Microglia M2A Polarization as Potential Link between Food Allergy and Autism Spectrum Disorders

1
Gänsbühlgartenweg 7, CH4132 Muttenz, Switzerland
2
Novartis Pharma AG, CH4002 Basel, Switzerland
*
Author to whom correspondence should be addressed.
Pharmaceuticals 2017, 10(4), 95; https://doi.org/10.3390/ph10040095
Submission received: 13 November 2017
/
Revised: 5 December 2017
/
Accepted: 5 December 2017
/
Published: 9 December 2017
(This article belongs to the Special Issue Understanding Inflammation-induced Mental Diseases: New Opportunities for Treatment)
{kind=link}
Abstract
:Atopic diseases are frequently co-morbid with autism spectrum disorders (ASD). Allergic responses are associated with an activation of mast cells, innate lymphoid cells, and Th2 cells. These cells produce type-2 cytokines (IL4 and IL13), which stimulate microglia and macrophages to adopt a phenotype referred to as ‘alternative activation’ or ‘M2A’. M2A-polarized macrophages and microglia play a physiological role in tissue repair by secreting growth factors such as brain-derived neurotrophic factor (BDNF) and insulin-like growth factor-1. In ASD there is evidence for increased type-2 cytokines, microglia activation, M2A polarization, and increased levels of growth factors. In neurons, these growth factors drive a signal transduction pathway that leads to activation of the enzyme mammalian Target of Rapamycin (mTOR), and thereby to the inhibition of autophagy. Activation of mTOR is an effect that is also common to several of the genetic forms of autism. In the central nervous system, redundant synapses are removed via an autophagic process. Activation of mTOR would diminish the pruning of redundant synapses, which in the context of ASD is likely to be undesired. Based on this line of reasoning, atopic diseases like food allergy, eczema or asthma would represent risk factors for autism spectrum disorders.
1. Introduction
Autism spectrum disorders (ASD) are a collection of neurodevelopmental conditions characterized by language deficits, social impairments, and repetitive behaviors [1,2]. These symptoms become apparent usually around 2–3 years of age and sometimes manifest with a regression in acquired language and behavioral skills. There are wide variations in clinical presentation and disease progression. Attempts to categorize ASD have resulted in the recognition of distinct subgroups: classic autism (which can entail general intellectual disability and language delay), Asperger syndrome (in which there is no language delay or intellectual disability), and several syndromic forms, in which high-impact gene mutations (e.g., Rett syndrome, Timothy syndrome, Fragile X or tuberous sclerosis) are important contributing factors. Cognitively, ASD is characterized by weakened executive function, reduced central coherence, and poorer mentalizing properties. Conversely, ASD patients may also have special ‘strengths’ (savant syndrome; [3]), such as attention to detail or systemizing (reviewed in [4]).
Data from multiple studies indicate that the synapse may play a central role in the pathology of ASD [5,6,7,8,9,10]. Pruning of synapses and spines by microglial cells [11] is critically dependent on intraneuronal autophagy [12,13]. Stimulation of mTOR (mammalian target of rapamycin) displays an inhibitory effect on autophagy [13], whereas multiple ASD syndromes are caused by disruptive mutations in genes that, if intact, function as inhibitors of mTOR (for recent reviews see [14,15]).
Whereas prevalence studies in twins with ASD indicate that the genetic factors play a dominant role in ASD prevalence [16,17], these studies similarly emphasize the importance of a shared environment [18,19]. Childhood disorders of the immune system (e.g., asthma, life-threatening food allergies) have reached “an epidemic level” over the past two decades [20] and the same is true for the ASD prevalence. As summarized by Estes and McAllister [20], in the United States the estimate in 1992 was 1 in 500 children, in 2007 this was 1 in 110, and recent estimates are 1 in 68 (n.b. 1 in 42 boys!). This drastic rise over such a short period of time cannot be explained by genetic changes and suggests that, apart from trivial reasons like increased awareness and diagnostic criteria that are more inclusive, recent environmental changes must contribute [20]. Unfortunately, the nature of environmental influences that are relevant for ASD remains largely undefined, although parallels have been drawn to the increasing prevalence of atopic disorders in the general population [21]. Food allergy in particular seems to be frequently comorbid with ASD [22,23]. In the present review, the hypothesis is put forward that allergies exacerbate ASD by an immunological mechanism that leads to activation of microglia. As a consequence, mTOR in neurons is turned on, leading to reduction of synaptic pruning.
2. Environmental Risk Factors for ASD
In an epidemiological study, a doubled risk of ASD was found when mothers were diagnosed with asthma or allergy during the second trimester of pregnancy [24]. Maternal stress has been proposed as a factor that could lead to allergy or asthma [25,26,27] and ASD [28,29]. Becker [21] has reviewed the epidemiological, morphometric, molecular and genetic similarities between autism and allergic disorders, and suggested that the increase in ASD-prevalence might relate to the increase in atopic disorders owing to improvement in hygiene. Over the years, numerous studies have appeared that point to an association between ASD and atopic disorders. For instance, in a small study of individuals with Asperger syndrome, two thirds of the individuals suffered from atopic diseases (dermatitis, asthma or rhinitis) and had high serum levels of IgE and eosinophils [30]. In a longitudinal study of children with ASD and a control group of typically developing children, food, environmental and seasonal allergies were present in a minority only, but were significantly more frequent in the cohort with ASD compared to controls [31]. Angelidou et al. [32] proposed that mast cell activation could be an important pathological factor in ASD. These authors suggest that mast cell activation by allergic, infectious, environmental and stress-related triggers might release pro-inflammatory and neurotoxic molecules that disrupt the gut–blood, and blood–brain barriers, thus contributing to brain inflammation and ASD pathogenesis [32]. Head size of ASD patients at birth is often smaller, but in the period between approximately 2 months of age until approximately the 12th month of age, a growth spurt can result in abnormally large head sizes [33,34,35,36]. Interestingly, the presence of asthma, hay fever or high serum levels of IgE in pregnant mothers were prospectively associated with an increased head circumference in the offspring [37,38,39]. In an investigation of children diagnosed with allergy (asthma, atopic dermatitis, food allergy, high IgE), the head sizes at mid-gestational age (determined in utero by an ultrasound scan) were significantly smaller than non-allergic controls [40]. Thus, the data collected in allergic children resemble those from autistic children: a small head size in utero is postnatally followed by an overshooting growth spurt. In contrast to the association between food allergy and ASD [22,23], the association between respiratory allergy and ASD did not reach significance [23,41], but nevertheless respiratory allergy seems to exacerbate the behavioral symptoms of ASD [23].
3. Mast Cells, Th2 Lymphocytes, and Cytokine Profiles in ASD
The role of immune factors in ASD has been summarized recently in several excellent reviews [20,42,43]. After cross-linking of the cell-bound IgE-FcεRI complex, human mast cells release inflammatory mediators such as histamine and eicosanoids. In addition, mast cells are the sources of several cytokines like pro-inflammatory interleukins (IL1β, TNFα and IL6), interleukins related to tissue repair and allergy (IL4, IL5 and IL13), chemokines (CXCL8 and CCL2) and the growth factor VEGF [44,45]. Elevated concentrations of IL4 and IL5 were found in a study of banked serum collected from women during the 15th and the 19th week of gestation who gave birth to children who were ultimately diagnosed with ASD [46]. Furthermore, elevated levels of IL4, IL10 and TNFα were measured in the amnion fluid of fetuses that later developed ASD [47]. This small summary shows that elevated levels of mast cell products have been observed in mothers of ASD patients. As the immune system continues to mature in the postnatal period, one would also expect evidence for differences in the immune system of children with ASD compared to control children. Indeed, in neonatal blood spots collected from ASD patients and normally developing control children, the presence of elevated concentrations of IL1β and IL4 were independently associated with ASD [48]. This study noted that whereas IL1β was associated with increased odd ratios of mild/moderate ASD, an elevated level of IL4 was associated with severe ASD. Furthermore, IL4 levels were inversely correlated with nonverbal cognitive ability in a group of male ASD subjects. The authors thus concluded that peripheral cytokine profiles at birth are not only associated with ASD later in childhood, but also differ in relation to symptom severity [48]. In a further study, blood serum from adults with Asperger syndrome and controls was investigated [49]. In this study, male and female individuals with Asperger syndrome displayed very different cytokine profiles, with males expressing increased amounts of IL4, IL5, IL10, TNFα and a few other cytokines, while females mainly expressed altered amounts of growth factors (e.g., insulin, brain derived neurotrophic factor [both increased] and growth hormone and endothelin [both decreased]). Notably, there was very little overlap between the factors altered in male and in female Asperger individuals [49]. Several other studies have investigated cytokine profiles in blood and blood cells of ASD patients. An early study by Gupta et al. [50] found increases in typical Th1 cytokines (IL2, IL6 and IFNγ), as well as typical Th2 cytokines (IL4 and IL10) and an increase in the number of Th2-cells. This result is in agreement with data from Molloy et al. [51], but somewhat at variance with data from Suzuki et al. [52]. Molloy et al. [51] investigated peripheral blood mononuclear cells (PBMCs) from children with ASD and found that these cells produced higher amounts of Th2 cytokines (IL4, IL5, IL10 and IL13) than PBMCs from matching control subjects. Th1 cytokines were only slightly elevated. In contrast, Suzuki et al. [52] noted that Th1 and Th17 cytokines (IL1β, IL8, IL12p70 and IL17) in blood plasma of male high-functioning ASD patients were significantly elevated, whereas IL4, IL5 and IL13 were increased only trend-wise. Also Careaga and colleagues [53] investigated PBMCs of male ASD patients. Upon phytohemagglutinin-challenge, different subgroups were distinguishable: a group with an increased Th2 profile (increase in IL13), a group with an increased Th1 profile (IFNγ increase), and a third group with no typical response. In contrast to the third group, patients in the Th1 and Th2 groups were developmentally impaired [53]. To complete this summary, also a study by Hu et al. [54] provided evidence for a distinct Th2-related immune phenotype within ASD. These investigators generated lymphoblastoid cell lines from ASD-, and/or language-discordant monozygotic twins. Differences in gene expression were studied by microarray, and the data were used to establish networks of similarly regulated genes. One of the identified networks included the Th2-genes, IL5RA, IL13 and IRF4 [54]. The conclusion from these cross-sectional studies is that maternal and neonatal immune factors related to mast cells and Th2-skewed lymphocytes could play a role in increasing the risk of ASD.
Quite recently, the first longitudinal study has become available, which did not substantiate the conclusion of the former cross-sectional studies. This study enrolled 104 children with ASD. The levels of 20 cytokines and 10 chemokines were assessed in serum but did not differ from the levels in an age-matched control group of 54 children. Moreover, the serum levels of analytes were poor predictors of the levels in the cerebrospinal fluid (CSF) [31]. This negative result conflicts with previous data, including those of the same research group [55]. Possible explanations involve a different compartment (brain tissue versus CSF), different assay techniques (multiplex bead assay versus immunostaining, Elisa or protein array) or a considerable temporal fluctuation in inflammatory process.
4. IL4 Induces M2A Polarization of Macrophages and Microglia
Cells belonging to the monocyte-macrophage lineage have long been recognized to be heterogeneous. The apparent diversity of macrophages is most likely a functional adaptation to cues in the microenvironment. Similar to the Th1 and Th2 polarization of the adaptive immune system, a nomenclature has been proposed for the polarization of macrophages, with “M1” reflecting a distinct activation program seen in response to bacterial and viral infections, Toll-like receptor agonists, and pro-inflammatory cytokines (IFNγ, IL1β, TNFα or IL6). A distinctly different activation program that takes place in response to parasites, during tissue repair and in response to the cytokines IL4, IL5 and IL13 has been termed “M2” [56]. Mantovani and co-authors [56] distinguish several M2 subtypes (M2A, M2B and M2C); however, this subdivision is not generally accepted. Notably, very similar phenotypic changes have been observed in microglia, and the M1/M2 nomenclature is used for microglia polarization states as well [57].
IL4 is produced by mast cells, by Th2 and TREG T-cells, as well as by innate lymphoid cells [44,58,59]. In B-lymphocytes, IL4 promotes immunoglobulin isotype-switching from IgG to IgE, and thereby contributes to the typical immune responses seen in allergy and asthma [60,61]. As an additional important activity, IL4 stimulates the differentiation of macrophages and microglia towards the M2A-phenotype (known as M2A “polarization”; see [58,62]). Similar to IL4, also IL13 is able to induce M2A polarization [63,64]. Whereas IL4 activates the IL4Rα receptor, IL13 activates the IL13Rα1 receptor. The two receptors can form heterodimers, which are responsive to both IL4 and IL13 [63]. IL5 activates IL5Rα receptor and this plays a role in eosinophilia [63]. The induction of an M2A phenotype by exposure to IL4 or IL13 provokes a near loss of the specific M1 marker, inducible nitric oxide synthase ‘iNOS’ [64]. Typical markers for M2A polarized macrophages and microglia are the arginase enzyme Arg1, scavenger receptors, and the mannose receptor [56,57,64]. The transcription of growth factors like VEGF, BDNF and PDGF is increased in IL4-induced M2A microglia [64]. In the M2A-polarized state, microglia cells also express insulin-like growth factor-1 [57,65]. As a consequence, allergy may indirectly lead to an increased release of a number of potent growth factors from microglia.
5. Changes in Post Mortem Brain of ASD
Direct evidence for microglia activation in the brain of ASD patients has been obtained with PET-radiotracer [11C](R)-PK11195 [66]. The tracer selectively labels the 18 kDa translocator protein (TSPO) in microglia. In activated microglia the expression of TSPO is strongly increased. The study by Suzuki and colleagues investigated adult male ASD patients and compared these to age- and IQ-matched controls. The binding values of the PET-ligand were significantly higher in multiple brain regions in ASD cases [66]. These results by Suzuki et al. [66] are complemented by a large longitudinal study in children with ASD by Pardo and colleagues [31]. Using repeated CSF sampling at different time points, it was noted that CSF levels of FLT3L, IL15, CX3CL1, CXCL8 and CCL2 were elevated [31]. Each of these immune mediators exerts a modulatory role on microglia and neuroglia–neuronal interactions. E.g., they promote cell differentiation, proliferation, and survival of microglia but also facilitate migration to areas of injury or play critical roles in the homeostatic function of microglia (see [31] for further details).
These in-life studies are further complemented by several post mortem studies. For instance, Morgan et al. [67] compared microglia in sections from the dorsolateral prefrontal cortex of 13 male ASD cases with that from control cases. The morphological alterations observed in ASD sections included somal enlargement, process retraction and thickening, and extension of filopodia from processes. Average microglial soma volume was significantly increased in white matter. These signs of microglia activation were seen in 5 out of 13 cases with ASD, whilst the effect was evident already at young age, since two cases were less than 6 years old [67]. Whereas such data provide evidence for microglia activation in ASD and corroborate well the TSPO study by Suzuki [66] mentioned above, they do not allow a distinction between the classical ‘infection-related’ M1-phenotype or the allergy and repair-related M2A phenotype [58]. Measurements of cytokine levels and microglia markers provide clear evidence for M1 polarization in frontal cortex samples. A study by Li et al. [68] found increased levels of TNFα, IL6, GM-CSF, IFNγ (Th1 cytokines), whereas IL4, IL5 and IL10 were not altered. However, the majority of post mortem studies in ASD suggest a mix of M1 and M2 states [55,69,70]. During infections, M1-polarized microglia produce several cytotoxic molecules, such as quinolinic acid, as well as oxygen and nitrogen radicals [71,72,73,74]. The radicals oxidize tetrahydrobiopterin (also known as BH4; an important cofactor for the biosynthesis of monoamines) to the functionally inactive metabolite neopterin [75]. In a remarkable study, Zimmerman et al. [76] noted that the levels of quinolinic acid and neopterin in the cerebrospinal fluid of ASD patients were lower, whereas the level of BH4 was higher than in healthy controls. This result is compatible with a decreased M1 polarization in ASD. The largest post mortem study thus far investigated cortical samples of 47 ASD cases and 57 controls [77]. The transcriptome of each sample was determined, and the data were analyzed for co-expression patterns. The authors made the significant discovery that one of the identified “modules” (clusters of co-expressed genes) was enriched for genes related to the microglial M2 state [77]. Taken together, these data provide support for an increase in microglial M1 polarization in ASD, but also support an increase in M2 polarization. This may either reflect distinct subtypes of ASD, but might also reflect different timely separated phases (e.g., when an infection first induces a defense response [M1] and then a repair [M2A]).
6. Growth Factors from M2A-Polarized Microglia Are Associated with ASD
In accordance with the role of M2A-polarized microglia and macrophages in repair and recovery, these cells produce growth factors that stimulate proliferation of stem cells (reviewed by [58,72]). Upon stimulation by IL4 or IL13, the expression of insulin-like growth factor-1 (IGF1) by microglia [57,65,78] and macrophages [78,79] was strongly increased. IGF1 is an important cytoprotective protein with a relevant function in tissue repair after inflammation [80,81]. Compared to controls, blood levels of IGF1 were significantly increased in male ASD patients who had a large head circumference, and also the head sizes correlated with blood level [82]. The contention that IGF1 is an important factor for head size is also supported by data from genetic studies. Thus, whereas hemizygosity for IGF1R (the receptor for IGF1 and IGF2) is associated with growth failure, individuals who carry three copies of the IGF1 receptor present with a pronounced macrocephaly [83]. Importantly, in a candidate gene-study, single nucleotide polymorphisms in the IGF1 gene were associated with Asperger syndrome [84].
In addition to IGF1, also BDNF levels may be increased in ASD patients. A meta-analysis of serum and plasma BDNF levels identified 14 studies involving 2707 participants (1131 ASD patients), and noted significantly elevated peripheral BDNF levels in young (n.b. not in adult) ASD patients [85]. The study by Schwarz et al. [49] mentioned before reported that BDNF levels in blood were increased in female, but not male Asperger patients. Finally, in a small post mortem study of mainly male adult ASD patients, BDNF-expression in the basal forebrain was threefold higher than in controls [86]. The possibility that BDNF influences brain development is indicated by an MRI study that found that a single nucleotide polymorphism in the BDNF gene affected cortical volume [87]. These findings in patients corroborate the aforementioned evidence that IL4-induced microglia are a source of a number of potent growth factors.
7. Mechanisms by Which Growth Factors May Cause ASD Symptoms
Microglia has a profound influence on neuronal and astroglial function and on brain development [88,89]. One current hypothesis of ASD proposes that glial dysfunction directly contributes to pathophysiology [90]. This hypothesis was formulated mainly on the basis of results from animal studies; for instance, in mice with a genetic deletion of the chemokine receptor CX3CR3 (a receptor that is exclusively expressed on microglia), which led to delayed synaptic pruning, and ultimately resulted in excessive numbers of immature synapses and a diminished performance in cognition tests [11,69,91]. A microglia-specific deletion of BDNF expression resulted in similar deleterious effect on cognitive function [92]. These data clearly indicate that a proper microglia function is important. Decreased BDNF signaling has been thoroughly studied in the context of depression, and numerous transgenic mouse models of impaired BDNF function have been generated (reviewed in [93]). On the other hand, BDNF-overexpression has rarely been investigated [93]. E.g., in female BDNF-overexpressing mice, working memory was impaired, while they displayed more anxiety and self-grooming, and were more susceptible to seizures than their wild-type littermates [94]. Although this phenotype is reminiscent of ASD, it is unclear whether this study is informative about ASD, because BDNF was overexpressed exclusively in excitatory neurons. Transgenic mice with IGF1-overexpression in neurons or in astrocytes have been generated, too [80]. Such mice exhibited marked increases in brain weight, in number of neurons, astrocytes and oligodendrocytes, as well as increases in myelination and synapse-to-neuron ratio [80,81]. IGF1 stimulates the proliferation of progenitor cells and drives their fate-selection towards neurogenesis [95,96]. A higher IGF1 level could be one of the causes for the increase in neuron numbers that is observed in ASD patients [97] and could represent a partial explanation for the large head circumference [33]. Both BDNF and IGF1 activate the PI3K–Akt pathway. This pathway is important for cell survival and cell growth, whereas apoptosis and macrophage/microglia M1 polarization are suppressed [43,78,93,98,99]. Taken together, the increased expression of BDNF and IGF1 could explain several aspects of ASD.
8. Discussion
The data summarized above indicate that allergy is associated with ASD. Allergy involves an increase in the activation of Th2 lymphocytes, mast cells, innate lymphoid cells and others [45,59,100]. These cell types are known to express and release IL4, IL5 and IL13 [44,59,98,101]. IL4 and IL13 are potent inducers of the M2A phenotype of macrophages and microglia [98]. In fact, not only allergy but also other potential environmental risk factors for ASD, like maternal prenatal stress or prenatal exposure to antidepressant medication, may lead to M2A skewing of phagocytes [25,102,103]. In the M2A-polarized form these cells express and secrete growth factors that activate vascular and neuronal stem cells, improve neurogenesis and neuronal survival, and thereby influence different domains of cognitive function [104]. In certain subtypes of the autism spectrum there is evidence for increases in blood levels of IL4 and/or IL13. There is also evidence for an M2A polarization of microglia, including findings of increased expression of M2A-markers and increased levels of the growth factors VEGF, IGF1 and BDNF. It is therefore conceivable that allergy contributes to the prevalence and severity of ASD via an increased release of growth factors in the brain. The next paragraph describes the putative mechanism of how elevated levels of growth factors could lead to ASD symptoms.
Numerous studies attempted to identify the genetic causes of ASD. Interestingly, such studies frequently detected alterations in genes that are critically important for synapse function [5,6,7,8,9,10]. In particular, Voineagu and Eapen [9] have suggested that immunological processes (either genetically or environmentally induced) contribute to ASD by exerting a negative influence on synapse function. Pruning of non-functional synapses involves an initial interaction between microglia and synapses [11], which is followed by the removal of the redundant synapses via an intra-neuronal autophagic process [12,13]. Stimulation of mTOR leads to an inhibition of autophagy [13], whereas multiple ASD syndromes are caused by mutations in genes that, if intact, function as inhibitors of mTOR. Importantly, via stimulation of the PI3K–Akt–mTOR pathway, both IGF1 and BDNF activate mTOR. In theory, this would hinder the autophagic removal of redundant synapses [80,105]. Following this line of reasoning, allergy-induced M2A polarization, with its increased production of IGF1, BDNF and further growth factors, would contribute to ASD via inhibition of normal pruning of synapses (see Figure 1). A dysfunction of normal pruning may help to explain alterations in cognition and behavior.
It seems that, during the last decades, the prevalence of allergic diseases has increased considerably [106]. As for ASD, this fast increase rather argues against a genetic mechanism, and is probably explainable by environmental factors in combination with an increased awareness [106,107]. Environmental factors that have been proposed include allergens (for instance exposure to house dust mites; [108]), air pollution and passive smoking [108], Western style of living, including an increase in hygiene [21] and industrialized methods for food production [109]. As these environmental factors are not mutually exclusive, it will be difficult to tease out which of these has the greatest impact. In this review, it is argued that allergy represents an environmental risk factor for autism spectrum disorders, which involves a cascade of events including the Th2-cytokines IL4 and IL13, an M2A polarization of microglia, the release of growth factors, and the inhibition of autophagy in neurons (see Figure 1). For this reason, the most logical therapeutic approach would be to reduce the exposure to allergens. The therapeutic response to gluten- or casein-free diets is often strongly advocated by parents [110]; however, results from objective clinical studies are less convincing [111]. Although development of therapies for very young children is particularly challenging, it might prove possible to interfere in the pathological cascade in a manner that the benefit exceeds the risks. For instance, specific antibodies against IL4 or IL13 might be explored in order to prevent an excessive M2A polarization [100]. Alternatively, one could try to interrupt signaling pathways that promote M2A polarization, e.g., by tyrosine kinase inhibitors that block STAT6 phosphorylation [112]. Since mTOR activation seems to be an effect common to several forms of ASD, including the allergy mechanism discussed in the current review, it might be useful to prevent or to inhibit the activation of mTOR [15,113,114]. So in summary, it might very well be possible that allergy represents a treatable risk factor for ASD.
Acknowledgments
This research did not receive any specific grant from funding agencies, commercial or not-for-profit sectors.
Conflicts of Interest
H.O.K. holds shares of Novartis pharma AG. D.F. is an employee of Novartis pharma AG and holds shares thereof.
References
- Happe, F.; Ronald, A. The ‘fractionable autism triad’: A review of evidence from behavioural, genetic, cognitive and neural research. Neuropsychol. Rev. 2008, 18, 287–304. [Google Scholar] [PubMed]
- King, B.H.; Navot, N.; Bernier, R.; Webb, S.J. Update on diagnostic classification in autism. Curr. Opin. Psychiatry 2014, 27, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.K. The savant syndrome: Intellectual impairment and exceptional skill. Psychol. Bull. 1999, 125, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Young, A.M.; Chakrabarti, B.; Roberts, D.; Lai, M.C.; Suckling, J.; Baron-Cohen, S. From molecules to neural morphology: Understanding neuroinflammation in autism spectrum condition. Mol. Autism 2016, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Gilman, S.R.; Iossifov, I.; Levy, D.; Ronemus, M.; Wigler, M.; Vitkup, D. Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron 2011, 70, 898–907. [Google Scholar] [CrossRef] [PubMed]
- Hussman, J.P.; Chung, R.H.; Griswold, A.J.; Jaworski, J.M.; Salyakina, D.; Ma, D.; Konidari, I.; Whitehead, P.L.; Vance, J.M.; Martin, E.R.; et al. A noise-reduction GWAS analysis implicates altered regulation of neurite outgrowth and guidance in autism. Mol. Autism 2011, 2, 1. [Google Scholar] [CrossRef] [PubMed]
- Toro, R.; Konyukh, M.; Delorme, R.; Leblond, C.; Chaste, P.; Fauchereau, F.; Coleman, M.; Leboyer, M.; Gillberg, C.; Bourgeron, T. Key role for gene dosage and synaptic homeostasis in autism spectrum disorders. Trends Genet. 2010, 26, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Tsai, N.P.; Wilkerson, J.R.; Guo, W.; Maksimova, M.A.; DeMartino, G.N.; Cowan, C.W.; Huber, K.M. Multiple autism-linked genes mediate synapse elimination via proteasomal degradation of a synaptic scaffold PSD-95. Cell 2012, 151, 1581–1594. [Google Scholar] [CrossRef] [PubMed]
- Voineagu, I.; Eapen, V. Converging pathways in autism spectrum disorders: Interplay between synaptic dysfunction and immune responses. Front. Hum. Neurosci. 2013, 7, 738. [Google Scholar] [CrossRef] [PubMed]
- Krey, J.F.; Pasca, S.P.; Shcheglovitov, A.; Yazawa, M.; Schwemberger, R.; Rasmusson, R.; Dolmetsch, R.E. Timothy syndrome is associated with activity-dependent dendritic retraction in rodent and human neurons. Nat. Neurosci. 2013, 16, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [PubMed]
- Poultney, C.S.; Goldberg, A.P.; Drapeau, E.; Kou, Y.; Harony-Nicolas, H.; Kajiwara, Y.; De Rubeis, S.; Durand, S.; Stevens, C.; Rehnstrom, K.; et al. Identification of small exonic CNV from whole-exome sequence data and application to autism spectrum disorder. Am. J. Hum. Genet. 2013, 93, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Gudsnuk, K.; Kuo, S.H.; Cotrina, M.L.; Rosoklija, G.; Sosunov, A.; Sonders, M.S.; Kanter, E.; Castagna, C.; Yamamoto, A.; et al. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 2014, 83, 1131–1143. [Google Scholar] [CrossRef] [PubMed]
- Huber, K.M.; Klann, E.; Costa-Mattioli, M.; Zukin, R.S. Dysregulation of mammalian target of rapamycin signaling in mouse models of autism. J. Neurosci. 2015, 35, 13836–13842. [Google Scholar] [CrossRef] [PubMed]
- Sato, A. mTOR, a potential target to treat autism spectrum disorder. CNS Neurol. Disord. Drug Targets 2016, 15, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Colvert, E.; Tick, B.; McEwen, F.; Stewart, C.; Curran, S.R.; Woodhouse, E.; Gillan, N.; Hallett, V.; Lietz, S.; Garnett, T.; et al. Heritability of autism spectrum disorder in a UK population-based twin sample. JAMA Psychiatry 2015, 72, 415–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tick, B.; Bolton, P.; Happe, F.; Rutter, M.; Rijsdijk, F. Heritability of autism spectrum disorders: A meta-analysis of twin studies. J. Child Psychol. Psychiatry 2016, 57, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Grabrucker, A.M. Environmental factors in autism. Front. Psychiatry 2012, 3, 118. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Asadi, S.; Patel, A.B. Focal brain inflammation and autism. J. Neuroinflamm. 2013, 10, 46. [Google Scholar] [CrossRef] [PubMed]
- Estes, M.L.; McAllister, A.K. Immune mediators in the brain and peripheral tissues in autism spectrum disorder. Nat. Rev. Neurosci. 2015, 16, 469–486. [Google Scholar] [PubMed]
- Becker, K.G. Autism, asthma, inflammation, and the hygiene hypothesis. Med. Hypotheses 2007, 69, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Gurney, J.G.; McPheeters, M.L.; Davis, M.M. Parental report of health conditions and health care use among children with and without autism: National survey of children’s health. Arch. Pediatr. Adolesc. Med. 2006, 160, 825–830. [Google Scholar] [CrossRef] [PubMed]
- Lyall, K.; Van de Water, J.; Ashwood, P.; Hertz-Picciotto, I. Asthma and allergies in children with autism spectrum disorders: Results from the CHARGE study. Autism Res. 2015, 8, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Croen, L.A.; Grether, J.K.; Yoshida, C.K.; Odouli, R.; Van de Water, J. Maternal autoimmune diseases, asthma and allergies, and childhood autism spectrum disorders: A case-control study. Arch. Pediatr. Adolesc. Med. 2005, 159, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Entringer, S.; Kumsta, R.; Nelson, E.L.; Hellhammer, D.H.; Wadhwa, P.D.; Wust, S. Influence of prenatal psychosocial stress on cytokine production in adult women. Dev. Psychobiol. 2008, 50, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Lim, R.; Fedulov, A.V.; Kobzik, L. Maternal stress during pregnancy increases neonatal allergy susceptibility: Role of glucocorticoids. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L141–L148. [Google Scholar] [CrossRef] [PubMed]
- von Hertzen, L.C. Maternal stress and T-cell differentiation of the developing immune system: Possible implications for the development of asthma and atopy. J. Allergy Clin. Immunol. 2002, 109, 923–928. [Google Scholar] [CrossRef] [PubMed]
- Kinney, D.K.; Munir, K.M.; Crowley, D.J.; Miller, A.M. Prenatal stress and risk for autism. Neurosci. Biobehav. Rev. 2008, 32, 1519–1532. [Google Scholar] [CrossRef] [PubMed]
- Ronald, A.; Pennell, C.E.; Whitehouse, A.J. Prenatal maternal stress associated with ADHD and autistic traits in early childhood. Front. Psychol. 2010, 1, 223. [Google Scholar] [CrossRef] [PubMed]
- Magalhaes, E.S.; Pinto-Mariz, F.; Bastos-Pinto, S.; Pontes, A.T.; Prado, E.A.; deAzevedo, L.C. Immune allergic response in Asperger syndrome. J. Neuroimmunol. 2009, 216, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Pardo, C.A.; Farmer, C.A.; Thurm, A.; Shebl, F.M.; Ilieva, J.; Kalra, S.; Swedo, S. Serum and cerebrospinal fluid immune mediators in children with autistic disorder: A longitudinal study. Mol. Autism 2017, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Angelidou, A.; Alysandratos, K.D.; Asadi, S.; Zhang, B.; Francis, K.; Vasiadi, M.; Kalogeromitros, D.; Theoharides, T.C. Brief report: “Allergic symptoms” in children with autism spectrum disorders. More than meets the eye? J. Autism Dev. Disord. 2011, 41, 1579–1585. [Google Scholar] [CrossRef] [PubMed]
- Chawarska, K.; Campbell, D.; Chen, L.; Shic, F.; Klin, A.; Chang, J. Early generalized overgrowth in boys with autism. Arch. Gen. Psychiatry 2011, 68, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Courchesne, E.; Carper, R.; Akshoomoff, N. Evidence of brain overgrowth in the first year of life in autism. JAMA 2003, 290, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Nordahl, C.W.; Lange, N.; Li, D.D.; Barnett, L.A.; Lee, A.; Buonocore, M.H.; Simon, T.J.; Rogers, S.; Ozonoff, S.; Amaral, D.G. Brain enlargement is associated with regression in preschool-age boys with autism spectrum disorders. Proc. Natl. Acad. Sci. USA 2011, 108, 20195–20200. [Google Scholar] [CrossRef] [PubMed]
- Sacco, R.; Militerni, R.; Frolli, A.; Bravaccio, C.; Gritti, A.; Elia, M.; Curatolo, P.; Manzi, B.; Trillo, S.; Lenti, C.; et al. Clinical, morphological, and biochemical correlates of head circumference in autism. Biol. Psychiatry 2007, 62, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Gregory, A.; Doull, I.; Pearce, N.; Cheng, S.; Leadbitter, P.; Holgate, S.; Beasley, R. The relationship between anthropometric measurements at birth: Asthma and atopy in childhood. Clin. Exp. Allergy 1999, 29, 330–333. [Google Scholar] [CrossRef]
- Katz, K.A.; Pocock, S.J.; Strachan, D.P. Neonatal head circumference, neonatal weight, and risk of hayfever, asthma and eczema in a large cohort of adolescents from Sheffield, England. Clin. Exp. Allergy 2003, 33, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Oryszczyn, M.P.; Annesi-Maesano, I.; Campagna, D.; Sahuquillo, J.; Huel, G.; Kauffmann, F. Head circumference at birth and maternal factors related to cord blood total IgE. Clin. Exp. Allergy 1999, 29, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Eviston, D.P.; Minasyan, A.; Mann, K.P.; Campbell, D.E.; Nanan, R.K. In utero head circumference is associated with childhood allergy. Front. Pediatr. 2015, 3, 73. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Zhang, L.; Zhu, T.; Huang, J.; Qu, Y.; Mu, D. Peripheral brain-derived neurotrophic factor in autism spectrum disorder: A systematic review and meta-analysis. Sci. Rep. 2016, 6, 31241. [Google Scholar] [CrossRef] [PubMed]
- Kern, J.K.; Geier, D.A.; Sykes, L.K.; Geier, M.R. Relevance of neuroinflammation and encephalitis in autism. Front. Cell. Neurosci. 2015, 9, 519. [Google Scholar] [CrossRef] [PubMed]
- Onore, C.; Careaga, M.; Ashwood, P. The role of immune dysfunction in the pathophysiology of autism. Brain Behav. Immun. 2012, 26, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Ishizuka, T.; Okayama, Y. Human mast cells and basophils as sources of cytokines. Clin. Exp. Allergy 2000, 30, 1205–1212. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Tsilioni, I.; Patel, A.B.; Doyle, R. Atopic diseases and inflammation of the brain in the pathogenesis of autism spectrum disorders. Transl. Psychiatry 2016, 6, e844. [Google Scholar] [CrossRef] [PubMed]
- Goines, P.E.; Croen, L.A.; Braunschweig, D.; Yoshida, C.K.; Grether, J.; Hansen, R.; Kharrazi, M.; Ashwood, P.; Van de Water, J. Increased midgestational IFN-gamma, IL-4 and IL-5 in women bearing a child with autism: A case-control study. Mol. Autism 2011, 2, 13. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, M.W.; Larsen, N.; Grove, J.; Norgaard-Pedersen, B.; Thorsen, P.; Mortensen, E.L.; Hougaard, D.M. Amniotic fluid inflammatory cytokines: Potential markers of immunologic dysfunction in autism spectrum disorders. World J. Biol. Psychiatry 2013, 14, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Krakowiak, P.; Goines, P.E.; Tancredi, D.J.; Ashwood, P.; Hansen, R.L.; Hertz-Picciotto, I.; Van de Water, J. Neonatal cytokine profiles associated with autism spectrum disorder. Biol. Psychiatry 2017, 81, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, E.; Guest, P.C.; Rahmoune, H.; Wang, L.; Levin, Y.; Ingudomnukul, E.; Ruta, L.; Kent, L.; Spain, M.; Baron-Cohen, S.; Bahn, S. Sex-specific serum biomarker patterns in adults with Asperger’s syndrome. Mol. Psychiatry 2011, 16, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Aggarwal, S.; Rashanravan, B.; Lee, T. Th1- and Th2-like cytokines in CD4+ and CD8+ T cells in autism. J. Neuroimmunol. 1998, 85, 106–109. [Google Scholar] [CrossRef]
- Molloy, C.A.; Morrow, A.L.; Meinzen-Derr, J.; Schleifer, K.; Dienger, K.; Manning-Courtney, P.; Altaye, M.; Wills-Karp, M. Elevated cytokine levels in children with autism spectrum disorder. J. Neuroimmunol. 2006, 172, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Matsuzaki, H.; Iwata, K.; Kameno, Y.; Shimmura, C.; Kawai, S.; Yoshihara, Y.; Wakuda, T.; Takebayashi, K.; Takagai, S.; et al. Plasma cytokine profiles in subjects with high-functioning autism spectrum disorders. PLoS ONE 2011, 6, e20470. [Google Scholar] [CrossRef] [PubMed]
- Careaga, M.; Murai, T.; Bauman, M.D. Maternal immune activation and autism spectrum disorder: From rodents to nonhuman and human primates. Biol. Psychiatry 2017, 81, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Hu, V.W.; Frank, B.C.; Heine, S.; Lee, N.H.; Quackenbush, J. Gene expression profiling of lymphoblastoid cell lines from monozygotic twins discordant in severity of autism reveals differential regulation of neurologically relevant genes. BMC Genom. 2006, 7, 118. [Google Scholar] [CrossRef]
- Vargas, D.L.; Nascimbene, C.; Krishnan, C.; Zimmerman, A.W.; Pardo, C.A. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. 2005, 57, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Chhor, V.; Le Charpentier, T.; Lebon, S.; Ore, M.V.; Celador, I.L.; Josserand, J.; Degos, V.; Jacotot, E.; Hagberg, H.; Savman, K.; et al. Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. Brain Behav. Immun. 2013, 32, 70–85. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Leak, R.K.; Shi, Y.; Suenaga, J.; Gao, Y.; Zheng, P.; Chen, J. Microglial and macrophage polarization-new prospects for brain repair. Nat. Rev. Neurol. 2015, 11, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Licona-Limon, P.; Kim, L.K.; Palm, N.W.; Flavell, R.A. TH2, allergy and group 2 innate lymphoid cells. Nat. Immunol. 2013, 14, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Georas, S.N.; Guo, J.; De Fanis, U.; Casolaro, V. T-helper cell type-2 regulation in allergic disease. Eur. Respir. J. 2005, 26, 1119–1137. [Google Scholar] [CrossRef] [PubMed]
- Goines, P.E.; Ashwood, P. Cytokine dysregulation in autism spectrum disorders (ASD): Possible role of the environment. Neurotoxicol. Teratol. 2013, 36, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, N.A.; Bennett, B.L.; Graham, N.M.; Pirozzi, G.; Stahl, N.; Yancopoulos, G.D. Targeting key proximal drivers of type 2 inflammation in disease. Nat. Rev. Drug Discov. 2016, 15, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Xu, Y.; Pearse, D.D. Cyclic AMP is a key regulator of M1 to M2a phenotypic conversion of microglia in the presence of Th2 cytokines. J. Neuroinflamm. 2016, 13, 9. [Google Scholar] [CrossRef] [PubMed]
- Butovsky, O.; Talpalar, A.E.; Ben-Yaakov, K.; Schwartz, M. Activation of microglia by aggregated beta-amyloid or lipopolysaccharide impairs MHC-II expression and renders them cytotoxic whereas IFN-gamma and IL-4 render them protective. Mol. Cell. Neurosci. 2005, 29, 381–393. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Sugihara, G.; Ouchi, Y.; Nakamura, K.; Futatsubashi, M.; Takebayashi, K.; Yoshihara, Y.; Omata, K.; Matsumoto, K.; Tsuchiya, K.J.; et al. Microglial activation in young adults with autism spectrum disorder. JAMA Psychiatry 2013, 70, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.T.; Chana, G.; Pardo, C.A.; Achim, C.; Semendeferi, K.; Buckwalter, J.; Courchesne, E.; Everall, I.P. Microglial activation and increased microglial density observed in the dorsolateral prefrontal cortex in autism. Biol. Psychiatry 2010, 68, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chauhan, A.; Sheikh, A.M.; Patil, S.; Chauhan, V.; Li, X.M.; Ji, L.; Brown, T.; Malik, M. Elevated immune response in the brain of autistic patients. J. Neuroimmunol. 2009, 207, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Edmonson, C.A.; Ziats, M.N.; Rennert, O.M. A Non-inflammatory role for microglia in autism spectrum disorders. Front. Neurol. 2016, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Pardo, C.A.; Vargas, D.L.; Zimmerman, A.W. Immunity, neuroglia and neuroinflammation in autism. Int. Rev. Psychiatry 2005, 17, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Campbell, B.M.; Charych, E.; Lee, A.W.; Moller, T. Kynurenines in CNS disease: Regulation by inflammatory cytokines. Front. Neurosci. 2014, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J. Neuroinflamm. 2014, 11, 98. [Google Scholar] [CrossRef] [PubMed]
- Kita, T.; Morrison, P.F.; Heyes, M.P.; Markey, S.P. Effects of systemic and central nervous system localized inflammation on the contributions of metabolic precursors to the L-kynurenine and quinolinic acid pools in brain. J. Neurochem. 2002, 82, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.; Schaible, U.E. Macrophage defense mechanisms against intracellular bacteria. Immunol. Rev. 2015, 264, 182–203. [Google Scholar] [PubMed]
- Neurauter, G.; Schrocksnadel, K.; Scholl-Burgi, S.; Sperner-Unterweger, B.; Schubert, C.; Ledochowski, M.; Fuchs, D. Chronic immune stimulation correlates with reduced phenylalanine turnover. Curr. Drug Metab. 2008, 9, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, A.W.; Jyonouchi, H.; Comi, A.M.; Connors, S.L.; Milstien, S.; Varsou, A.; Heyes, M.P. Cerebrospinal fluid and serum markers of inflammation in autism. Pediatr. Neurol. 2005, 33, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Ellis, S.E.; Ashar, F.N.; Moes, A.; Bader, J.S.; Zhan, J.; West, A.B.; Arking, D.E. Transcriptome analysis reveals dysregulation of innate immune response genes and neuronal activity-dependent genes in autism. Nat. Commun. 2014, 5, 5748. [Google Scholar] [CrossRef] [PubMed]
- Suh, H.S.; Zhao, M.L.; Derico, L.; Choi, N.; Lee, S.C. Insulin-like growth factor 1 and 2 (IGF1, IGF2) expression in human microglia: Differential regulation by inflammatory mediators. J. Neuroinflamm. 2013, 10, 37. [Google Scholar] [CrossRef] [PubMed]
- Wynes, M.W.; Riches, D.W. Induction of macrophage insulin-like growth factor-I expression by the Th2 cytokines IL-4 and IL-13. J. Immunol. 2003, 171, 3550–3559. [Google Scholar] [CrossRef] [PubMed]
- O’Kusky, J.; Ye, P. Neurodevelopmental effects of insulin-like growth factor signaling. Front. Neuroendocrinol. 2012, 33, 230–251. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.; Popken, G.J.; Kemper, A.; McCarthy, K.; Popko, B.; D’Ercole, A.J. Astrocyte-specific overexpression of insulin-like growth factor-I promotes brain overgrowth and glial fibrillary acidic protein expression. J. Neurosci. Res. 2004, 78, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Mills, J.L.; Hediger, M.L.; Molloy, C.A.; Chrousos, G.P.; Manning-Courtney, P.; Yu, K.F.; Brasington, M.; England, L.J. Elevated levels of growth-related hormones in autism and autism spectrum disorder. Clin. Endocrinol. 2007, 67, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Faivre, L.; Gosset, P.; Cormier-Daire, V.; Odent, S.; Amiel, J.; Giurgea, I.; Nassogne, M.C.; Pasquier, L.; Munnich, A.; Romana, S.; et al. Overgrowth and trisomy 15q26.1-qter including the IGF1 receptor gene: Report of two families and review of the Literature. Eur. J. Hum. Genet. 2002, 10, 699–706. [Google Scholar] [PubMed]
- Chakrabarti, B.; Dudbridge, F.; Kent, L.; Wheelwright, S.; Hill-Cawthorne, G.; Allison, C.; Banerjee-Basu, S.; Baron-Cohen, S. Genes related to sex steroids, neural growth, and social-emotional behavior are associated with autistic traits, empathy, and Asperger syndrome. Autism Res. 2009, 2, 157–177. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Zhang, L.; Zhu, T.; Huang, J.; Qu, Y.; Mu, D. Association between asthma and autism spectrum disorder: A meta-analysis. PLoS ONE 2016, 11, e0156662. [Google Scholar] [CrossRef] [PubMed]
- Perry, E.K.; Lee, M.L.; Martin-Ruiz, C.M.; Court, J.A.; Volsen, S.G.; Merrit, J.; Folly, E.; Iversen, P.E.; Bauman, M.L.; Perry, R.H.; et al. Cholinergic activity in autism: Abnormalities in the cerebral cortex and basal forebrain. Am. J. Psychiatry 2001, 158, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Raznahan, A.; Toro, R.; Proitsi, P.; Powell, J.; Paus, T.; Bolton, F.P.; Murphy, D.G. A functional polymorphism of the brain derived neurotrophic factor gene and cortical anatomy in autism spectrum disorder. J. Neurodev. Disord. 2009, 1, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Antony, J.M.; Paquin, A.; Nutt, S.L.; Kaplan, D.R.; Miller, F.D. Endogenous microglia regulate development of embryonic cortical precursor cells. J. Neurosci. Res. 2011, 89, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Gross, C.T. Microglia in development: Linking brain wiring to brain environment. Neuron Glia Biol. 2011, 7, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Petrelli, F.; Pucci, L.; Bezzi, P. Astrocytes and microglia and their potential link with autism spectrum disorders. Front. Cell. Neurosci. 2016, 10, 21. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R., III; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef] [PubMed]
- Lindholm, J.S.; Castren, E. Mice with altered BDNF signaling as models for mood disorders and antidepressant effects. Front. Behav. Neurosci. 2014, 8, 143. [Google Scholar] [CrossRef] [PubMed]
- Papaleo, F.; Silverman, J.L.; Aney, J.; Tian, Q.; Barkan, C.L.; Chadman, K.K.; Crawley, J.N. Working memory deficits, increased anxiety-like traits, and seizure susceptibility in BDNF overexpressing mice. Learn. Mem. 2011, 18, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Ajo, R.; Cacicedo, L.; Navarro, C.; Sanchez-Franco, F. Growth hormone action on proliferation and differentiation of cerebral cortical cells from fetal rat. Endocrinology 2003, 144, 1086–1097. [Google Scholar] [CrossRef] [PubMed]
- Pathania, M.; Yan, L.D.; Bordey, A. A symphony of signals conducts early and late stages of adult neurogenesis. Neuropharmacology 2010, 58, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Courchesne, E.; Mouton, P.R.; Calhoun, M.E.; Semendeferi, K.; Ahrens-Barbeau, C.; Hallet, M.J.; Barnes, C.C.; Pierce, K. Neuron number and size in prefrontal cortex of children with autism. JAMA 2011, 306, 2001–2010. [Google Scholar] [CrossRef] [PubMed]
- Gadani, S.P.; Cronk, J.C.; Norris, G.T.; Kipnis, J. IL-4 in the brain: A cytokine to remember. J. Immunol. 2012, 189, 4213–4219. [Google Scholar] [CrossRef] [PubMed]
- Peltier, J.; O’Neill, A.; Schaffer, D.V. PI3K/Akt and CREB regulate adult neural hippocampal progenitor proliferation and differentiation. Dev. Neurobiol. 2007, 67, 1348–1361. [Google Scholar] [CrossRef] [PubMed]
- Bosnjak, B.; Stelzmueller, B.; Erb, K.J.; Epstein, M.M. Treatment of allergic asthma: Modulation of Th2 cells and their responses. Respir. Res. 2011, 12, 114. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A.; Medzhitov, R. Control of adaptive immunity by the innate immune system. Nat. Immunol. 2015, 16, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Elenkov, I.J. Glucocorticoids and the Th1/Th2 balance. Ann. N. Y. Acad. Sci. 2004, 1024, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Kalkman, H.O.; Feuerbach, D. Antidepressant therapies inhibit inflammation and microglial M1-polarization. Pharmacol. Ther. 2016, 163, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Derecki, N.C.; Quinnies, K.M.; Kipnis, J. Alternatively activated myeloid (M2) cells enhance cognitive function in immune compromised mice. Brain Behav. Immun. 2011, 25, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, A.; Constantine-Paton, M. Postsynaptic BDNF-TrkB signaling in synapse maturation, plasticity, and disease. Dev. Neurobiol. 2010, 70, 304–322. [Google Scholar] [PubMed]
- Branum, A.M.; Lukacs, S.L. Food allergy among children in the United States. Pediatrics 2009, 124, 1549–1555. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Sheikh, A.; Strachan, D.P.; Anderson, H.R. Time trends in allergic disorders in the UK. Thorax 2007, 62, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Charpin, D.; Gouitaa, M. Why is the prevalence of allergic diseases increasing? A critical assessment of some classical risk factors. Mediat. Inflamm. 2001, 10, 292–294. [Google Scholar] [CrossRef] [PubMed]
- Hadley, C. Food allergies on the rise? Determining the prevalence of food allergies, and how quickly it is increasing, is the first step in tackling the problem. EMBO Rep. 2006, 7, 1080–1083. [Google Scholar] [CrossRef] [PubMed]
- Pennesi, C.M.; Klein, L.C. Effectiveness of the gluten-free, casein-free diet for children diagnosed with autism spectrum disorder: Based on parental report. Nutr. Neurosci. 2012, 15, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Ou, J.J.; Li, Y.M.; Xiang, D.X. Dietary supplement for core symptoms of autism spectrum disorder: Where are we now and where should we go? Front. Psychiatry 2017, 8, 155. [Google Scholar] [CrossRef] [PubMed]
- Chiba, Y.; Todoroki, M.; Nishida, Y.; Tanabe, M.; Misawa, M. A novel STAT6 inhibitor AS1517499 ameliorates antigen-induced bronchial hypercontractility in mice. Am. J. Respir. Cell Mol. Biol. 2009, 41, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.B.; Tsilioni, I.; Leeman, S.E.; Theoharides, T.C. Neurotensin stimulates sortilin and mTOR in human microglia inhibitable by methoxyluteolin, a potential therapeutic target for autism. Proc. Natl. Acad. Sci. USA 2016. [Google Scholar] [CrossRef]
- Wu, J.; de Theije, C.G.; da Silva, S.L.; Abbring, S.; van der Horst, H.; Broersen, L.M.; Willemsen, L.; Kas, M.; Garssen, J.; Kraneveld, A.D. Dietary interventions that reduce mTOR activity rescue autistic-like behavioral deficits in mice. Brain Behav. Immun. 2017, 59, 273–287. [Google Scholar] [CrossRef] [PubMed]
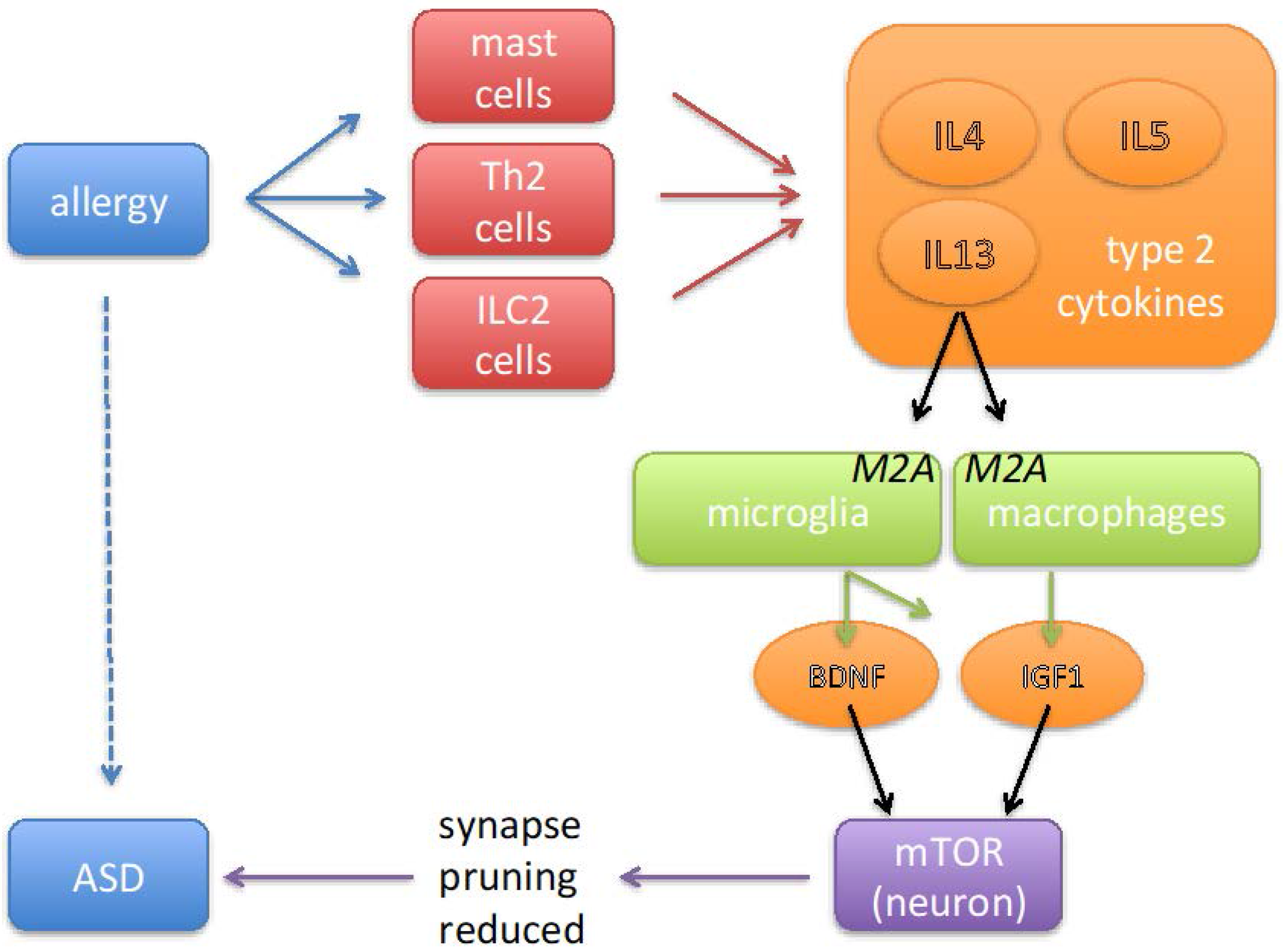
Figure 1.
Cartoon summarizing the sequence of events via which allergic disorders may contribute to the pathology of autism spectrum disorders (ASD). Allergy is associated with an activation of mast cells, innate lymphoid cells (ILC) and Th2 T cells. These cells produce type-2 cytokines (IL4, IL5 and IL13), which stimulate microglia and macrophages to adopt a phenotype referred to as ‘alternative activation’ or ‘M2A’. By secreting a variety of growth factors, including brain-derived neurotrophic factor (BDNF) and insulin-like growth factor-1 (IGF1), M2A-polarized macrophages and microglia cells play a physiological role in tissue repair. In neurons these growth factors activate a signal transduction pathway that leads to activation of the enzyme mammalian target of rapamycin (mTOR) and thereby to the inhibition of autophagy. Inhibition of autophagy results in diminished removal of redundant synapses, which in the context of ASD is likely to be undesired, as the results from genetic studies indicate that insufficient synaptic pruning is an effect that is common to several syndromic forms of ASD.
Figure 1.
Cartoon summarizing the sequence of events via which allergic disorders may contribute to the pathology of autism spectrum disorders (ASD). Allergy is associated with an activation of mast cells, innate lymphoid cells (ILC) and Th2 T cells. These cells produce type-2 cytokines (IL4, IL5 and IL13), which stimulate microglia and macrophages to adopt a phenotype referred to as ‘alternative activation’ or ‘M2A’. By secreting a variety of growth factors, including brain-derived neurotrophic factor (BDNF) and insulin-like growth factor-1 (IGF1), M2A-polarized macrophages and microglia cells play a physiological role in tissue repair. In neurons these growth factors activate a signal transduction pathway that leads to activation of the enzyme mammalian target of rapamycin (mTOR) and thereby to the inhibition of autophagy. Inhibition of autophagy results in diminished removal of redundant synapses, which in the context of ASD is likely to be undesired, as the results from genetic studies indicate that insufficient synaptic pruning is an effect that is common to several syndromic forms of ASD.

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kalkman, H.O.; Feuerbach, D. Microglia M2A Polarization as Potential Link between Food Allergy and Autism Spectrum Disorders. Pharmaceuticals 2017, 10, 95. https://doi.org/10.3390/ph10040095
AMA Style
Kalkman HO, Feuerbach D. Microglia M2A Polarization as Potential Link between Food Allergy and Autism Spectrum Disorders. Pharmaceuticals. 2017; 10(4):95. https://doi.org/10.3390/ph10040095
Chicago/Turabian StyleKalkman, Hans O., and Dominik Feuerbach. 2017. "Microglia M2A Polarization as Potential Link between Food Allergy and Autism Spectrum Disorders" Pharmaceuticals 10, no. 4: 95. https://doi.org/10.3390/ph10040095
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.