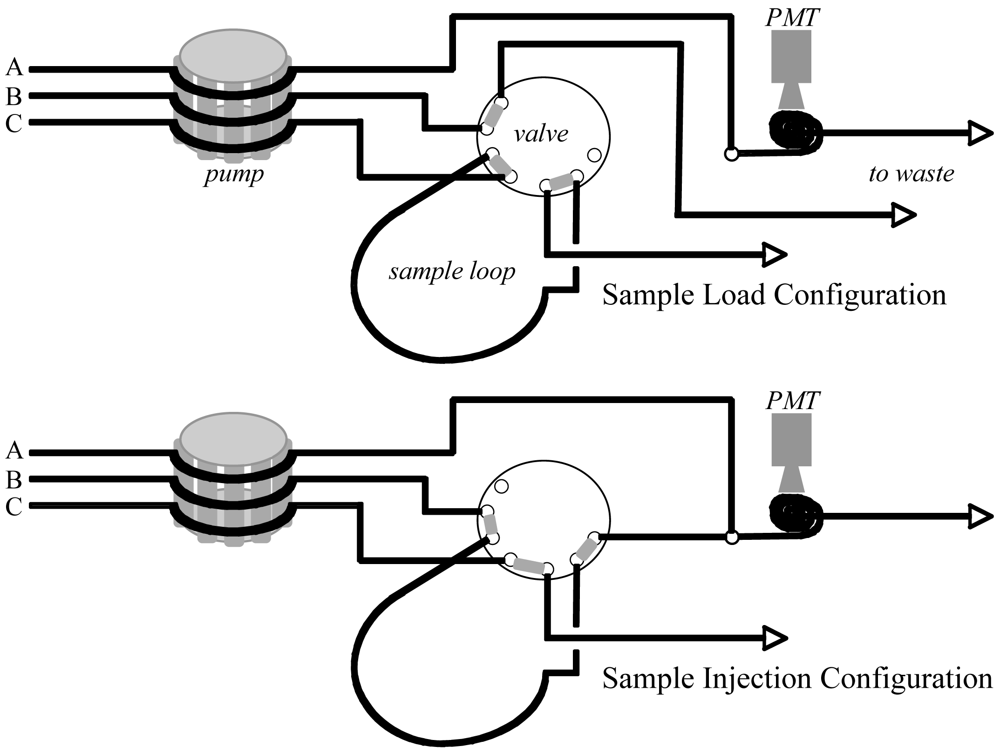
2.2. Instrumentation and Glassware
3. Results and Discussion
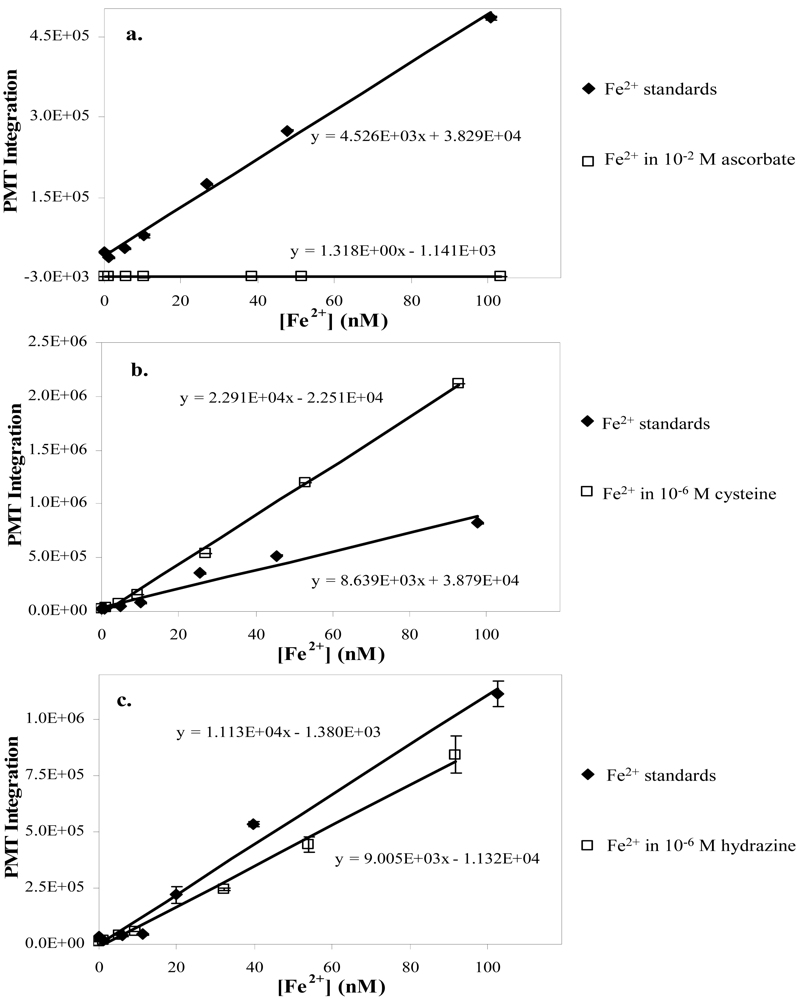
3.1. Chelators and Reductants
3.1.1. Ascorbate and oxalate
3.1.2. Cysteine and glycine
3.1.3. Hydroxylamine and hydrazine
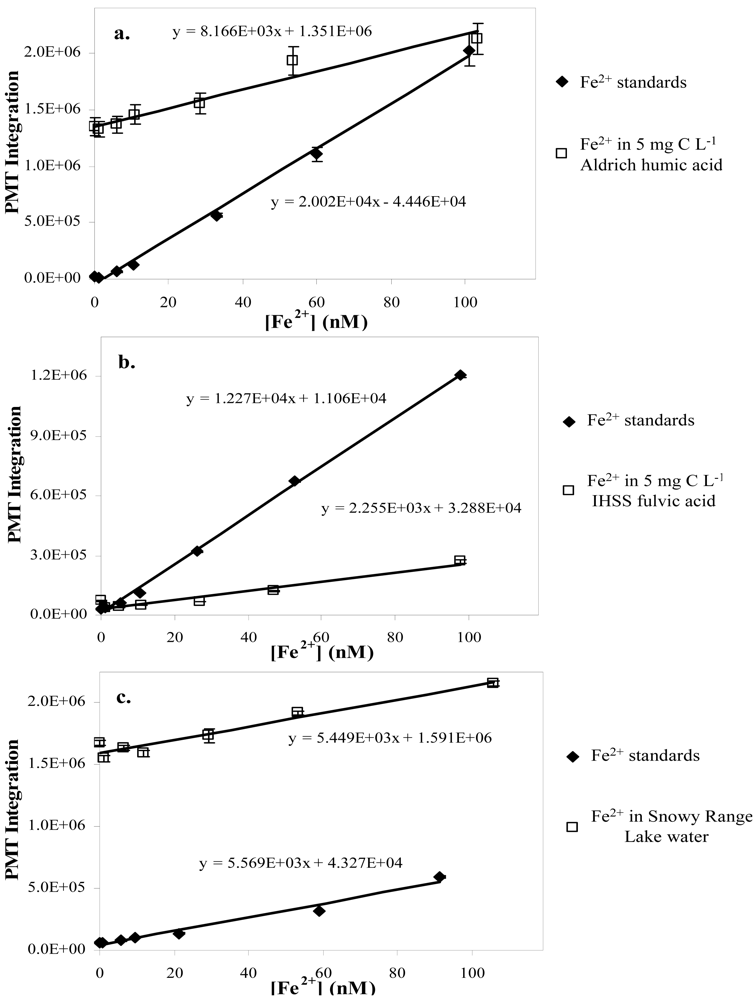
3.2. Natural Waters, Humic and Fulvic Acids
3.2.1. Humic and fulvic acids
3.2.2. Natural water samples
3.3. Method Evaluation
3.4. Recommendations
Acknowledgments
References
- Millward, G.E.; Moore, R.M. The adsorption of Cu, Mn and Zn by iron oxyhydroxide in model estuarine solutions. Water Res. 1982, 16, 981–985. [Google Scholar]
- Tessier, A.; Rapin, F.; Carignan, R. Trace metals in oxic lake sediments: Possible adsorption onto iron oxyhydroxides. Geochim. Cosmochim. Acta 1985, 49, 183–194. [Google Scholar]
- Ehrlich, H.L. Geomicrobiology; Marcel Dekker, Inc.: New York, NY, USA, 1990. [Google Scholar]
- Cox, C.D. Importance of Iron in Bacterial Virulence. In Metal Ions and Bacteria; Beveridge, T.J., Doyle, R.J., Eds.; John Wiley & Sons: New York, NY, USA, 1989. [Google Scholar]
- Harrison, P.M.; Hoare, R.J. Metals in Biochemistry; Chapman and Hall: New York, NY, USA, 1980. [Google Scholar]
- Afonso, M.D.S.; Morando, P.J.; Blesa, M.A.; Banwart, S.; Stumm, W. The reductive dissolution of iron oxides by ascorbate. The role of carboxylate anions in accelerating reductive dissolution. J. Colloid Interface Sci. 1990, 138, 74–82. [Google Scholar]
- Suter, D.; Siffert, C.; Sulzberger, B.; Stumm, W. Catalytic dissolution of iron(III) (hydr)oxides by oxalic acid in the presence of Fe(II). Naturwissenschaften 1988, 75, 571–573. [Google Scholar]
- Suter, D.; Banwart, S.; Stumm, W. Dissolution of hydrous iron(III) oxides by reductive mechanisms. Langmuir 1991, 7, 809–813. [Google Scholar]
- Torres, R.; Blesa, M.A.; Matijevic, E. Interactions of metal oxides with chelating agents. IX. Reductive dissolution of hematite and magnetite by aminocarboxylic acids. J. Colloid Interface Sci. 1990, 134, 475–485. [Google Scholar]
- Zinder, B.; Gerhard, F.; Stumm, W. The coordination chemistry of weathering: II. Dissolution of Fe(III) oxides. Geochim. Cosmochim. Acta 1986, 50, 1861–1869. [Google Scholar]
- Butcher, S.S.; Charlson, R.J.; Orians, G.H.; Wolfe, G.V. Global Biogeochemical Cycles; Academic Press Limited: London, UK, 1992. [Google Scholar]
- Brown, D.A.; Sherriff, B.L.; Sawicki, J.A.; Sparling, R. Precipitation of iron minerals by a natural microbial consortium. Geochim. Cosmochim. Acta 1999, 63, 2163–2169. [Google Scholar]
- Greenwood, N.N.; Earnshaw, A. Chemistry of the Elements, 2nd ed; Butterworth-Heinemann: Oxford, UK, 2002. [Google Scholar]
- Stumm, W.; Morgan, J.J. Aquatic Chemistry - An Introduction Emphasizing Chemical Equilibria in Natural Waters, 2nd ed; John Wiley & Sons: New York, NY, USA, 1981. [Google Scholar]
- Pehkonen, S. Determination of the oxidation states of iron in natural waters: A review. Analyst 1995, 120, 2655–2663. [Google Scholar]
- Figueiredo, A.A. Determination of iron by EDTA titrimetry of thiocyanate complex. Analyt. Chem. 1971, 43, 484–485. [Google Scholar]
- Sun, B.; Ye, Y.; Huang, H.; Bai, Y. Potentiometric determination of iron using a fluoride ion-selective electrode - the application of the Apple II-ISE intelligent ion analyzer. Talanta 1993, 40, 891–895. [Google Scholar]
- Jagner, D.; Renman, L.; Stefansdottir, S.H. Determination of iron(III) and titanium(IV) as their solchrome violet RS complexes by constant-current stripping voltammetry. Part 1. Automated single point calibration method for iron(III). Analyt. Chim. Acta 1993, 281, 305–314. [Google Scholar]
- Florence, T.M. Trace element speciation by anodic stripping voltammetry. Analyst 1992, 117, 551–553. [Google Scholar]
- Badocco, D.; Pastore, P.; Favaro, G.; Macca, C. Effect of eluent composition and pH and chemiluminescent reagent pH on ion chromatographic selectivity and luminol-based chemiluminescence detection of Co2+, Mn2+ and Fe2+ at trace levels. Talanta 2007, 72, 249–255. [Google Scholar]
- Schlosser, C.; Croot, P.L. Application of cross-flow filtration for determining the solubility of iron species in open ocean seawater. Limnol. Oceanogr. Methods 2008, 6, 630–642. [Google Scholar]
- Hase, U.; Yoshimura, K. Determination of trace amounts of iron in highly purified water by ion-exchanger phase absorptiometry combined with flow analysis. Analyst 1992, 117, 1501–1506. [Google Scholar]
- Stookey, L.L. Ferrozine - A new spectrophotometric reagent for iron. Analyt. Chem. 1970, 42, 779–781. [Google Scholar]
- Andac, M.; Asan, A.; Isildak, I. A simple flow injection spectrophotometric determination method for iron(III) based on o-acetylsalicylhydroxamic acid complexation. Chem. Papers 2009, 63, 268–273. [Google Scholar]
- Xue, H.; Lurdes, M.D.; Goncalves, S.; Reutlinger, M.; Sigg, L.; Stumm, W. Copper(I) in fogwater: Determination and interactions with sulfite. Environ. Sci. Technol. 1991, 25, 1716–1722. [Google Scholar]
- Zhuang, G.; Yi, Z.; Duce, R.A.; Brown, P.A. Link between iron and sulphur cycles suggested by detection of Fe(II) in remote marine aerosols. Nature 1992, 355, 537–539. [Google Scholar]
- Andersen, J.E. A novel method for the filterless preconcentration of iron. Analyst 2005, 130, 385–390. [Google Scholar]
- Waterbury, R.D.; Yao, W.; Byrne, R.H. Long pathlength absorbance spectroscopy: Trace analysis of Fe(II) using a 4.5 m liquid core waveguide. Analyt. Chim. Acta 1997, 357, 99–102. [Google Scholar]
- Bowie, A.R.; Achterberg, E.P.; Sedwick, P.N.; Ussher, S.; Worsford, P.J. Real-time monitoring of picomolar concentrations of iron(II) in marine waters using automated flow injection-chemiluminescence instrumentation. Environ. Sci. Technol. 2002, 36, 4600–4607. [Google Scholar]
- Croot, P.L.; Laan, P. Continuous shipboard determination of Fe(II) in polar waters using flow injection analysis with chemiluminescence detection. Analyt. Chim. Acta 2002, 466, 261–273. [Google Scholar]
- de Jong, J.T.M.; den Das, J.; Bathmann, U.; Stoll, M.H.C.; Kattner, G.; Nolting, R.F.; de Baar, H.J.W. Dissolved iron at subnanomolar levels in the Southern Ocean as determined by ship-board analysis. Analyt. Chim. Acta 1998, 377, 113–124. [Google Scholar]
- O' Sullivan, D.W.; Hanson, A.K., Jr.; Kester, D.R. Stopped flow luminol chemiluminescence determination of Fe(II) and reducible iron in seawater at subnanomolar levels. Mar. Chem. 1995, 49, 65–77. [Google Scholar]
- Qin, W.; Zhang, Z.J.; Wang, F.C. Chemiluminescence flow system for the determination of Fe(II) and Fe(III) in water. Fresenius J. Anal. Chem. 1998, 360, 130–132. [Google Scholar]
- Rose, A.L.; Waite, T.D. Chemiluminescence of luminol in the presence of iron(II) and oxygen: Oxidation mechanism and implications for its use. Analyt. Chem. 2001, 73, 5909–5920. [Google Scholar]
- Nussbaum, M.A.; Nekimken, H.L.; Nieman, T.A. Luminol chemiluminescence for determination of iron(II) in ferrioxalate chemical actinometry. Analyt. Chem. 1987, 59, 211–212. [Google Scholar]
- Ussher, S.J.; Yaqoob, M.; Achterberg, E.P.; Nabi, A.; Worsfold, P.J. Effect of model ligands on iron redox speciation in natural waters using flow injection with luminol chemiluminescence detection. Analyt. Chem. 2005, 77, 1971–1978. [Google Scholar]
- Albrecht, H.O. Chemiluminescence of aminophthalic hydrazide. Z. Physikal. Chem. 1928, 135, 321–330. [Google Scholar]
- Stross, F.H.; Branch, G.E.K. The chemiluminescence of 3-aminophthalhydrazide. J. Org. Chem. 1938, 3, 385–404. [Google Scholar]
- White, E.H.; Roswell, D.F. Luminol Chemiluminescence. In Chemi- and Bioluminescence; Burr, J. G., Ed.; Marcel Dekker, Inc.: New York, NY, USA, 1985; pp. 215–244. [Google Scholar]
- Wheatley, R.A.; Sariahmetoglu, M.; Cakici, I. Enhancement of luminol chemiluminescence by cysteine and glutathione. Analyst 2000, 125, 1902–1904. [Google Scholar]
- Seitz, W.R.; Hercules, D.M. Determination of trace amounts of iron(III) using chemiluminescence analysis. Analyt. Chem. 1972, 44, 2143–2149. [Google Scholar]
- King, D.W.; Lounsbury, H.A.; Millero, F.J. Rates and mechanism of Fe(II) oxidation at nanomolar total iron concentrations. Environ. Sci. Technol. 1995, 29, 818–824. [Google Scholar]
- Emmenegger, L.; King, D.W.; Sigg, L.; Sulzberger, B. Oxidation kinetics of Fe(II) in a eutrophic Swiss lake. Environ. Sci. Technol. 1998, 32, 2990–2996. [Google Scholar]
- Pullin, M.J.; Cabaniss, S.E. Colorimetric flow-injection analysis of dissolved iron in high DOC waters. Water Res. 2001, 35, 363–372. [Google Scholar]
- Hopkinson, B.M.; Barbeau, K.A. Organic and redox speciation of iron in the eastern tropical North Pacific suboxic zone. Mar. Chem. 2007, 106, 2–17. [Google Scholar]
- Geng, H.; Meng, Z. Inhibition of superoxide dismutase, vitamin C and glutathione on chemiluminescence produced by luminol and the mixture of sulfite and bisulfite. Spectrochim. Acta A: Mol. Biomol. Spectrosc. 2006, 64, 87–92. [Google Scholar]
- Theis, T.L.; Singer, P.C. Complexation of iron(II) by organic matter and its effect on iron(II) oxygenation. Environ. Sci. Technol. 1974, 8, 596–573. [Google Scholar]
- Croot, P.L.; Frew, R.D.; Sander, S.; Hunter, K.A.; Ellwood, M.J.; Pickmere, S.E.; Abraham, E.R.; Law, C.S.; Smith, M.J.; Boyd, P.W. Physical mixing effects on iron biogeochemical cycling: FeCycle experiment. J. Geophys. Res. Oceans 2007, 112, C06015. [Google Scholar] [CrossRef]
- Santana-Casiano, J.M.; Davila, M.G.; Rodriguez, M.J.; Millero, F.J. The effect of organic compounds in the oxidation kinetics of Fe(II). Mar. Chem. 2000, 70, 211–222. [Google Scholar]
- King, D.W. Personal communication. 2005. [Google Scholar]
- Cotton, F.A.; Wilkinson, G.; Murillo, C.A.; Bochmann, M. Advanced Inorganic Chemistry, 6th ed; John Wiley & Sons, Inc.: New York, NY, USA, 1999. [Google Scholar]
- Barry, R.C.; Schnoor, J.L.; Sulzberger, B.; Sigg, L.; Stumm, W. Iron oxidation kinetics in an acidic alpine lake. Water Res. 1994, 28, 323–333. [Google Scholar]
- Amirbahman, A.; Sigg, L.; von Gunten, U. Reductive dissolution of Fe(III) (hydr)oxides by cysteine: kinetics and mechanism. J. Colloid Interface Sci. 1997, 194, 194–206. [Google Scholar]
- Holmen, B.A.; Casey, W.H. Hydroxamate ligands, surface chemistry, and the mechanism of ligand-promoted dissolution of geothite [a-FeOOH(s)]. Geochim. Cosmochim. Acta 1996, 60, 4403–4416. [Google Scholar]
- Sulzberger, B.; Suter, D.; Siffert, C.; Banwart, S.; Stumm, W. Dissolution of Fe(III) (hydr)oxides in natural waters; Laboratory assessment on the kinetics controlled by surface coordination. Mar. Chem. 1989, 28, 127–144. [Google Scholar]
- Drever, J.I. The Geochemistry of Natural Waters: Surface and Groundwaters Environments, 3rd ed; Prentice-Hall, Inc.: Upper Saddle River, NJ, USA, 1982. [Google Scholar]
- Borman, C.J.; Colberg, P.J.S.; Eggleston, C.M.; Sullivan, B.P. Is iron redox cycling in a high altitude watershed photochemically or thermally driven? Chem. Geol. 2009. in revision. [Google Scholar]
- Kubo, H.; Toriba, A. Chemiluminescence flow injection analysis of reducing agents based on the luminol reaction. Analyt. Chim. Acta 1997, 353, 345–349. [Google Scholar]
- Rose, R.C. Ascorbic acid metabolism in protection against free radicals: a radiation model. Biophys. Res. Commun. 1990, 169, 430–436. [Google Scholar]
- Soares, D.G.; Andreazza, A.C.; Salvador, M. Sequestering ability of butylated hydroxytoluene, propyl gallate, resveratol and vitamins C and E against ABTS, DPPH, and hydroxyl free radicals in chemical and biological systems. J. Agric. Food Chem. 2003, 51, 1077–1080. [Google Scholar]
- Demas, J.N.; Bowman, W.D.; Zalewski, E.F.; Velapoldi, R.A. Determination of the quantum yield of the ferrioxalate actinometer with electrically calibrated radiometers. J. Phys. Chem. 1981, 85, 2766–2771. [Google Scholar]
- Hatchard, C.G.; Parker, C.A. A new sensitive chemical actinometer. II. Potassium ferrioxalate as a standard chemical actinometer. Proc. Royal Soc. 1956, 235, 518–536. [Google Scholar]
- Zuo, Y.; Hoigne, J. Formation of hydrogen peroxide and depletion of oxalic acid in atmospheric water by photolysis of iron(III)-oxalato complexes. Environ. Sci. Technol. 1992, 26, 1014–1022. [Google Scholar]
- Zuo, Y.G.; Deng, Y.W. Iron(II)-catalyzed photochemical decomposition of oxalic acid and generation of H2O2 in atmospheric liquid phases. Chemosphere 1997, 35, 2051–2058. [Google Scholar]
- Zuo, Y.G.; Hoigne, J. Photochemical decomposition of oxalic, glyoxalic and pyruvic acid catalysed by iron in atmospheric waters. Atmos. Environ. 1994, 28, 1231–1239. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amendment | Concentration | mN a | Effectiveness |
|---|---|---|---|
| Ascorbate | 10-6 M | < 0.1 | NR |
| 10-4 M | < 0.1 | NR | |
| 10-2 M | < 0.1 | NR | |
| Oxalate | 10-6 M | 0.9(0) | Yes |
| 10-4 M | 1.1 | Yes | |
| 10-2 M | 2.(0) | Yes | |
| Ascorbate/oxalate | 10-6 M | 0.6(4) | LS |
| 10-4 M | < 0.1 | NR | |
| 10-2 M | < 0.1 | NR | |
| Cysteine | 10-6 M | 2.(6) | Yes |
| 10-4 M | 0.3(7) | LS | |
| 10-2 M | < 0.1 | NR | |
| Glycine | 10-6 M | 1.(8) | Yes |
| 10-4 M | 0.8(8) | LS | |
| 10-2 M | 1.3 | Yes | |
| Hydroxylamine | 10-6 M | 0.4(5) | LS |
| 10-4 M | 0.4(2) | LS | |
| 10-2 M | < 0.1 | NR | |
| Hydrazine | 10-6 M | 0.7(5) | LS |
| 10-4 M | 0.2(5) | LS | |
| 10-2 M | < 0.1 | NR |
| Amendment | Concentration | mN a | Effectiveness |
|---|---|---|---|
| Aldrich Humic Acid | 1 mg C L-1 | 0.7(0) | LS |
| 5 mg C L-1 | 0.3(6) | LS | |
| 10 mg C L-1 | 0.3(7) | LS | |
| IHSS Humic Acid | 1 mg C L-1 | 0.8(4) | LS |
| 5 mg C L-1 | 0.1(7) | LS | |
| 10 mg C L-1 | 0.1(9) | LS | |
| IHSS Fulvic Acid | 1 mg C L-1 | 0.7(7) | LS |
| 5 mg C L-1 | 0.1(9) | LS | |
| 10 mg C L-1 | 0.1(7) | LS | |
| MCC water | ∼ 5 mg C L-1 * | <0.1 | NR |
| SR lake water | ∼ 10 mg C L-1 * | 0.8(6) | LS |
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Borman, C.J.; Sullivan, B.P.; Eggleston, C.M.; Colberg, P.J.S. The Use of Flow-Injection Analysis with Chemiluminescence Detection of Aqueous Ferrous Iron in Waters Containing High Concentrations of Organic Compounds. Sensors 2009, 9, 4390-4406. https://doi.org/10.3390/s90604390
Borman CJ, Sullivan BP, Eggleston CM, Colberg PJS. The Use of Flow-Injection Analysis with Chemiluminescence Detection of Aqueous Ferrous Iron in Waters Containing High Concentrations of Organic Compounds. Sensors. 2009; 9(6):4390-4406. https://doi.org/10.3390/s90604390
Chicago/Turabian StyleBorman, Christopher J., B. Patrick Sullivan, Carrick M. Eggleston, and Patricia J. S. Colberg. 2009. "The Use of Flow-Injection Analysis with Chemiluminescence Detection of Aqueous Ferrous Iron in Waters Containing High Concentrations of Organic Compounds" Sensors 9, no. 6: 4390-4406. https://doi.org/10.3390/s90604390
APA StyleBorman, C. J., Sullivan, B. P., Eggleston, C. M., & Colberg, P. J. S. (2009). The Use of Flow-Injection Analysis with Chemiluminescence Detection of Aqueous Ferrous Iron in Waters Containing High Concentrations of Organic Compounds. Sensors, 9(6), 4390-4406. https://doi.org/10.3390/s90604390