Exploiting Surface Plasmon Resonance (SPR) Technology for the Identification of Fibroblast Growth Factor-2 (FGF2) Antagonists Endowed with Antiangiogenic Activity


Abstract
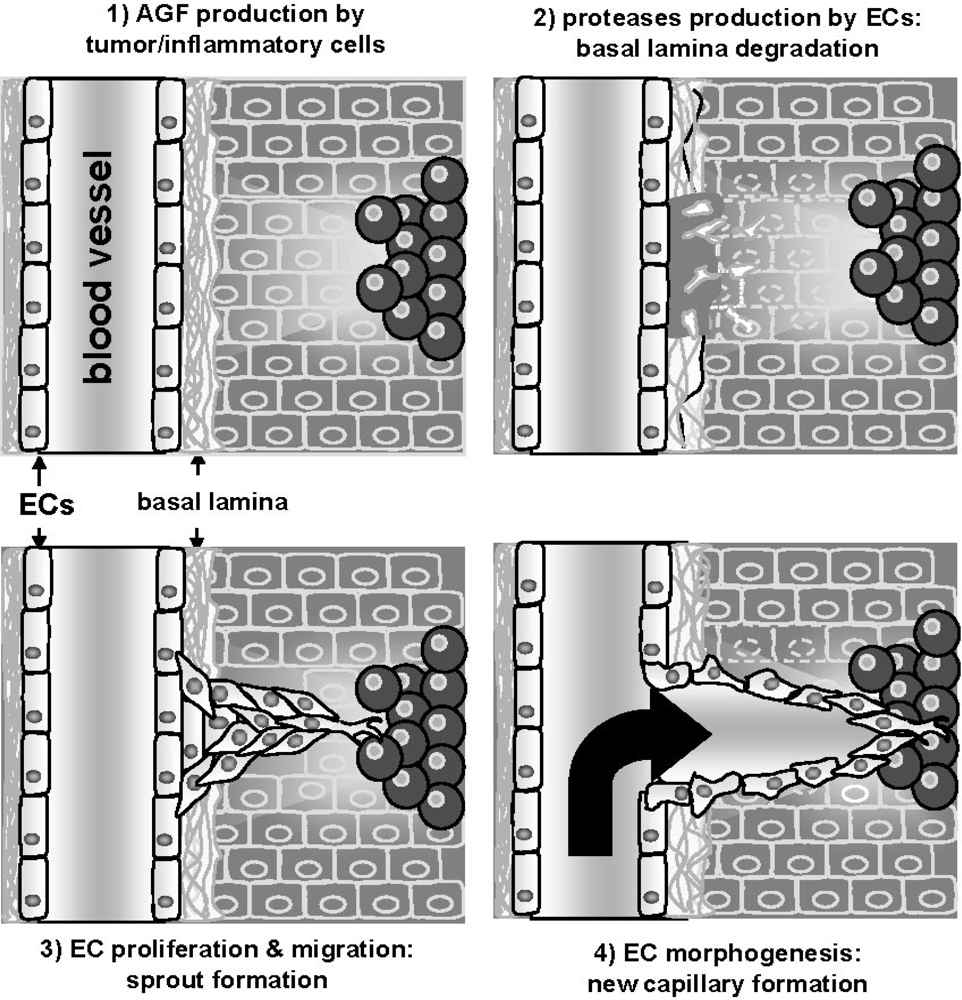
:1. The Angiogenesis Process
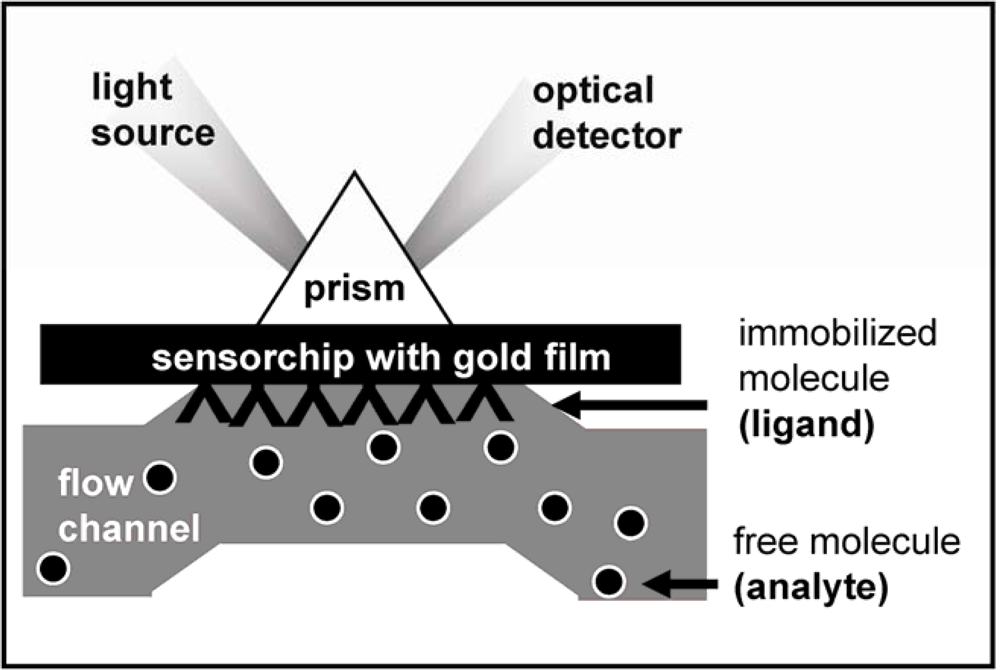
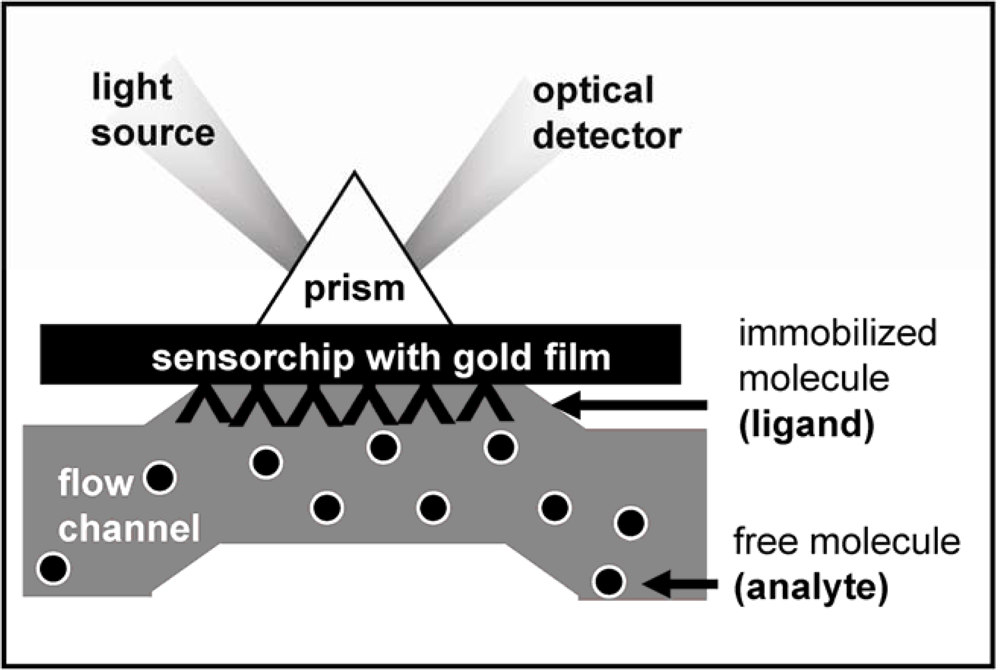
2. Surface Plasmon Resonance (SPR) Spectroscopy for the Study of the Angiogenesis Process
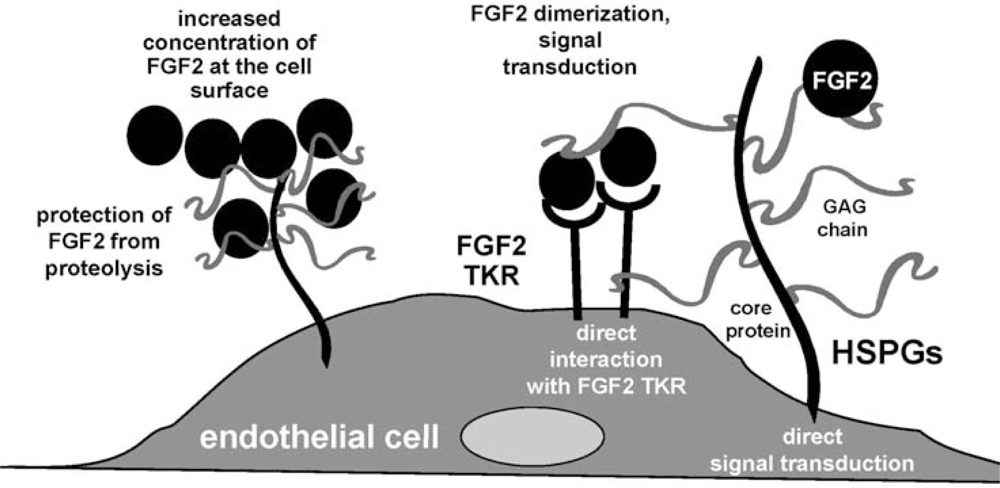
3. Fibroblast Growth Factor-2 (FGF2) and Its Receptors
4. Extracellular FGF2 Antagonists: Exploiting SPR for the Identification of Antiangiogenic Compounds
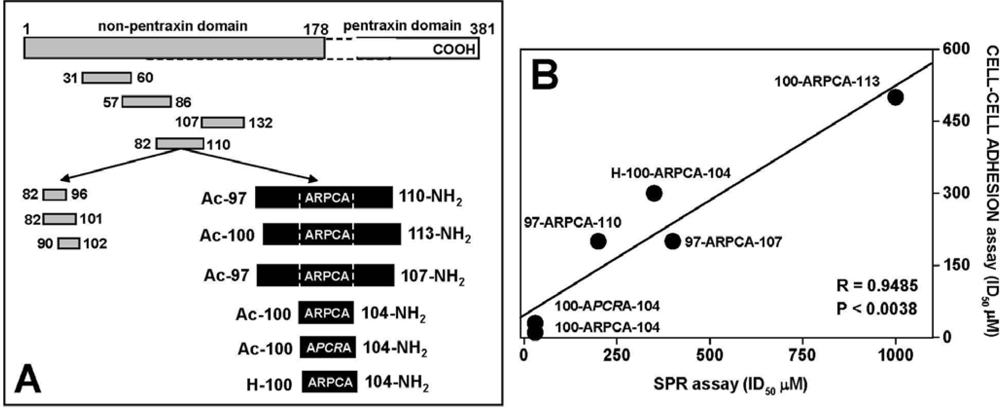
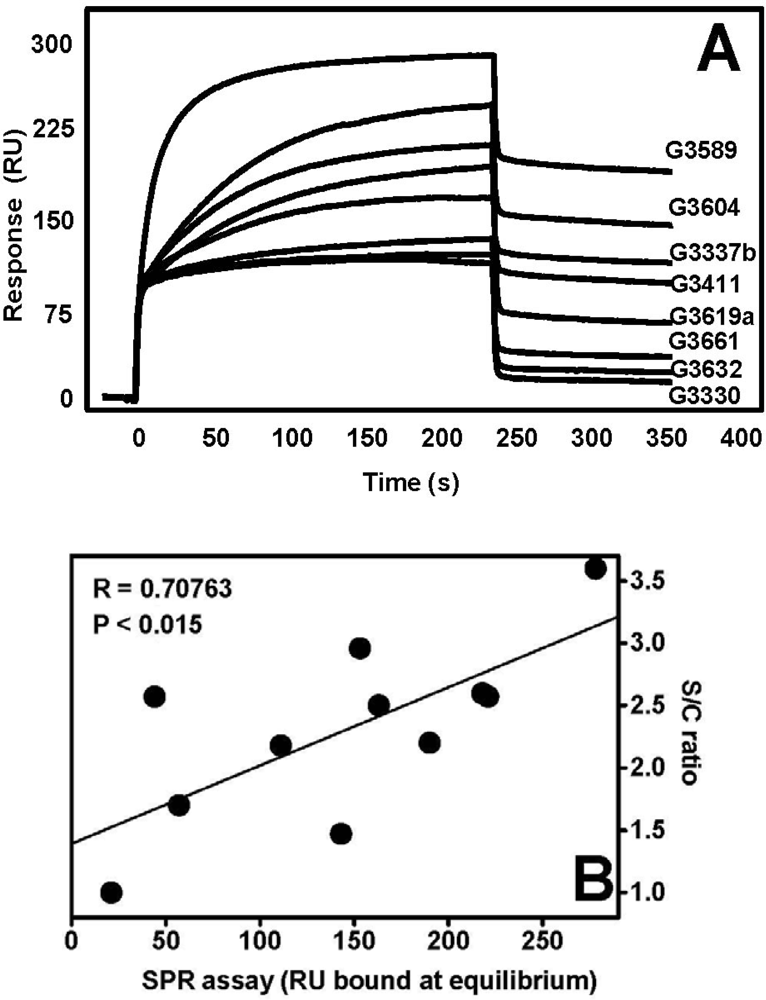
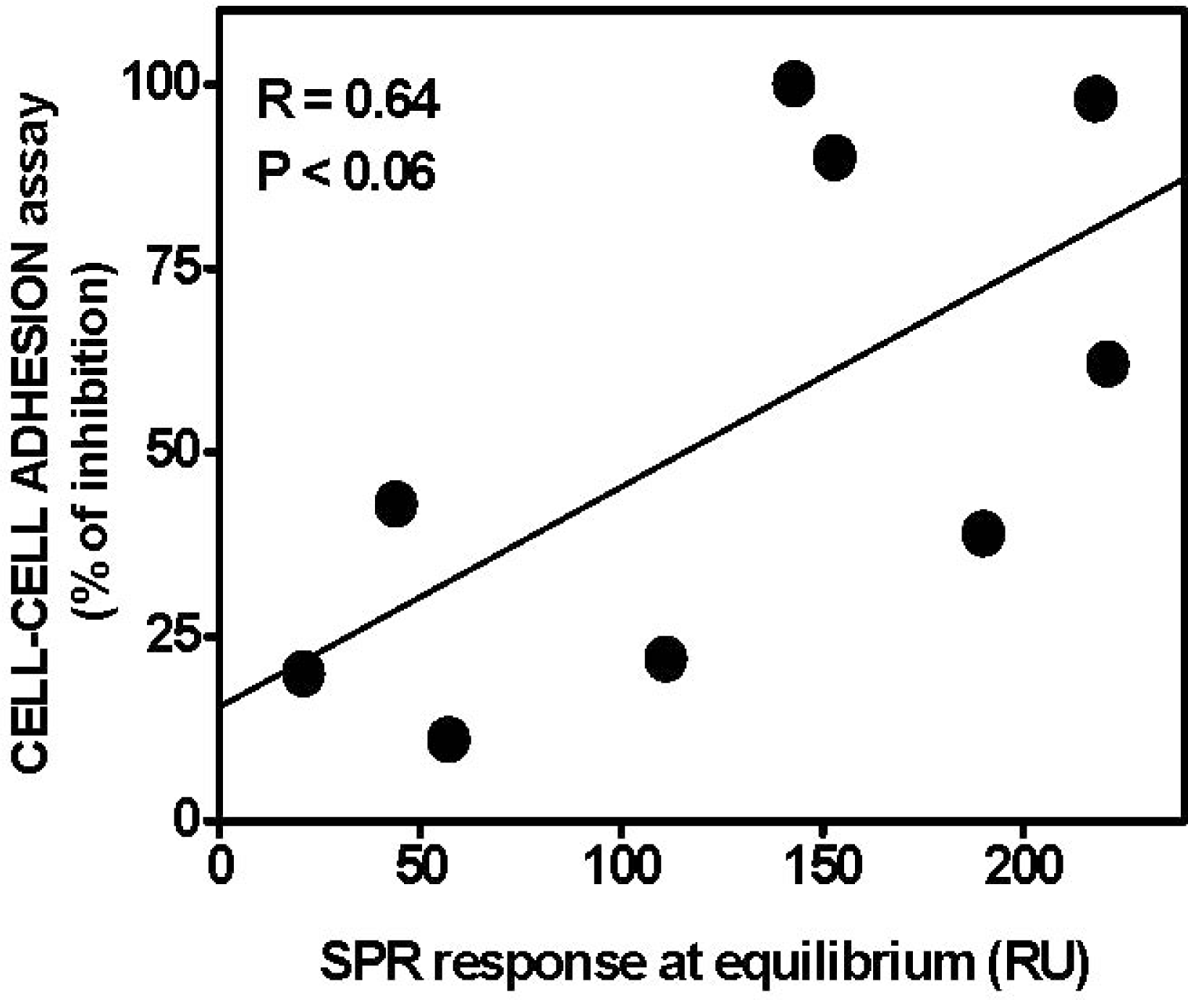
4.1. Anti-FGF2 Peptides
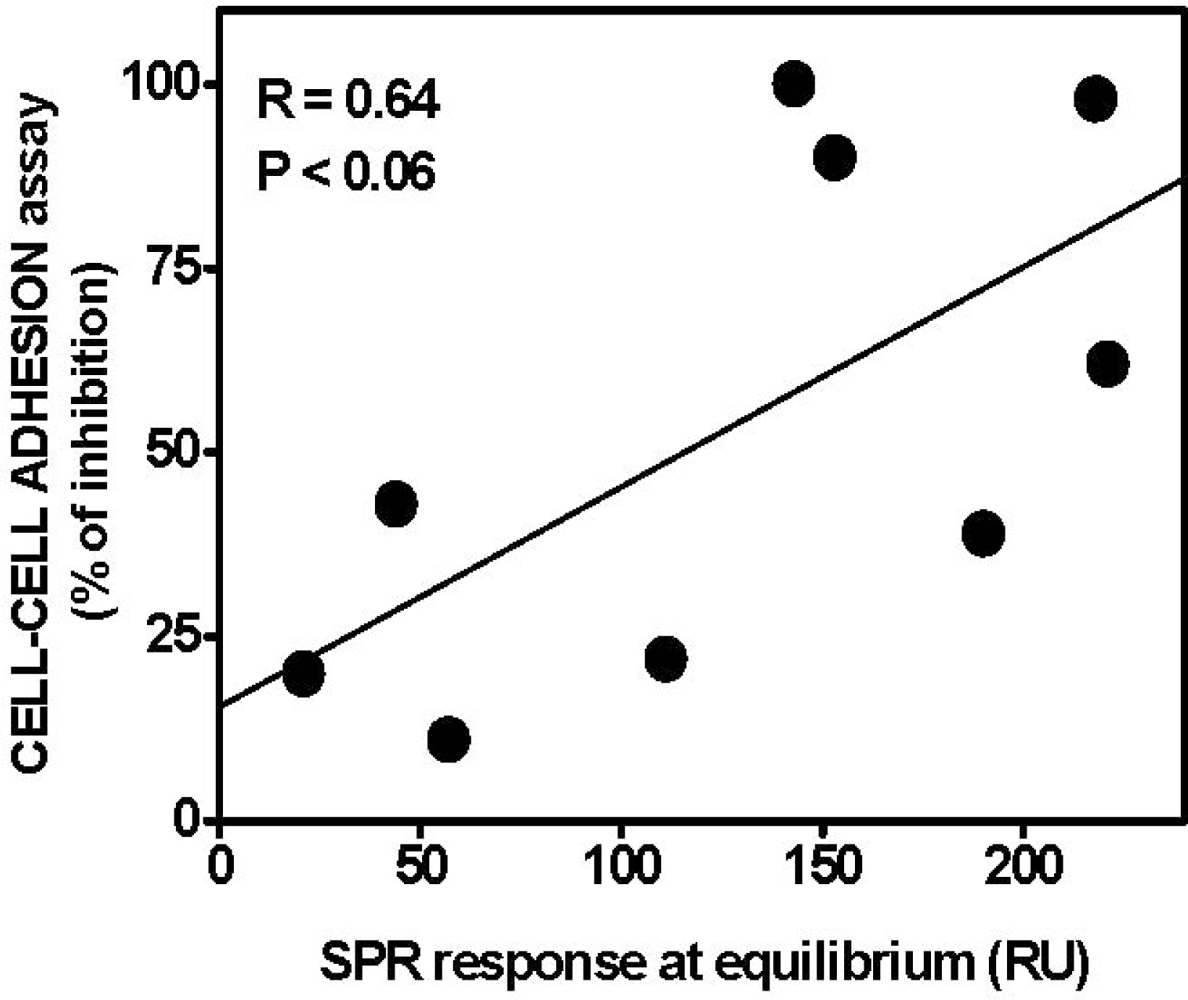
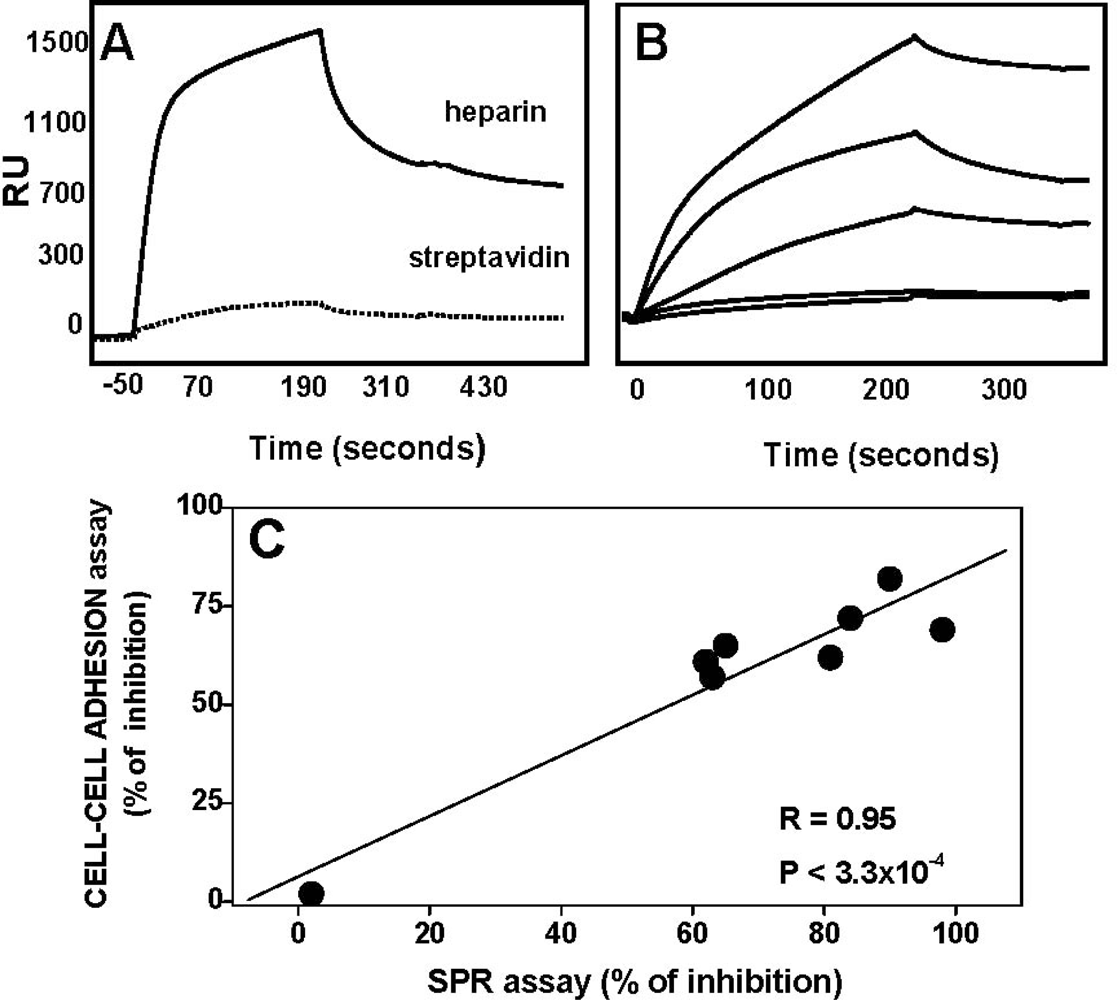
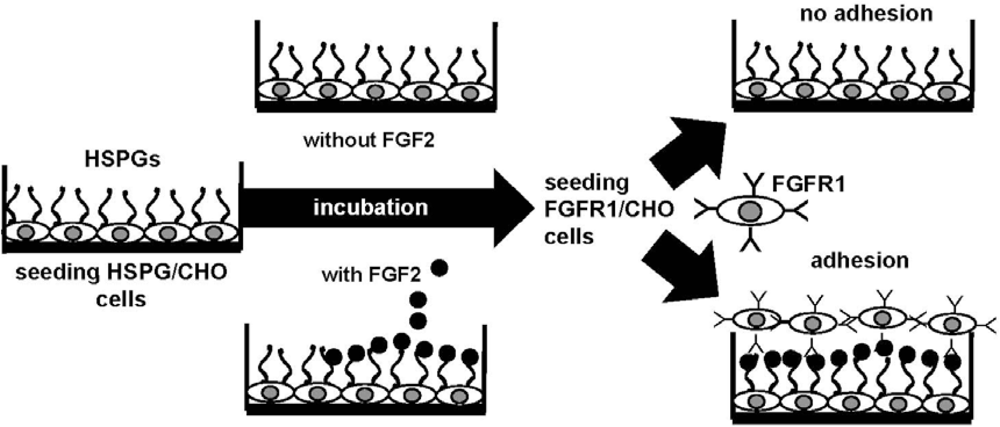
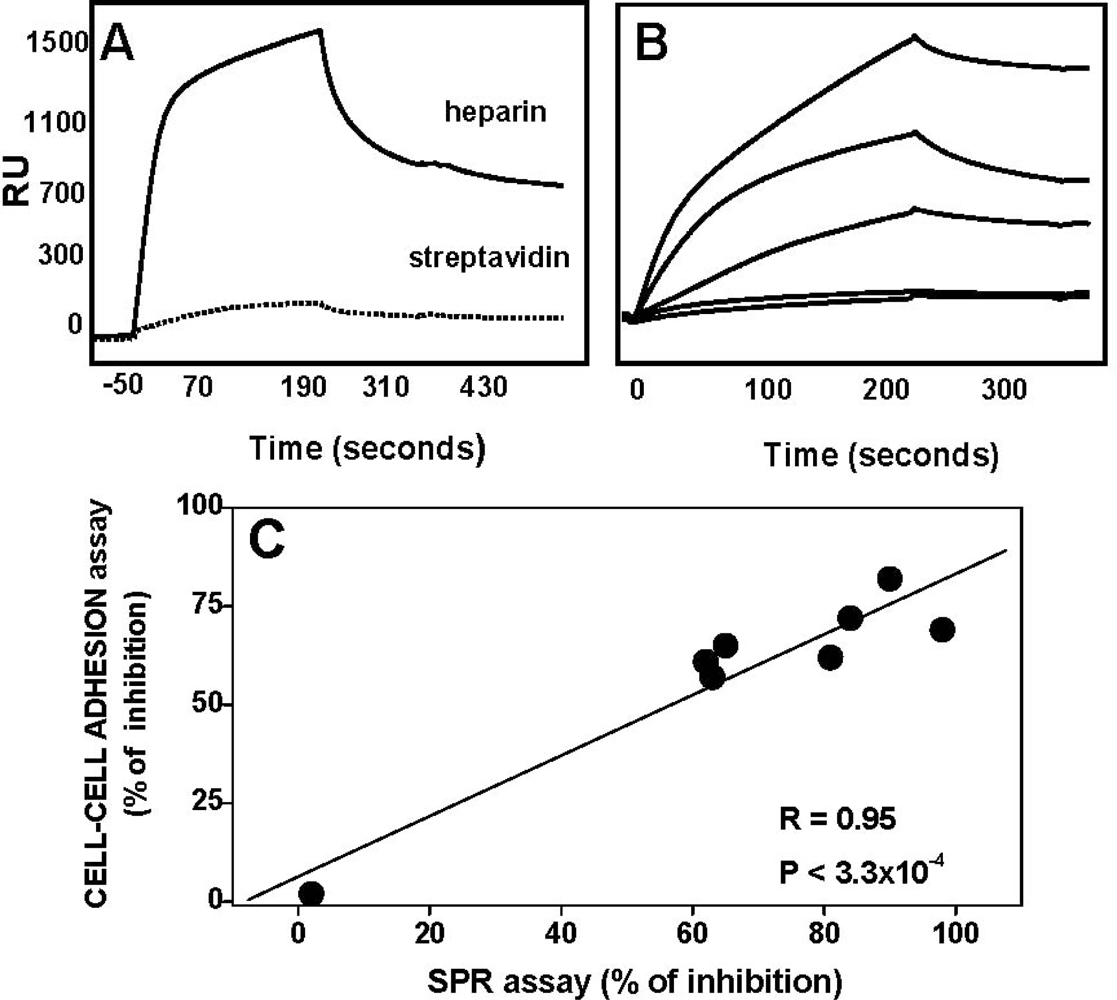
4.2. Anti-FGF2 Polyanionic Heparin-Like Molecules
5. Understanding the Surface-Confined Molecular Recognition: towards an Integrated Biosensing Strategy
- suboptimal data and/or over-interpreted results. A long list of these flaws, that are outside the target of this review, has been reported in details in the survey of commercial optical biosensor literature series [15–23]. It is important to note that a broad and detailed array of “guidelines” for the interpretation of SPR results is reported in these reviews. If correctly applied, these guidelines would reduce by a great extent the misinterpretations of SPR data, with great benefits for SPR studies in general and angiogenesis in particular.
- “lead up” or “lead down” experiments (e.g., the alternative choice to immobilize the AGF or its receptor onto the sensorchip surface). According to the state-of-the-art theory, the alternative choice of immobilization should lead to the same binding parameters. However, many “immobilization-driven” artefacts may affect the binding results under the different experimental conditions. Among them, mass transport effect may greatly (and differently) impact the lead up or lead down experiments. The comparison among binding data obtained at the solid-solution interface and in bulk solution may provide a valuable help in the interpretation of the results.
- different immobilization procedures that can mask or alter the accessibility of binding sites present in the ligand molecule;
- the use of different molecular forms of the ligand and/or of the analyte (e.g., different receptor isoforms, different AGF variants, GAGs of different chemical structure and charge).
6. Conclusions
Acknowledgments
References
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar]
- Folkman, J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med 1995, 1, 27–31. [Google Scholar]
- Shing, Y.; Folkman, J.; Sullivan, R.; Butterfield, C.; Murray, J.; Klagsbrun, M. Heparin affinity: purification of a tumor-derived capillary endothelial cell growth factor. Science 1984, 223, 1296–1299. [Google Scholar]
- Maciag, T.; Mehlman, T.; Friesel, R.; Schreiber, A.B. Heparin binds endothelial cell growth factor, the principal endothelial cell mitogen in bovine brain. Science 1984, 225, 932–935. [Google Scholar]
- Carmeliet, P. Mechanisms of angiogenesis and arteriogenesis. Nat. Med 2000, 6, 389–395. [Google Scholar]
- Underwood, P.A.; Bean, P.A.; Gamble, J.R. Rate of endothelial expansion is controlled by cell:cell adhesion. Int. J. Biochem. Cell Biol 2002, 34, 55–69. [Google Scholar]
- Rusnati, M.; Presta, M. Extracellular angiogenic growth factor interactions: an angiogenesis interactome survey. Endothelium 2006, 13, 93–111. [Google Scholar]
- Klein, S.; Giancotti, F.G.; Presta, M.; Albelda, S.M.; Buck, C.A.; Rifkin, D.B. Basic fibroblast growth factor modulates integrin expression in microvascular endothelial cells. Mol. Biol. Cell 1993, 4, 973–982. [Google Scholar]
- Gerritsen, M.E.; Soriano, R.; Yang, S.; Zlot, C.; Ingle, G.; Toy, K.; Williams, P.M. Branching out: a molecular fingerprint of endothelial differentiation into tube-like structures generated by Affymetrix oligonucleotide arrays. Microcirculation 2003, 10, 63–81. [Google Scholar]
- Schnaper, H.W.; Barnathan, E.S.; Mazar, A.; Maheshwari, S.; Ellis, S.; Cortez, S.L.; Baricos, W.H.; Kleinman, H.K. Plasminogen activators augment endothelial cell organization in vitro by two distinct pathways. J. Cell Physiol 1995, 165, 107–118. [Google Scholar]
- Davis, G.E.; Camarillo, C.W. Regulation of endothelial cell morphogenesis by integrins, mechanical forces, and matrix guidance pathways. Exp. Cell Res 1995, 216, 113–123. [Google Scholar]
- Sheibani, N.; Newman, P.J.; Frazier, W.A. Thrombospondin-1, a natural inhibitor of angiogenesis, regulates platelet-endothelial cell adhesion molecule-1 expression and endothelial cell morphogenesis. Mol. Biol. Cell 1997, 8, 1329–1341. [Google Scholar]
- Kanda, S.; Miyata, Y.; Kanetake, H. Fibroblast growth factor-2-mediated capillary morphogenesis of endothelial cells requires signals via Flt-1/vascular endothelial growth factor receptor-1: possible involvement of c-Akt. J. Biol. Chem 2004, 279, 4007–4016. [Google Scholar]
- Rusnati, M.; Urbinati, C.; Presta, M. Glycomics and angiogenesis: biological implications and therapeutical exploiting. In New Development in Therapeutic Glycomics; Delehedde, M., Lortat-Jacob, H., Eds.; Research Signpost: Trivandum, India, 2006; pp. 33–77. [Google Scholar]
- Rich, R.L.; Myszka, D.G. Survey of the year 2001 commercial optical biosensor literature. J. Mol. Recognit 2002, 15, 352–376. [Google Scholar]
- Rich, R.L.; Myszka, D.G. Survey of the year 2000 commercial optical biosensor literature. J. Mol. Recognit 2001, 14, 273–294. [Google Scholar]
- Rich, R.L.; Myszka, D.G. Survey of the 1999 surface plasmon resonance biosensor literature. J. Mol. Recognit 2000, 13, 388–407. [Google Scholar]
- Rich, R.L.; Myszka, D.G. A survey of the year 2002 commercial optical biosensor literature. J. Mol. Recognit 2005, 18, 431–478. [Google Scholar]
- Rich, R.L.; Myszka, D.G. Survey of the year 2003 commercial optical biosensor literature. J. Mol. Recognit 2005, 18, 1–39. [Google Scholar]
- Rich, R.L.; Myszka, D.G. Survey of the year 2005 commercial optical biosensor literature. J. Mol. Recognit 2006, 19, 478–534. [Google Scholar]
- Rich, R.L.; Myszka, D.G. Survey of the year 2006 commercial optical biosensor literature. J. Mol. Recognit 2007, 20, 300–366. [Google Scholar]
- Rich, R.L.; Myszka, D.G. Survey of the year 2007 commercial optical biosensor literature. J. Mol. Recognit 2008, 21, 355–400. [Google Scholar]
- Lin, X.; Takahashi, K.; Campion, S.L.; Liu, Y.; Gustavsen, G.G.; Pena, L.A.; Zamora, P.O. Synthetic peptide F2A4-K-NS mimics fibroblast growth factor-2 in vitro and is angiogenic in vivo. Int. J. Mol. Med 2006, 17, 833–839. [Google Scholar]
- Ibrahimi, O.A.; Zhang, F.; Hrstka, S.C.; Mohammadi, M.; Linhardt, R.J. Kinetic model for FGF, FGFR, and proteoglycan signal transduction complex assembly. Biochemistry 2004, 43, 4724–4730. [Google Scholar]
- Ibrahimi, O.A.; Zhang, F.; Eliseenkova, A.V.; Linhardt, R.J.; Mohammadi, M. Proline to arginine mutations in FGF receptors 1 and 3 result in Pfeiffer and Muenke craniosynostosis syndromes through enhancement of FGF binding affinity. Hum. Mol. Genet 2004, 13, 69–78. [Google Scholar]
- Anderson, J.; Burns, H.D.; Enriquez-Harris, P.; Wilkie, A.O.; Heath, J.K. Apert syndrome mutations in fibroblast growth factor receptor 2 exhibit increased affinity for FGF ligand. Hum. Mol. Genet 1998, 7, 1475–1483. [Google Scholar]
- Li, S.; Christensen, C.; Kiselyov, V.V.; Kohler, L.B.; Bock, E.; Berezin, V. Fibroblast growth factor-derived peptides: functional agonists of the fibroblast growth factor receptor. J. Neurochem 2008, 104, 667–682. [Google Scholar]
- Mori, S.; Wu, C.Y.; Yamaji, S.; Saegusa, J.; Shi, B.; Ma, Z.; Kuwabara, Y.; Lam, K.S.; Isseroff, R.R.; Takada, Y.K.; Takada, Y. Direct binding of integrin alphavbeta3 to FGF1 plays a role in FGF1 signaling. J. Biol. Chem 2008, 283, 18066–18075. [Google Scholar]
- Olsen, S.K.; Ibrahimi, O.A.; Raucci, A.; Zhang, F.; Eliseenkova, A.V.; Yayon, A.; Basilico, C.; Linhardt, R.J.; Schlessinger, J.; Mohammadi, M. Insights into the molecular basis for fibroblast growth factor receptor autoinhibition and ligand-binding promiscuity. Proc. Natl. Acad. Sci. USA 2004, 101, 935–940. [Google Scholar]
- Cunningham, S.A.; Tran, T.M.; Arrate, M.P.; Brock, T.A. Characterization of vascular endothelial cell growth factor interactions with the kinase insert domain-containing receptor tyrosine kinase. A real time kinetic study. J. Biol. Chem 1999, 274, 18421–18427. [Google Scholar]
- Shinkai, A.; Ito, M.; Anazawa, H.; Yamaguchi, S.; Shitara, K.; Shibuya, M. Mapping of the sites involved in ligand association and dissociation at the extracellular domain of the kinase insert domain-containing receptor for vascular endothelial growth factor. J. Biol. Chem 1998, 273, 31283–31288. [Google Scholar]
- Chen, Y.L.; Tsai, I.H.; Hong, T.M.; Tsai, S.H. Crotalid venom vascular endothelial growth factors has preferential affinity for VEGFR-1. Characterization of Protobothrops mucrosquamatus venom VEGF. Thromb Haemost 2005, 93, 331–338. [Google Scholar]
- Huang, X.; Gottstein, C.; Brekken, R.A.; Thorpe, P.E. Expression of soluble VEGF receptor 2 and characterization of its binding by surface plasmon resonance. Biochem. Biophys. Res. Commun 1998, 252, 643–648. [Google Scholar]
- von Tiedemann, B.; Bilitewski, U. Characterization of the vascular endothelial growth factor-receptor interaction and determination of the recombinant protein by an optical receptor sensor. Biosens. Bioelectron 2002, 17, 983–991. [Google Scholar]
- Herve, M.A.; Buteau-Lozano, H.; Vassy, R.; Bieche, I.; Velasco, G.; Pla, M.; Perret, G.; Mourah, S.; Perrot-Applanat, M. Overexpression of vascular endothelial growth factor 189 in breast cancer cells leads to delayed tumor uptake with dilated intratumoral vessels. Am. J. Pathol 2008, 172, 167–178. [Google Scholar]
- Fuh, G.; Garcia, K.C.; de Vos, A.M. The interaction of neuropilin-1 with vascular endothelial growth factor and its receptor flt-1. J. Biol. Chem 2000, 275, 26690–26695. [Google Scholar]
- Jeltsch, M.; Karpanen, T.; Strandin, T.; Aho, K.; Lankinen, H.; Alitalo, K. Vascular endothelial growth factor (VEGF)/VEGF-C mosaic molecules reveal specificity determinants and feature novel receptor binding patterns. J. Biol. Chem 2006, 281, 12187–12195. [Google Scholar]
- Stamos, J.; Lazarus, R.A.; Yao, X.; Kirchhofer, D.; Wiesmann, C. Crystal structure of the HGF beta-chain in complex with the Sema domain of the Met receptor. Embo J 2004, 23, 2325–2335. [Google Scholar]
- Merkulova-Rainon, T.; England, P.; Ding, S.; Demerens, C.; Tobelem, G. The N-terminal domain of hepatocyte growth factor inhibits the angiogenic behavior of endothelial cells independently from binding to the c-met receptor. J. Biol. Chem 2003, 278, 37400–37408. [Google Scholar]
- Tiran, Z.; Oren, A.; Hermesh, C.; Rotman, G.; Levine, Z.; Amitai, H.; Handelsman, T.; Beiman, M.; Chen, A.; Landesman-Milo, D.; Dassa, L.; Peres, Y.; Koifman, C.; Glezer, S.; Vidal-Finkelstein, R.; Bahat, K.; Pergam, T.; Israel, C.; Horev, J.; Tsarfaty, I.; Ayalon-Soffer, M. A novel recombinant soluble splice variant of Met is a potent antagonist of the hepatocyte growth factor/scatter factor-Met pathway. Clin. Cancer Res 2008, 14, 4612–4621. [Google Scholar]
- Urbinati, C.; Bugatti, A.; Giacca, M.; Schlaepfer, D.; Presta, M.; Rusnati, M. Alpha(v)beta3-integrin-dependent activation of focal adhesion kinase mediates NF-kappaB activation and motogenic activity by HIV-1 Tat in endothelial cells. J. Cell Sci 2005, 118, 3949–2958. [Google Scholar]
- Lin, X.; Takahashi, K.; Liu, Y.; Derrien, A.; Zamora, P.O. A synthetic, bioactive PDGF mimetic with binding to both alpha-PDGF and beta-PDGF receptors. Growth Factors 2007, 25, 87–93. [Google Scholar]
- Cotman, S.L.; Halfter, W.; Cole, G.J. Identification of extracellular matrix ligands for the heparan sulfate proteoglycan agrin. Exp. Cell Res 1999, 249, 54–64. [Google Scholar]
- Deepa, S.S.; Yamada, S.; Zako, M.; Goldberger, O.; Sugahara, K. Chondroitin sulfate chains on syndecan-1 and syndecan-4 from normal murine mammary gland epithelial cells are structurally and functionally distinct and cooperate with heparan sulfate chains to bind growth factors. A novel function to control binding of midkine, pleiotrophin, and basic fibroblast growth factor. J. Biol. Chem 2004, 279, 37368–37376. [Google Scholar]
- Sugaya, N.; Habuchi, H.; Nagai, N.; Ashikari-Hada, S.; Kimata, K. 6-O-sulfation of heparan sulfate differentially regulates various fibroblast growth factor-dependent signalings in culture. J. Biol. Chem 2008, 283, 10366–10376. [Google Scholar]
- Vincent, T.L.; McLean, C.J.; Full, L.E.; Peston, D.; Saklatvala, J. FGF-2 is bound to perlecan in the pericellular matrix of articular cartilage, where it acts as a chondrocyte mechanotransducer. Osteoarthritis Cartilage 2007, 15, 752–763. [Google Scholar]
- Kirkpatrick, C.A.; Knox, S.M.; Staatz, W.D.; Fox, B.; Lercher, D.M.; Selleck, S.B. The function of a Drosophila glypican does not depend entirely on heparan sulfate modification. Dev. Biol 2006, 300, 570–582. [Google Scholar]
- Nandini, C.D.; Mikami, T.; Ohta, M.; Itoh, N.; Akiyama-Nambu, F.; Sugahara, K. Structural and functional characterization of oversulfated chondroitin sulfate/dermatan sulfate hybrid chains from the notochord of hagfish. Neuritogenic and binding activities for growth factors and neurotrophic factors. J. Biol. Chem 2004, 279, 50799–50809. [Google Scholar]
- Goretzki, L.; Burg, M.A.; Grako, K.A.; Stallcup, W.B. High-affinity binding of basic fibroblast growth factor and platelet-derived growth factor-AA to the core protein of the NG2 proteoglycan. J. Biol. Chem 1999, 274, 16831–16837. [Google Scholar]
- Melrose, J.; Roughley, P.; Knox, S.; Smith, S.; Lord, M.; Whitelock, J. The structure, location, and function of perlecan, a prominent pericellular proteoglycan of fetal, postnatal, and mature hyaline cartilages. J. Biol. Chem 2006, 281, 36905–36914. [Google Scholar]
- Knox, S.; Melrose, J.; Whitelock, J. Electrophoretic, biosensor, and bioactivity analyses of perlecans of different cellular origins. Proteomics 2001, 1, 1534–1541. [Google Scholar]
- Maeda, N.; He, J.; Yajima, Y.; Mikami, T.; Sugahara, K.; Yabe, T. Heterogeneity of the chondroitin sulfate portion of phosphacan/6B4 proteoglycan regulates its binding affinity for pleiotrophin/heparin binding growth-associated molecule. J. Biol. Chem 2003, 278, 35805–35811. [Google Scholar]
- Gohring, W.; Sasaki, T.; Heldin, C. H.; Timpl, R. Mapping of the binding of platelet-derived growth factor to distinct domains of the basement membrane proteins BM-40 and perlecan and distinction from the BM-40 collagen-binding epitope. Eur. J. Biochem 1998, 255, 60–66. [Google Scholar]
- Nayeri, F.; Nayeri, T.; Aili, D.; Brudin, L.; Liedberg, B. Clinical impact of real-time evaluation of the biological activity and degradation of hepatocyte growth factor. Growth Factors 2008, 26, 163–171. [Google Scholar]
- Nayeri, F.; Xu, J.; Abdiu, A.; Nayeri, T.; Aili, D.; Liedberg, B.; Carlsson, U. Autocrine production of biologically active hepatocyte growth factor (HGF) by injured human skin. J. Dermatol. Sci 2006, 43, 49–56. [Google Scholar]
- Nayeri, F.; Aili, D.; Nayeri, T.; Xu, J.; Almer, S.; Lundstrom, I.; Akerlind, B.; Liedberg, B. Hepatocyte growth factor (HGF) in fecal samples: rapid detection by surface plasmon resonance. BMC Gastroenterol 2005, 5, 13. [Google Scholar]
- Zhu, G.; Yang, B.; Jennings, R.N. Quantitation of basic fibroblast growth factor by immunoassay using BIAcore 2000. J. Pharm. Biomed. Anal 2000, 24, 281–290. [Google Scholar]
- Margosio, B.; Rusnati, M.; Bonezzi, K.; Cordes, B.L.; Annis, D.S.; Urbinati, C.; Giavazzi, R.; Presta, M.; Ribatti, D.; Mosher, D.F.; Taraboletti, G. Fibroblast growth factor-2 binding to the thrombospondin-1 type III repeats, a novel antiangiogenic domain. Int. J. Biochem. Cell Biol 2008, 40, 700–709. [Google Scholar]
- Chen, C.; Patel, S.; Corisdeo, S.; Liu, X.; Micolochick, H.; Xue, J.; Yang, Q.; Lei, Y.; Wang, B.; Soltis, D. Generation and characterization of a panel of monoclonal antibodies specific for human fibroblast growth factor receptor 4 (FGFR4). Hybridoma (Larchmt) 2005, 24, 152–159. [Google Scholar]
- Ueda, Y.; Yamagishi, T.; Samata, K.; Ikeya, H.; Hirayama, N.; Takashima, H.; Nakaike, S.; Tanaka, M.; Saiki, I. A novel low molecular weight antagonist of vascular endothelial growth factor receptor binding: VGA1155. Mol. Cancer Ther 2003, 2, 1105–1111. [Google Scholar]
- Erdag, B.; Balcioglu, K.B.; Kumbasar, A.; Celikbicak, O.; Zeder-Lutz, G.; Altschuh, D.; Salih, B.; Baysal, K. Novel short peptides isolated from phage display library inhibit vascular endothelial growth factor activity. Mol. Biotechnol 2007, 35, 51–63. [Google Scholar]
- Maynard, H.D.; Hubbell, J.A. Discovery of a sulfated tetrapeptide that binds to vascular endothelial growth factor. Acta Biomater 2005, 1, 451–459. [Google Scholar]
- Leonard, P.; Scotney, P.D.; Jabeen, T.; Iyer, S.; Fabri, L.J.; Nash, A.D.; Acharya, K.R. Crystal structure of vascular endothelial growth factor-B in complex with a neutralising antibody Fab fragment. J. Mol. Biol 2008, 384, 1203–1217. [Google Scholar]
- Lowe, J.; Araujo, J.; Yang, J.; Reich, M.; Oldendorp, A.; Shiu, V.; Quarmby, V.; Lowman, H.; Lien, S.; Gaudreault, J.; Maia, M. Ranibizumab inhibits multiple forms of biologically active vascular endothelial growth factor in vitro and in vivo. Exp. Eye Res 2007, 85, 425–430. [Google Scholar]
- Di Benedetto, M.; Starzec, A.; Vassy, R.; Perret, G.Y.; Crepin, M. Distinct heparin binding sites on VEGF165 and its receptors revealed by their interaction with a non sulfated glycoaminoglycan (NaPaC). Biochim. Biophys. Acta 2008, 1780, 723–732. [Google Scholar]
- Hasegawa, H.; Sode, K.; Ikebukuro, K. Selection of DNA aptamers against VEGF165 using a protein competitor and the aptamer blotting method. Biotechnol. Lett 2008, 30, 829–834. [Google Scholar]
- Lake, A.C.; Vassy, R.; Di Benedetto, M.; Lavigne, D.; Le Visage, C.; Perret, G.Y.; Letourneur, D. Low molecular weight fucoidan increases VEGF165-induced endothelial cell migration by enhancing VEGF165 binding to VEGFR-2 and NRP1. J. Biol. Chem 2006, 281, 37844–37852. [Google Scholar]
- Urbinati, C.; Bugatti, A.; Oreste, P.; Zoppetti, G.; Waltenberger, J.; Mitola, S.; Ribatti, D.; Presta, M.; Rusnati, M. Chemically sulfated Escherichia coli K5 polysaccharide derivatives as extracellular HIV-1 Tat protein antagonists. FEBS Lett 2004, 568, 171–177. [Google Scholar]
- Bugatti, A.; Urbinati, C.; Ravelli, C.; De Clercq, E.; Liekens, S.; Rusnati, M. Heparin-mimicking sulfonic acid polymers as multitarget inhibitors of human immunodeficiency virus type 1 Tat and gp120 proteins. Antimicrob. Agents Chemother 2007, 51, 2337–2345. [Google Scholar]
- Ostergren-Lunden, G.; Olivas, R.G.; Eftekhari, P.; Krettek, A.; Sanjuan, X.; Fager, G.; Vilaro, S.; Lustig, F.; Hoebeke, J. Characterisation and application of antibodies specific for the long platelet-derived growth factor A and B chains. Int. J. Biochem. Cell Biol 2004, 36, 2226–2241. [Google Scholar]
- Cochran, S.; Li, C.P.; Ferro, V. A surface plasmon resonance-based solution affinity assay for heparan sulfate-binding proteins. Glycoconj. J 2008, 26, 577–587. [Google Scholar]
- Freeman, I.; Kedem, A.; Cohen, S. The effect of sulfation of alginate hydrogels on the specific binding and controlled release of heparin-binding proteins. Biomaterials 2008, 29, 3260–3268. [Google Scholar]
- Terada, T.; Mizobata, M.; Kawakami, S.; Yabe, Y.; Yamashita, F.; Hashida, M. Basic fibroblast growth factor-binding peptide as a novel targeting ligand of drug carrier to tumor cells. J. Drug Target 2006, 14, 536–545. [Google Scholar]
- Wang, L.; Geng, M.; Li, J.; Guan, H.; Ding, J. Studies of marine sulfated polymannuroguluronate on endothelial cell proliferation and endothelial immunity and related mechanisms. J. Pharmacol. Sci 2003, 92, 367–373. [Google Scholar]
- Yu, L.; Wu, X.; Cheng, Z.; Lee, C.V.; LeCouter, J.; Campa, C.; Fuh, G.; Lowman, H.; Ferrara, N. Interaction between bevacizumab and murine VEGF-A: a reassessment. Invest. Ophthalmol. Vis. Sci 2008, 49, 522–527. [Google Scholar]
- Potty, A.S.; Kourentzi, K.; Fang, H.; Jackson, G.W.; Zhang, X.; Legge, G.B.; Willson, R.C. Biophysical characterization of DNA aptamer interactions with vascular endothelial growth factor. Biopolymers 2009, 91, 145–156. [Google Scholar]
- Varey, A.H.; Rennel, E.S.; Qiu, Y.; Bevan, H.S.; Perrin, R.M.; Raffy, S.; Dixon, A.R.; Paraskeva, C.; Zaccheo, O.; Hassan, A.B.; Harper, S.J.; Bates, D.O. VEGF 165 b, an antiangiogenic VEGF-A isoform, binds and inhibits bevacizumab treatment in experimental colorectal carcinoma: balance of pro- and antiangiogenic VEGF-A isoforms has implications for therapy. Br. J. Cancer 2008, 98, 1366–1379. [Google Scholar]
- Bock, F.; Onderka, J.; Dietrich, T.; Bachmann, B.; Kruse, F.E.; Paschke, M.; Zahn, G.; Cursiefen, C. Bevacizumab as a potent inhibitor of inflammatory corneal angiogenesis and lymphangiogenesis. Invest. Ophthalmol. Vis. Sci 2007, 48, 2545–2552. [Google Scholar]
- Martinez-Torrecuadrada, J.; Cifuentes, G.; Lopez-Serra, P.; Saenz, P.; Martinez, A.; Casal, J.I. Targeting the extracellular domain of fibroblast growth factor receptor 3 with human single-chain Fv antibodies inhibits bladder carcinoma cell line proliferation. Clin. Cancer Res 2005, 11, 6280–6290. [Google Scholar]
- Ricard-Blum, S.; Feraud, O.; Lortat-Jacob, H.; Rencurosi, A.; Fukai, N.; Dkhissi, F.; Vittet, D.; Imberty, A.; Olsen, B.R.; van der Rest, M. Characterization of endostatin binding to heparin and heparan sulfate by surface plasmon resonance and molecular modeling: role of divalent cations. J. Biol. Chem 2004, 279, 2927–2936. [Google Scholar]
- Kono, K.; Ueba, T.; Takahashi, J.A.; Murai, N.; Hashimoto, N.; Myoumoto, A.; Itoh, N.; Fukumoto, M. In vitro growth suppression of human glioma cells by a 16-mer oligopeptide: a potential new treatment modality for malignant glioma. J. Neurooncol 2003, 63, 163–171. [Google Scholar]
- Fitz, L.J.; Morris, J.C.; Towler, P.; Long, A.; Burgess, P.; Greco, R.; Wang, J.; Gassaway, R.; Nickbarg, E.; Kovacic, S.; Ciarletta, A.; Giannotti, J.; Finnerty, H.; Zollner, R.; Beier, D.R.; Leak, L.V.; Turner, K.J.; Wood, C.R. Characterization of murine Flt4 ligand/VEGF-C. Oncogene 1997, 15, 613–618. [Google Scholar]
- Fujisawa, D.; Yamazaki, Y.; Lomonte, B.; Morita, T. Catalytically inactive phospholipase A2 homologue binds to vascular endothelial growth factor receptor-2 via a C-terminal loop region. Biochem. J 2008, 411, 515–522. [Google Scholar]
- Zhu, Z.; Rockwell, P.; Lu, D.; Kotanides, H.; Pytowski, B.; Hicklin, D.J.; Bohlen, P.; Witte, L. Inhibition of vascular endothelial growth factor-induced receptor activation with anti-kinase insert domain-containing receptor single-chain antibodies from a phage display library. Cancer Res 1998, 58, 3209–3214. [Google Scholar]
- Jendreyko, N.; Popkov, M.; Beerli, R.R.; Chung, J.; McGavern, D.B.; Rader, C.; Barbas, C.F. Intradiabodies, bispecific, tetravalent antibodies for the simultaneous functional knockout of two cell surface receptors. J. Biol. Chem 2003, 278, 47812–47819. [Google Scholar]
- Yu, H.; Wang, Z.; Zhang, L.; Zhang, J.; Huang, Q. Pharmacophore modeling and in silico screening for new KDR kinase inhibitors. Bioorg. Med. Chem. Lett 2007, 17, 2126–2133. [Google Scholar]
- Ma, J.; Xin, X.; Meng, L.; Tong, L.; Lin, L.; Geng, M.; Ding, J. The marine-derived oligosaccharide sulfate (MdOS), a novel multiple tyrosine kinase inhibitor, combats tumor angiogenesis both in vitro and in vivo. PLoS ONE 2008, 3, e3774. [Google Scholar]
- Boldicke, T.; Tesar, M.; Griesel, C.; Rohde, M.; Grone, H.J.; Waltenberger, J.; Kollet, O.; Lapidot, T.; Yayon, A.; Weich, H. Anti-VEGFR-2 scFvs for cell isolation. Single-chain antibodies recognizing the human vascular endothelial growth factor receptor-2 (VEGFR-2/flk-1) on the surface of primary endothelial cells and preselected CD34+ cells from cord Blood. Stem Cells 2001, 19, 24–36. [Google Scholar]
- Cochran, S.; Li, C.P.; Bytheway, I. An experimental and molecular-modeling study of the binding of linked sulfated tetracyclitols to FGF-1 and FGF-2. Chem. Biochem 2005, 6, 1882–1890. [Google Scholar]
- Karoli, T.; Liu, L.; Fairweather, J.K.; Hammond, E.; Li, C.P.; Cochran, S.; Bergefall, K.; Trybala, E.; Addison, R.S.; Ferro, V. Synthesis, biological activity, and preliminary pharmacokinetic evaluation of analogues of a phosphosulfomannan angiogenesis inhibitor (PI-88). J. Med. Chem 2005, 48, 8229–8236. [Google Scholar]
- Camozzi, M.; Rusnati, M.; Bugatti, A.; Bottazzi, B.; Mantovani, A.; Bastone, A.; Inforzato, A.; Vincenti, S.; Bracci, L.; Mastroianni, D.; Presta, M. Identification of an antiangiogenic FGF2-binding site in the N terminus of the soluble pattern recognition receptor PTX3. J. Biol. Chem 2006, 281, 22605–22613. [Google Scholar]
- Liu, L.; Ping Li, C.; Cochran, S.; Ferro, V. Application of the four-component Ugi condensation for the preparation of sulfated glycoconjugate libraries. Bioorg. Med. Chem. Lett 2004, 14, 2221–2226. [Google Scholar]
- Presta, M.; Oreste, P.; Zoppetti, G.; Belleri, M.; Tanghetti, E.; Leali, D.; Urbinati, C.; Bugatti, A.; Ronca, R.; Nicoli, S.; Moroni, E.; Stabile, H.; Camozzi, M.; Hernandez, G.A.; Mitola, S.; Dell’Era, P.; Rusnati, M.; Ribatti, D. Antiangiogenic activity of semisynthetic biotechnological heparins: low-molecular-weight-sulfated Escherichia coli K5 polysaccharide derivatives as fibroblast growth factor antagonists. Arterioscler Thromb Vasc Biol 2005, 25, 71–76. [Google Scholar]
- Rusnati, M.; Urbinati, C.; Caputo, A.; Possati, L.; Lortat-Jacob, H.; Giacca, M.; Ribatti, D.; Presta, M. Pentosan polysulfate as an inhibitor of extracellular HIV-1 Tat. J. Biol. Chem 2001, 276, 22420–22425. [Google Scholar]
- Pisano, C.; Aulicino, C.; Vesci, L.; Casu, B.; Naggi, A.; Torri, G.; Ribatti, D.; Belleri, M.; Rusnati, M.; Presta, M. Undersulfated, low-molecular-weight glycol-split heparin as an antiangiogenic VEGF antagonist. GlycoBiology. 2005, 15, 1C–6C. [Google Scholar]
- Cochran, S.; Li, C.; Fairweather, J.K.; Kett, W.C.; Coombe, D.R.; Ferro, V. Probing the interactions of phosphosulfomannans with angiogenic growth factors by surface plasmon resonance. J. Med. Chem 2003, 46, 4601–4608. [Google Scholar]
- Garcia-Olivas, R.; Hoebeke, J.; Castel, S.; Reina, M.; Fager, G.; Lustig, F.; Vilaro, S. Differential binding of platelet-derived growth factor isoforms to glycosaminoglycans. Histochem. Cell Biol 2003, 120, 371–382. [Google Scholar]
- Johnson, D.E.; Williams, L.T. Structural and functional diversity in the FGF receptor multigene family. Adv. Cancer Res 1993, 60, 1–41. [Google Scholar]
- Mohammadi, M.; Olsen, S.K.; Ibrahimi, O.A. Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev 2005, 16, 107–137. [Google Scholar]
- Yoon, S.Y.; Tefferi, A.; Li, C.Y. Cellular distribution of platelet-derived growth factor, transforming growth factor-beta, basic fibroblast growth factor, and their receptors in normal bone marrow. Acta Haematol 2000, 104, 151–157. [Google Scholar]
- Ribatti, D.; Gualandris, A.; Bastaki, M.; Vacca, A.; Iurlaro, M.; Roncali, L.; Presta, M. New model for the study of angiogenesis and antiangiogenesis in the chick embryo chorioallantoic membrane: the gelatin sponge/chorioallantoic membrane assay. J. Vasc Res 1997, 34, 455–463. [Google Scholar]
- Dell’Era, P.; Belleri, M.; Stabile, H.; Massardi, M.L.; Ribatti, D.; Presta, M. Paracrine and autocrine effects of fibroblast growth factor-4 in endothelial cells. Oncogene 2001, 20, 2655–2663. [Google Scholar]
- Eswarakumar, V.P.; Lax, I.; Schlessinger, J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev 2005, 16, 139–149. [Google Scholar]
- Rusnati, M.; Tanghetti, E.; Dell’Era, P.; Gualandris, A.; Presta, M. alphavbeta3 integrin mediates the cell-adhesive capacity and biological activity of basic fibroblast growth factor (FGF-2) in cultured endothelial cells. Mol. Biol. Cell 1997, 8, 2449–2461. [Google Scholar]
- Rusnati, M.; Urbinati, C.; Tanghetti, E.; Dell’Era, P.; Lortat-Jacob, H.; Presta, M. Cell membrane GM1 ganglioside is a functional coreceptor for fibroblast growth factor 2. Proc. Natl. Acad. Sci. USA 2002, 99, 4367–4372. [Google Scholar]
- Marcum, J.A.; Rosenberg, R.D. Heparinlike molecules with anticoagulant activity are synthesized by cultured endothelial cells. Biochem. Biophys. Res. Commun 1985, 126, 365–372. [Google Scholar]
- Kojima, T.; Leone, C.W.; Marchildon, G.A.; Marcum, J.A.; Rosenberg, R.D. Isolation and characterization of heparan sulfate proteoglycans produced by cloned rat microvascular endothelial cells. J. Biol. Chem 1992, 267, 4859–4869. [Google Scholar]
- Hayashi, K.; Madri, J.A.; Yurchenco, P.D. Endothelial cells interact with the core protein of basement membrane perlecan through beta 1 and beta 3 integrins: an adhesion modulated by glycosaminoglycan. J. Cell Biol 1992, 119, 945–959. [Google Scholar]
- Marcum, J.A.; McKenney, J.B.; Rosenberg, R.D. Acceleration of thrombin-antithrombin complex formation in rat hindquarters via heparinlike molecules bound to the endothelium. J. Clin. Invest 1984, 74, 341–350. [Google Scholar]
- Danielsson, A.; Raub, E.; Lindahl, U.; Bjork, I. Role of ternary complexes, in which heparin binds both antithrombin and proteinase, in the acceleration of the reactions between antithrombin and thrombin or factor Xa. J. Biol. Chem 1986, 261, 15467–15473. [Google Scholar]
- Rusnati, M.; Presta, M. Interaction of angiogenic basic fibroblast growth factor with endothelial cell heparan sulfate proteoglycans. Biological implications in neovascularization. Int. J. Clin. Lab Res 1996, 26, 15–23. [Google Scholar]
- Horowitz, A.; Tkachenko, E.; Simons, M. Fibroblast growth factor-specific modulation of cellular response by syndecan-4. J. Cell Biol 2002, 157, 715–725. [Google Scholar]
- Rusnati, M.; Urbinati, C.; Presta, M. Internalization of basic fibroblast growth factor (bFGF) in cultured endothelial cells: role of the low affinity heparin-like bFGF receptors. J. Cell Physiol 1993, 154, 152–161. [Google Scholar]
- Coltrini, D.; Rusnati, M.; Zoppetti, G.; Oreste, P.; Grazioli, G.; Naggi, A.; Presta, M. Different effects of mucosal, bovine lung and chemically modified heparin on selected biological properties of basic fibroblast growth factor. Biochem. J 1994, 303, 583–590. [Google Scholar]
- Presta, M.; Maier, J.A.; Rusnati, M.; Ragnotti, G. Basic fibroblast growth factor is released from endothelial extracellular matrix in a biologically active form. J. Cell Physiol 1989, 140, 68–74. [Google Scholar]
- Conrad, H. E. Heparin binding proteins. Academic Press: New York, NY, USA, 1998. [Google Scholar]
- Lindahl, U.; Lidholt, K.; Spillmann, D.; Kjellen, L. More to “heparin” than anticoagulation. Thromb Res 1994, 75, 1–32. [Google Scholar]
- Gospodarowicz, D.; Cheng, J. Heparin protects basic and acidic FGF from inactivation. J. Cell Physiol 1986, 128, 475–484. [Google Scholar]
- Sommer, A.; Rifkin, D.B. Interaction of heparin with human basic fibroblast growth factor: protection of the angiogenic protein from proteolytic degradation by a glycosaminoglycan. J. Cell Physiol 1989, 138, 215–220. [Google Scholar]
- Flaumenhaft, R.; Moscatelli, D.; Rifkin, D.B. Heparin and heparan sulfate increase the radius of diffusion and action of basic fibroblast growth factor. J. Cell Biol 1990, 111, 1651–1659. [Google Scholar]
- Casu, B.; Guerrini, M.; Naggi, A.; Perez, M.; Torri, G.; Ribatti, D.; Carminati, P.; Giannini, G.; Penco, S.; Pisano, C.; Belleri, M.; Rusnati, M.; Presta, M. Short heparin sequences spaced by glycol-split uronate residues are antagonists of fibroblast growth factor 2 and angiogenesis inhibitors. Biochemistry 2002, 41, 10519–10528. [Google Scholar]
- Klagsbrun, M.; Baird, A. A dual receptor system is required for basic fibroblast growth factor activity. Cell 1991, 67, 229–231. [Google Scholar]
- Guimond, S.; Maccarana, M.; Olwin, B.B.; Lindahl, U.; Rapraeger, A.C. Activating and inhibitory heparin sequences for FGF-2 (basic FGF). Distinct requirements for FGF-1, FGF-2, and FGF-4. J. Biol. Chem 1993, 268, 23906–23914. [Google Scholar]
- Richard, C.; Liuzzo, J.P.; Moscatelli, D. Fibroblast growth factor-2 can mediate cell attachment by linking receptors and heparan sulfate proteoglycans on neighboring cells. J. Biol. Chem 1995, 270, 24188–24196. [Google Scholar]
- Liekens, S.; Leali, D.; Neyts, J.; Esnouf, R.; Rusnati, M.; Dell’Era, P.; Maudgal, P.C.; De Clercq, E.; Presta, M. Modulation of fibroblast growth factor-2 receptor binding, signaling, and mitogenic activity by heparin-mimicking polysulfonated compounds. Mol. Pharmacol 1999, 56, 204–213. [Google Scholar]
- Presta, M.; Leali, D.; Stabile, H.; Ronca, R.; Camozzi, M.; Coco, L.; Moroni, E.; Liekens, S.; Rusnati, M. Heparin derivatives as angiogenesis inhibitors. Curr. Pharm. Des 2003, 9, 553–566. [Google Scholar]
- Leali, D.; Belleri, M.; Urbinati, C.; Coltrini, D.; Oreste, P.; Zoppetti, G.; Ribatti, D.; Rusnati, M.; Presta, M. Fibroblast growth factor-2 antagonist activity and angiostatic capacity of sulfated Escherichia coli K5 polysaccharide derivatives. J. Biol. Chem 2001, 276, 37900–37908. [Google Scholar]
- Presta, M.; Dell’era, P.; Mitola, S.; Moroni, E.; Ronca, R.; Rusnati, M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev 2005, 16, 159–178. [Google Scholar]
- Rusnati, M.; Presta, M. Fibroblast growth factors/fibroblast growth factor receptors as targets for the development of anti-angiogenesis strategies. Curr. Pharm. Des 2007, 13, 2025–2044. [Google Scholar]
- Bornstein, P.; Armstrong, L. C.; Hankenson, K. D.; Kyriakides, T. R.; Yang, Z. Thrombospondin 2, a matricellular protein with diverse functions. Matrix Biol 2000, 19, 557–568. [Google Scholar]
- Taraboletti, G.; Belotti, D.; Borsotti, P.; Vergani, V.; Rusnati, M.; Presta, M.; Giavazzi, R. The 140-kilodalton antiangiogenic fragment of thrombospondin-1 binds to basic fibroblast growth factor. Cell Growth Differ 1997, 8, 471–479. [Google Scholar]
- Margosio, B.; Marchetti, D.; Vergani, V.; Giavazzi, R.; Rusnati, M.; Presta, M.; Taraboletti, G. Thrombospondin 1 as a scavenger for matrix-associated fibroblast growth factor 2. Blood 2003, 102, 4399–4406. [Google Scholar]
- Bossard, C.; van den Berghe, L.; Laurell, H.; Castano, C.; Cerutti, M.; Prats, A.C.; Prats, H. Antiangiogenic properties of fibstatin, an extracellular FGF-2-binding polypeptide. Cancer Res 2004, 64, 7507–7512. [Google Scholar]
- Asplin, I.R.; Wu, S.M.; Mathew, S.; Bhattacharjee, G.; Pizzo, S.V. Differential regulation of the fibroblast growth factor (FGF) family by alpha (2)-macroglobulin: evidence for selective modulation of FGF-2-induced angiogenesis. Blood 2001, 97, 3450–3457. [Google Scholar]
- Dennis, P.A.; Saksela, O.; Harpel, P.; Rifkin, D.B. Alpha 2-macroglobulin is a binding protein for basic fibroblast growth factor. J. Biol. Chem 1989, 264, 7210–7216. [Google Scholar]
- Presta, M.; Camozzi, M.; Salvatori, G.; Rusnati, M. Role of the soluble pattern recognition receptor PTX3 in vascular biology. J. Cell Mol. Med 2007, 11, 723–738. [Google Scholar]
- Rusnati, M.; Camozzi, M.; Moroni, E.; Bottazzi, B.; Peri, G.; Indraccolo, S.; Amadori, A.; Mantovani, A.; Presta, M. Selective recognition of fibroblast growth factor-2 by the long pentraxin PTX3 inhibits angiogenesis. Blood 2004, 104, 92–99. [Google Scholar]
- De Marchis, F.; Ribatti, D.; Giampietri, C.; Lentini, A.; Faraone, D.; Scoccianti, M.; Capogrossi, M.C.; Facchiano, A. Platelet-derived growth factor inhibits basic fibroblast growth factor angiogenic properties in vitro and in vivo through its alpha receptor. Blood 2002, 99, 2045–2053. [Google Scholar]
- Perollet, C.; Han, Z.C.; Savona, C.; Caen, J.P.; Bikfalvi, A. Platelet factor 4 modulates fibroblast growth factor 2 (FGF-2) activity and inhibits FGF-2 dimerization. Blood 1998, 91, 3289–3299. [Google Scholar]
- Spinetti, G.; Camarda, G.; Bernardini, G.; Romano Di Peppe, S.; Capogrossi, M.C.; Napolitano, M. The chemokine CXCL13 (BCA-1) inhibits FGF-2 effects on endothelial cells. Biochem. BioPhys. Res. Commun 2001, 289, 19–24. [Google Scholar]
- Facchiano, A.; Russo, K.; Facchiano, A. M.; De Marchis, F.; Facchiano, F.; Ribatti, D.; Aguzzi, M.S.; Capogrossi, M.C. Identification of a novel domain of fibroblast growth factor 2 controlling its angiogenic properties. J. Biol. Chem 2003, 278, 8751–8760. [Google Scholar]
- Presta, M.; Rusnati, M.; Urbinati, C.; Sommer, A.; Ragnotti, G. Biologically active synthetic fragments of human basic fibroblast growth factor (bFGF): identification of two Asp-Gly-Arg-containing domains involved in the mitogenic activity of bFGF in endothelial cells. J. Cell Physiol 1991, 149, 512–524. [Google Scholar]
- Ray, J.; Baird, A.; Gage, F.H. A 10-amino acid sequence of fibroblast growth factor 2 is sufficient for its mitogenic activity on neural progenitor cells. Proc. Natl. Acad. Sci. USA 1997, 94, 7047–7052. [Google Scholar]
- Schubert, D.; Ling, N.; Baird, A. Multiple influences of a heparin-binding growth factor on neuronal development. J. Cell Biol 1987, 104, 635–643. [Google Scholar]
- Baird, A.; Schubert, D.; Ling, N.; Guillemin, R. Receptor- and heparin-binding domains of basic fibroblast growth factor. Proc. Natl. Acad. Sci. USA 1988, 85, 2324–2328. [Google Scholar]
- Walicke, P.A.; Feige, J.J.; Baird, A. Characterization of the neuronal receptor for basic fibroblast growth factor and comparison to receptors on mesenchymal cells. J. Biol. Chem 1989, 264, 4120–4126. [Google Scholar]
- Oyama, S., Jr.; Miranda, M.T.; Toma, I.N.; Viviani, W.; Gambarini, A.G. Mitogenic activity of peptides related to the sequence of human fibroblast growth factor-1. Biochem. Mol. Biol. Int 1996, 39, 1237–1244. [Google Scholar]
- Kiyota, S.; Franzoni, L.; Nicastro, G.; Benedetti, A.; Oyama, S., Jr.; Viviani, W.; Gambarini, A. G.; Spisni, A.; Miranda, M. T. Introduction of a chemical constraint in a short peptide derived from human acidic fibroblast growth factor elicits mitogenic structural determinants. J. Med. Chem 2003, 46, 2325–2333. [Google Scholar]
- Williams, E.J.; Williams, G.; Howell, F.V.; Skaper, S.D.; Walsh, F.S.; Doherty, P. Identification of an N-cadherin motif that can interact with the fibroblast growth factor receptor and is required for axonal growth. J. Biol. Chem 2001, 276, 43879–43886. [Google Scholar]
- Ito, C.; Saitoh, Y.; Fujita, Y.; Yamazaki, Y.; Imamura, T.; Oka, S.; Suzuki, S. Decapeptide with fibroblast growth factor (FGF)-5 partial sequence inhibits hair growth suppressing activity of FGF-5. J. Cell Physiol 2003, 197, 272–283. [Google Scholar]
- Hagedorn, M.; Zilberberg, L.; Lozano, R.M.; Cuevas, P.; Canron, X.; Redondo-Horcajo, M.; Gimenez-Gallego, G.; Bikfalvi, A. A short peptide domain of platelet factor 4 blocks angiogenic key events induced by FGF-2. Faseb. J 2001, 15, 550–552. [Google Scholar]
- Tzeng, S.F.; Deibler, G.E.; DeVries, G.H. Myelin basic protein and myelin basic protein peptides induce the proliferation of Schwann cells via ganglioside GM1 and the FGF receptor. Neurochem. Res 1999, 24, 255–260. [Google Scholar]
- Kanda, S.; Shono, T.; Tomasini-Johansson, B.; Klint, P.; Saito, Y. Role of thrombospondin-1-derived peptide, 4N1K, in FGF-2-induced angiogenesis. Exp. Cell Res 1999, 252, 262–272. [Google Scholar]
- Kiselyov, V.V.; Skladchikova, G.; Hinsby, A.M.; Jensen, P.H.; Kulahin, N.; Soroka, V.; Pedersen, N.; Tsetlin, V.; Poulsen, F.M.; Berezin, V.; Bock, E. Structural basis for a direct interaction between FGFR1 and NCAM and evidence for a regulatory role of ATP. Structure 2003, 11, 691–701. [Google Scholar]
- Berezin, V.; Bock, E. NCAM mimetic peptides: An update. Neurochem. Res 2008. [Google Scholar] [CrossRef]
- Neiiendam, J.L.; Kohler, L.B.; Christensen, C.; Li, S.; Pedersen, M.V.; Ditlevsen, D.K.; Kornum, M.K.; Kiselyov, V.V.; Berezin, V.; Bock, E. An NCAM-derived FGF-receptor agonist, the FGL-peptide, induces neurite outgrowth and neuronal survival in primary rat neurons. J. Neurochem 2004, 91, 920–935. [Google Scholar]
- Hansen, S. M.; Li, S.; Bock, E.; Berezin, V. Synthetic NCAM-derived Ligands of the Fibroblast Growth Factor Receptor. Neurochem. Res 2008. [Google Scholar] [CrossRef]
- Anderson, A.A.; Kendal, C.E.; Garcia-Maya, M.; Kenny, A.V.; Morris-Triggs, S.A.; Wu, T.; Reynolds, R.; Hohenester, E.; Saffell, J.L. A peptide from the first fibronectin domain of NCAM acts as an inverse agonist and stimulates FGF receptor activation, neurite outgrowth and survival. J. Neurochem 2005, 95, 570–583. [Google Scholar]
- Jacobsen, J.; Kiselyov, V.; Bock, E.; Berezin, V. A peptide motif from the second fibronectin module of the neural cell adhesion molecule, NCAM, NLIKQDDGGSPIRHY, is a binding site for the FGF receptor. Neurochem. Res 2008. [Google Scholar] [CrossRef]
- Hansen, S.M.; Kohler, L.B.; Li, S.; Kiselyov, V.; Christensen, C.; Owczarek, S.; Bock, E.; Berezin, V. NCAM-derived peptides function as agonists for the fibroblast growth factor receptor. J. Neurochem 2008, 106, 2030–2041. [Google Scholar]
- Yayon, A.; Aviezer, D.; Safran, M.; Gross, J.L.; Heldman, Y.; Cabilly, S.; Givol, D.; Katchalski-Katzir, E. Isolation of peptides that inhibit binding of basic fibroblast growth factor to its receptor from a random phage-epitope library. Proc. Natl. Acad. Sci. USA 1993, 90, 10643–10647. [Google Scholar]
- Wu, X.; Yan, Q.; Huang, Y.; Huang, H.; Su, Z.; Xiao, J.; Zeng, Y.; Wang, Y.; Nie, C.; Yang, Y.; Li, X. Isolation of a novel basic FGF-binding peptide with potent antiangiogenetic activity. J. Cell Mol. Med 2008. [Google Scholar] [CrossRef]
- Ballinger, M.D.; Shyamala, V.; Forrest, L.D.; Deuter-Reinhard, M.; Doyle, L.V.; Wang, J.X.; Panganiban-Lustan, L.; Stratton, J.R.; Apell, G.; Winter, J.A.; Doyle, M.V.; Rosenberg, S.; Kavanaugh, W.M. Semirational design of a potent, artificial agonist of fibroblast growth factor receptors. Nat. Biotechnol 1999, 17, 1199–1204. [Google Scholar]
- Maruta, F.; Parker, A.L.; Fisher, K.D.; Hallissey, M.T.; Ismail, T.; Rowlands, D.C.; Chandler, L.A.; Kerr, D.J.; Seymour, L.W. Identification of FGF receptor-binding peptides for cancer gene therapy. Cancer Gene Ther 2002, 9, 543–552. [Google Scholar]
- McConnell, S.J.; Thon, V.J.; Spinella, D.G. Isolation of fibroblast growth factor receptor binding sequences using evolved phage display libraries. Comb. Chem High Throughput Screen 1999, 2, 155–163. [Google Scholar]
- Fan, H.; Duan, Y.; Zhou, H.; Li, W.; Li, F.; Guo, L.; Roeske, R.W. Selection of peptide ligands binding to fibroblast growth factor receptor 1. IUBMB Life 2002, 54, 67–72. [Google Scholar]
- Cosic, I.; Drummond, A.E.; Underwood, J.R.; Hearn, M.T. In vitro inhibition of the actions of basic FGF by a novel 16 amino acid peptide. Mol. Cell Biochem 1994, 130, 1–9. [Google Scholar]
- Leali, D.; Bianchi, R.; Bugatti, A.; Nicoli, S.; Mitola, S.; Ragona, L.; Tomaselli, S.; Gallo, G.; Catello, S.; Riviecco, V.; Zetta, L.; Presta, M. Fibroblast growth factor 2-aantagonist activity of a long pentraxin-3-derived antiangiogenic penta peptide. J. Cell Mol. Med 2009, in press.. [Google Scholar]
- Urbinati, C.; Chiodelli, P.; Rusnati, M. Polyanionic drugs and viral oncogenesis: a novel approach to control infection, tumor-associated inflammation and angiogenesis. Molecules 2008, 13, 2758–2785. [Google Scholar]
- Rusnati, M.; Oreste, P.; Zoppetti, G.; Presta, M. Biotechnological engineering of heparin/heparan sulphate: a novel area of multi-target drug discovery. Curr. Pharm. Des 2005, 11, 2489–2499. [Google Scholar]
- Casu, B.; Petitou, M.; Provasoli, M.; Sinay, P. Conformational flexibility: a new concept for explaining binding and biological properties of iduronic acid-containing glycosaminoglycans. Trends Biochem. Sci 1988, 13, 221–225. [Google Scholar]
- Marquette, C.A.; Blum, L.J. State of the art and recent advances in immunoanalytical systems. Biosens. Bioelectr 2006, 21, 1424–1433. [Google Scholar]
- Homola, J. Present and future of surface plasmon biosensor. Anal. Bioanal. Chem 2003, 377, 528–539. [Google Scholar]
- Maccarana, M.; Casu, B.; Lindahl, U. Minimal sequence in heparin/heparan sulfaate required for binding of basic fibroblast growth factor. J. Biol. Chem 1993, 268, 23898–23905. [Google Scholar]
- Levicky, R.; Horgan, A. Physicochemical perspectives on DNA microarray and biosensor technologies. Trends Biotechnol 2005, 23, 143–149. [Google Scholar]
- Wild, D. The Immunoassay Handbook; Elsevier: London, UK, 2005. [Google Scholar]
- Bergese, P.; Cretich, M.; Oldani, C.; Oliviero, G.; Di Carlo, G.; Depero, L.E.; Chiari, M. Advances in parallel screening of drug candidates. Curr. Med. Chem 2008, 15, 1706–1719. [Google Scholar]
- Bergese, P.; Oliviero, G.; Colombo, I.; Depero, L.E. Molecular recognition by contact angle: proof of concept with DNA hybridization. Langmuir 2009, 25, 4271–4273. [Google Scholar]
- Keshet, E.; Ben-Sasson, S.A. Anticancer drug targets: approaching angiogenesis. J. Clin. Invest 1999, 104, 1497–1501. [Google Scholar]
- Eggert, A.; Ikegaki, N.; Kwiatkowski, J.; Zhao, H.; Brodeur, G.M.; Himelstein, B.P. High-level expression of angiogenic factors is associated with advanced tumor stage in human neuroblastomas. Clin. Cancer Res 2000, 6, 1900–1908. [Google Scholar]
- Ashikari-Hada, S.; Habuchi, H.; Kariya, Y.; Itoh, N.; Reddi, A.H.; Kimata, K. Characterization of growth factor-binding structures in heparin/heparan sulfate using an octasaccharide library. J. Biol. Chem 2004, 279, 12346–12354. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AGF | Immobilized receptor | Binding parameters | Reference | ||
|---|---|---|---|---|---|
| Kon (M−1 s−1) | Koff (s−1) | Kd (nM) | |||
| FGF2 | FGFR1-IIIc (D1–D3) | 1.1 × 105 | 1.1 × 10−2 | 99.0 | [24] |
| FGFR1-IIIc (D2–D3) | 9.6 × 104 | 5.9 × 10−3 | 62.0 | [25] * | |
| FGFR1-IIIc (D2–D3) | 9.6 × 104 | 6.0 × 10−3 | 62.0 | [26]* | |
| FGFR1-IIIc (D1–D3) | 3.0 × 105 | 1.4 × 10−6 | 0.005 | [A. Bugatti,UD] | |
| FGFR2-IIIb | 1.3 × 106 | 6.5 × 10−4 | 0.5 | [27] | |
| FGFR3-IIIc (D2–D3) | No binding | No binding | No binding | [26] * | |
| αvβ3 integrin receptor | 5.1 × 104 | 3.3 × 10−4 | 6.5 | [A. Bugatti,UD]* | |
| FGF1 | FGFR1-IIIc | 2.4 × 106 | 4.1 × 10−3 | 35.0 | [28] |
| FGFR1-IIIc (D2–D3) | NR | NR | 0.03 | [29] | |
| FGFR1-IIIc (D2–D3) | 2.2 × 105 | 3.0 × 10−2 | 136.0 | [26]* | |
| FGFR2-IIIb (D2–D3) | 1.4 × 105 | 4.7 × 10−3 | 59.6 | [28] | |
| FGFR2-IIIb | 8.0 × 105 | 6.4 × 10−4 | 0.8 | [27] | |
| FGFR3-IIIc (D2–D3) | 8.8 × 105 | 2.0 × 10−1 | 230.0 | [30] * | |
| FGFR3-IIIc (D2–D3) | 8.8 × 105 | 2.0 × 10−1 | 230.0 | [26] * | |
| FGFR3-IIIc (D1–D3) | 2.0 × 105 | 1.8 × 10−1 | 916.0 | [30] * | |
| αvβ3 integrin receptor | NR | NR | 1,100.0 | [29] | |
| FGF4 | FGFR1-IIIc (D2–D3) | 2.6 × 105 | 4.3 × 10−2 | 165.0 | [26] * |
| FGFR2-IIIb | 1.5 × 106 | 6.1 × 10−4 | 0.42 | [27] | |
| FGFR3-IIIc (D2–D3) | No binding | No binding | No binding | [26] * | |
| VEGF-A165 | VEGFR2/KDR | 3.6 × 106 | 1.3 × 10−4 | 0.037 | [31] |
| VEGF-A165 | VEGFR2/KDR | 6.6 × 104 | 1.3 × 10−5 | 0.19 | [32] * |
| VEGF-A165 | VEGFR2/KDR | 8.4 × 104 | 3.2 × 10−5 | 0.38 | [33] * |
| VEGF-A165 | VEGFR2/KDR | 0.5–2.2 × 106 | 2.0–4.0 × 10−4 | 0.2–0.6 | [34] |
| VEGF-A165 | VEGFR2/KDR | 5.7 × 104 | 2.3 × 10−6 | 0.041 | [A. Bugatti, UD] |
| VEGF-A165 | VEGFR1/Flt | 4.0 × 106 | 3.0 × 10−5 | 0.007 | [35] |
| VEGF-A165 | VEGFR1/Flt | 5.7 × 105 | 1.7 × 10−5 | 0.03 | [33] * |
| VEGF-A165 | Neuropilin-1 | NR | NR | NR | [36] |
| VEGF-A165 | Neuropilin-1 | 1–10 × 105 | 1.0 × 10−2 | 2,000.0 | [37] |
| VEGF-A189 | Neuropilin-1 | NR | NR | NR | [38] |
| VEGF-A121 | Neuropilin-1 | No binding | No binding | No binding | [36] |
| VEGF-A109 | Neuropilin-1 | No binding | No binding | No binding | [37] |
| VEGF-C | VEGFR1/Flt | NR | NR | NR | [38] |
| VEGFR2/KDR | NR | NR | NR | ||
| VEGFR3 | NR | NR | NR | ||
| VEGF ** | VEGFR1/Flt | 8.7 × 105 | 1.5 × 10−5 | 0.017 | [33] * |
| VEGFR2/KDR | 4.2 × 104 | 2.7 × 10−4 | 6.5 | ||
| HGF | c-MET | 1.2 × 105 | 1.1 × 10−2 | 90.0 | [39] |
| c-MET | 3.0 × 104 | 6.2 × 10−3 | 50.0 | [40] * | |
| c-MET | NR | NR | NR | [40] | |
| c-MET | NR | NR | NR | [41] * | |
| HIV-Tat | VEGFR2/KDR | 1.7 × 105 | 1.2 × 10−5 | 0.07 | [A. Bugatti, UD] |
| αvβ3 integrin receptor | 1.2 × 107 | 3.8 × 10−1 | 32.0 | [42] * | |
| PDGF-BB | PDGFRα | 8.3 × 103 | 1.2 × 10−3 | 150.0 | [43] |
| PDGF-BB | PDGFRβ | 9.5 × 105 | 1.5 × 10−3 | 1.6 | |
| PDGF-AA | PDGFRα | 1.1 × 105 | 1.5 × 10−3 | 13.4 | |
| PDGF-AA | PDGFRβ | 3.5 × 103 | 1.6 × 10−3 | 453.0 | |
| AGF | Immobilized proteoglycan | Binding parameters | Reference | ||
|---|---|---|---|---|---|
| Kon (M−1 s−1) | Koff (s−1) | Kd (nM) | |||
| FGF2 | agrin | 1.8 × 105 | 4.6 × 10−4 | 2.5 | [44] |
| syndecan 1/4 | 1.6 × 107 | 4.4 × 10−2 | 2.5 | [45] | |
| HSPG | 8.5 × 105 | 1.3 × 10−2 | 14.7 | [46] | |
| HSPG (perlecan) | NR | NR | NR | [47] * | |
| HSPG (glypican) | NR | NR | NR | [48] | |
| CSPG | 7.7 × 105 | 2.3 × 10−2 | 30.5 | [49] | |
| CSPG | 1.5 × 105 | 2.0 × 10−4 | 12.7 | [50]* | |
| CSPG | No binding | No binding | No binding | [45] | |
| FGF1 | HSPG (perlecan) | NR | NR | NR | [51] |
| HSPG (perlecan) | NR | NR | NR | [52] | |
| CSPG | No binding | No binding | No binding | [49] | |
| FGF4 | HSPG | 1.7 × 105 | 1.5 | NR | [46] |
| VEGF-A165 | CSPG | 7.0 × 105 | 1.7 × 10−2 | 24.0 | [49] |
| MK | CSPG | 8.2 × 104 | 8.9 × 10−5 | 1.5 | [49] |
| CSPG | 1.3 × 104 | 4.8 × 10−3 | 367.0 | [45] | |
| HSPG (syndecan 1/4) | 6.9 × 104 | 1.7 × 10−3 | 25.9 | [45] | |
| PTN | CSPG | 4.2 × 105 | 7.4 × 10−5 | 0.2 | [49] |
| CSPG | 6.6 × 103 | 3.5 × 10−2 | 5210.0 | [45] | |
| CSPG | 2.0 × 106 | 2.7 × 10−4 | 0.14 | [53] * | |
| HSPG | 7.6 × 105 | 8.9 × 10−3 | 11.9 | [45] | |
| HB-EGF | CSPG | 1.1 × 106 | 9.1 × 10−3 | 10.0 | [49] |
| PDGF-BB | HSPG | 2.4 × 105 | 7.8 × 10−4 | 3.0 | [54] |
| PDGF-AA | HSPG | 3.4 × 104 | 7.8 × 10−4 | 23.0 | |
| PDGF-AA | CSPG | 8.5 × 104 | 2.2 × 10−3 | 25.9 | [50] * |
| Experimental model | AGF (references) | |
|---|---|---|
| AFG/receptor interaction | Ligand: receptor Analyte: AGF | FGF2 [24,25,27,44–46,48,49]; FGF1 [27–29,49,51,52]; FGF4 [26,27,46,49]; VEGF [31,34–38,49]; MK, PTN [45,49]; HB-EGF [49]; HGF [39,40]; HIV-Tat [A. Bugatti, UD]; PDGF [54] |
| Ligand: AGF Analyte: receptor | FGF2 [25,26,47,50]; FGF1 [26,30]; FGF4 [27]; VEGF [32,33]; PTN [53]; HIV-1 Tat [42]; HGF [40,41]; PDGF [50] | |
| AGF/inhibitor or receptor/inhibitor interactions | Ligand: AGF Analyte: inhibitor | FGF2 [59]; FGF4[60]; VEGF [61–68]; HIV-Tat [69,70]; PDGF [71] |
| Ligand: inhibitor Analyte: AGF | FGF1 [72,73]; FGF2 [25,59,72–75]; VEGF [72,73,76–79]; IL-8 [72]; PDGF [73]; HGF [73] | |
| Ligand: receptor Analyte: inhibitor | FGF1 [80]; FGF2 [74,81,82]; VEGF [61,66,68,83–87]; PDGF [43]; angiopoietin [86] | |
| Ligand: inhibitor Analyte: receptor | FGF1 [28]; FGF2 [74,88]; VEGF [89] | |
| competition experiments: inhibitor vs analyte | Ligand: receptor Analyte: AGF | FGF1 [80]; FGF2 [48] [A. Bugatti, UD]; VEGF [34]; HB-EGF [49]; MK & PTN [45] |
| Ligand: AGF binder (e.g., heparin) Analyte: AGF | FGF1 [90,91]; FGF2 [72,74,90–94]; FGF8 [A. Bugatti, UD]; HIV-Tat [70,95]; VEGF [91,96,97]; PDGF [98] | |
| Protein of origin | Peptides | Target | References |
|---|---|---|---|
| FGF2 | FGF2(48–58) (FREG) | FGF2 | [142] |
| FGF2(38–61) | ? | [143] | |
| FGF2(82–101) | ? | [143] | |
| FGF2(119–126) | FGF2 | [74]* | |
| FGF2-derived DGR-containing peptides (4 peptides studied) | ? | [143] | |
| FGF2(68–77) | FGFR | [144] | |
| FGF2(24–68) (Peptide D) | FGFR | [145,146] | |
| FGF2(93–120) (Peptide N) | FGFR | [145] | |
| FGF2(106–115) | FGFR | [145,146] | |
| FGF2(103–146) | FGFR | [147] | |
| F2A4-K-NS | FGFR | [24] | |
| FGFs (β10-β11 loop) | dekafins (homologous to the NCAM FGFR-binding region) | FGFR | [28]* |
| FGF1 | FGF1(112–147) and related peptides | FGFR | [148] |
| FGF1(99–108) | FGFR | [149] | |
| FGF1 mimetics (6 peptides studied) | FGFR | [150] | |
| FGF5 | FGF5(95–104) (peptide P3) | FGFR | [151] |
| N-cadherin | EDC4 mimetics (2 peptides studied) | FGFR | [150] |
| PTX3 N-terminus | PTX3(97–110) (and 28 related peptides) | FGF2 | [92]* |
| PF4 | PF4(47–70) | FGF2 | [152] |
| Myelin Basic Protein | MBP(152–167) | FGFR | [153] |
| TSP-1 | 4N1K | ? | [154] |
| (type III repeats-derived peptides) (6 peptides studied) | FGF2 | [59]* | |
| NCAM(681–695) | FGL | FGFR | [155–158] |
| FRM-10 | FGFR | [156,158,159] | |
| FRM-10 cyclic | FGFR | [156,158,159] | |
| FRM-13 | FGFR | [156,158,159] | |
| DekaCAM | FGFR | [28,156,158] | |
| BCL | FGFR | [156,158,160] | |
| Encamin A | FGFR | [156,161] | |
| Encamin C | FGFR | [156,161] | |
| Encamin E | FGFR | [156,161] | |
| Random phage epitope library screening | Epitope sequence | FGFR | [162] |
| FGF2(13–18) | FGFR | [162] | |
| FGF2(119–126) | FGFR | [162] | |
| FGF2(120–125) | FGFR | [162] | |
| Peptide P7 (hydrophobic) | FGF2 | [163] | |
| C19 (3 peptides studied) | FGFR | [164–166] | |
| Peptide P2 (hydrophobic) | FGFR | [167] | |
| Molecular modelling | 16–24 mer peptides | FGFR | [82,168]* |
| Heparin-like compounds | AGF | Reference |
|---|---|---|
| K5 derivatives suramin analogs | FGF2 | [94] [M. Presta, UD] |
| K5 derivatives | FGF8 | [M. Presta, UD] |
| Glycol-split heparins phosphosulfomannan derivatives | VEGF | [96] [97] |
| pentosan polysulfate sulfonic acid polymers K5 derivatives | HIV-Tat | [95] [70] [A. Bugatti, UD] |
| partially digested CS* | HB-EGF | [49] |
© 2009 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rusnati, M.; Bugatti, A.; Mitola, S.; Leali, D.; Bergese, P.; Depero, L.E.; Presta, M. Exploiting Surface Plasmon Resonance (SPR) Technology for the Identification of Fibroblast Growth Factor-2 (FGF2) Antagonists Endowed with Antiangiogenic Activity. Sensors 2009, 9, 6471-6503. https://doi.org/10.3390/s90806471
Rusnati M, Bugatti A, Mitola S, Leali D, Bergese P, Depero LE, Presta M. Exploiting Surface Plasmon Resonance (SPR) Technology for the Identification of Fibroblast Growth Factor-2 (FGF2) Antagonists Endowed with Antiangiogenic Activity. Sensors. 2009; 9(8):6471-6503. https://doi.org/10.3390/s90806471
Chicago/Turabian StyleRusnati, Marco, Antonella Bugatti, Stefania Mitola, Daria Leali, Paolo Bergese, Laura E. Depero, and Marco Presta. 2009. "Exploiting Surface Plasmon Resonance (SPR) Technology for the Identification of Fibroblast Growth Factor-2 (FGF2) Antagonists Endowed with Antiangiogenic Activity" Sensors 9, no. 8: 6471-6503. https://doi.org/10.3390/s90806471
APA StyleRusnati, M., Bugatti, A., Mitola, S., Leali, D., Bergese, P., Depero, L. E., & Presta, M. (2009). Exploiting Surface Plasmon Resonance (SPR) Technology for the Identification of Fibroblast Growth Factor-2 (FGF2) Antagonists Endowed with Antiangiogenic Activity. Sensors, 9(8), 6471-6503. https://doi.org/10.3390/s90806471