In Silico SAR Studies of HIV-1 Inhibitors

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
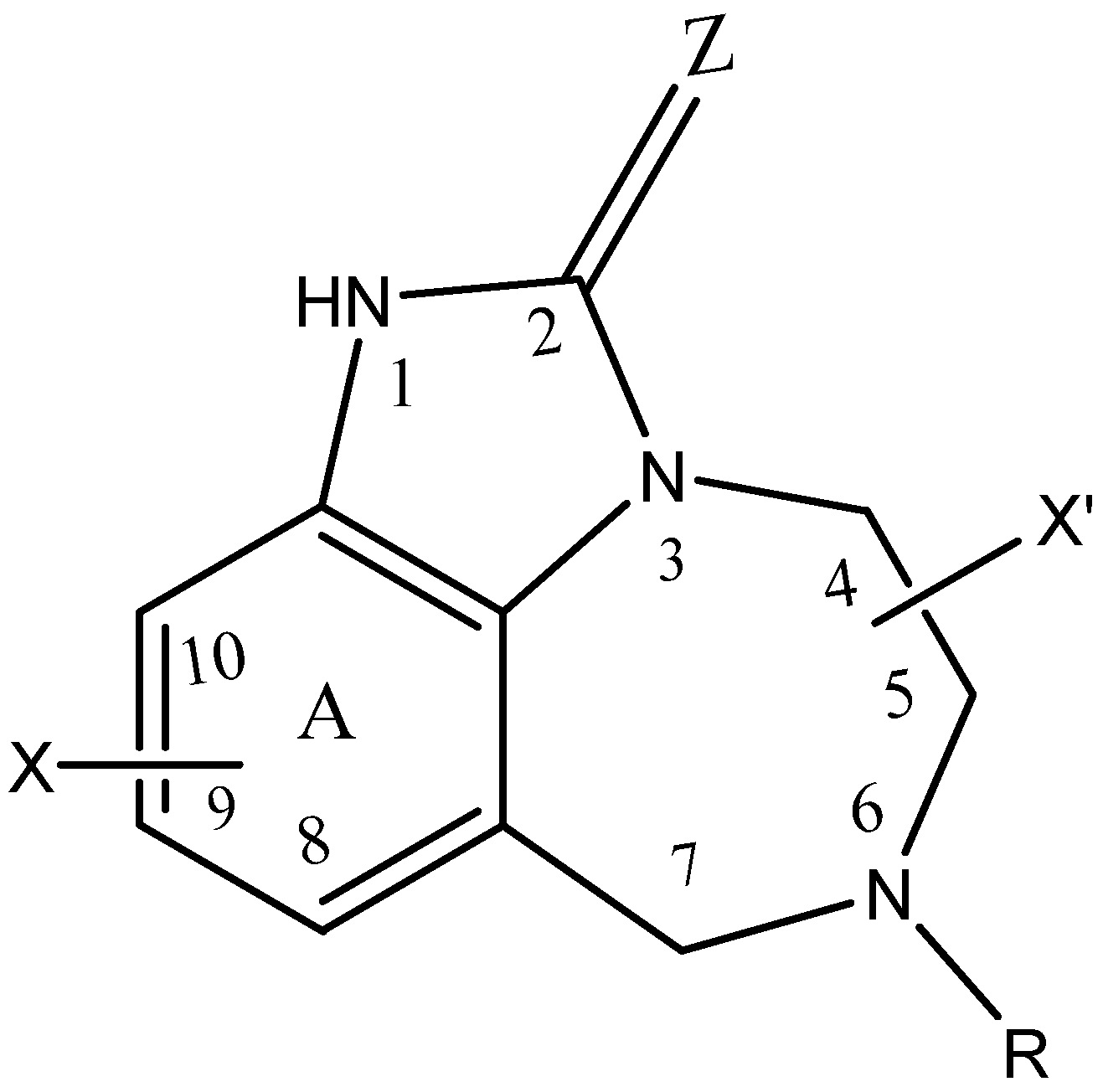
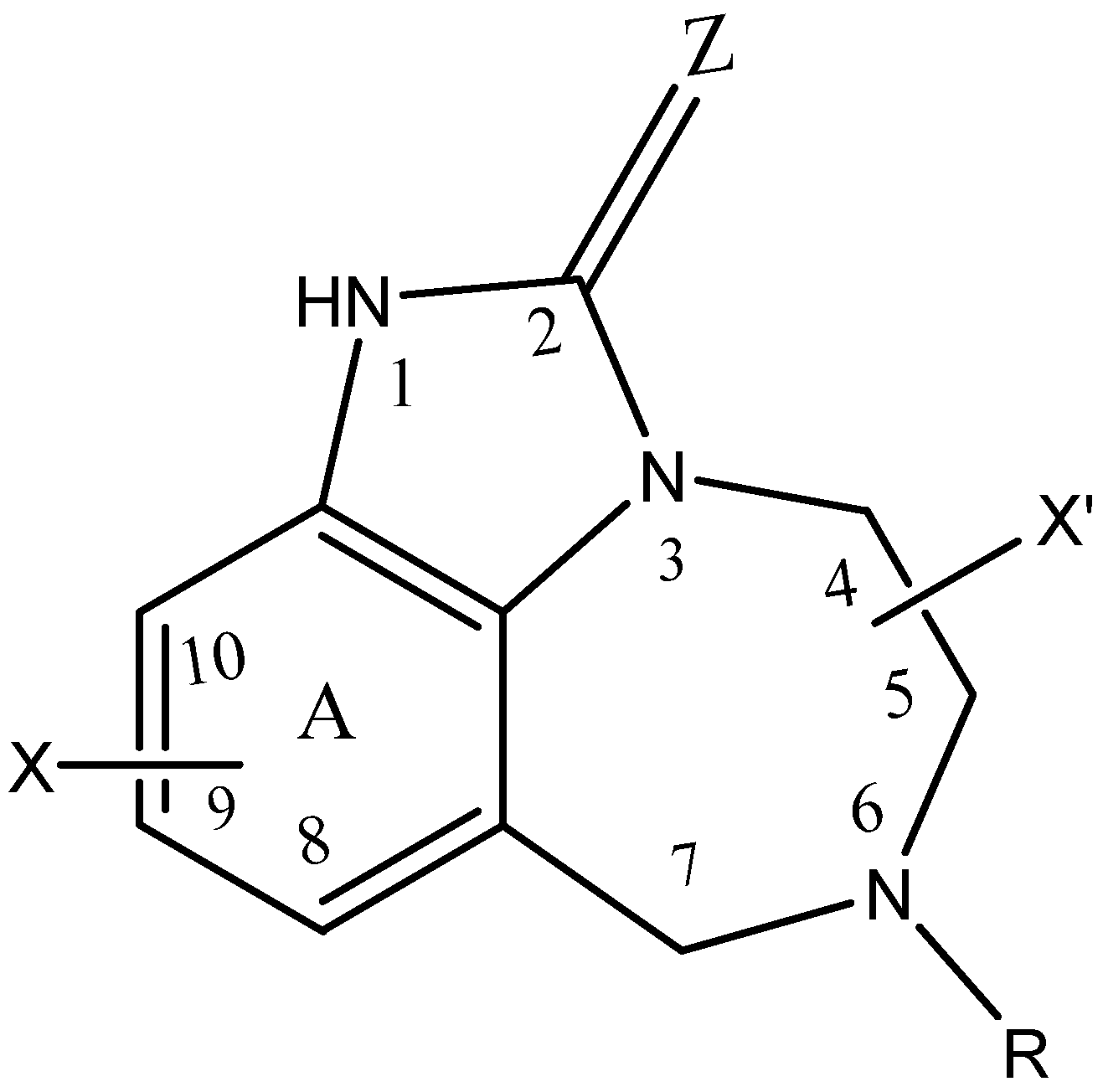
2.1. Selection of Data Set
- -
- Class H includes compounds with high activities (i.e., log (1/IC50) ≥ 5.79).
- -
- Class L contains compounds with low activities (i.e., log (1/IC50) < 5.79).
2.2. Molecular Descriptors
3. Results and Discussion
3.1. Support Vector Machines
3.2. Artificial Neural Networks
- ✓
- The input layer contains ten neurons, representing the ten parameters described previously;
- ✓
- The output layer contains a single neuron describing the class of the compound (Low or High HIV inhibitor)
- ✓
- The hidden layer contains a variable number of neurons. This layer allows ANN to model nonlinear relationships between inputs and outputs.
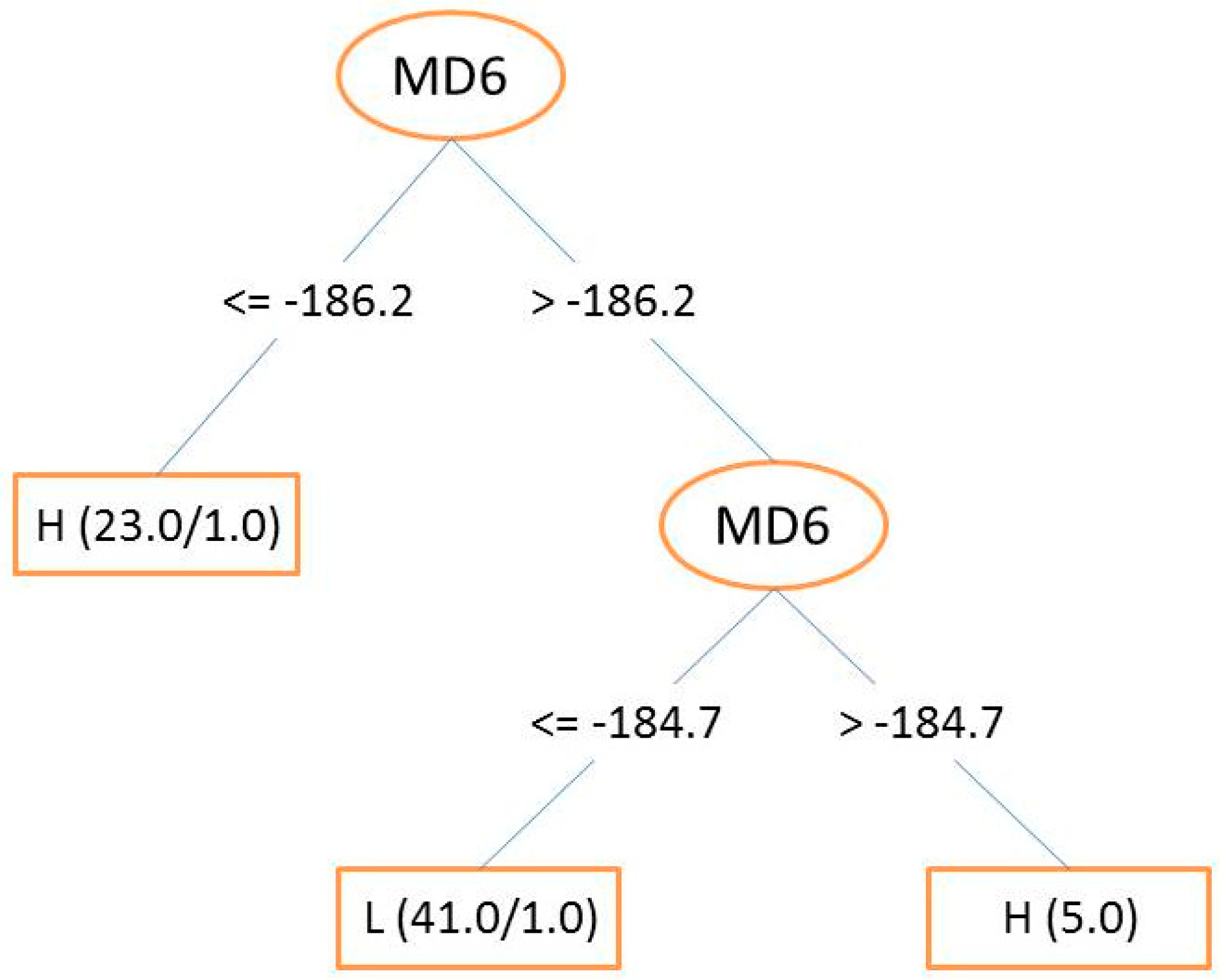
3.3. Decision Trees (DT) and Random Forest (RF)
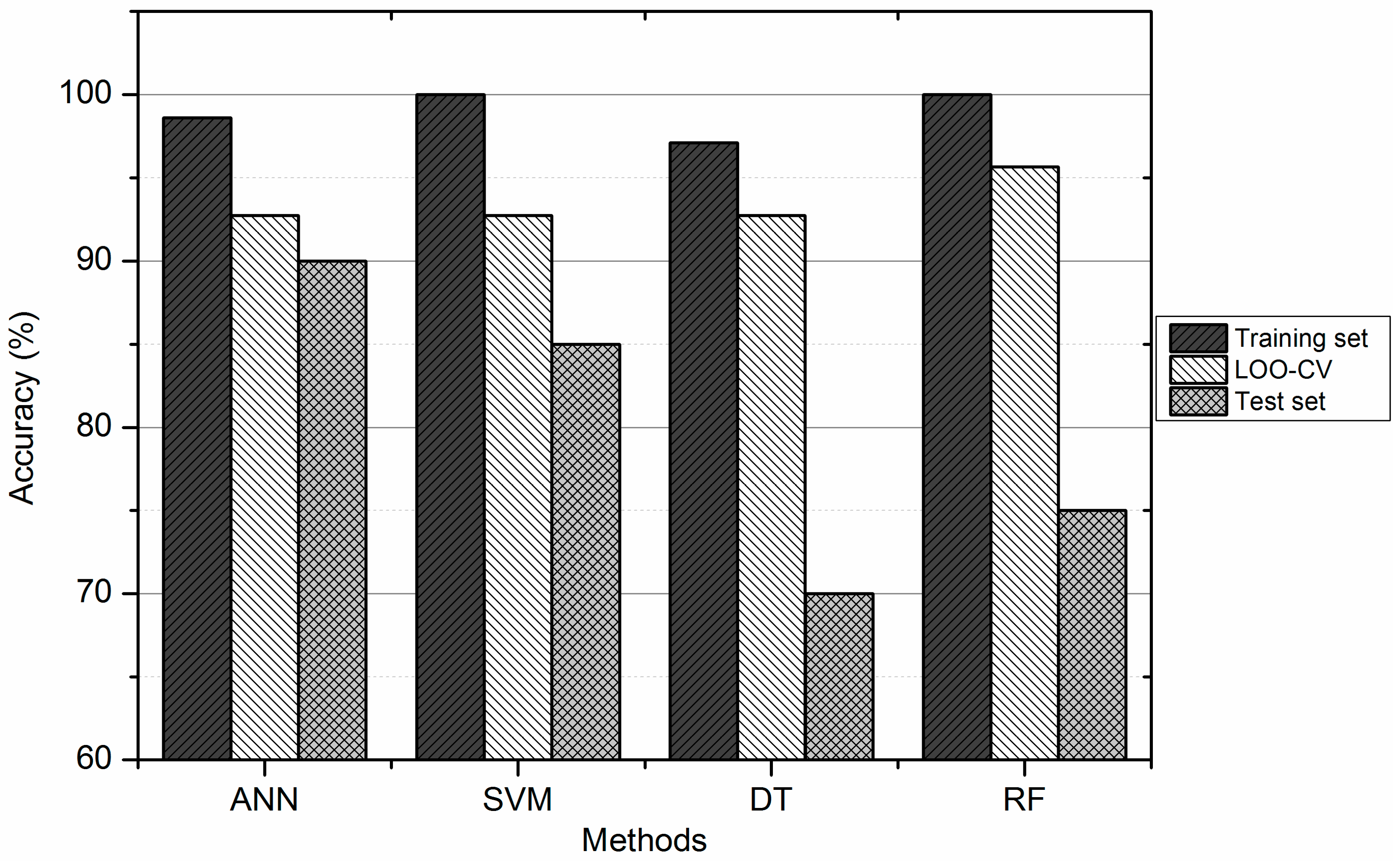
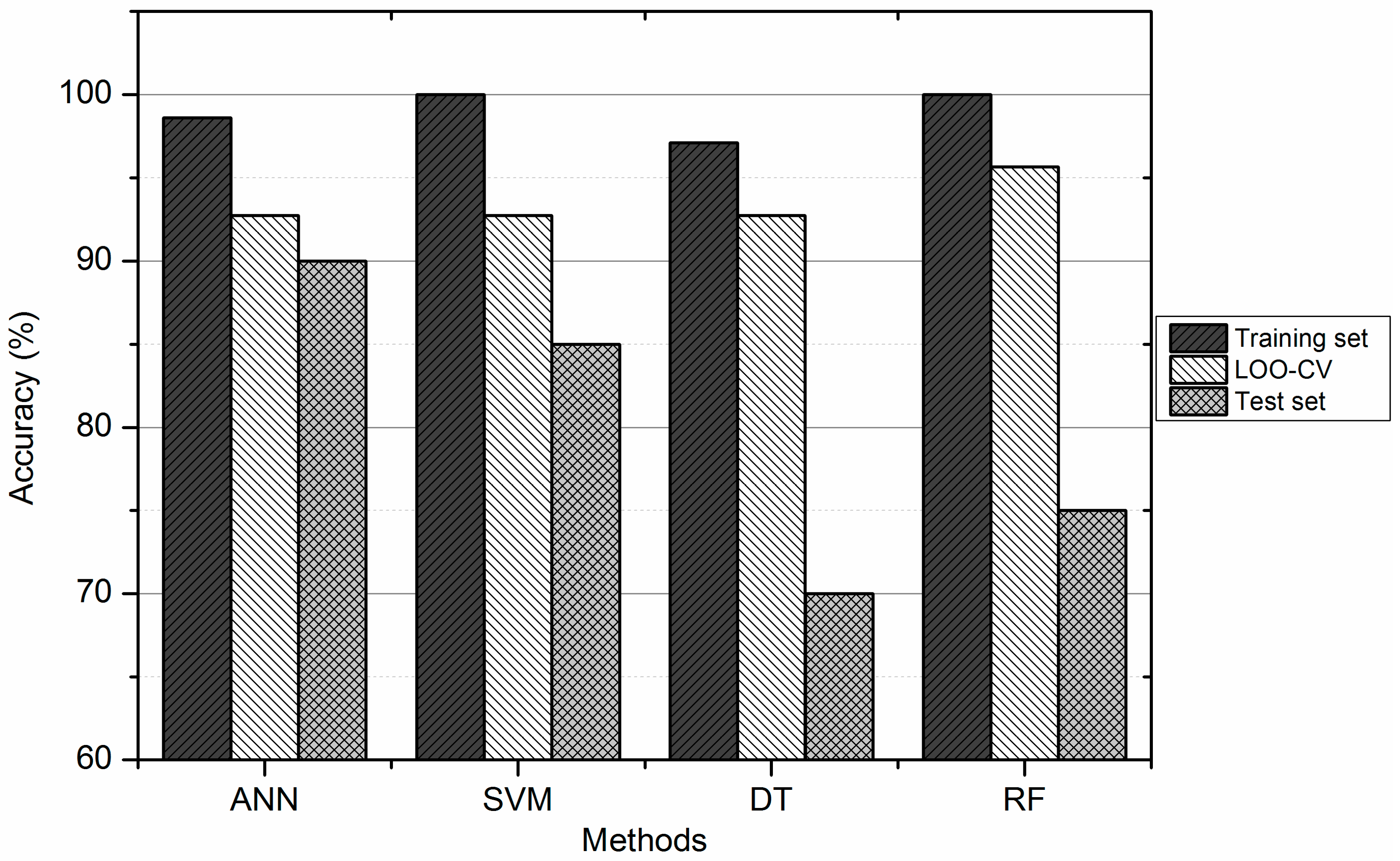
3.4. Comparison between ANN, DT, SVM, and RF
3.5. Descriptor Contributions
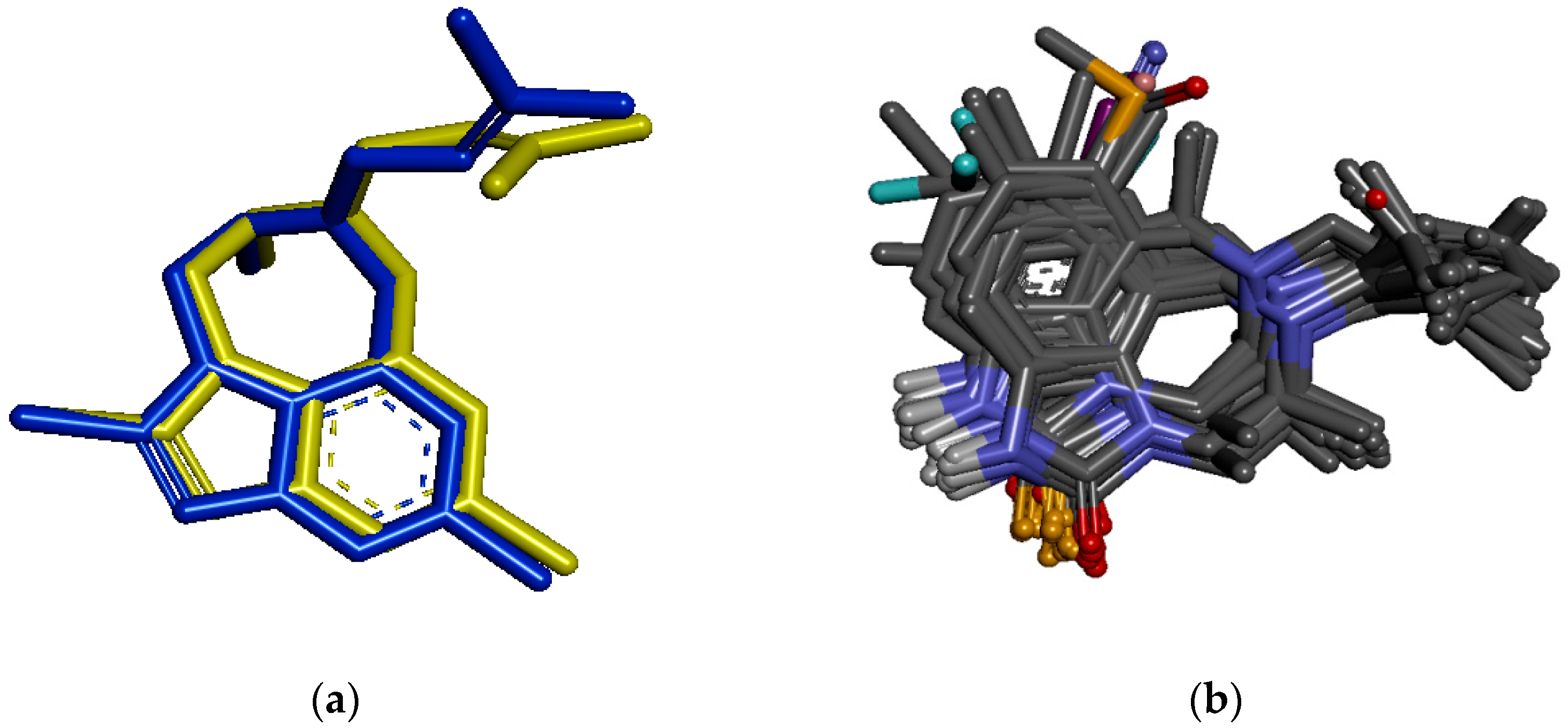
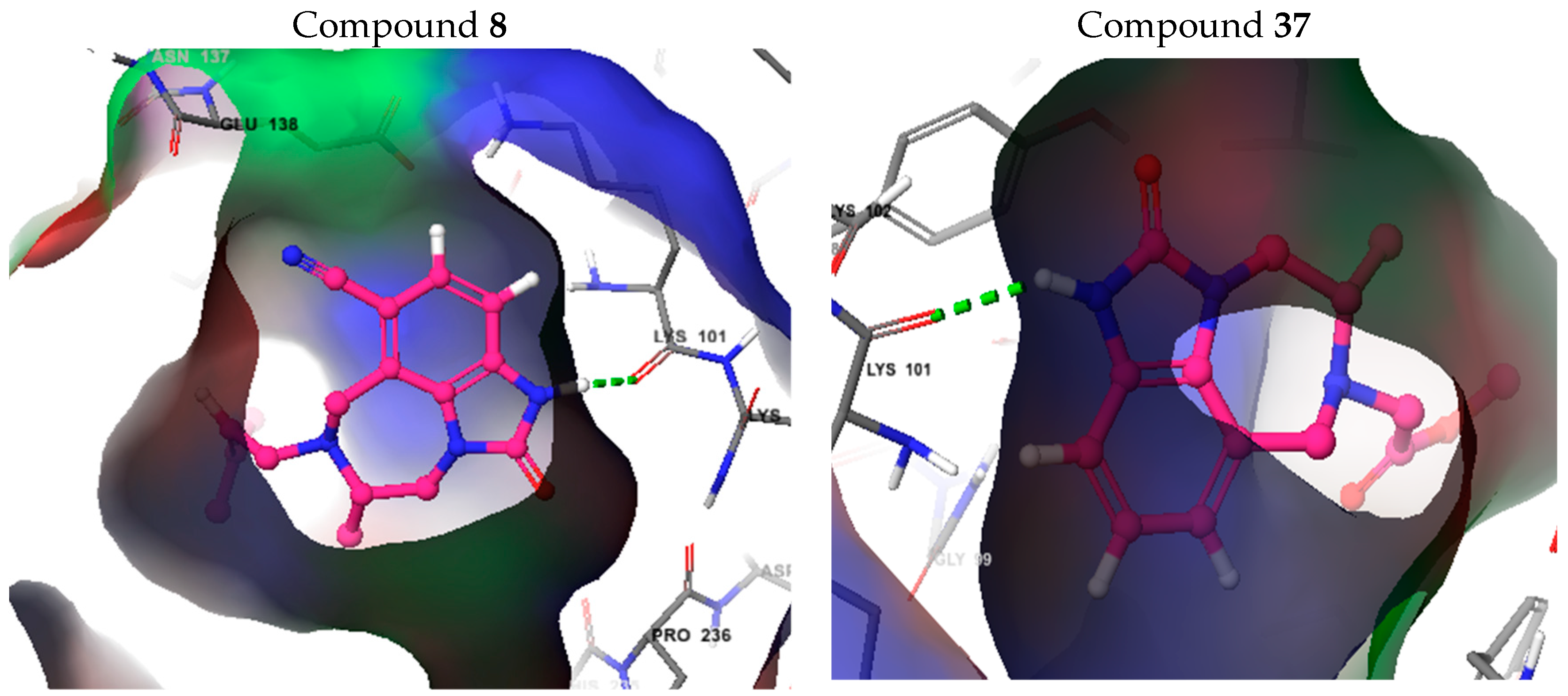
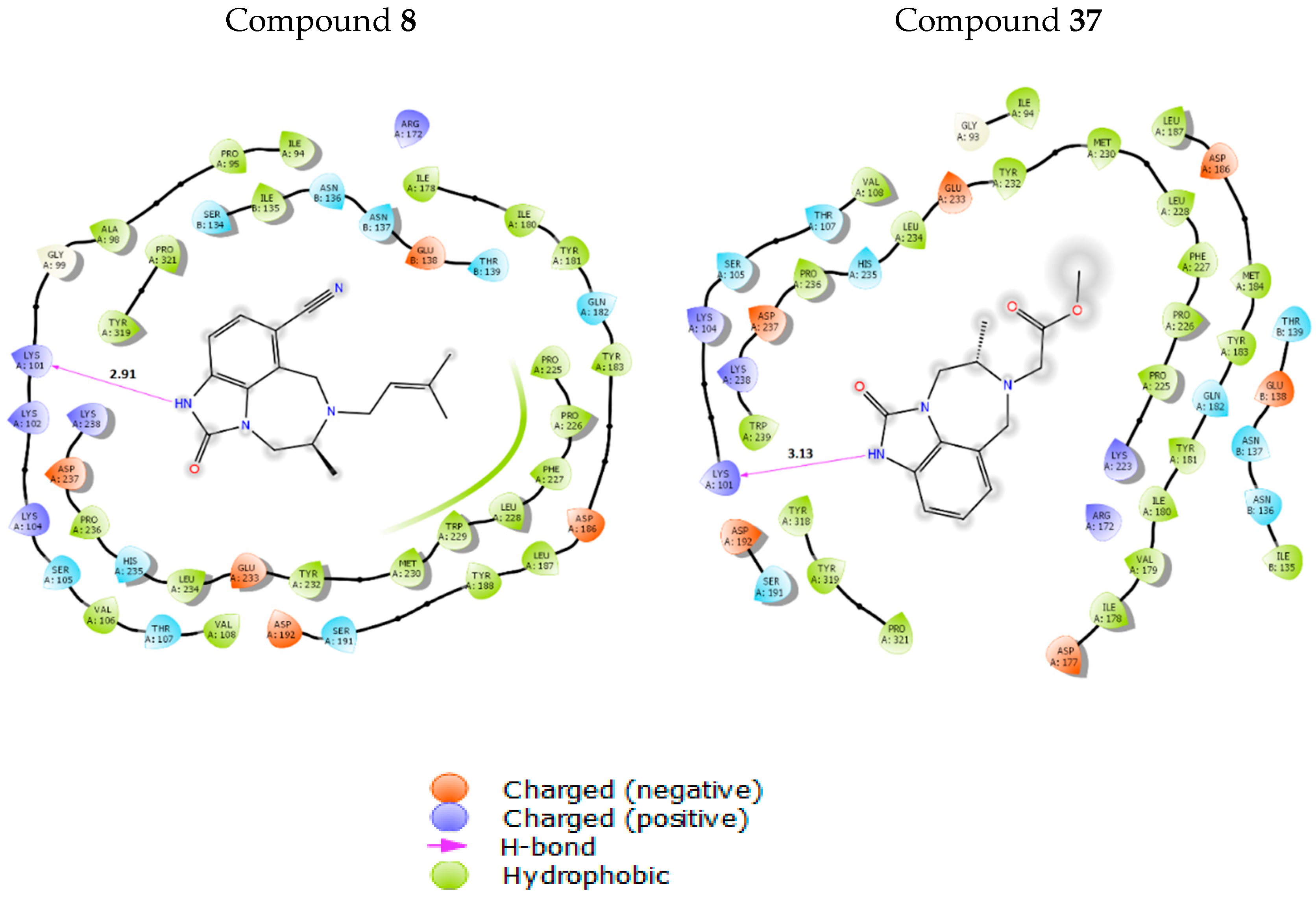
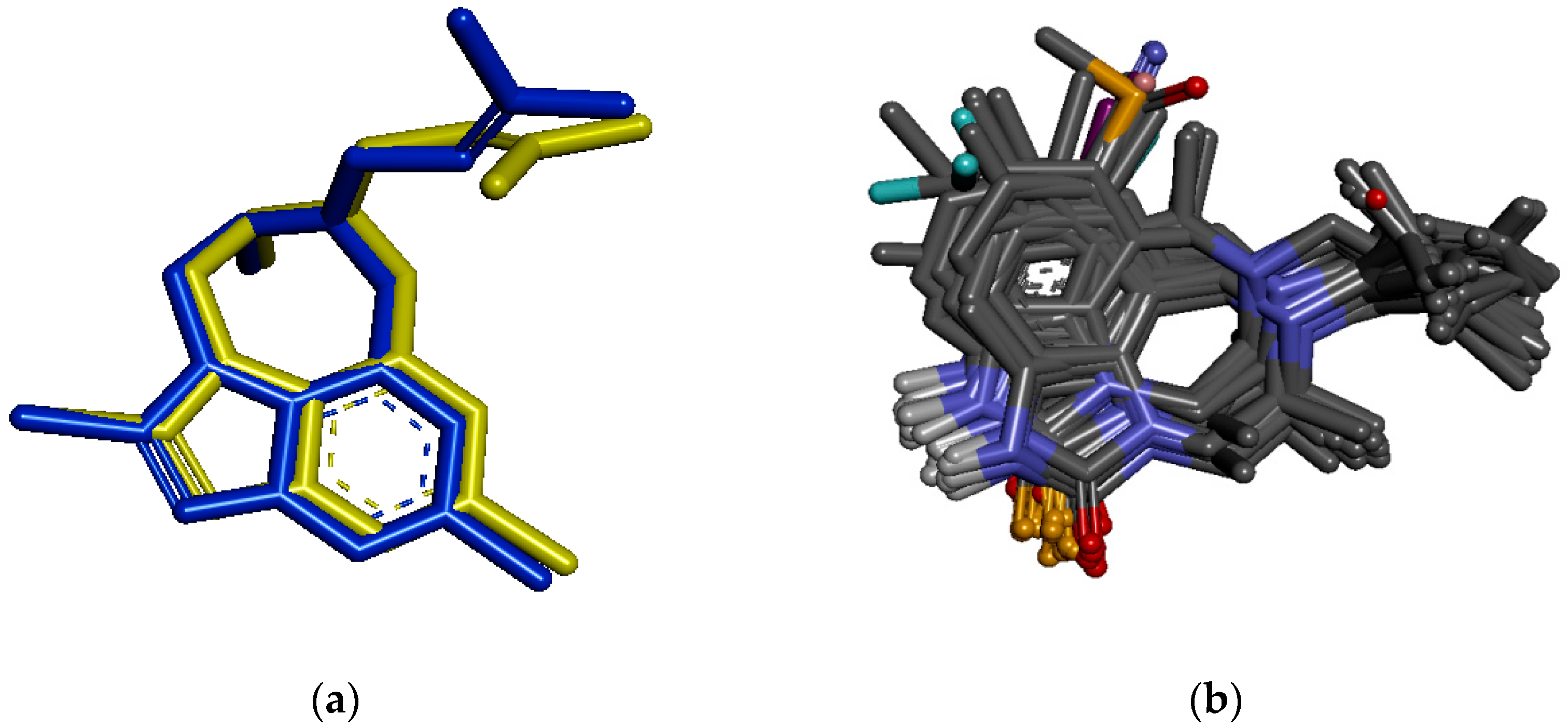
3.6. Molecular Docking Study
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sarafianos, S.G.; Marchand, B.; Das, K.; Himmel, D.M.; Parniak, M.A.; Hughes, S.H.; Arnold, E. Structure and function of HIV-1 reverse transcriptase: Molecular mechanisms of polymerization and inhibition. J. Mol. Biol. 2009, 385, 693–713. [Google Scholar] [CrossRef] [PubMed]
- Che, Z.; Liu, S.; Tian, Y.; Hu, Z.; Chen, Y.; Chen, G. Design and synthesis of novel N-arylsulfonyl-3-(2-yl-ethanone)-6-methylindole derivatives as inhibitors of HIV-1 replication. Pharmaceuticals 2015, 8, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Adams, C.P.; Brantner, V.V. Spending on new drug development. Health Econ. 2010, 19, 130–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darnag, R.; Schmitzer, A.; Belmiloud, Y.; Villemin, D.; Jarid, A.; Chait, A.; Mazouz, E.; Cherqaoui, D. Quantitative structure-activity relationship studies of TIBO derivatives using support vector machines. SAR QSAR Environ. Res. 2010, 21, 231–246. [Google Scholar] [CrossRef] [PubMed]
- Darnag, R.; Mazouz, E.; Schmitzer, A.; Villemin, D.; Jarid, A.; Cherqaoui, D. Support vector machines: Development of QSAR models for predicting anti-HIV-1 activity of TIBO derivatives. Eur. J. Med. Chem. 2010, 45, 1590–1597. [Google Scholar] [CrossRef] [PubMed]
- Douali, L.; Villemin, D.; Cherqaoui, D. Exploring QSAR of non-nucleoside reverse transcriptase inhibitors by neural networks: TIBO derivatives. Int. J. Mol. Sci. 2004, 5, 48–55. [Google Scholar] [CrossRef]
- Mandal, A.S.; Roy, K. Predictive QSAR modeling of HIV reverse transcriptase inhibitor TIBO derivatives. Eur. J. Med. Chem. 2009, 44, 1509–1524. [Google Scholar] [CrossRef] [PubMed]
- Hannongbua, S.; Pungpo, P.; Limtrakul, J.; Wolschann, P. Quantitative structure-activity relationships and comparative molecular field analysis of TIBO derivatised HIV-1 reverse transcriptase inhibitors. J. Comput. Aided Mol. Des. 1999, 13, 563–577. [Google Scholar] [CrossRef] [PubMed]
- Bjij, I.; Hdoufane, I.; Jarid, A.; Cherqaoui, D.; Villemin, D. Molecular modeling: Application of Support Vector Machines and Decision trees for anti-HIV activity prediction of organic compounds. In Proceedings of the 2016 IEEE 5th International Conference on Multimedia Computing and Systems (ICMCS), Marrakech, Morocco, 29 September–1 October 2016; pp. 169–173. [Google Scholar]
- Garg, R.; Gupta, S.P.; Gao, H.; Babu, M.S.; Debnath, A.K.; Hansch, C. Comparative Quantitative structure-Activity relationship studies on anti-HIV Drugs. Chem. Rev. 1999, 99, 3525–3601. [Google Scholar] [CrossRef] [PubMed]
- Pirhadi, S.; Shiri, F.; Ghasemi, J.B. Multivariate statistical analysis methods in QSAR. RSC Adv. 2015, 5, 104635–104665. [Google Scholar] [CrossRef]
- Belmiloud, Y. Theoritical Modeling of Enantiomeric Separation in a Racemic Mixture. Ph.D. Thesis, Science and Technology University of Alger, Algiers, Algeria, 2009. [Google Scholar]
- Gramatica, P. Principles of QSAR models validation: Internal and external. QSAR Comb. Sci. 2007, 26, 694–701. [Google Scholar] [CrossRef]
- Vapnik, V.N. Statistical Learning Theory; Wiley & Sons: New York, NY, USA, 1998; ISBN 0-471-03003-1. [Google Scholar]
- Cristianini, N.; Shawe-Taylor, J. An Introduction to Support. Vector Machines and Other Kernel-Based Learning Methods; Cambrige University Press: Cambridge, UK, 2000; ISBN 0-521-78019-5. [Google Scholar]
- Schölkopf, B.; Smola, A.J. Learning with Kernels; MIT Press: Cambridge, MA, USA, 2001; ISBN 0262194759. [Google Scholar]
- Wang, W.; Xu, Z.; Lu, W.; Zhang, X. Determination of the spread parameter in the Gaussian kernel for classification and regression. Neurocomputing 2003, 55, 643–663. [Google Scholar] [CrossRef]
- Chang, C.C.; Lin, C.J. LIBSVM: A library for support vector machines. ACM Trans. Intell. Syst. Technol. 2011, 2, 1–27. [Google Scholar] [CrossRef]
- Tropsha, A.; Gramatica, P.; Gombar, V. The importance of being earnest: Validation is the absolute essential for successful application and interpretation of QSPR Models. QSAR Comb. Sci. 2003, 22, 69–77. [Google Scholar] [CrossRef]
- Zupan, J.; Gasteiger, J. Neural networks in chemistry and drug design. Angew. Chem. Int. Ed. 1993, 32, 503–527. [Google Scholar]
- Zupan, J.; Gasteiger, J. Neural Networks for Chemists. An Introduction; Wiley-VCH: Weinheim, Germany, 1993; ISBN 352728592X. [Google Scholar]
- Cherqaoui, D.; Villemin, D. Use of a neural network to determine the boiling point of alkanes. J. Chem. Soc. Faraday Trans. 1994, 90, 97. [Google Scholar] [CrossRef]
- Freeman, J.A.; Skapura, D.A. Neural Networks: Algorithms, Applications, and Programming Techniques; Addison-Wesley Publishing Company: Boston, MA, USA, 1991; ISBN 0-201-51376-5. [Google Scholar]
- Andrea, T.A.; Kalayeh, H. Applications of neural networks in quantitative structure-activity relationships of dihydrofolate reductase inhibitors. J. Med. Chem. 1991, 34, 2824–2836. [Google Scholar] [CrossRef] [PubMed]
- So, S.S.; Richards, W.G. Application of neural networks: Quantitative structure-Activity relationships of the derivatives of 2,4-diamino-5-(substituted-benzyl)pyrimidines as DHFR inhibitors. J. Med. Chem. 1992, 35, 3201–3207. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.A.; Frank, E.; Holmes, G.; Pfahringer, B.; Reutemann, P.; Witten, I.H. The WEKA data mining software: An update. SIGKDD Explor. 2009, 11, 10–18. [Google Scholar] [CrossRef]
- Quinlan, J.R. C4.5: Programs for Machine Learning. Mach. Learn. 1994, 16, 235–240. [Google Scholar]
- Frank, E.; Hall, M.A.; Witten, I.H. The WEKA Workbench, 4th ed.; Online Appendix for “Data Mining: Practical Machine Learning Tools and Techniques”; Morgan Kaufmann: Burlington, MA, USA, 2016; pp. 553–571. ISBN 0123748569. [Google Scholar]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Jaworska, J.S.; Comber, M.; Van Leeuwen, C.J.; Auer, C. Summary of the workshop on regulatory acceptance of QSARs. Environ. Health Perpect. 2003, 111, 1358–1360. [Google Scholar] [CrossRef]
- Afantitis, A.; Melagraki, G.; Sarimveis, H.; Koutentis, P.A.; Markopoulos, J.; Igglessi-Markopoulou, O. A novel QSAR model for predicting induction of apoptosis by 4-aryl-4H-chromenes. Bioorg. Med. Chem. 2006, 14, 6686–6694. [Google Scholar] [CrossRef] [PubMed]
- Novakovic, J.; Strbac, P.; Bulatovic, D. Toward optimal feature selection using ranking methods and classification algorithms. Yugosl. J. Oper. Res. 2011, 21, 119–135. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A. NIH Public Access. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dassault Systèmes BIOVIA. Discovery Studio Modeling Environment; Release 2017; Dassault Systèmes: San Diego, LA, USA, 2016. [Google Scholar]
- Schrödinger Release 2018-1: Maestro; Schrödinger, LLC: New York, NY, USA, 2018.
- Ren, J.; Esnouf, R.; Hopkins, A.; Ross, C.; Jones, Y.; Stammers, D.; Stuart, D. The structure of HIV-1 reverse transcriptase complexed with 9-chloro-TIBO: Lessons for inhibitor design. Structure 1995, 3, 915–926. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substituents | Classes | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| N | X | Z | R | X’ | a Exp | b SVM | c ANN | d DT | e RF |
| 1 | H | S | DMA | 5-Me(S) | H | H | H | H | H |
| 2 | 9-Cl | S | DMA | 5-Me(S) | H | H | H | H | H |
| t 3 | 8-Cl | S | DMA | 5-Me(S) | H | H | H | H | H |
| 4 | 8-F | S | DMA | 5-Me(S) | H | H | H | H | H |
| 5 | 8-SMe | S | DMA | 5-Me(S) | H | H | H | H | H |
| t 6 | 8-OMe | S | DMA | 5-Me(S) | H | H | H | H | H |
| 7 | 8-OC2H5 | S | DMA | 5-Me(S) | H | H | H | H | H |
| 8 | 8-CN | O | DMA | 5-Me(S) | H | H | H | H | H |
| t 9 | 8-CN | S | DMA | 5-Me(S) | H | H | H | H | H |
| 10 | 8-CHO | S | DMA | 5-Me(S) | H | H | H | H | H |
| 11 | 8-CONH2 | O | DMA | 5-Me(S) | L | L | L | L | L |
| 12 | 8-Br | O | DMA | 5-Me(S) | H | H | H | H | H |
| t 13 | 8-Br | S | DMA | 5-Me(S) | H | H | H | H | H |
| 14 | 8-I | O | DMA | 5-Me(S) | H | H | H | H | H |
| t 15 | 8-I | S | DMA | 5-Me(S) | H | H | H | H | H |
| 16 | 8-C=CH | O | DMA | 5-Me(S) | H | H | H | H | H |
| t 17 | 8-C=CH | S | DMA | 5-Me(S) | H | H | H | H | H |
| 18 | 8-Me | O | DMA | 5-Me(S) | H | H | H | H | H |
| 19 | 8-Me | S | DMA | 5-Me(S) | H | H | H | H | H |
| 20 | 9-NO2 | O | CPM | 5-Me(S) | L | L | L | L | L |
| t 21 | 8-NH2 | O | CPM | 5-Me(S) | L | L | L | L | L |
| 22 | 8-NMe2 | O | CPM | 5-Me(S) | L | L | L | L | L |
| 23 | 9-NH2 | O | CPM | 5-Me(S) | L | L | L | L | L |
| t 24 | 9-NMe2 | O | CPM | 5-Me(S) | L | L | L | L | L |
| 25 | 9-NHCOMe | O | CPM | 5-Me(S) | L | L | L | L | L |
| t 26 | 9-NO2 | S | CPM | 5-Me(S) | L | H | L | H | H |
| 27 | 9-F | S | DMA | 5-Me(S) | H | H | H | H | H |
| 28 | 9-CF3 | O | DMA | 5-Me(S) | L | L | L | L | L |
| t 29 | 9-CF3 | S | DMA | 5-Me(S) | H | H | H | H | H |
| t 30 | 9-Me | O | DEA | 5-Me(S) | H | L | L | L | L |
| 31 | 10-OMe | O | DMA | 5-Me(S) | L | L | L | L | L |
| t 32 | 10-OMe | S | DMA | 5-Me(S) | L | H | L | H | H |
| 33 | 9,10-di-Cl | S | DMA | 5-Me(S) | H | H | H | H | H |
| 34 | 10-Br | S | DMA | 5-Me(S) | H | H | H | H | H |
| 35 | H | O | CH2CH=CH2 | 5-Me(S) | L | L | L | L | L |
| 36 | H | O | 2-MA | 5-Me(S) | L | L | L | L | L |
| 37 | H | O | CH2CO2Me | 5-Me(S) | L | L | L | L | L |
| t 38 | H | O | CH2C≡CH | 5-Me(S) | L | L | L | L | L |
| 39 | H | O | CH2-2-furanyl | 5-Me(S) | L | L | L | L | L |
| 40 | H | O | CH2CH=CH2[S(+)] | 5-Me(S) | L | L | L | L | L |
| 41 | H | O | CH2CH2CH=CH2 | 5-Me(S) | L | L | L | L | L |
| 42 | H | O | CH2CH2CH3 | 5-Me(S) | L | L | L | L | L |
| 43 | H | O | 2-MA[S(+)] | 5-Me(S) | L | L | L | L | L |
| 44 | H | O | CPM | 5-Me(S) | L | L | L | L | L |
| t 45 | H | O | CH2CH=CHMe(E) | 5-Me(S) | L | L | L | L | L |
| 46 | H | O | CH2CH=CHMe(Z) | 5-Me(S) | L | L | L | L | L |
| 47 | H | O | CH2CH2CH2Me | 5-Me(S) | L | L | L | L | L |
| 48 | H | O | DMA | 5-Me(S) | L | L | L | L | L |
| 49 | H | O | CH2C(Br)=CH2 | 5-Me(S) | L | L | L | L | L |
| 50 | H | O | CH2C(Me)=CHMe(E) | 5-Me(S) | L | L | L | L | L |
| 51 | H | O | DMA[R(+)] | 5-Me(S) | L | L | L | L | L |
| 52 | H | O | DMA[S(+)] | 5-Me(S) | L | L | L | L | L |
| t 53 | H | O | CH2C(C2H5)=CH2 | 5-Me(S) | L | L | L | L | L |
| 54 | H | O | CH2CH=CHC6H5(Z) | 5-Me(S) | L | L | L | L | L |
| 55 | H | O | CH2C(CH=CH2)=CH2 | 5-Me(S) | L | L | L | L | L |
| 56 | 8-Cl | S | DMA | H | H | H | H | H | H |
| 57 | 9-Cl | S | DMA | H | H | H | H | H | H |
| 58 | H | O | 2-MA | 5,5-di-Me | L | L | L | L | L |
| 59 | H | O | 2-MA | 4-Me | L | L | L | L | L |
| 60 | 9-Cl | S | 2-MA | 4-Me(S) | H | H | H | L | H |
| 61 | 9-Cl | S | CPM | 4-Me(R) | L | L | L | L | L |
| 62 | H | O | C3H7 | 4-CHMe2 | L | L | L | L | L |
| 63 | H | O | 2-MA | 4-CHMe2 | L | L | L | L | L |
| 64 | H | O | 2-MA | 4-C3H7 | L | L | L | L | L |
| 65 | H | O | DMA | 7-Me | L | L | L | H | L |
| t 66 | 8-Cl | O | DMA | 7-Me | H | H | H | L | H |
| t 67 | 9-Cl | O | DMA | 7-Me | H | H | H | L | H |
| 68 | H | S | C3H7 | 7-Me | L | L | L | L | L |
| 69 | H | S | DMA | 7-Me | H | H | H | H | H |
| 70 | 8-Cl | S | DMA | 7-Me | H | H | H | H | H |
| 71 | 9-Cl | S | DMA | 7-Me | H | H | H | H | H |
| 72 | H | O | DMA | 4,5-di-Me(cis) | L | L | L | L | L |
| 73 | H | S | DMA | 4,5-di-Me(cis) | L | L | L | L | L |
| t 74 | H | S | CPM | 4,5-di-Me(trans) | L | L | L | L | L |
| 75 | H | S | DMA | 4,5-di-Me(trans) | L | L | L | L | L |
| 76 | H | S | DMA | 5,7-di-Me(trans) | H | H | H | H | H |
| 77 | H | S | DMA | 5,7-di-Me(cis) | H | H | H | H | H |
| 78 | 9-Cl | O | DMA | 5,7-di-Me(R,R-trans) | H | H | H | H | H |
| 79 | 9-Cl | S | DMA | 5,7-di-Me(R,R-trans) | H | H | H | H | H |
| 80 | H | S | DMA | 4,7-di-Me(trans) | L | L | L | L | L |
| t 81 | 9-Cl | O | DMA | 5-Me(S) | H | H | H | L | L |
| 82 | 9-Cl | S | CPM | 5-Me(S) | H | H | H | H | H |
| t 83 | H | S | CPM | 5-Me(S) | H | H | L | H | L |
| 84 | H | O | C3H7 | 5-Me | L | L | L | L | L |
| 85 | H | S | C3H7 | 5-Me | L | L | L | L | L |
| 86 | H | O | 2-MA | 5-Me | L | L | L | L | L |
| 87 | H | S | DMA | 5-Me | H | H | H | H | H |
| 88 | H | O | DMA | 5-Me(S) | L | L | L | L | L |
| 89 | H | S | 2-MA | 5-Me(S) | H | H | L | H | H |
| Descriptors | Chemical Meaning |
|---|---|
| MD1 | logP: Octanol/water partition coefficient for the compound studied |
| MD2 | Average nucleophilic reaction index for a N atom |
| MD3 | Minimum total interaction for a H-N bond |
| MD4 | Minimum (>0.1) bond order of a N atom |
| MD5 | ESP-HBSA H-bonding surface area |
| MD6 | Maximum atomic state energy for a N atom |
| MD7 | 3χ: Molecular connectivity index to the third order |
| Methods | Training Set (%) | Test Set (%) | ||||
|---|---|---|---|---|---|---|
| Total Accuracy | Sn(H) | Sn(L) | Total Accuracy | Sn(H) | Sn(L) | |
| SVM | 100.00 | 100.00 | 100.00 | 85.00 | 91.67 | 75.00 |
| ANN | 98.55 | 96.43 | 100.00 | 90.00 | 83.33 | 100.00 |
| DT | 97.10 | 96.43 | 97.56 | 70.00 | 66.67 | 75.00 |
| RF | 100.00 | 100.00 | 100.00 | 75.00 | 75.00 | 75.00 |
| Method | Sets | Misclassified Compounds |
|---|---|---|
| SVM | Training set | |
| Test set | 26,30,32 | |
| ANN | Training set | 89 |
| Test set | 30,83 | |
| DT | Training set | 60,65 |
| Test set | 26,30,32,66,67,81 | |
| RF | Training set | |
| Test set | 26,30,32,81,83 |
| MD1 | MD2 | MD3 | MD4 | MD5 | MD6 | MD7 | MD8 | MD9 | MD10 |
|---|---|---|---|---|---|---|---|---|---|
| Info Gain Attribute Eval-Ranker (%) | |||||||||
| 12.96 | 0.00 | 13.88 | 6.70 | 10.20 | 22.18 | 8.80 | 8.66 | 8.80 | 7.82 |
| Gain Ratio Attribute Eval-Ranker (%) | |||||||||
| 11.44 | 0.00 | 10.55 | 6.21 | 11.06 | 18.91 | 9.78 | 14.09 | 9.60 | 8.35 |
| Symmetrical Uncert Attribute Eval-Ranker (%) | |||||||||
| 12.02 | 0.00 | 13.63 | 6.34 | 10.70 | 20.52 | 9.35 | 10.01 | 9.27 | 8.15 |
| Average (%) | |||||||||
| 12.14 | 0.00 | 12.69 | 6.42 | 10.65 | 20.54 | 9.31 | 10.92 | 9.22 | 8.11 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hdoufane, I.; Bjij, I.; Soliman, M.; Tadjer, A.; Villemin, D.; Bogdanov, J.; Cherqaoui, D. In Silico SAR Studies of HIV-1 Inhibitors. Pharmaceuticals 2018, 11, 69. https://doi.org/10.3390/ph11030069
Hdoufane I, Bjij I, Soliman M, Tadjer A, Villemin D, Bogdanov J, Cherqaoui D. In Silico SAR Studies of HIV-1 Inhibitors. Pharmaceuticals. 2018; 11(3):69. https://doi.org/10.3390/ph11030069
Chicago/Turabian StyleHdoufane, Ismail, Imane Bjij, Mahmoud Soliman, Alia Tadjer, Didier Villemin, Jane Bogdanov, and Driss Cherqaoui. 2018. "In Silico SAR Studies of HIV-1 Inhibitors" Pharmaceuticals 11, no. 3: 69. https://doi.org/10.3390/ph11030069