
Targeting Protein Kinase CK2: Evaluating CX-4945 Potential for GL261 Glioblastoma Therapy in Immunocompetent Mice

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
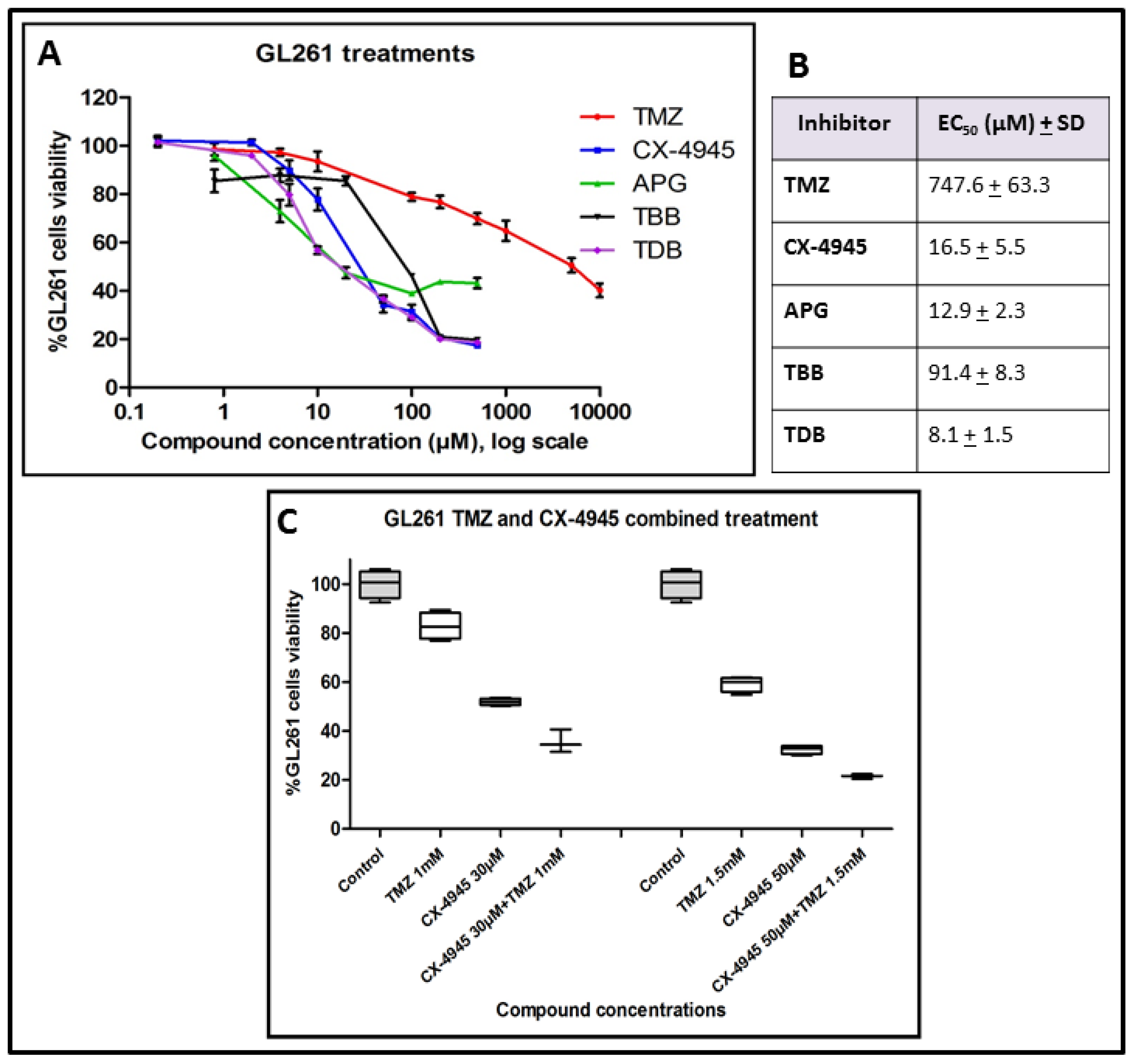
2.1. GL261 Cell Viability under CK2 Inhibition Treatment
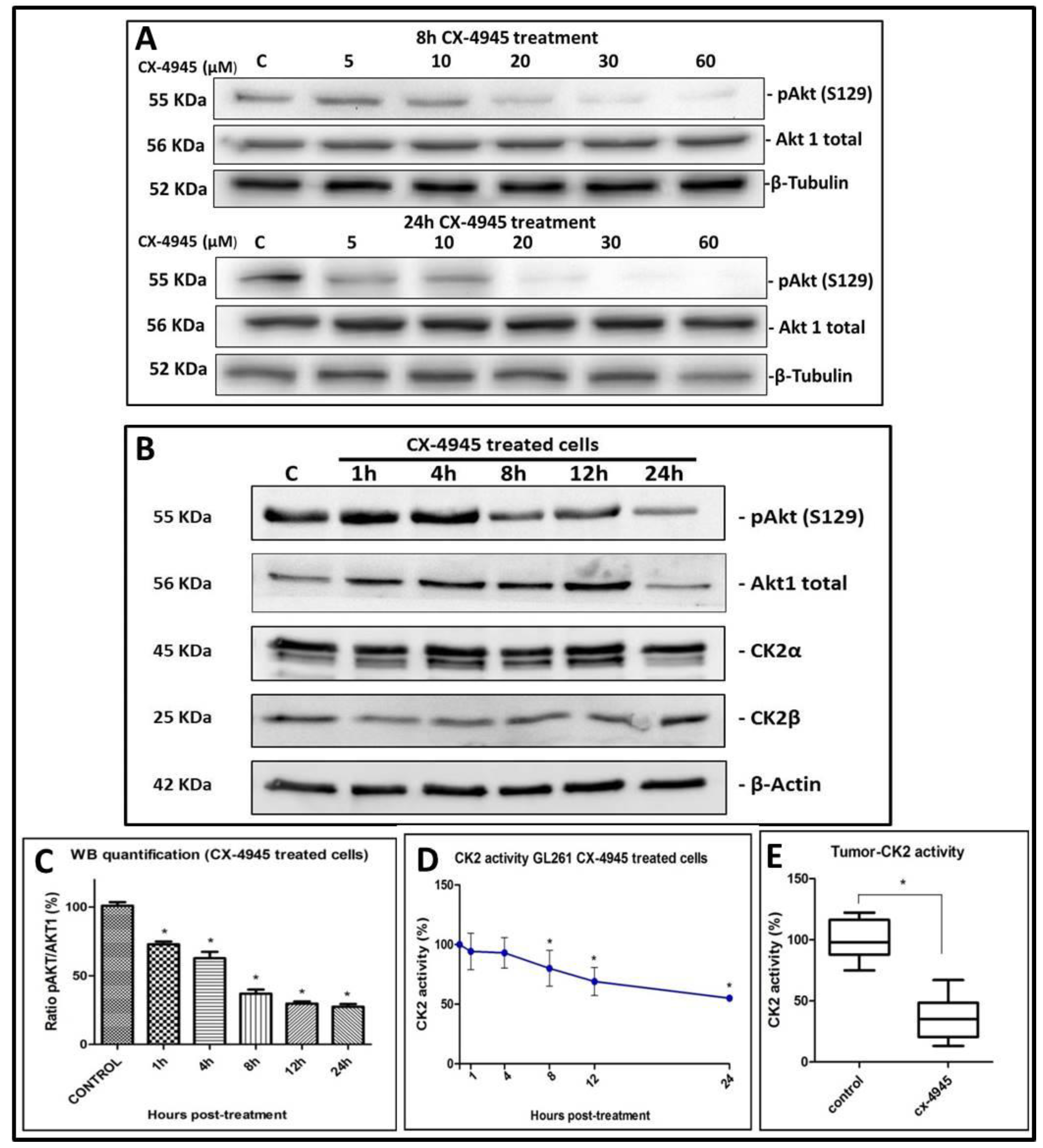
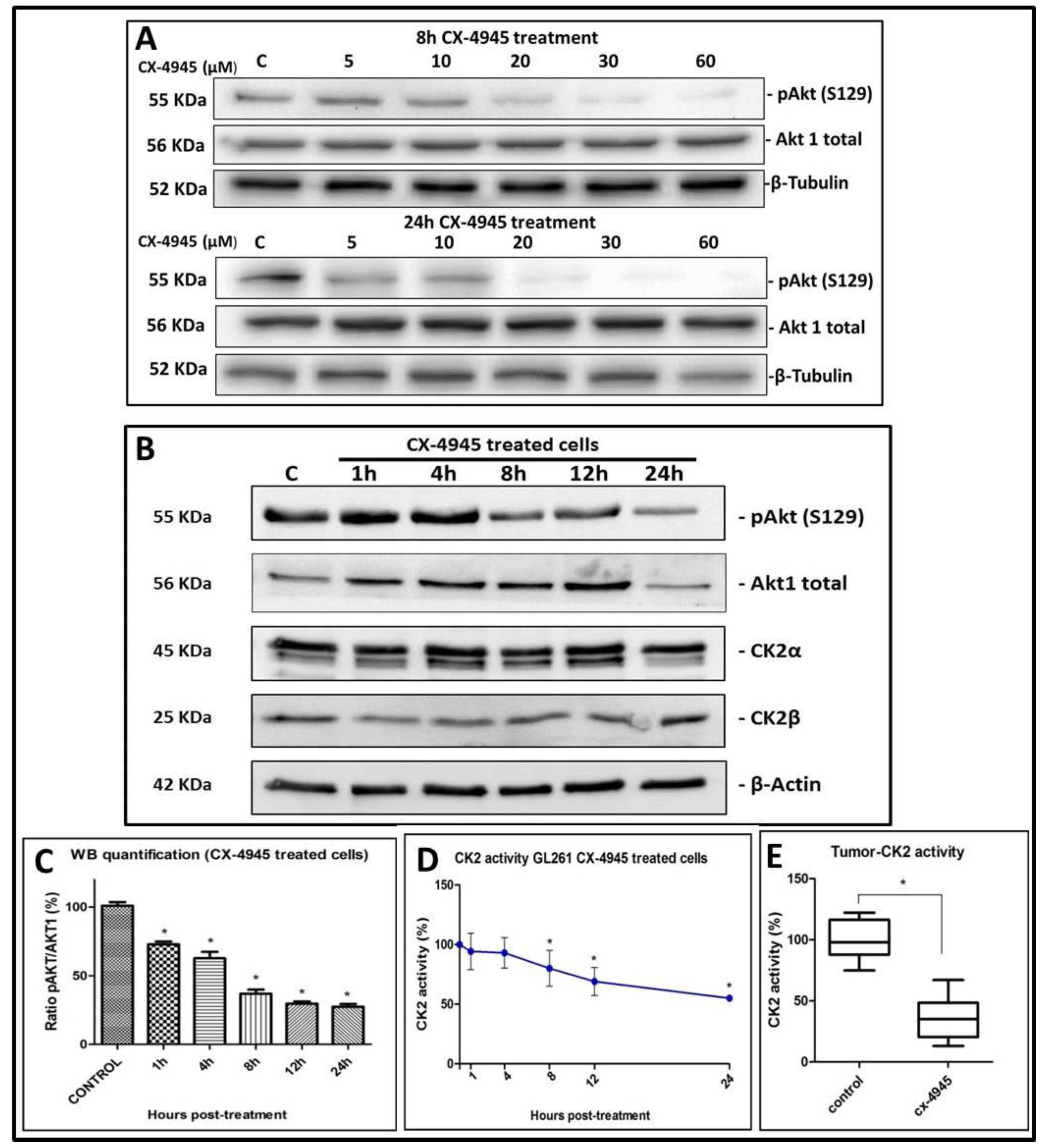
2.2. CK2 Activity in GL261 Cells Treated with CX-4945
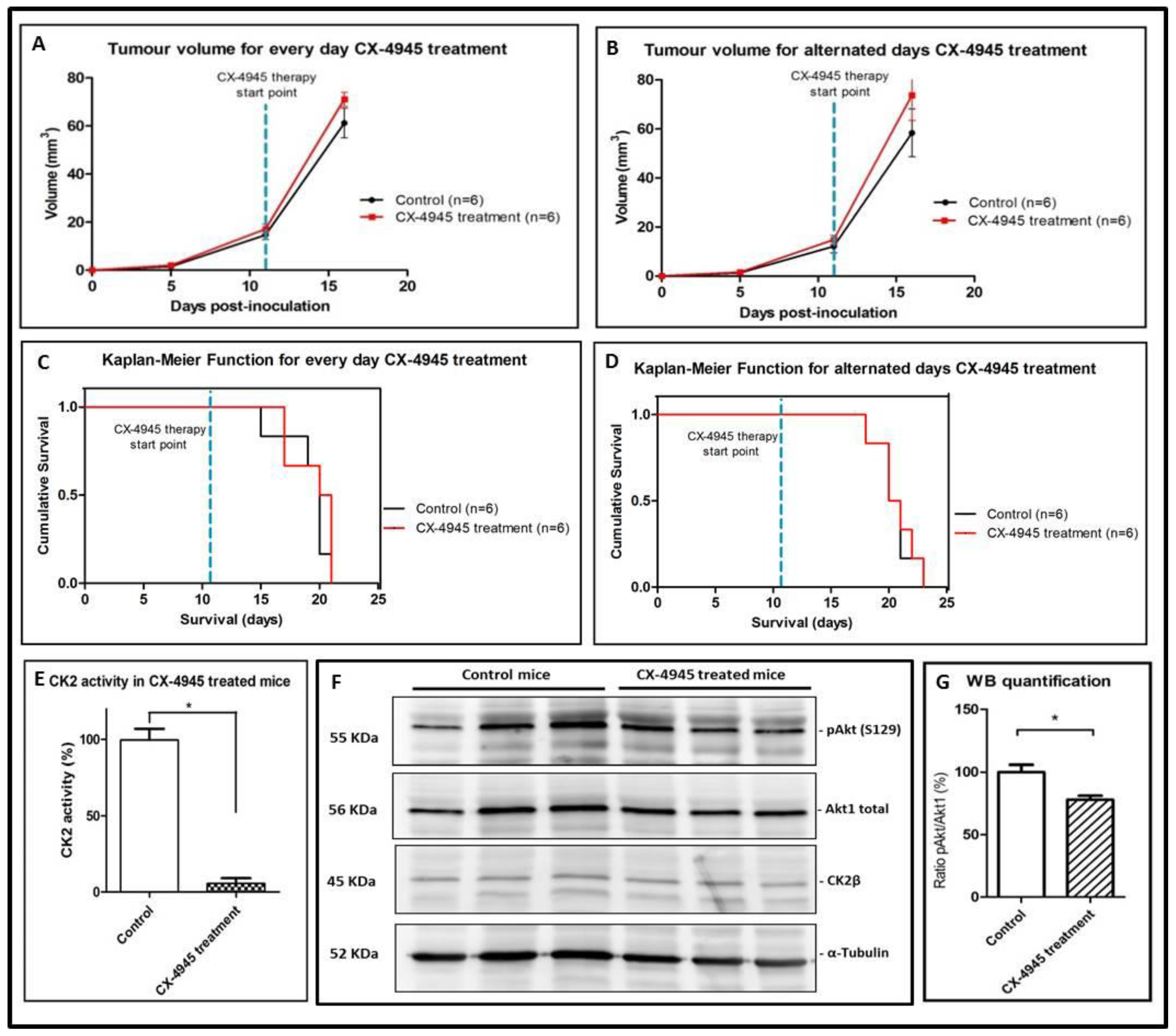
2.3. CX-4945 Mice Tolerability
2.4. CK2 Activity in CX-4945 Treated Mice
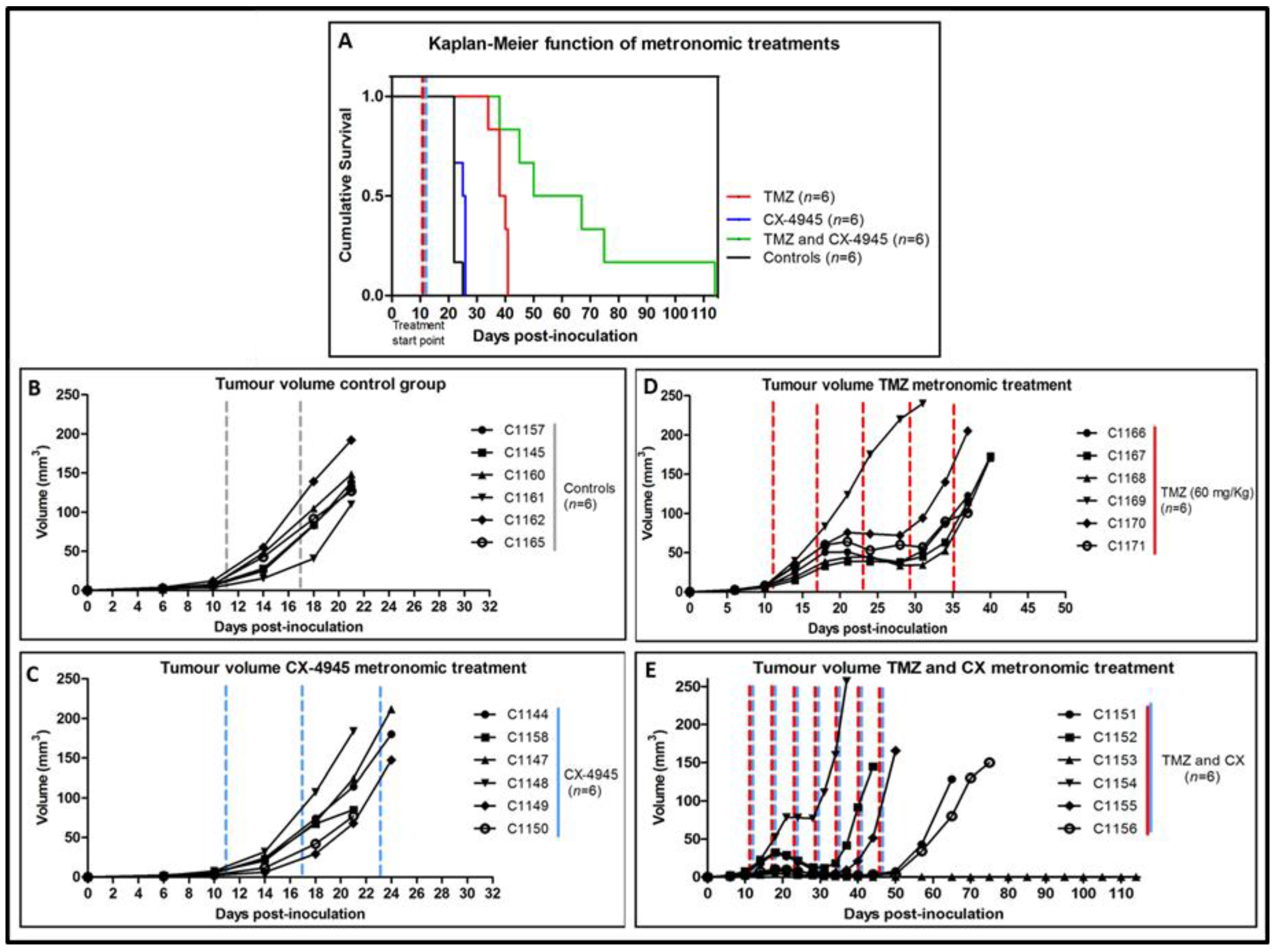
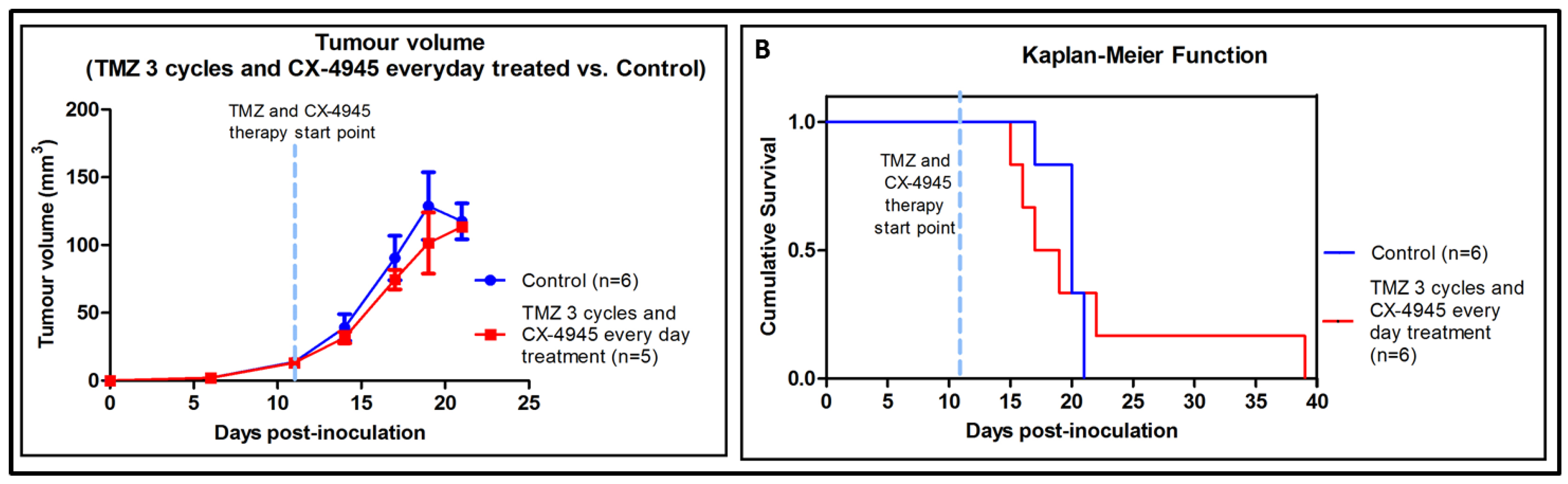
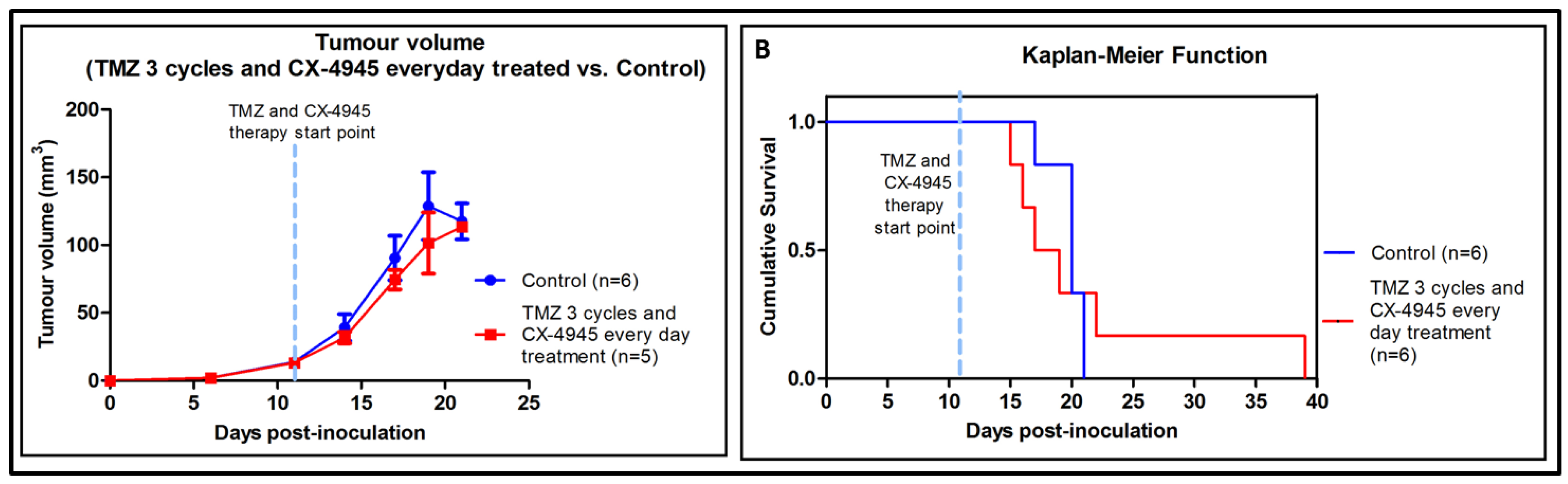
2.5. Metronomic Longitudinal Treatments with CX-4945 and/or TMZ in Tumour-Bearing Mice
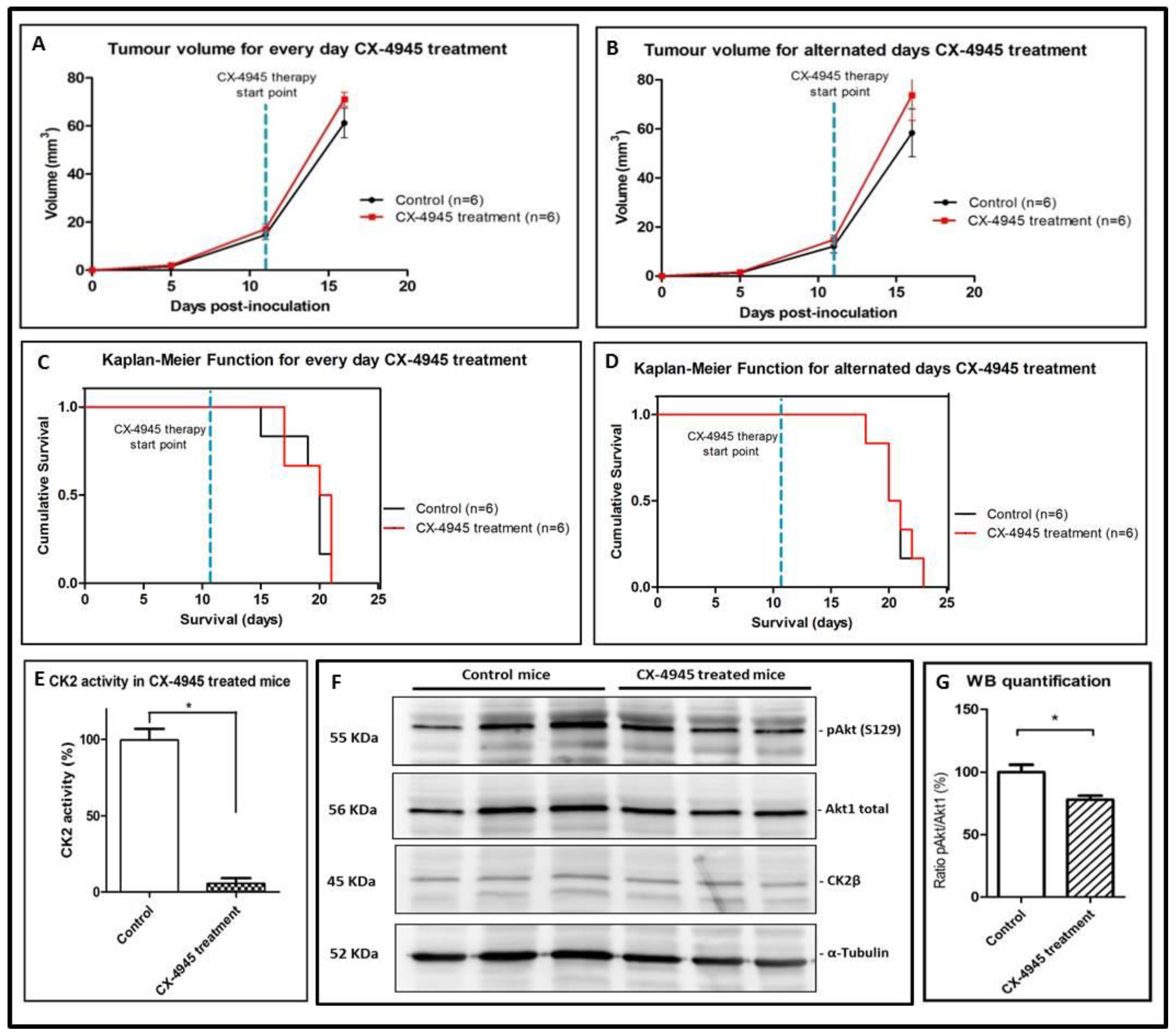
2.6. Non-Metronomic Longitudinal Treatments with CX-4945 and/or TMZ in Tumour-Bearing Mice
3. Discussion
3.1. Effect of iCK2 on GL261 Cultured Cells
3.2. Effects of CX-4945 in In Vivo Studies
3.3. Non-Metronomic CX-4945 Longitudinal In Vivo Studies
3.4. Non-Metronomic Combined CX-4945 and TMZ Longitudinal In Vivo Studies
3.5. Metronomic CX-4945 and/or TMZ Longitudinal In Vivo Studies and Possible Implication of the Immune System
4. Materials and Methods
4.1. GL261 Cells
4.2. Cell Viability Assay
4.3. Antibodies
4.4. Target Evaluation in GL261 Cultured Cells with CX-4945 Treatment
4.5. Animal Model for In Vivo Studies
4.6. CX-4945 and TMZ Tolerability Assay
4.7. Use of iCK2 in Tumour-Bearing Animals
4.7.1. Preliminary Studies of Target Validation
4.7.2. Longitudinal Studies with iCK2 (alone or in combination)
4.8. CK2 Activity Assay
4.9. MRI Acquisition
4.10. Tissue Homogenization and Protein Extraction/Western Blot Analysis
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviation
| APG | Apigenin |
| CK2 | Protein Kinase CK2 |
| CSCs | Cancer stem cells |
| CX-4945 | 5-(3-Chlorophenylamino)benzo[c][2,6] naphthyridine-8-carboxylic acid |
| CPA | Cyclophosphamide |
| DC50 | Half maximal cell death concentration |
| DMSO | Dimethyl sulfoxide |
| EC50 | Half maximal effective concentration, |
| GABRMN | Grup d’Aplicacions Biomèdiques de la RMN |
| GBM | Glioblastoma |
| IC50 | Half maximal inhibitory concentration |
| iCK2 | Protein Kinase CK2 inhibitors |
| JAK | Janus kinase |
| MGMT | O(6)-methylguanine-DNA methyltransferase |
| MTD | Maximum tolerated dose |
| p.i. | Post-inoculation |
| PIM-1 | Proviral Integration of Moloney virus 1 |
| STAT 3 | Signal transducer and activator of transcription 3 |
| TBB | 4,5,6,7-Tetrabromobenzotriazole |
| TDB | Tetrabromodeoxyribofuranosyl-benzimidazole |
| TMZ | Temozolomide |
| WB | Western blot. |
References
- Buckner, J.C. Factors influencing survival in high-grade gliomas. Semin. Oncol. 2003, 30, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Kleihues, P. Genetic profile of astrocytic and oligodendroglial gliomas. Brain Tumour Pathol. 2011, 28, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, K.; Mizoguchi, M.; Hata, N.; Murata, H.; Hatae, R.; Amano, T.; Nakamizo, A.; Sasaki, T. Complex DNA repair pathways as possible therapeutic targets to overcome temozolomide resistance in glioblastoma. Front. Oncol. 2012, 2, 186. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Goni, T.; Julia-Sape, M.; Candiota, A.P.; Pumarola, M.; Arus, C. Molecular imaging coupled to pattern recognition distinguishes response to temozolomide in preclinical glioblastoma. NMR Biomed. 2014, 27, 1333–1345. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Cheng, L.; Guryanova, O.A.; Wu, Q.; Bao, S. Cancer stem cells in glioblastoma—Molecular signaling and therapeutic targeting. Protein Cell 2010, 1, 638–655. [Google Scholar] [CrossRef] [PubMed]
- Kaina, B.; Ochs, K.; Grösch, S.; Fritz, G.; Lips, J.; Tomicic, M.; Dunkern, T.; Christmann, M. BER, MGMT, and MMR in defense against alkylation-induced genotoxicity and apoptosis. Prog. Nucleic Acid Res. Mol. Biol. 2001, 68, 41–54. [Google Scholar] [PubMed]
- Duncan, J.S.; Litchfield, D.W. Too much of a good thing: The role of protein kinase CK2 in tumourigenesis and prospects for therapeutic inhibition of CK2. Biochim. Biophys. 2008, 1784, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Ruzzene, M.; Pinna, L.A. Addiction to protein kinase CK2: A common denominator of diverse cancer cells? Biochim. Biophys. Acta 2010, 1804, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Münstermann, U.; Fritz, G.; Seitz, G.; Lu, Y.P.; Schneider, H.R.; Issinger, O.-G.G. Casein kinase II is elevated in solid human tumours and rapidly proliferating non-neoplastic tissue. Eur. J. Biochem. 1990, 189, 251–257. [Google Scholar]
- Ortega, C.E.; Seidner, Y.; Dominguez, I. Mining CK2 in cancer. PLoS ONE 2014, 9, e115609. [Google Scholar] [CrossRef] [PubMed]
- Dixit, D.; Sharma, V.; Ghosh, S.; Mehta, V.S.; Sen, E. Inhibition of Casein kinase-2 induces p53-dependent cell cycle arrest and sensitizes glioblastoma cells to tumour necrosis factor (TNFα)-induced apoptosis through SIRT1 inhibition. Cell Death Dis. 2012, 3, e271. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Lu, Z. The Role of Protein Kinase CK2 in Glioblastoma Development. Clin. Cancer Res. 2013, 19, 6335–6337. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Font, L.; Alcaraz, E.; Plana, M.; Candiota, A.P.; Itarte, E.; Arús, C. Protein Kinase CK2 Content in GL261 Mouse Glioblastoma. Pathol. Oncol. Res. 2016, 22, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Tapia, J.C.; Torres, V.A.; Rodriguez, D.A.; Leyton, L.; Quest, A.F.G. Casein kinase 2 (CK2) increases survivin expression via enhanced beta-catenin-T cell factor/lymphoid enhancer binding factor-dependent transcription. Proc. Natl. Acad. Sci. USA 2006, 103, 15079–15084. [Google Scholar] [CrossRef] [PubMed]
- Dixit, D.; Ahmad, F.; Ghildiyal, R.; Joshi, S.D.; Sen, E. CK2 inhibition induced PDK4-AMPK axis regulates metabolic adaptation and survival responses in glioma. Exp. Cell Res. 2016, 344, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Romieu-Mourez, R.; Landesman-Bollag, E.; Seldin, D.C.; Traish, A.M.; Mercurio, F.; Sonenshein, G.E. Roles of IKK Kinases and Protein Kinase CK2 in Activation of Nuclear Factor-{{kappa}}B in Breast Cancer. Cancer Res. 2001, 61, 3810–3818. [Google Scholar] [PubMed]
- Das, A.; Banik, N.L.; Ray, S.K. Flavonoids activated caspases for apoptosis in human glioblastoma T98G and U87MG cells but not in human normal astrocytes. Cancer 2010, 116, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Zanin, S.; Borgo, C.; Girardi, C.; O’Brien, S.E.; Miyata, Y.; Pinna, L.A.; Donella-Deana, A.; Ruzzene, M. Effects of the CK2 inhibitors CX-4945 and CX-5011 on drug-resistant cells. PLoS ONE 2012, 7, e49193. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Hwan Kim, S. CK2 inhibitor CX-4945 blocks TGF-β1-induced epithelial-to-mesenchymal transition in A549 human lung adenocarcinoma cells. PLoS ONE 2013, 8, e74342. [Google Scholar]
- Siddiqui-Jain, A.; Drygin, D.; Streiner, N.; Chua, P.; Pierre, F.; O’Brien, S.E.; Bliesath, J.; Omori, M.; Huser, N.; Ho, C.; et al. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumour efficacy. Cancer Res. 2010, 70, 10288–10298. [Google Scholar] [CrossRef] [PubMed]
- Pierre, F.; Chua, P.C.; O’Brien, S.E.; Siddiqui-Jain, A.; Bourbon, P.; Haddach, M.; Michaux, J.; Nagasawa, J.; Schwaebe, M.K. Pre-clinical characterization of CX-4945, a potent and selective small molecule inhibitor of CK2 for the treatment of cancer. Mol. Cell. Biochem. 2011, 356, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; McFarland, B.C.; Drygin, D.; Yu, H.; Bellis, S.L.; Kim, H.; Bredel, M.; Benveniste, E.N. Targeting protein kinase CK2 suppresses prosurvival signaling pathways and growth of glioblastoma. Clin. Cancer Res. 2013, 19, 6484–6494. [Google Scholar] [CrossRef] [PubMed]
- Dose-escalation Study of Oral CX-4945. Available online: http://www.cancer.gov/clinicaltrials/search/view?cdrid=642699&version=HealthProfessional (accessed on 12 February 2017).
- Cylene Presents Encouraging Clinical Data for Oral CK2 Inhibitor at ASCO. Available online: http://www.prnewswire.com/news-releases/cylene-presents-encouraging-clinical-data-for-oral-ck2-inhibitor-at-asco-123219423.html (accessed on 12 February 2017).
- Nitta, R.T.; Gholamin, S.; Feroze, A.H.; Agarwal, M.; Cheshier, S.H.; Mitra, S.S.; Li, G. Casein kinase 2α regulates glioblastoma brain tumour-initiating cell growth through the β-catenin pathway. Oncogene 2015, 34, 3688–3699. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Girardi, C.; Ranchio, A.; Lolli, G.; Sarno, S.; Orzeszko, A.; Kazimierczuk, Z.; Battistutta, R.; Ruzzene, M.; Pinna, L.A. Cell-permeable dual inhibitors of protein kinases CK2 and PIM-1: Structural features and pharmacological potential. Cell. Mol. Life Sci. 2014, 71, 3173–3185. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Bergers, G.; Bergsland, E. Less is more, regularly: Metronomic dosing of cytotoxic drugs can target tumour angiogenesis in mice. J. Clin. Investig. 2000, 105, 1045–1047. [Google Scholar] [CrossRef] [PubMed]
- Gnoni, A.; Silvestris, N.; Licchetta, A.; Santini, D.; Scartozzi, M.; Ria, R.; Pisconti, S.; Petrelli, F.; Vacca, A.; Lorusso, V. Metronomic chemotherapy from rationale to clinical studies: A dream or reality? Crit. Rev. Oncol. Hematol. 2015, 95, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Waxman, D.J. Metronomic cyclophosphamide schedule-dependence of innate immune cell recruitment and tumour regression in an implanted glioma model. Cancer Lett. 2014, 353, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Ausman, J.I.; Shapiro, W.R.; Rall, D.P. Studies on the chemotherapy of experimental brain tumours: Development of an experimental model. Cancer Res. 1970, 30, 2394–2400. [Google Scholar] [PubMed]
- Szatmári, T.; Lumniczky, K.; Désaknai, S.; Trajcevski, S.; Hídvégi, E.J.; Hamada, H.; Sáfrány, G. Detailed characterization of the mouse glioma 261 tumour model for experimental glioblastoma therapy. Cancer Sci. 2006, 97, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Di Maira, G.; Salvi, M.; Arrigoni, G.; Marin, O.; Sarno, S.; Brustolon, F.; Pinna, L.A.; Ruzzene, M. Protein kinase CK2 phosphorylates and upregulates Akt/PKB. Cell Death Differ. 2005, 12, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Ruzzene, M.; Di Maira, G.; Tosoni, K.; Pinna, L.A. Assessment of CK2 constitutive activity in cancer cells. Methods Enzymol. 2010, 484, 495–514. [Google Scholar] [PubMed]
- Kusabe, Y.; Kawashima, H.; Ogose, A.; Sasaki, T.; Ariizumi, T.; Hotta, T.; Endo, N. Effect of temozolomide on the viability of musculoskeletal sarcoma cells. Oncol. Lett. 2015, 10, 2511–2518. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Han, J.; Kannabiran, V.; Mohan, S.; Cheng, H.; Friedman, J.; Zhang, L.; VanWaes, C.; Chen, Z. MEK inhibitor PD-0325901 overcomes resistance to CK2 inhibitor CX-4945 and exhibits anti-tumour activity in head and neck cancer. Int. J. Biol. Sci. 2015, 11, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Pierre, F.; Chua, P.C.; O’Brien, S.E.; Siddiqui-Jain, A.; Bourbon, P.; Haddach, M.; Michaux, J.; Nagasawa, J.; Schwaebe, M.K.; Stefan, E.; et al. Discovery and SAR of 5-(3-chlorophenylamino)benzo[c][2,6] naphthyridine-8-carboxylic acid (CX-4945), the first clinical stage inhibitor of protein kinase CK2 for the treatment of cancer. J. Med. Chem. 2011, 54, 635–654. [Google Scholar] [CrossRef] [PubMed]
- McConville, P.; Hambardzumyan, D.; Moody, J.B.; Leopold, W.R.; Kreger, A.R.; Woolliscroft, M.J.; Rehemtulla, A.; Ross, B.D.; Holland, E.C. Magnetic resonance imaging determination of tumour grade and early response to temozolomide in a genetically engineered mouse model of glioma. Clin. Cancer Res. 2007, 13, 2897–2904. [Google Scholar] [CrossRef] [PubMed]
- Plowman, J.; Waud, W.R.; Koutsoukos, A.D.; Rubinstein L, V.; Moore, T.D.; Grever, M.R. Preclinical antitumor activity of temozolomide in mice: Efficacy against human brain tumor xenografts and synergism with 1,3-bis(2-chloroethyl)-1-nitrosourea. Cancer Res. 1994, 54, 3793–3799. [Google Scholar] [PubMed]
- Siddiqui-Jain, A.; Bliesath, J.; Macalino, D.; Omori, M.; Huser, N.; Streiner, N.; Ho, C.B.; Anderes, K.; Proffitt, C.; O’Brien, S.E.; et al. CK2 inhibitor CX-4945 suppresses DNA repair response triggered by DNA-targeted anticancer drugs and augments efficacy: mechanistic rationale for drug combination therapy. Mol. Cancer Ther. 2012, 11, 994–1005. [Google Scholar] [CrossRef] [PubMed]
- Drablos, F.; Feyzi, E.; Aas, P.A.; Vaagbø, C.B.; Kavli, B.; Bratlie, M.S.; Peña-Diaz, J.; Otterlei, M.; Slupphaug, G.; Krokan, H.E. Alkylation damage in DNA and RNA-repair mechanisms and medical significance. DNA Repair (Amst.) 2004, 3, 1389–1407. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.-C.; Godard, S.; Dietrich, P.-Y.; Regli, L.; Ostermann, S.; Otten, P.; Van Melle, G.; de Tribolet, N.; Stupp, R. Clinical trial substantiates the predictive value of O-6-methylguanine-DNA methyltransferase promoter methylation in glioblastoma patients treated with temozolomide. Clin. Cancer Res. 2004, 10, 1871–1874. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Zheng, Y.; Qin, H.; Frank, S.J.; Deng, L.; Litchfield, D.W.; Tefferi, A.; Pardanani, A.; Lin, F.-T.; Li, J.; Sha, B.; et al. A CK2-dependent mechanism for activation of the JAK-STAT signaling pathway. Blood 2011, 118, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Kohsaka, S.; Wang, L.; Yachi, K.; Mahabir, R.; Narita, T.; Itoh, T.; Tanino, M.; Kimura, T.; Nishihara, H.; Tanaka, S. STAT3 inhibition overcomes temozolomide resistance in glioblastoma by downregulating MGMT expression. Mol. Cancer Ther. 2012, 11, 1289–1299. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Choi, K.; Kang, H.; Lee, S.Y.; Chi, S.W.; Lee, M.S.; Song, J.; Im, D.; Choi, Y.; Cho, S. Identification of a novel function of CX-4945 as a splicing regulator. PLoS ONE 2014, 9, e94978. [Google Scholar] [CrossRef] [PubMed]
- Vacchelli, E.; Aranda, F.; Eggermont, A.; Galon, J.; Sautès-Fridman, C.; Cremer, I.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial Watch: Chemotherapy with immunogenic cell death inducers. Oncoimmunology 2014, 3, e27878. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Waxman, D.J. Metronomic cyclophosphamide eradicates large implanted GL261 gliomas by activating antitumour Cd8(+) T-cell responses and immune memory. Oncoimmunology 2015, 4, e1005521. [Google Scholar] [CrossRef] [PubMed]
- Karman, J.; Ling, C.; Sandor, M.; Fabry, Z. Initiation of immune responses in brain is promoted by local dendritic cells. J. Immunol. 2004, 173, 2353–2361. [Google Scholar] [CrossRef] [PubMed]
- Tabbekh, M.; Mokrani-Hammani, M.; Bismuth, G.; Mami-Chouaib, F. T-cell modulatory properties of CD5 and its role in antitumour immune responses. Oncoimmunology 2013, 2, e22841. [Google Scholar] [CrossRef] [PubMed]
- Sestero, C.M.; McGuire, D.J.; De Sarno, P.; Brantley, E.C.; Soldevila, G.; Axtell, R.C.; Raman, C. CD5-dependent CK2 activation pathway regulates threshold for T cell anergy. J. Immunol. 2012, 189, 2918–2930. [Google Scholar] [CrossRef] [PubMed]
- Alonso, M.N.; Wong, M.T.; Zhang, A.L.; Winer, D.; Suhoski, M.M.; Tolentino, L.L.; Gaitan, J.; Davidson, M.G.; Kung, T.H.; Galel, D.M.; et al. T(H)1, T(H)2, and T(H)17 cells instruct monocytes to differentiate into specialized dendritic cell subsets. Blood 2011, 118, 3311–3320. [Google Scholar] [CrossRef] [PubMed]
- Ulges, A.; Witsch, E.J.; Pramanik, G.; Klein, M.; Birkner, K.; Bühler, U.; Wasser, B.; Luessi, F.; Stergiou, N.; Dietzen, S.; et al. Protein kinase CK2 governs the molecular decision between encephalitogenic TH17 cell and Treg cell development. Proc. Natl. Acad. Sci. USA 2016, 113, 10145–10150. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.-G.; Kim, C.-H.; Park, J.-S.; Park, S.-D.; Kim, C.K.; Chung, D.-S.; Hong, Y.-K. Immunological factors relating to the antitumour effect of temozolomide chemoimmunotherapy in a murine glioma model. Clin. Vaccine Immunol. 2010, 17, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Fritzell, S.; Sandén, E.; Eberstål, S.; Visse, E.; Darabi, A.; Siesjö, P. Intratumoural temozolomide synergizes with immunotherapy in a T cell-dependent fashion. Cancer Immunol. Immunother. 2013, 62, 1463–1474. [Google Scholar] [CrossRef] [PubMed]
- Martins, L.R.; Lúcio, P.; Silva, M.C.; Anderes, K.L.; Gameiro, P.; Silva, M.G.; Anderes, K.L.; Gameiro, P.; Silva, M.G.; Barata, J.T. Targeting CK2 overexpression and hyperactivation as a novel therapeutic tool in chronic lymphocytic leukemia. Blood 2010, 116, 2724–2731. [Google Scholar] [CrossRef] [PubMed]
- Simões R, V.; Delgado-Goñi, T.; Lope-Piedrafita, S.; Arús, C. 1H-MRSI pattern perturbation in a mouse glioma: the effects of acute hyperglycemia and moderate hypothermia. NMR Biomed. 2010, 23, 23–33. [Google Scholar] [CrossRef] [PubMed]
- GraphPad. Available online: http://www.graphpad.com/ (accessed on 12 February 2017).
- Martins, L.R.; Lúcio, P.; Melão, A.; Antunes, I.; Cardoso, B.; Stansfield, R.; Bertilaccio, M.T.S.; Ghia, P.; Drygin, D.; Silva, M.G.; et al. Activity of the clinical-stage CK2-specific inhibitor CX-4945 against chronic lymphocytic leukemia. Leukemia 2014, 28, 179–182. [Google Scholar] [CrossRef] [PubMed]
- Web CEEAH. Available online: http://www.recerca.uab.es/ceeah (accessed on 12 February 2017).
- Simões, R.V.; García-Martín, M.L.; Cerdán, S.; Arús, C. Perturbation of mouse glioma MRS pattern by induced acute hyperglycemia. NMR Biomed. 2008, 21, 251–264. [Google Scholar]
- Saldaña-Ruíz, S.; Soler-Martín, C.; Llorens, J. Role of CYP2E1-mediated metabolism in the acute and vestibular toxicities of nineteen nitriles in the mouse. Toxicol. Lett. 2012, 208, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrer-Font, L.; Villamañan, L.; Arias-Ramos, N.; Vilardell, J.; Plana, M.; Ruzzene, M.; Pinna, L.A.; Itarte, E.; Arús, C.; Candiota, A.P. Targeting Protein Kinase CK2: Evaluating CX-4945 Potential for GL261 Glioblastoma Therapy in Immunocompetent Mice. Pharmaceuticals 2017, 10, 24. https://doi.org/10.3390/ph10010024
Ferrer-Font L, Villamañan L, Arias-Ramos N, Vilardell J, Plana M, Ruzzene M, Pinna LA, Itarte E, Arús C, Candiota AP. Targeting Protein Kinase CK2: Evaluating CX-4945 Potential for GL261 Glioblastoma Therapy in Immunocompetent Mice. Pharmaceuticals. 2017; 10(1):24. https://doi.org/10.3390/ph10010024
Chicago/Turabian StyleFerrer-Font, Laura, Lucia Villamañan, Nuria Arias-Ramos, Jordi Vilardell, Maria Plana, Maria Ruzzene, Lorenzo A. Pinna, Emilio Itarte, Carles Arús, and Ana Paula Candiota. 2017. "Targeting Protein Kinase CK2: Evaluating CX-4945 Potential for GL261 Glioblastoma Therapy in Immunocompetent Mice" Pharmaceuticals 10, no. 1: 24. https://doi.org/10.3390/ph10010024