Mitochondrial Targeting in Neurodegeneration: A Heme Perspective


Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center, University of Torino, 10126 Torino, Italy
*
Author to whom correspondence should be addressed.
†
These authors equally contributed.
Pharmaceuticals 2018, 11(3), 87; https://doi.org/10.3390/ph11030087
Submission received: 18 July 2018
/
Revised: 7 September 2018
/
Accepted: 14 September 2018
/
Published: 18 September 2018
(This article belongs to the Special Issue Iron as Therapeutic Targets in Human Diseases)
Abstract
:Mitochondrial dysfunction has achieved an increasing interest in the field of neurodegeneration as a pathological hallmark for different disorders. The impact of mitochondria is related to a variety of mechanisms and several of them can co-exist in the same disease. The central role of mitochondria in neurodegenerative disorders has stimulated studies intended to implement therapeutic protocols based on the targeting of the distinct mitochondrial processes. The review summarizes the most relevant mechanisms by which mitochondria contribute to neurodegeneration, encompassing therapeutic approaches. Moreover, a new perspective is proposed based on the heme impact on neurodegeneration. The heme metabolism plays a central role in mitochondrial functions, and several evidences indicate that alterations of the heme metabolism are associated with neurodegenerative disorders. By reporting the body of knowledge on this topic, the review intends to stimulate future studies on the role of heme metabolism in neurodegeneration, envisioning innovative strategies in the struggle against neurodegenerative diseases.
1. Implication of Heme in Neurodegeneration
Heme is a molecule composed by protoporphyrin IX and iron produced by all the cells in the organism, including neurons. Heme mediates a series of functions that encompass oxygen transport, the regulation of gene expression and the modulation of enzyme activity, just to cite the most relevant ones. Moreover, heme can also promote oxidative stress, thus performing as a double-face molecule with both positive and negative properties [1]. This concept is also true for neuronal cells. Indeed, on one hand heme is required for the survival and differentiation of neuronal cells, as demonstrated by the observation that heme deficiency interferes with neurite outgrowth in nerve growth factor (NGF)-induced PC12 cells [2,3] and results in apoptosis in PC12 pheochromocytoma cells, SHSY5Y neuroblastoma cells and U373 astrocytoma cells, as well as in rat primary hippocampal neurons [2,3,4]. However, on the other hand, an excess of free-heme is associated with neurodegeneration. The large amount of hemoglobin and heme released in the brain during intracerebral or subarachnoid hemorrhages promotes oxidative stress, lipid peroxidation, inflammatory response and finally, neuronal cell death [5,6,7]. Moreover, loss of the heme scavenger hemopexin (Hx) causes defective myelination in mice [8,9,10]. Furthermore, impairment of cellular heme export reduces SHSY5Y cells survival [11]. Together, these data indicate that both heme deficiency and excess are deleterious for the survival of neuronal cells (Figure 1), thus suggesting that heme levels must be finely controlled both at the systemic and cellular level. At the systemic level, circulating free-heme is scavenged by the plasma proteins haptoglobin and hemopexin [6,7,8,10,12,13,14]. However, at cellular level, the amount of intracellular free-heme (labile heme or heme regulatory pool) is regulated at multiple steps: heme synthesis, incorporation into hemoproteins, catabolism, import and export [1,15]. Although this tight regulation has been extensively studied in non-neuronal cells, similar mechanisms likely occur in the nervous system. Indeed, the main proteins involved in the control of labile heme are also expressed in the nervous system [16].
Neurodegenerative disorders are a common and growing cause of mortality and morbidity worldwide [17]. Recently, a series of rare neurodegenerative disorders have been directly linked to alterations of heme metabolism (see Table 1). Defective heme synthesis causes porphyrias, some of which are associated with a wide array of neurological disturbances involving both the central and peripheral nervous systems (neuropathic porphyria). Neuropathic porphyria includes acute intermittent porphyria (AIP), hereditary coproporphyria (HCP), variegate porphyria (VP) and 5-aminolevulinate dehydratase deficiency (ALAD deficiency) [18,19,20]. Furthermore, reduced heme synthesis has been observed in Friederich Ataxia (FRDA), an autosomal recessive disorder caused by mutations in Frataxin (FXN), a mitochondrial iron chaperone involved in iron-sulfur (Fe-S) clusters and heme biosynthesis [21,22,23,24]. Finally, reduced heme synthesis has been observed during aging [4,25].
In addition, other rare neurodegenerative disorders have been associated with defective heme transport across membranes. Several proteins are involved in this process [1,15]. Among them, Feline Leukemia Virus Subgroup C Receptor 1 (FLVCR1) and 2 (FLVCR2) are implicated in heme export and import, respectively [11,26,27,28,29,30,31]. Mutations in the heme exporter FLVCR1 are associated with three distinct disorders affecting the sensory nervous system: posterior column ataxia and retinitis pigmentosa (PCARP) [32,33,34], non-syndromic retinitis pigmentosa (RP) [35,36] and hereditary sensory and autonomic neuropathy (HSAN) [11,37]. Mutations in the heme importer FLVCR2 are responsible for the Fowler syndrome, a proliferative glomerular vasculopathy [30,38,39].
Furthermore, several lines of evidence suggest that heme may also contribute to the pathogenesis of common neurodegenerative disorders. The deregulation of enzymes critically involved in heme synthesis has been reported in both Alzheimer’s disease (AD) and Parkinson’s disease (PD). Reduced 5-aminolevulinate synthase 1 (ALAS1) and porphobilinogen deaminase (PBGD) mRNA were observed in AD brains [40], suggesting decreased heme synthesis rates in AD. Moreover, heme deficiency has been reported in the brain of patients with AD [41]. It has been proposed that heme deficiency may arise from either decreased heme synthesis rates or heme depletion as a consequence of heme binding to amyloid-β [41,42,43]. However, increased Ferrochelatase (FECH) levels were reported in another study [41].
Heme binding to α-Synuclein has also been reported [44], suggesting that heme depletion may also occur in PD. In addition, blood transcriptomic meta-analysis showed downregulation of 5-aminolevulinate synthase 2 (ALAS2) and FECH in PD [45]. However, within PD erythroid cells, α-synuclein gene (SNCA) was co-expressed with crucial enzymes involved in heme metabolism, including ALAS2, FECH and biliverdin reductase B (BLVRB) [46]. Moreover, increased striatal 5-aminolevulinate dehydratase (ALAD) activity was observed in the MPTP-induced mouse model of PD, indicative of increased heme synthesis rates [47]. It is difficult to conclude from these studies whether heme synthesis is increased or reduced in AD and PD. Data are still controversial due to the low amount of human samples analyzed and the different experimental approaches adopted. Further studies are required to definitively determine heme synthesis rates in these pathological conditions. Mouse models of AD and PD will be extremely useful to analyze in detail the role of heme metabolism in these disorders.
A role for heme derived from extracellular sources in the pathogenesis of neurodegenerative disorders has also been proposed. The heme scavenger Hx has been found strongly increased in the cerebrospinal fluid of AD patients [48,49,50]. Similarly, altered expression of the hemoglobin scavenger haptoglobin was shown in AD [51,52], PD [53,54] and Huntington’s disease (HD) [55]. The induction of heme oxygenase 1 (HO1), the enzyme responsible for heme degradation, is common in patients affected by neurodegenerative conditions. Although HO1 is induced by a plethora of stimuli, it has been proposed that HO1 overexpression in AD and PD may be a consequence of increased brain-blood barrier permeability and hemoglobin-derived heme levels in AD and PD [16]. Moreover, increased expression of HO1 was reported in a mouse model of amyotrophic lateral sclerosis (ALS) [56] and ALS patients [57].
All the reported examples highlight the importance of the maintenance of heme homeostasis in the context of neurodegeneration. However, the molecular mechanisms underlying these disorders and the precise mechanism through which heme participates to them remains elusive and requires further investigations.
2. Role of Mitochondria in Neurodegenerative Diseases
Neurodegeneration can be elicited by several systems. Among them, mitochondrial-dependent processes have become increasingly relevant [58,59,60,61,62,63]. Several mechanisms account for mitochondria-dependent neurodegeneration (see Figure 2 for a graphic list of them) and below we attempt to summarize the most important ones.
2.1. Mutations on Mitochondrial DNA (mtDNA) Genes
Mitochondria contain their own genome, which is made of multiple copies of a circular double stranded molecule, which is 16.6 kb long in humans. It comprises 37 genes, 13 encoding for proteins involved in adenosine triphosphate (ATP) production and the other 24 encoding for two rRNAs and 22 tRNAs. Cells contain thousands of molecules of mtDNA and the majority of them have the same sequence, a condition known as homoplasmy. Inefficient mtDNA repair, localized oxidative environment and increased replication, however, can promote mtDNA mutations that, due to the polyploidy nature of mtDNA, often co-exist with their wildtype counterpart in various proportions (a condition termed heteroplasmy). mtDNA mutations are usually responsible for defects in the respiratory chain functions, but only if they are present above a certain threshold level.
The replication of mtDNA occurs independently on cell cycle, and a particular mtDNA molecule may be strongly replicated (or not at all) during cell division. Moreover, replication occurs also in postmitotic cells. These phenomena account for the clonal expansion of mutated mtDNA molecules and in association with heteroplasmy, result in mosaicism, with the levels of mutated mtDNA varying dramatically between tissues in the same organism and in different regions of the same tissue.
Somatic mtDNA mutations accumulate during a person’s lifetime and undergo clonal expansion, so aging is typically associated with mosaic occurrence of respiratory chain-deficient cells in tissues [64].
Mitochondrial reactive oxygen species (ROS) production is the major cause for the higher mtDNA nucleotide instability when compared with nDNA.
Mitochondrial DNA mutations potently affect tissue that require a large amount of ATP to function, such as heart and brain. Some haplogroups [65] (evolutionary selected population subgroups carrying neutral single-base pair variants of mtDNA) have been associated with susceptibility to a variety of human diseases, including age-related neurodegenerative disorders such as PD and AD. Moreover, inherited point mutations and sporadic rearrangements on mtDNA have been described in association with neurodegeneration [65,66,67,68,69].
Although the presence of mtDNA deletions below a certain threshold is not sufficient to induce PD, small changes inside the genome of mitochondria could represent a risk factor for this pathology [70,71,72,73,74]. In addition, the accumulation of mutations in mtDNA over the course of PD has been observed to correlate with severity and burden of the disease [75]. Accumulation of mtDNA damage is also considered a possible mechanism of neurodegeneration over the course of HD [76]. Moreover, mtDNA mutations have been associated with AD [77], although the degree of mtDNA damage does not seem to correlate with the severity of AD symptoms [78].
As stated before, mtDNA alterations often result in defects on a particular component of the electron transport chain (ETC). The three main mechanisms through which mtDNA damage can contribute to neurodegeneration are therefore consequences of ETC alteration and include the decrease of ATP synthesis, the increase of ROS production and the enhanced sensitivity to neurotoxins associated to ETC disruption.
2.2. Mutations on Nuclear DNA Genes Encoding Proteins Crucial for Mitochondrial Functionality
Besides mutations on mitochondrial DNA, mutations in nuclear DNA (nDNA) at the level of genes encoding for mitochondrial proteins have also been associated with neurodegenerative disorders. For example, many of the identified ALS genes have a role in mitochondrial-associated functions (see [79] for a comprehensive description on this topic). In addition, mitochondrial dysfunction and oxidative stress in PD have been linked to mutations in genes encoding for parkin RBR E3 ubiquitin protein ligase (PRKN, commonly referred to as parkin), PTEN induced putative kinase 1 (PINK1, a protein that acts in the same pathway of parkin) and parkinsonism associated deglycase (DJ-1) [80], just to cite some of them. Parkin promotes autophagy of damaged mitochondria [81] and its deficiency is associated with defects in mitochondria morphology [82] and low levels of proteins involved in mitochondrial functions, thus resulting in decreased mitochondrial respiration [83]. Similarly, mutations in PINK1 gene lead to decreased mitochondrial respiration [84] and alterations in mitochondria functions [85,86,87,88]. Regarding DJ-1, this protein localizes into mitochondria [89] and exerts crucial antioxidant functions [80,90,91,92]. Mutations in DJ-1 impair mitochondrial respiration, reduce mitochondrial membrane potential, increase ROS within the mitochondria and alter mitochondrial morphology [93]. Also, nDNA encoded proteins implicated in AD, like presenilin-1 and presenilin 2 (PSEN1 and 2) are related to mitochondria [94,95].
2.3. Alterations of Mitochondrial Dynamics (Fusion, Fission, Motility)
Mitochondria are not rigidly structured. They form a complex reticulum that undergoes regulated processes of fusion (the combination of two smaller mitochondria into a single organelle) and fission (the division of one large mitochondrion into two smaller fragments). Fusion allows mitochondria to mix their contents, enabling protein complementation, mtDNA repair and equal distribution of metabolites. Fission facilitates equal segregation of mitochondria into daughter cells during cellular division, enhances the distribution of mitochondria along cytoskeletal tracts and participates in the targeting of damaged segments of mitochondria to the autophagic process. Fusion and fission also contribute to the movement of mitochondria necessary for mitochondria distribution along neuronal axons and dendrites. Mitochondrial dynamics are crucial for neurotransmission, synaptic maintenance and neuronal survival. Proper mitochondrial trafficking is particularly important in neurons compared to other cell types, due to their exceptional cellular morphology. Indeed, neurons extend their axons and dendrites for very long distances that, in the case of human peripheral nerves or corticospinal tracts, extend up to a meter. Thus, the neuron represents an extreme case of mitochondrial distribution: dysfunctions in mitochondrial distribution that are not dangerous for other cells could be fatal for neuronal survival [96].
Alterations in mitochondria motility have been reported in several neurodegenerative disorders and neuropathies [97,98]. Aberrant activity of the fission-fusion machinery contributes to the pathogenesis of PD [99,100,101,102]. Moreover, alterations of mitochondrial dynamics have been observed in AD [61,102] and HD [100,103]. Particularly in HD, mitochondrial fission is promoted and mitochondrial fusion proteins are downregulated as the severity of the pathology increases [104]. Finally, defects in mitochondrial dynamics and disruption of the axonal transport of mitochondria have been reported in ALS [60,105,106,107].
2.4. Inappropriate Activation of Cell Apoptosis by Mitochondria
Mitochondria are pivotal organelles for the execution of apoptosis. The inappropriate activation of apoptosis leads to the disruption of the cellular proliferation-death balance. Neurodegenerative disorders are believed to partly depend on alterations of this equilibrium. PINK1 loss-of-function mutations lead to early signals for apoptosis, promoting neurodegeneration in the context of PD [108]. Moreover, the low levels of PTPA (phosphotyrosyl phosphatase activator) observed in AD affected-people contribute to induce cell apoptosis in the brain of these patients [109]. Furthermore, caspase-6, an effector of the caspase-dependent apoptotic pathway, is known to be involved in the cleavage of mutant huntingtin resulting in neurodegeneration in HD patients [110]. Finally, in ALS the mutant SOD1 can trigger cytochrome c release from mitochondria to operate apoptosis [111].
2.5. Alteration of Mitochondria-Dependent Ca2+ Homeostasis
An additional neurodegenerative mechanism related to dysfunctions of mitochondria concerns the modulation of calcium. Mitochondria are involved in Ca2+ homeostasis as they are able to both accumulate and release Ca2+. Mitochondrial Ca2+ concentration is fundamental for the regulation of specific mitochondrial key functions, such as the apoptotic process and the activity of several mitochondrial enzymes. The deregulation of Ca2+ homeostasis is a hallmark of different neurodegenerative diseases including PD, AD, HD and ALS [112,113]. Moreover, alterations of calcium levels have been observed in neuropathies. Neuropathic pain phenotypes include chemotherapy induced neuropathy, diabetic neuropathy, human immunodeficiency virus (HIV)-associated neuropathy and Charcot-Marie-Tooth neuropathy. Neuropathies have been associated with mitochondrial dysfunctions [63], and particularly in diabetic neuropathy, impaired cellular calcium homeostasis, including alterations in mitochondrial Ca2+ concentration, has been reported. Indeed, in the context of diabetes, sensory neurons, above all the lumbar dorsal root ganglia neurons (which have the longest axons), show an increased intracellular Ca2+ concentration that triggers elevated mitochondrial Ca2+ levels. This condition induces mitochondrial membrane depolarization and can favor the generation of reactive oxygen species (ROS) and oxidative stress, as well as alterations in mitochondrial functionality that can ultimately lead to neuronal damage [114].
2.6. Additional Alterations of Mitochondrial-Related Processes: Biogenesis, Mitophagy, Mdvs Exchange, Interaction with Mams, Control of Cellular Metabolism
Besides the mechanisms reported above, a series of additional mechanisms involving mitochondria have been described. Among them, neurodegeneration has been associated with the impairment of mitochondrial biogenesis. Particularly, the deficit of peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1α), a key regulator of mitochondrial biogenesis, has been associated with HD, PD and AD [115,116,117,118,119,120].
Mitophagy is also linked to neurodegenerative disorders [121,122,123]. Mitophagy is the selective autophagic process responsible for the elimination of damaged or excess mitochondria. In this process, a peculiar role is played by PINK1, that recruits parkin to dysfunctional mitochondria, where it induces their degradation by mitophagy [124]. Thus, it is not surprising that in addition to other neurodegenerative disorders, defective mitophagy is highly implicated in PD and is considered one of the major pathological mechanisms of mitochondrial dysfunction in autosomal recessive forms of PD [125].
Mitophagy is a cellular process that eliminates whole mitochondria, but other mechanisms exist to partially eliminate portions or components of mitochondria [122]. Mitochondria-derived vesicles (MDVs) exchange between mitochondria and peroxisomes represent one of these possible mechanisms. MDVs are crucial for the transport of cargo from mitochondria to peroxisomes. This process is regulated by the retromer complex. Studies on vacuolar protein sorting 35 (VPS35), a component of the retromer complex, indicate that mutations or alterations in VPS35 expression are associated to PD [126,127]. Moreover, it has been demonstrated that Parkin and PINK1, two genes highly implicated in PD, play crucial roles in the control of this process [128,129].
Other than with peroxisomes, mitochondria also physically interact with other subcellular organelles to ensure efficient and rapid metabolism and signaling. For example, the interaction between mitochondria and the endoplasmic reticulum (ER) occurs at the level of MAMs (mitochondrial associated membranes), a subdomain of the ER. The proteins involved in neurodegenerative diseases such as DRP1 (dynamin related protein 1) and MFN2 (mitofusin 2) are enriched in MAMs [130,131] and the perturbation of mitochondria-ER contacts has been described in neurodegenerative disorders, including PD, AD and ALS [132].
A further mitochondrial-dependent mechanism has been highlighted in neurodegeneration. This is the impairment of cell metabolism [133]. Mitochondria are the main energy-producing organelles of the cell, thus any process impairing mitochondrial functionality may lead to metabolic switching aimed to compensate for their decreased ATP production. Among the different neurodegenerative disorders, many lines of evidence suggest that mitochondria-dependent mechanisms are responsible for the metabolic changes observed in dopaminergic neurons in the context of PD. Mitochondrial ROS are particularly abundant in dopaminergic neurons due to dopamine oxidative metabolism, enhanced Fenton’s reaction initiated by the high iron content of these cells, and the high rate of ATP production required to sustain the activity of a particular L-type voltage dependent Ca2+ channel expressed by these neurons. An excess of mitochondrial ROS can induce a series of cellular modifications, including hypoxia inducible factor 1α (HIF1α)-dependent up-regulation of glucose transporters [134], favoring the switch of energy metabolism towards glycolysis. When sustained for a long period, this metabolic change can be deleterious for dopaminergic neurons. Indeed, neurons need to deliver glucose in the pentose phosphate pathway (PPP), a process that produces NADPH (nicotinamide adenine dinucleotide phosphate), crucial for the recycling of the antioxidant glutathione. PPP is especially important in neurons as these cells show less robust antioxidant systems and are more vulnerable to oxidative stress than other cell types. Switching from PPP to glycolysis promotes oxidative stress and, consequently, neurodegeneration [135].
3. Heme and Mitochondrial Dysfunction Related to Neurodegenerative Diseases
The examples reported above sustain the notion that mitochondrial dysfunctions play a critical role in neurodegenerative disorders. The mitochondrion is a critical organelle for cells, representing a crossroad for a plethora of reactions contributing to a variety of metabolic processes, including heme metabolism. Considering the impact of impaired heme homeostasis in several neurodegenerative disorders and the interesting potential that heme takes on for the research in the context of neurodegeneration, it is curious to notice that heme is an under-investigated molecule in the field. This discrepancy could be due to the lack of knowledge on the possible systems through which heme can influence pivotal processes implicated in neurodegeneration. Among the possible ways, it is tempting to speculate that heme could directly or indirectly affect many of the mitochondrial-dependent mechanisms of neurodegeneration described in the previous paragraphs. Indeed, the relationship between heme and mitochondria is based on several elements (Figure 3): heme is produced through a series of reactions that occur partly in the mitochondria and partly in the cytosol [136]; heme acts as a cofactor for cytochromes c and cytochromes in complexes II-III-IV of the mitochondrial ETC [137]; heme has been reported to directly or indirectly influence ATP translocation between mitochondria and cytosol [138,139,140] mediated by adenine nucleotide translocases (ANTs); finally, heme biosynthesis is considered a cataplerotic pathway for the Kreb’s cycle due to the fact that the first step of heme production consumes succynil-CoA [141,142]. Thus, modulation of heme homeostasis can affect mitochondrial functions.
Considering the mechanisms by which mitochondria contribute to neurodegeneration, it is interesting to note that heme levels can influence iron homeostasis, and both iron deficiency and iron excess are reported to cause damage on mitochondria [143,144] and on mitochondrial DNA [145,146]. Moreover, a decrease in heme itself leads to mitochondrial decay [4,147].
Furthermore, a connection exists between heme and nDNA genes encoding mitochondrial proteins typically implicated in neurodegenerative disorders. Indeed, it has been demonstrated that amyloid precursor protein, particularly when mutated, interacts and negatively regulates the heme-degrading enzyme HO1 [148]. Also PINK1 mutation is related to alterations in HO1 expression [149]. Moreover, DJ1 regulates nuclear factor-E2-related factor 2 (NRF2) [150], a key transcription factor for the induction of HO1 expression [151].
In addition, heme and the heme-degrading enzyme HO1 are implicated in the regulation of mitophagy, mitochondrial biogenesis and morphology [152,153,154].
In endothelial cells, it has been demonstrated that alterations in heme metabolism, in addition to promoting lipid peroxidation and activation of autophagy, induce mitophagy and apoptosis, indicating mitochondrial dysfunction [155]. Similarly, FLVCR1 loss is associated with alterations in mitochondrial morphology in human microvascular endothelial cells [26].
Furthermore, FLVCR1-deficient HeLa cells show impaired mitochondrial calcium uptake [156].
These data have been obtained in non-neuronal cells. However, similar mechanisms could also occur in neuronal cells.
Finally, compromised ETC complexes activity has been observed in the brain of a mouse model for acute intermittent porphyria, a type of porphyric neuropathy caused by alterations of heme biosynthesis [157]. Moreover, in three cases of Fowler syndrome it was suggested to be the presence of a defect in complex III and IV of the ETC [158,159].
These examples directly suggest that a connection between heme-mitochondria-neurodegeneration exists and open the possibility that future studies on this topic will further strengthen this notion.
4. Current Therapies and Potential Future Approaches to Face Mitochondrial Dysfunction in Neurodegenerative Diseases
Currently, there is no cure for reversing neurodegeneration and the treatment of neurodegenerative disorders is mainly symptomatic [160]. In order to face neurodegeneration, several pathways could be targeted to improve and/or restore mitochondrial functions, including mitochondrial biogenesis and metabolic flexibility, mitochondrial dynamics and mitophagy [161].
There are several pharmacological approaches to induce mitochondrial biogenesis and metabolic flexibility, that is the ability to switch from one fuel source to another. Several compounds have been generated to target the upstream sensors of energy production, including AMP-activated protein kinase (AMPK), mammalian target of rapamycin (mTOR) and sirtuins, or downstream transcriptional factors and co-factors, such as nuclear receptors, nuclear respiratory factor 1 (NRF1) and mitochondrial transcription factor A (TFAM) [161]. The therapeutic potential of these drugs has been evaluated mostly in the context of metabolic diseases, but also seems promising for neurodegeneration. For example, resveratrol is a natural compound that mimics caloric restriction and activates the sirtuin family of histone deacetylases. In humans, resveratrol improves mitochondrial function in obese patients and type 2 diabetes [162]. Furthermore, resveratrol counteracts neurodegeneration in worms and mice [163].
As described above, the disruption of the balance between mitochondrial fusion and fission contributes to neurodegeneration. Therefore, targeting mitochondrial dynamics represents another important strategy to improve mitochondrial function in neurodegenerative diseases. Strategies aimed at increasing mitochondrial fusion or inhibiting mitochondrial fission might improve mitochondrial function and are therapeutically interesting. The promotion of mitochondrial fusion by the overexpression of key components of the fusion machinery, like MFN2 or OPA1, rescues ATP production and mitochondrial morphology in a cellular model of PD [164]. Similar results were obtained with the inhibition of fission through the genetic deletion of DRP1 [164,165]. Although the understanding of the regulation of mitochondrial dynamics is still in its infancy, novel compounds have been identified to promote fusion or inhibit fission [166]. M1 hydrazone [167] and S3-derivative [168] promote mitochondrial fusion in cells deficient for mitofusin 1 (MFN1) and MFN2. Mdivi-1 (mitochondrial division inhibitor) attenuates fission in yeast and mammalian cells by inhibiting DRP1. In vitro, Mdivi-1 delays apoptosis by inhibiting mitochondrial outer membrane permeabilization and blocking cytochrome c release from mitochondria [169]. The therapeutic potential of Mdivi-1 seems promising for neurodegenerative disorders. Indeed, the administration of Mdivi-1 in mouse and cellular models of PD attenuates disease-associated phenotypes [165,170]. Although initially reported as an inhibitor of fission, Mdivi-1 was recently reported to reversibly inhibit complex I in a DRP1-independent manner [171]. The complete inhibition of complex I in vivo would be expected to cause neurodegeneration. Indeed, rotenone completely inhibits complex I, induces ROS levels and causes parkinsonian neurodegeneration in mice [172]. In contrast, Mdivi-1 lacks neuronal toxicity in vivo and is neuroprotective. This is likely due to the ability of Mdivi-1 to attenuate complex I-dependent reverse electron transfer (RET)-mediated ROS production. Indeed, Mdivi-1 fails to increase ROS levels in intact neurons and in isolated brain mitochondria [171].
The accumulation of dysfunctional mitochondria is another key event in several neurodegenerative conditions [173,174]. In this context, mitophagy is essential for the maintenance of mitochondrial integrity. As stated before in this review, the impairment of autophagy/mitophagy is common in neurodegenerative disorders. Therefore, mitophagy may be an additional pathway amenable for therapeutic intervention to ameliorate mitochondria function and counteract neurodegeneration [161,175]. Interestingly, both genetic and pharmacological induction of the mitochondrial autophagy receptor Nip3-like protein X (NIX) restores mitophagy in patient-derived fibroblasts [176].
Considering the crucial role of heme in maintaining mitochondrial function, it is tempting to speculate that targeting heme metabolism might be a promising strategy for the treatment of neurodegenerative diseases. Multiple approaches can be used to target heme metabolism (Figure 3); theoretically, targeting any of the enzymes involved in the heme biosynthetic pathway or proteins involved in the control of the intracellular heme pool may be a good strategy. Among these methods, HO1 represents a potentially interesting target. Due to its anti-oxidant and anti-inflammatory properties, HO1 plays a well-established neuroprotective role. The improvement of HO1 expression has been initially proposed for neurodegenerative conditions [177]. However, it has been reported that the overexpression of HO1 induces oxidative mitochondrial damage [178,179] and macroautophagy [179] in cultured astroglia. More importantly, HO1 induction has been associated with the later phases of neurodegeneration [180] and the deletion of HO1 has been proposed as a therapeutic option [181]. HO1 activity can be suppressed by synthetic metalloporphyrin compounds that unfortunately present important limitations [182]. However, novel HO1 inhibitors have been synthesized to overcome these side effects. Interestingly, these inhibitors confer neuroprotection in a mouse model of AD [182]. Considering the complex role of HO1 in neurodegeneration and the still controversial data reported in literature [180], further work is needed to fully elucidate the therapeutic potential of HO1 targeting. Recently, long-term 5–aminolevulinic acid (ALA) treatment has been exploited as a therapeutic approach in a mouse model of AD. Omori C. et al. reported that the oral administration of ALA increased cytochrome c oxidase (COX) activity and protein expression as well as mitochondrial membrane potential in the brain of treated mice [183]. Additional studies are required to understand more in detail the functional consequences of ALA administration in AD pathogenesis and the translation of this therapeutic approach to other neurodegenerative disorders. Considering that ALA formulations are already used for photodynamic therapy in a variety of cancer types [184], results obtained by Omori C. et al. are extremely encouraging for therapeutic purposes and further research in this direction is desirable.
5. Conclusions
The information reported over the course of the present review showed that mitochondria participate to neurodegenerative disorders by different mechanisms, encompassing DNA mutations, mitobiogenesis, mitophagy, mitochondrial dynamics, metabolism and mitochondrial interactions with other organelles. The literary contributions on the role of mitochondria in neurodegeneration are constantly growing and the present review attempted to make an excursus on the most important mechanisms by which these crucial organelles contribute to neurodegenerative diseases, with the awareness that not all the wide literature on this topic has been covered. Some of these mechanisms are currently considered as strategic targets for pharmacological interventions to counteract neurodegeneration. However, the investigation on additional elements contributing to the control of mitochondrial functions in neuronal cells will offer a wider window of intervention. In this perspective, heme metabolism provides an interesting opportunity. Indeed, as reported above in the review, heme participates to crucial processes occurring in mitochondria, influencing their functions and properties. The tight relationship between heme and the mitochondrion has been curiously underestimated and poorly investigated in the context of neurodegeneration. However, the implication of heme in crucial mitochondrial functions and the involvement of heme in a subset of neurodegenerative diseases strongly suggest an implication of heme in these disorders.
The comprehension of these mechanisms will allow the consideration of possible therapies based on the targeting of heme metabolism as an additional option to promote mitochondrial function. Alternatively, targeting heme metabolism may improve the efficacy of other drugs targeting mitochondria. The hope is that the understanding of the role of heme in mitochondria and its implication in neurodegeneration will open new perspectives in the struggle against neurodegenerative diseases.
Funding
This research was funded by University of Torino (Bando Ricerca Locale ex-60%).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Chiabrando, D.; Vinchi, F.; Fiorito, V.; Mercurio, S.; Tolosano, E. Heme in pathophysiology: A matter of scavenging, metabolism and trafficking across cell membranes. Front. Pharmacol. 2014, 5, 61. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Hon, T.; Ye, W.; Zhang, L. Heme deficiency interferes with the Ras-mitogen-activated protein kinase signaling pathway and expression of a subset of neuronal genes. Cell Growth Differ. 2002, 13, 431–439. [Google Scholar] [PubMed]
- Sengupta, A.; Hon, T.; Zhang, L. Heme deficiency suppresses the expression of key neuronal genes and causes neuronal cell death. Brain Res. Mol. Brain Res. 2005, 137, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Atamna, H.; Killilea, D.W.; Killilea, A.N.; Ames, B.N. Heme deficiency may be a factor in the mitochondrial and neuronal decay of aging. Proc. Natl. Acad. Sci. USA 2002, 99, 14807–14812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Righy, C.; Bozza, M.T.; Oliveira, M.F.; Bozza, F.A. Molecular, Cellular and Clinical Aspects of Intracerebral Hemorrhage: Are the Enemies Within? Curr. Neuropharmacol. 2016, 14, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Day, J.P.; Phillips, H.; Slootsky, B.; Tolosano, E.; Doré, S. Deletion of the hemopexin or heme oxygenase-2 gene aggravates brain injury following stroma-free hemoglobin-induced intracerebral hemorrhage. J. Neuroinflamm. 2016, 13, 26. [Google Scholar] [CrossRef] [PubMed]
- Hahl, P.; Davis, T.; Washburn, C.; Rogers, J.T.; Smith, A. Mechanisms of neuroprotection by hemopexin: Modeling the control of heme and iron homeostasis in brain neurons in inflammatory states. J. Neurochem. 2013, 125, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Morello, N.; Tonoli, E.; Logrand, F.; Fiorito, V.; Fagoonee, S.; Turco, E.; Silengo, L.; Vercelli, A.; Altruda, F.; Tolosano, E. Haemopexin affects iron distribution and ferritin expression in mouse brain. J. Cell. Mol. Med. 2009, 13, 4192–4204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morello, N.; Bianchi, F.T.; Marmiroli, P.; Tonoli, E.; Rodriguez Menendez, V.; Silengo, L.; Cavaletti, G.; Vercelli, A.; Altruda, F.; Tolosano, E. A role for hemopexin in oligodendrocyte differentiation and myelin formation. PLoS ONE 2011, 6, e20173. [Google Scholar] [CrossRef] [PubMed]
- Tolosano, E.; Fagoonee, S.; Morello, N.; Vinchi, F.; Fiorito, V. Heme scavenging and the other facets of hemopexin. Antioxid. Redox Signal. 2010, 12, 305–320. [Google Scholar] [CrossRef] [PubMed]
- Chiabrando, D.; Castori, M.; di Rocco, M.; Ungelenk, M.; Gießelmann, S.; Di Capua, M.; Madeo, A.; Grammatico, P.; Bartsch, S.; Hübner, C.A.; et al. Mutations in the Heme Exporter FLVCR1 Cause Sensory Neurodegeneration with Loss of Pain Perception. PLoS Genet. 2016, 12, e1006461. [Google Scholar] [CrossRef] [PubMed]
- Chiabrando, D.; Vinchi, F.; Fiorito, V.; Tolosano, E. Haptoglobin and Hemopexin in Heme Detoxification and Iron Recycling. In Acute Phase Proteins Francisco Veas; IntechOpen: London, UK, 2011; pp. 262–288. [Google Scholar]
- Blackburn, S.L.; Kumar, P.T.; McBride, D.; Zeineddine, H.A.; Leclerc, J.; Choi, H.A.; Dash, P.K.; Grotta, J.; Aronowski, J.; Cardenas, J.C.; et al. Unique Contribution of Haptoglobin and Haptoglobin Genotype in Aneurysmal Subarachnoid Hemorrhage. Front. Physiol. 2018, 9, 592. [Google Scholar] [CrossRef] [PubMed]
- Marro, S.; Barisani, D.; Chiabrando, D.; Fagoonee, S.; Muckenthaler, M.U.; Stolte, J.; Meneveri, R.; Haile, D.; Silengo, L.; Altruda, F.; et al. Lack of haptoglobin affects iron transport across duodenum by modulating ferroportin expression. Gastroenterology 2007, 133, 1261–1271. [Google Scholar] [CrossRef] [PubMed]
- Reddi, A.R.; Hamza, I. Heme Mobilization in Animals: A Metallolipid’s Journey. Acc. Chem. Res. 2016, 49, 1104–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gozzelino, R. The Pathophysiology of Heme in the Brain. Curr. Alzheimer Res. 2016, 13, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Erkkinen, M.G.; Kim, M.O.; Geschwind, M.D. Clinical Neurology and Epidemiology of the Major Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2018, 10, a033118. [Google Scholar] [CrossRef] [PubMed]
- Tracy, J.A.; Dyck, P.J. Porphyria and its neurologic manifestations. Handb. Clin. Neurol. 2014, 120, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Albers, J.W.; Fink, J.K. Porphyric neuropathy. Muscle Nerve 2004, 30, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Simon, N.G.; Herkes, G.K. The neurologic manifestations of the acute porphyrias. J. Clin. Neurosci. 2011, 18, 1147–1153. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, V.; Montermini, L.; Moltò, M.D.; Pianese, L.; Cossée, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef] [PubMed]
- Yoon, T.; Cowan, J.A. Frataxin-mediated iron delivery to ferrochelatase in the final step of heme biosynthesis. J. Biol. Chem. 2004, 279, 25943–25946. [Google Scholar] [CrossRef] [PubMed]
- Lange, H.; Mühlenhoff, U.; Denzel, M.; Kispal, G.; Lill, R. The heme synthesis defect of mutants impaired in mitochondrial iron-sulfur protein biogenesis is caused by reversible inhibition of ferrochelatase. J. Biol. Chem. 2004, 279, 29101–29108. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, R.A.; Napoli, E.; Wong, A.; Zhan, S.; Reutenauer, L.; Morin, D.; Buckpitt, A.R.; Taroni, F.; Lonnerdal, B.; Ristow, M.; et al. Frataxin deficiency alters heme pathway transcripts and decreases mitochondrial heme metabolites in mammalian cells. Hum. Mol. Genet. 2005, 14, 3787–3799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chernova, T.; Nicotera, P.; Smith, A.G. Heme deficiency is associated with senescence and causes suppression of N-methyl-d-aspartate receptor subunits expression in primary cortical neurons. Mol. Pharmacol. 2006, 69, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Petrillo, S.; Chiabrando, D.; Genova, T.; Fiorito, V.; Ingoglia, G.; Vinchi, F.; Mussano, F.; Carossa, S.; Silengo, L.; Altruda, F.; et al. Heme accumulation in endothelial cells impairs angiogenesis by triggering paraptosis. Cell Death Differ. 2018, 25, 573–588. [Google Scholar] [CrossRef] [PubMed]
- Vinchi, F.; Ingoglia, G.; Chiabrando, D.; Mercurio, S.; Turco, E.; Silengo, L.; Altruda, F.; Tolosano, E. Heme exporter FLVCR1a regulates heme synthesis and degradation and controls activity of cytochromes P450. Gastroenterology 2014, 146, 1325–1338. [Google Scholar] [CrossRef] [PubMed]
- Fiorito, V.; Neri, F.; Pala, V.; Silengo, L.; Oliviero, S.; Altruda, F.; Tolosano, E. Hypoxia controls Flvcr1 gene expression in Caco2 cells through HIF2α and ETS1. Biochim. Biophys. Acta 2014, 1839, 259–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorito, V.; Forni, M.; Silengo, L.; Altruda, F.; Tolosano, E. Crucial Role of FLVCR1a in the Maintenance of Intestinal Heme Homeostasis. Antioxid. Redox Signal. 2015, 23, 1410–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffy, S.P.; Shing, J.; Saraon, P.; Berger, L.C.; Eiden, M.V.; Wilde, A.; Tailor, C.S. The Fowler syndrome-associated protein FLVCR2 is an importer of heme. Mol. Cell. Biol. 2010, 30, 5318–5324. [Google Scholar] [CrossRef] [PubMed]
- Quigley, J.G.; Yang, Z.; Worthington, M.T.; Phillips, J.D.; Sabo, K.M.; Sabath, D.E.; Berg, C.L.; Sassa, S.; Wood, B.L.; Abkowitz, J.L. Identification of a human heme exporter that is essential for erythropoiesis. Cell 2004, 118, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Rajadhyaksha, A.M.; Elemento, O.; Puffenberger, E.G.; Schierberl, K.C.; Xiang, J.Z.; Putorti, M.L.; Berciano, J.; Poulin, C.; Brais, B.; Michaelides, M.; et al. Mutations in FLVCR1 cause posterior column ataxia and retinitis pigmentosa. Am. J. Hum. Genet. 2010, 87, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Ishiura, H.; Fukuda, Y.; Mitsui, J.; Nakahara, Y.; Ahsan, B.; Takahashi, Y.; Ichikawa, Y.; Goto, J.; Sakai, T.; Tsuji, S. Posterior column ataxia with retinitis pigmentosa in a Japanese family with a novel mutation in FLVCR1. Neurogenetics 2011, 12, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Shaibani, A.; Wong, L.J.; Wei Zhang, V.; Lewis, R.A.; Shinawi, M. Autosomal recessive posterior column ataxia with retinitis pigmentosa caused by novel mutations in the FLVCR1 gene. Int. J. Neurosci. 2015, 125, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A.; Bahr, A.; Bähr, L.; Fleischhauer, J.; Zinkernagel, M.S.; Winkler, N.; Barthelmes, D.; Berger, L.; Gerth-Kahlert, C.; Neidhardt, J.; et al. Next generation sequencing based identification of disease-associated mutations in Swiss patients with retinal dystrophies. Sci. Rep. 2016, 6, 28755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yusuf, I.H.; Shanks, M.E.; Clouston, P.; MacLaren, R.E. A splice-site variant in FLVCR1 produces retinitis pigmentosa without posterior column ataxia. Ophthalmic Genet. 2018, 39, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Castori, M.; Morlino, S.; Ungelenk, M.; Pareyson, D.; Salsano, E.; Grammatico, P.; Tolosano, E.; Kurth, I.; Chiabrando, D. Posterior column ataxia with retinitis pigmentosa coexisting with sensory-autonomic neuropathy and leukemia due to the homozygous p.Pro221Ser FLVCR1 mutation. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2017, 174, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Meyer, E.; Ricketts, C.; Morgan, N.V.; Morris, M.R.; Pasha, S.; Tee, L.J.; Rahman, F.; Bazin, A.; Bessières, B.; Déchelotte, P.; et al. Mutations in FLVCR2 are associated with proliferative vasculopathy and hydranencephaly-hydrocephaly syndrome (Fowler syndrome). Am. J. Hum. Genet. 2010, 86, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Kvarnung, M.; Taylan, F.; Nilsson, D.; Albåge, M.; Nordenskjöld, M.; Anderlid, B.M.; Nordgren, A.; Syk Lundberg, E. Mutations in FLVCR2 associated with Fowler syndrome and survival beyond infancy. Clin. Genet. 2016, 89, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, B.E.; Smith, M.A.; Richardson, S.L.; Perry, G.; Zhu, X. Down-regulation of aminolevulinate synthase, the rate-limiting enzyme for heme biosynthesis in Alzheimer’s disease. Neurosci. Lett. 2009, 460, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Atamna, H.; Frey, W.H. A role for heme in Alzheimer’s disease: Heme binds amyloid beta and has altered metabolism. Proc. Natl. Acad. Sci. USA 2004, 101, 11153–11158. [Google Scholar] [CrossRef] [PubMed]
- Atamna, H. Heme binding to Amyloid-beta peptide: Mechanistic role in Alzheimer’s disease. J. Alzheimers Dis. 2006, 10, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.G.; Raven, E.L.; Chernova, T. The regulatory role of heme in neurons. Metallomics 2011, 3, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Hayden, E.Y.; Kaur, P.; Williams, T.L.; Matsui, H.; Yeh, S.R.; Rousseau, D.L. Heme Stabilization of α-Synuclein Oligomers during Amyloid Fibril Formation. Biochemistry 2015, 54, 4599–4610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santiago, J.A.; Potashkin, J.A. Blood Transcriptomic Meta-analysis Identifies Dysregulation of Hemoglobin and Iron Metabolism in Parkinson’ Disease. Front. Aging Neurosci. 2017, 9, 73. [Google Scholar] [CrossRef] [PubMed]
- Scherzer, C.R.; Grass, J.A.; Liao, Z.; Pepivani, I.; Zheng, B.; Eklund, A.C.; Ney, P.A.; Ng, J.; McGoldrick, M.; Mollenhauer, B.; et al. GATA transcription factors directly regulate the Parkinson’s disease-linked gene alpha-synuclein. Proc. Natl. Acad. Sci. USA 2008, 105, 10907–10912. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, T.B.; Marcondes Sari, M.H.; Pesarico, A.P.; Nogueira, C.W. δ-Aminolevulinate Dehydratase Activity is Stimulated in a MPTP Mouse Model of Parkinson’s Disease: Correlation with Myeloperoxidase Activity. Cell. Mol. Neurobiol. 2017, 37, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Ringman, J.M.; Schulman, H.; Becker, C.; Jones, T.; Bai, Y.; Immermann, F.; Cole, G.; Sokolow, S.; Gylys, K.; Geschwind, D.H.; et al. Proteomic changes in cerebrospinal fluid of presymptomatic and affected persons carrying familial Alzheimer disease mutations. Arch. Neurol. 2012, 69, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Roher, A.E.; Maarouf, C.L.; Sue, L.I.; Hu, Y.; Wilson, J.; Beach, T.G. Proteomics-derived cerebrospinal fluid markers of autopsy-confirmed Alzheimer’s disease. Biomarkers 2009, 14, 493–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castaño, E.M.; Roher, A.E.; Esh, C.L.; Kokjohn, T.A.; Beach, T. Comparative proteomics of cerebrospinal fluid in neuropathologically-confirmed Alzheimer’s disease and non-demented elderly subjects. Neurol. Res. 2006, 28, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Song, I.U.; Kim, Y.D.; Chung, S.W.; Cho, H.J. Association between serum haptoglobin and the pathogenesis of Alzheimer’s disease. Intern. Med. 2015, 54, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Cocciolo, A.; Di Domenico, F.; Coccia, R.; Fiorini, A.; Cai, J.; Pierce, W.M.; Mecocci, P.; Butterfield, D.A.; Perluigi, M. Decreased expression and increased oxidation of plasma haptoglobin in Alzheimer disease: Insights from redox proteomics. Free Radic. Biol. Med. 2012, 53, 1868–1876. [Google Scholar] [CrossRef] [PubMed]
- de Farias, C.C.; Maes, M.; Bonifacio, K.L.; Matsumoto, A.K.; Bortolasci, C.C.; Nogueira, A.S.; Brinholi, F.F.; Morimoto, H.K.; de Melo, L.B.; Moreira, E.G.; et al. Parkinson’s Disease is Accompanied by Intertwined Alterations in Iron Metabolism and Activated Immune-inflammatory and Oxidative Stress Pathways. CNS Neurol. Disord. Drug Targets 2017, 16, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Argüelles, S.; Venero, J.L.; García-Rodriguez, S.; Tomas-Camardiel, M.; Ayala, A.; Cano, J.; Machado, A. Use of haptoglobin and transthyretin as potential biomarkers for the preclinical diagnosis of Parkinson’s disease. Neurochem. Int. 2010, 57, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.C.; Wu, Y.R.; Tseng, M.Y.; Chen, Y.C.; Hsieh, S.Y.; Chen, C.M. Increased prothrombin, apolipoprotein A-IV, and haptoglobin in the cerebrospinal fluid of patients with Huntington’s disease. PLoS ONE 2011, 6, e15809. [Google Scholar] [CrossRef] [PubMed]
- Gajowiak, A.; Styś, A.; Starzyński, R.R.; Bednarz, A.; Lenartowicz, M.; Staroń, R.; Lipiński, P. Mice Overexpressing Both Non-Mutated Human SOD1 and Mutated SOD1(G93A) Genes: A Competent Experimental Model for Studying Iron Metabolism in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2015, 8, 82. [Google Scholar] [CrossRef] [PubMed]
- Calingasan, N.Y.; Chen, J.; Kiaei, M.; Beal, M.F. Beta-amyloid 42 accumulation in the lumbar spinal cord motor neurons of amyotrophic lateral sclerosis patients. Neurobiol. Dis. 2005, 19, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Arun, S.; Liu, L.; Donmez, G. Mitochondrial Biology and Neurological Diseases. Curr. Neuropharmacol. 2016, 14, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Bose, A.; Beal, M.F. Mitochondrial dysfunction in Parkinson’s disease. J. Neurochem. 2016, 139 (Suppl. 1), 216–231. [Google Scholar] [CrossRef]
- Shi, P.; Gal, J.; Kwinter, D.M.; Liu, X.; Zhu, H. Mitochondrial dysfunction in amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2010, 1802, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Cabezas-Opazo, F.A.; Vergara-Pulgar, K.; Pérez, M.J.; Jara, C.; Osorio-Fuentealba, C.; Quintanilla, R.A. Mitochondrial Dysfunction Contributes to the Pathogenesis of Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2015, 2015, 509654. [Google Scholar] [CrossRef] [PubMed]
- Costa, V.; Scorrano, L. Shaping the role of mitochondria in the pathogenesis of Huntington’s disease. EMBO J. 2012, 31, 1853–1864. [Google Scholar] [CrossRef] [PubMed]
- Bennett, G.J.; Doyle, T.; Salvemini, D. Mitotoxicity in distal symmetrical sensory peripheral neuropathies. Nat. Rev. Neurol. 2014, 10, 326–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsson, N.G. Somatic mitochondrial DNA mutations in mammalian aging. Annu. Rev. Biochem. 2010, 79, 683–706. [Google Scholar] [CrossRef] [PubMed]
- Chinnery, P.F.; Hudson, G. Mitochondrial genetics. Br. Med. Bull. 2013, 106, 135–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keogh, M.J.; Chinnery, P.F. Mitochondrial DNA mutations in neurodegeneration. Biochim. Biophys. Acta 2015, 1847, 1401–1411. [Google Scholar] [CrossRef] [PubMed]
- Cha, M.Y.; Kim, D.K.; Mook-Jung, I. The role of mitochondrial DNA mutation on neurodegenerative diseases. Exp. Mol. Med. 2015, 47, e150. [Google Scholar] [CrossRef] [PubMed]
- Nissanka, N.; Moraes, C.T. Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease. FEBS Lett. 2018, 592, 728–742. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.; Moraes, C.T. Mitochondrial genome changes and neurodegenerative diseases. Biochim. Biophys. Acta 2014, 1842, 1198–1207. [Google Scholar] [CrossRef] [PubMed]
- Kraytsberg, Y.; Kudryavtseva, E.; McKee, A.C.; Geula, C.; Kowall, N.W.; Khrapko, K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat. Genet. 2006, 38, 518–520. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T.; et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006, 38, 515–517. [Google Scholar] [CrossRef] [PubMed]
- Ikebe, S.; Tanaka, M.; Ozawa, T. Point mutations of mitochondrial genome in Parkinson’s disease. Brain Res. Mol. Brain Res. 1995, 28, 281–295. [Google Scholar] [CrossRef]
- Perier, C.; Bender, A.; García-Arumí, E.; Melià, M.J.; Bové, J.; Laub, C.; Klopstock, T.; Elstner, M.; Mounsey, R.B.; Teismann, P.; et al. Accumulation of mitochondrial DNA deletions within dopaminergic neurons triggers neuroprotective mechanisms. Brain 2013, 136, 2369–2378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, Y.; Clark, J.; Zheng, K.; Kujoth, G.C.; Prolla, T.A.; Simon, D.K. Somatic mitochondrial DNA mutations do not increase neuronal vulnerability to MPTP in young POLG mutator mice. Neurotoxicol. Teratol. 2014, 46, 62–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parkinson, G.M.; Dayas, C.V.; Smith, D.W. Increased mitochondrial DNA deletions in substantia nigra dopamine neurons of the aged rat. Curr. Aging Sci. 2014, 7, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, A.; Rivera-Sánchez, S.; Castro, M.E.R.; Acevedo-Torres, K.; Rane, A.; Torres-Ramos, C.A.; Nicholls, D.G.; Andersen, J.K.; Ayala-Torres, S. Mitochondrial DNA damage is associated with reduced mitochondrial bioenergetics in Huntington’s disease. Free Radic. Biol. Med. 2012, 53, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Phillips, N.R.; Simpkins, J.W.; Roby, R.K. Mitochondrial DNA deletions in Alzheimer’s brains: A review. Alzheimers Dement. 2014, 10, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Gerschütz, A.; Heinsen, H.; Grünblatt, E.; Wagner, A.K.; Bartl, J.; Meissner, C.; Fallgatter, A.J.; Al-Sarraj, S.; Troakes, C.; Ferrer, I.; et al. Neuron-specific mitochondrial DNA deletion levels in sporadic Alzheimer’s disease. Curr. Alzheimer Res. 2013, 10, 1041–1046. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2017. [Google Scholar] [CrossRef] [PubMed]
- Bekris, L.M.; Mata, I.F.; Zabetian, C.P. The genetics of Parkinson disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 228–242. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greene, J.C.; Whitworth, A.J.; Kuo, I.; Andrews, L.A.; Feany, M.B.; Pallanck, L.J. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc. Natl. Acad. Sci. USA 2003, 100, 4078–4083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palacino, J.J.; Sagi, D.; Goldberg, M.S.; Krauss, S.; Motz, C.; Wacker, M.; Klose, J.; Shen, J. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J. Biol. Chem. 2004, 279, 18614–18622. [Google Scholar] [CrossRef] [PubMed]
- Gautier, C.A.; Kitada, T.; Shen, J. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc. Natl. Acad. Sci. USA 2008, 105, 11364–11369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heeman, B.; Van den Haute, C.; Aelvoet, S.A.; Valsecchi, F.; Rodenburg, R.J.; Reumers, V.; Debyser, Z.; Callewaert, G.; Koopman, W.J.; Willems, P.H.; et al. Depletion of PINK1 affects mitochondrial metabolism, calcium homeostasis and energy maintenance. J. Cell Sci. 2011, 124, 1115–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, I.E.; Dodson, M.W.; Jiang, C.; Cao, J.H.; Huh, J.R.; Seol, J.H.; Yoo, S.J.; Hay, B.A.; Guo, M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 2006, 441, 1162–1166. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lee, S.B.; Lee, S.; Kim, Y.; Song, S.; Kim, S.; Bae, E.; Kim, J.; Shong, M.; Kim, J.M.; et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 2006, 441, 1157–1161. [Google Scholar] [CrossRef] [PubMed]
- Truban, D.; Hou, X.; Caulfield, T.R.; Fiesel, F.C.; Springer, W. PINK1, Parkin, and Mitochondrial Quality Control: What can we Learn about Parkinson’s Disease Pathobiology? J. Parkinsons Dis. 2017, 7, 13–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Shimoji, M.; Thomas, B.; Moore, D.J.; Yu, S.W.; Marupudi, N.I.; Torp, R.; Torgner, I.A.; Ottersen, O.P.; Dawson, T.M.; et al. Mitochondrial localization of the Parkinson’s disease related protein DJ-1: Implications for pathogenesis. Hum. Mol. Genet. 2005, 14, 2063–2073. [Google Scholar] [CrossRef] [PubMed]
- Taira, T.; Saito, Y.; Niki, T.; Iguchi-Ariga, S.M.; Takahashi, K.; Ariga, H. DJ-1 has a role in antioxidative stress to prevent cell death. EMBO Rep. 2004, 5, 213–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Freed, C.R. DJ-1 up-regulates glutathione synthesis during oxidative stress and inhibits A53T alpha-synuclein toxicity. J. Biol. Chem. 2005, 280, 43150–43158. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Sugawara, K.; Ito, K.; Takahashi, R.; Ariga, H.; Mizusawa, H. Down regulation of DJ-1 enhances cell death by oxidative stress, ER stress, and proteasome inhibition. Biochem. Biophys. Res. Commun. 2003, 312, 1342–1348. [Google Scholar] [CrossRef] [PubMed]
- Krebiehl, G.; Ruckerbauer, S.; Burbulla, L.F.; Kieper, N.; Maurer, B.; Waak, J.; Wolburg, H.; Gizatullina, Z.; Gellerich, F.N.; Woitalla, D.; et al. Reduced basal autophagy and impaired mitochondrial dynamics due to loss of Parkinson’s disease-associated protein DJ-1. PLoS ONE 2010, 5, e9367. [Google Scholar] [CrossRef] [PubMed]
- Ankarcrona, M.; Hultenby, K. Presenilin-1 is located in rat mitochondria. Biochem. Biophys. Res. Commun. 2002, 295, 766–770. [Google Scholar] [CrossRef]
- Area-Gomez, E.; de Groof, A.J.; Boldogh, I.; Bird, T.D.; Gibson, G.E.; Koehler, C.M.; Yu, W.H.; Duff, K.E.; Yaffe, M.P.; Pon, L.A.; et al. Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am. J. Pathol. 2009, 175, 1810–1816. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, T.L. Mitochondrial trafficking in neurons. Cold Spring Harb. Perspect. Biol. 2013, 5, a011304. [Google Scholar] [CrossRef] [PubMed]
- Burté, F.; Carelli, V.; Chinnery, P.F.; Yu-Wai-Man, P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat. Rev. Neurol. 2015, 11, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, L. Mitochondrial quality control in neurodegenerative diseases. Biochimie 2014, 100, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Ryan, B.J.; Hoek, S.; Fon, E.A.; Wade-Martins, R. Mitochondrial dysfunction and mitophagy in Parkinson’s: From familial to sporadic disease. Trends Biochem. Sci. 2015, 40, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Franco-Iborra, S.; Vila, M.; Perier, C. Mitochondrial Quality Control in Neurodegenerative Diseases: Focus on Parkinson’s Disease and Huntington’s Disease. Front. Neurosci. 2018, 12, 342. [Google Scholar] [CrossRef] [PubMed]
- Van Laar, V.S.; Berman, S.B. The interplay of neuronal mitochondrial dynamics and bioenergetics: Implications for Parkinson’s disease. Neurobiol. Dis. 2013, 51, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.H.; Wang, X.; Zhu, X. Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic. Biol. Med. 2013, 62, 90–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmo, C.; Naia, L.; Lopes, C.; Rego, A.C. Mitochondrial Dysfunction in Huntington’s Disease. Adv. Exp. Med. Biol. 2018, 1049, 59–83. [Google Scholar] [CrossRef] [PubMed]
- Shirendeb, U.; Reddy, A.P.; Manczak, M.; Calkins, M.J.; Mao, P.; Tagle, D.A.; Reddy, P.H. Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington’s disease: Implications for selective neuronal damage. Hum. Mol. Genet. 2011, 20, 1438–1455. [Google Scholar] [CrossRef] [PubMed]
- De Vos, K.J.; Chapman, A.L.; Tennant, M.E.; Manser, C.; Tudor, E.L.; Lau, K.F.; Brownlees, J.; Ackerley, S.; Shaw, P.J.; McLoughlin, D.M.; et al. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum. Mol. Genet. 2007, 16, 2720–2728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Wang, L.; Liu, J.; Xie, F.; Su, B.; Wang, X. Abnormalities of Mitochondrial Dynamics in Neurodegenerative Diseases. Antioxidants 2017, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Carrì, M.T.; Cozzolino, M. SOD1 and mitochondria in ALS: A dangerous liaison. J. Bioenerg. Biomembr. 2011, 43, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Wood-Kaczmar, A.; Gandhi, S.; Yao, Z.; Abramov, A.Y.; Abramov, A.S.; Miljan, E.A.; Keen, G.; Stanyer, L.; Hargreaves, I.; Klupsch, K.; et al. PINK1 is necessary for long term survival and mitochondrial function in human dopaminergic neurons. PLoS ONE 2008, 3, e2455. [Google Scholar] [CrossRef]
- Luo, D.J.; Feng, Q.; Wang, Z.H.; Sun, D.S.; Wang, Q.; Wang, J.Z.; Liu, G.P. Knockdown of phosphotyrosyl phosphatase activator induces apoptosis via mitochondrial pathway and the attenuation by simultaneous tau hyperphosphorylation. J. Neurochem. 2014, 130, 816–825. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.J.; Cao, Q.; Zhang, Y.; Su, X.D. Activation and regulation of caspase-6 and its role in neurodegenerative diseases. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 553–572. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H.; Kobayashi, Y.; Ishigaki, S.; Doyu, M.; Sobue, G. Mitochondrial localization of mutant superoxide dismutase 1 triggers caspase-dependent cell death in a cellular model of familial amyotrophic lateral sclerosis. J. Biol. Chem. 2002, 277, 50966–50972. [Google Scholar] [CrossRef] [PubMed]
- Calì, T.; Ottolini, D.; Brini, M. Mitochondrial Ca2+ and neurodegeneration. Cell Calcium 2012, 52, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Pelizzoni, I.; Macco, R.; Zacchetti, D.; Grohovaz, F.; Codazzi, F. Iron and calcium in the central nervous system: A close relationship in health and sickness. Biochem. Soc. Trans. 2008, 36, 1309–1312. [Google Scholar] [CrossRef] [PubMed]
- Fernyhough, P.; Roy Chowdhury, S.K.; Schmidt, R.E. Mitochondrial stress and the pathogenesis of diabetic neuropathy. Expert Rev. Endocrinol. Metab. 2010, 5, 39–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, A.W.; Yao, Z.; Vicencio, J.M.; Karkucinska-Wieckowska, A.; Szabadkai, G. PGC-1 family coactivators and cell fate: Roles in cancer, neurodegeneration, cardiovascular disease and retrograde mitochondria-nucleus signalling. Mitochondrion 2012, 12, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Weydt, P.; Pineda, V.V.; Torrence, A.E.; Libby, R.T.; Satterfield, T.F.; Lazarowski, E.R.; Gilbert, M.L.; Morton, G.J.; Bammler, T.K.; Strand, A.D.; et al. Thermoregulatory and metabolic defects in Huntington’s disease transgenic mice implicate PGC-1alpha in Huntington’s disease neurodegeneration. Cell Metab. 2006, 4, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Jeong, H.; Borovecki, F.; Parkhurst, C.N.; Tanese, N.; Krainc, D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 2006, 127, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Haroutunian, V.; Katsel, P.; Cardozo, C.P.; Ho, L.; Buxbaum, J.D.; Pasinetti, G.M. PGC-1alpha expression decreases in the Alzheimer disease brain as a function of dementia. Arch. Neurol. 2009, 66, 352–361. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jäger, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Liao, Z.; Locascio, J.J.; Lesniak, K.A.; Roderick, S.S.; Watt, M.L.; Eklund, A.C.; Zhang-James, Y.; Kim, P.D.; Hauser, M.A.; et al. PGC-1α, a potential therapeutic target for early intervention in Parkinson’s disease. Sci. Transl. Med. 2010, 2, 52ra73. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.T. Mechanisms of selective autophagy and mitophagy: Implications for neurodegenerative diseases. Neurobiol. Dis. 2018. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Vicente, M. Neuronal Mitophagy in Neurodegenerative Diseases. Front. Mol. Neurosci. 2017, 10, 64. [Google Scholar] [CrossRef] [PubMed]
- Rodolfo, C.; Campello, S.; Cecconi, F. Mitophagy in neurodegenerative diseases. Neurochem. Int. 2018, 117, 156–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deak, M.; et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012, 2, 120080. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.E.; Paek, S.H. Mitochondrial Dysfunction in Parkinson’s Disease. Exp. Neurobiol. 2015, 24, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Vilariño-Güell, C.; Wider, C.; Ross, O.A.; Dachsel, J.C.; Kachergus, J.M.; Lincoln, S.J.; Soto-Ortolaza, A.I.; Cobb, S.A.; Wilhoite, G.J.; Bacon, J.A.; et al. VPS35 mutations in Parkinson disease. Am. J. Hum. Genet. 2011, 89, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Zimprich, A.; Benet-Pagès, A.; Struhal, W.; Graf, E.; Eck, S.H.; Offman, M.N.; Haubenberger, D.; Spielberger, S.; Schulte, E.C.; Lichtner, P.; et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am. J. Hum. Genet. 2011, 89, 168–175. [Google Scholar] [CrossRef] [PubMed]
- McLelland, G.L.; Soubannier, V.; Chen, C.X.; McBride, H.M.; Fon, E.A. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 2014, 33, 282–295. [Google Scholar] [CrossRef] [PubMed]
- Shlevkov, E.; Schwarz, T.L. Have you seen? For parkin, it’s not all or nothing. EMBO J. 2014, 33, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER tubules mark sites of mitochondrial division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef] [PubMed]
- de Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhu, X. Endoplasmic reticulum-mitochondria tethering in neurodegenerative diseases. Transl. Neurodegener. 2017, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Camandola, S.; Mattson, M.P. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017, 36, 1474–1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Requejo-Aguilar, R.; Lopez-Fabuel, I.; Fernandez, E.; Martins, L.M.; Almeida, A.; Bolaños, J.P. PINK1 deficiency sustains cell proliferation by reprogramming glucose metabolism through HIF1. Nat. Commun. 2014, 5, 4514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrero-Mendez, A.; Almeida, A.; Fernández, E.; Maestre, C.; Moncada, S.; Bolaños, J.P. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat. Cell Biol. 2009, 11, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Chiabrando, D.; Mercurio, S.; Tolosano, E. Heme and erythropoieis: More than a structural role. Haematologica 2014, 99, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Khalimonchuk, O.; Smith, P.M.; Winge, D.R. Structure, function, and assembly of heme centers in mitochondrial respiratory complexes. Biochim. Biophys. Acta 2012, 1823, 1604–1616. [Google Scholar] [CrossRef] [PubMed]
- Sabová, L.; Zeman, I.; Supek, F.; Kolarov, J. Transcriptional control of AAC3 gene encoding mitochondrial ADP/ATP translocator in Saccharomyces cerevisiae by oxygen, heme and ROX1 factor. Eur. J. Biochem. 1993, 213, 547–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giraud, S.; Bonod-Bidaud, C.; Wesolowski-Louvel, M.; Stepien, G. Expression of human ANT2 gene in highly proliferative cells: GRBOX, a new transcriptional element, is involved in the regulation of glycolytic ATP import into mitochondria. J. Mol. Biol. 1998, 281, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Azuma, M.; Kabe, Y.; Kuramori, C.; Kondo, M.; Yamaguchi, Y.; Handa, H. Adenine nucleotide translocator transports haem precursors into mitochondria. PLoS ONE 2008, 3, e3070. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C.; Zheng, L.; Folger, O.; Rajagopalan, K.N.; MacKenzie, E.D.; Jerby, L.; Micaroni, M.; Chaneton, B.; Adam, J.; Hedley, A.; et al. Haem oxygenase is synthetically lethal with the tumour suppressor fumarate hydratase. Nature 2011, 477, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, Y.; Wang, Y.; Lian, S.; Lynch, J.; Nagai, S.; Fanshawe, B.; Kandilci, A.; Janke, L.J.; Neale, G.; Fan, Y.; et al. Upregulated heme biosynthesis, an exploitable vulnerability in MYCN-driven leukemogenesis. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, B.T.; Manz, D.H.; Torti, F.M.; Torti, S.V. Mitochondria and Iron: Current questions. Expert Rev. Hematol. 2017, 10, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, M.P.; Greenamyre, J.T. Mitochondrial iron metabolism and its role in neurodegeneration. J. Alzheimers Dis. 2010, 20 (Suppl. 2), S551–S568. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.B.; Knutson, M.D.; Paler-Martinez, A.; Lee, S.; Xu, Y.; Viteri, F.E.; Ames, B.N. Iron deficiency and iron excess damage mitochondria and mitochondrial DNA in rats. Proc. Natl. Acad. Sci. USA 2002, 99, 2264–2269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Campian, J.L.; Qian, M.; Sun, X.F.; Eaton, J.W. Mitochondrial DNA damage in iron overload. J. Biol. Chem. 2009, 284, 4767–4775. [Google Scholar] [CrossRef] [PubMed]
- Atamna, H.; Newberry, J.; Erlitzki, R.; Schultz, C.S.; Ames, B.N. Biotin deficiency inhibits heme synthesis and impairs mitochondria in human lung fibroblasts. J. Nutr. 2007, 137, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Doré, S.; Ferris, C.D.; Tomita, T.; Sawa, A.; Wolosker, H.; Borchelt, D.R.; Iwatsubo, T.; Kim, S.H.; Thinakaran, G.; et al. Amyloid precursor proteins inhibit heme oxygenase activity and augment neurotoxicity in Alzheimer’s disease. Neuron 2000, 28, 461–473. [Google Scholar] [CrossRef]
- Sheng, X.J.; Tu, H.J.; Chien, W.L.; Kang, K.H.; Lu, D.H.; Liou, H.H.; Lee, M.J.; Fu, W.M. Antagonism of proteasome inhibitor-induced heme oxygenase-1 expression by PINK1 mutation. PLoS ONE 2017, 12, e0183076. [Google Scholar] [CrossRef] [PubMed]
- Clements, C.M.; McNally, R.S.; Conti, B.J.; Mak, T.W.; Ting, J.P. DJ-1, a cancer- and Parkinson’s disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proc. Natl. Acad. Sci. USA 2006, 103, 15091–15096. [Google Scholar] [CrossRef] [PubMed]
- Alam, J.; Stewart, D.; Touchard, C.; Boinapally, S.; Choi, A.M.; Cook, J.L. Nrf2, a Cap’n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J. Biol. Chem. 1999, 274, 26071–26078. [Google Scholar] [CrossRef] [PubMed]
- Hull, T.D.; Boddu, R.; Guo, L.; Tisher, C.C.; Traylor, A.M.; Patel, B.; Joseph, R.; Prabhu, S.D.; Suliman, H.B.; Piantadosi, C.A.; et al. Heme oxygenase-1 regulates mitochondrial quality control in the heart. JCI Insight 2016, 1, e85817. [Google Scholar] [CrossRef] [PubMed]
- Atamna, H.; Walter, P.B.; Ames, B.N. The role of heme and iron-sulfur clusters in mitochondrial biogenesis, maintenance, and decay with age. Arch. Biochem. Biophys. 2002, 397, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Suliman, H.B.; Keenan, J.E.; Piantadosi, C.A. Mitochondrial quality-control dysregulation in conditional HO-1. JCI Insight 2017, 2, e89676. [Google Scholar] [CrossRef] [PubMed]
- Higdon, A.N.; Benavides, G.A.; Chacko, B.K.; Ouyang, X.; Johnson, M.S.; Landar, A.; Zhang, J.; Darley-Usmar, V.M. Hemin causes mitochondrial dysfunction in endothelial cells through promoting lipid peroxidation: The protective role of autophagy. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1394–H1409. [Google Scholar] [CrossRef] [PubMed]
- Chiabrando, D.; Marro, S.; Mercurio, S.; Giorgi, C.; Petrillo, S.; Vinchi, F.; Fiorito, V.; Fagoonee, S.; Camporeale, A.; Turco, E.; et al. The mitochondrial heme exporter FLVCR1b mediates erythroid differentiation. J. Clin. Investig. 2012, 122, 4569–4579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homedan, C.; Laafi, J.; Schmitt, C.; Gueguen, N.; Lefebvre, T.; Karim, Z.; Desquiret-Dumas, V.; Wetterwald, C.; Deybach, J.C.; Gouya, L.; et al. Acute intermittent porphyria causes hepatic mitochondrial energetic failure in a mouse model. Int. J. Biochem. Cell Biol. 2014, 51, 93–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro-Gago, M.; Alonso, A.; Pintos-Martínez, E.; Beiras-Iglesias, A.; Campos, Y.; Arenas, J.; Novo-Rodríguez, M.I.; Eirís-Puñal, J. Congenital hydranencephalic-hydrocephalic syndrome associated with mitochondrial dysfunction. J. Child Neurol. 1999, 14, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Castro-Gago, M.; Pintos-Martínez, E.; Forteza-Vila, J.; Iglesias-Diz, M.; Ucieda-Somoza, R.; Silva-Villar, I.; Codesido-López, J.; Viso-Lorenzo, A.; Campos, Y.; Arenas, J.; et al. Congenital hydranencephalic-hydrocephalic syndrome with proliferative vasculopathy: A possible relation with mitochondrial dysfunction. J. Child Neurol. 2001, 16, 858–862. [Google Scholar] [CrossRef] [PubMed]
- Kiaei, M. New hopes and challenges for treatment of neurodegenerative disorders: Great opportunities for young neuroscientists. Basic Clin. Neurosci. 2013, 4, 3–4. [Google Scholar] [PubMed]
- Andreux, P.A.; Houtkooper, R.H.; Auwerx, J. Pharmacological approaches to restore mitochondrial function. Nat. Rev. Drug Discov. 2013, 12, 465–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timmers, S.; Konings, E.; Bilet, L.; Houtkooper, R.H.; van de Weijer, T.; Goossens, G.H.; Hoeks, J.; van der Krieken, S.; Ryu, D.; Kersten, S.; et al. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab. 2011, 14, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Douglas, P.M.; Dillin, A. Protein homeostasis and aging in neurodegeneration. J. Cell Biol. 2010, 190, 719–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutz, A.K.; Exner, N.; Fett, M.E.; Schlehe, J.S.; Kloos, K.; Lämmermann, K.; Brunner, B.; Kurz-Drexler, A.; Vogel, F.; Reichert, A.S.; et al. Loss of parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. J. Biol. Chem. 2009, 284, 22938–22951. [Google Scholar] [CrossRef] [PubMed]
- Rappold, P.M.; Cui, M.; Grima, J.C.; Fan, R.Z.; de Mesy-Bentley, K.L.; Chen, L.; Zhuang, X.; Bowers, W.J.; Tieu, K. Drp1 inhibition attenuates neurotoxicity and dopamine release deficits in vivo. Nat. Commun. 2014, 5, 5244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lackner, L.L.; Nunnari, J. Small molecule inhibitors of mitochondrial division: Tools that translate basic biological research into medicine. Chem. Biol. 2010, 17, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, J.; Bonamy, G.M.; Meeusen, S.; Brusch, R.G.; Turk, C.; Yang, P.; Schultz, P.G. A small molecule promotes mitochondrial fusion in mammalian cells. Angew. Chem. Int. Ed. Engl. 2012, 51, 9302–9305. [Google Scholar] [CrossRef] [PubMed]
- Yue, W.; Chen, Z.; Liu, H.; Yan, C.; Chen, M.; Feng, D.; Wu, H.; Du, L.; Wang, Y.; Liu, J.; et al. A small natural molecule promotes mitochondrial fusion through inhibition of the deubiquitinase USP30. Cell Res. 2014, 24, 482–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassidy-Stone, A.; Chipuk, J.E.; Ingerman, E.; Song, C.; Yoo, C.; Kuwana, T.; Kurth, M.J.; Shaw, J.T.; Hinshaw, J.E.; Green, D.R.; et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell 2008, 14, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Tang, X.; Christian, W.V.; Yoon, Y.; Tieu, K. Perturbations in mitochondrial dynamics induced by human mutant PINK1 can be rescued by the mitochondrial division inhibitor mdivi-1. J. Biol. Chem. 2010, 285, 11740–11752. [Google Scholar] [CrossRef] [PubMed]
- Bordt, E.A.; Clerc, P.; Roelofs, B.A.; Saladino, A.J.; Tretter, L.; Adam-Vizi, V.; Cherok, E.; Khalil, A.; Yadava, N.; Ge, S.X.; et al. The Putative Drp1 Inhibitor mdivi-1 Is a Reversible Mitochondrial Complex I Inhibitor that Modulates Reactive Oxygen Species. Dev. Cell 2017, 40, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Sherer, T.B.; Betarbet, R.; Testa, C.M.; Seo, B.B.; Richardson, J.R.; Kim, J.H.; Miller, G.W.; Yagi, T.; Matsuno-Yagi, A.; Greenamyre, J.T. Mechanism of toxicity in rotenone models of Parkinson’s disease. J. Neurosci. 2003, 23, 10756–10764. [Google Scholar] [CrossRef] [PubMed]
- Lezi, E.; Swerdlow, R.H. Mitochondria in neurodegeneration. Adv. Exp. Med. Biol. 2012, 942, 269–286. [Google Scholar] [CrossRef] [PubMed]
- Witte, M.E.; Mahad, D.J.; Lassmann, H.; van Horssen, J. Mitochondrial dysfunction contributes to neurodegeneration in multiple sclerosis. Trends Mol. Med. 2014, 20, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011, 333, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Koentjoro, B.; Park, J.S.; Sue, C.M. Nix restores mitophagy and mitochondrial function to protect against PINK1/Parkin-related Parkinson’s disease. Sci. Rep. 2017, 7, 44373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J. Heme oxygenase in neuroprotection: From mechanisms to therapeutic implications. Rev. Neurosci. 2014, 25, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Su, H.; Song, S.; Paudel, H.K.; Schipper, H.M. Over-expression of heme oxygenase-1 promotes oxidative mitochondrial damage in rat astroglia. J. Cell Physiol. 2006, 206, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Zukor, H.; Song, W.; Liberman, A.; Mui, J.; Vali, H.; Fillebeen, C.; Pantopoulos, K.; Wu, T.D.; Guerquin-Kern, J.L.; Schipper, H.M. HO-1-mediated macroautophagy: A mechanism for unregulated iron deposition in aging and degenerating neural tissues. J. Neurochem. 2009, 109, 776–791. [Google Scholar] [CrossRef] [PubMed]
- Nitti, M.; Piras, S.; Brondolo, L.; Marinari, U.M.; Pronzato, M.A.; Furfaro, A.L. Heme Oxygenase 1 in the Nervous System: Does It Favor Neuronal Cell Survival or Induce Neurodegeneration? Int. J. Mol. Sci. 2018, 19, 2260. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M.; Gupta, A.; Szarek, W.A. Suppression of glial HO-1 activity as a potential neurotherapeutic intervention in AD. Curr. Alzheimer Res. 2009, 6, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Lacoste, B.; Pistell, P.J.; Pistel, P.J.; Ingram, D.K.; Hamel, E.; Alaoui-Jamali, M.A.; Szarek, W.A.; Vlahakis, J.Z.; Jie, S.; et al. Neurotherapeutic effects of novel HO-1 inhibitors in vitro and in a transgenic mouse model of Alzheimer’s disease. J. Neurochem. 2014, 131, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Omori, C.; Motodate, R.; Shiraki, Y.; Chiba, K.; Sobu, Y.; Kimura, A.; Nakaya, T.; Kondo, H.; Kurumiya, S.; Tanaka, T.; et al. Facilitation of brain mitochondrial activity by 5-aminolevulinic acid in a mouse model of Alzheimer’s disease. Nutr. Neurosci. 2017, 20, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.T.P.C.; Neves, M.G.P.M.; Cavaleiro, J.A.S. Cancer, Photodynamic Therapy and Porphyrin-Type Derivatives. An. Acad. Bras. Cienc. 2018, 90, 993–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Implication of heme in neurodegeneration. Both heme excess and heme deficiency contribute to neurodegeneration. Heme released during hemorrhages leads to inflammation, lipid peroxidation and oxidative stress; the loss of the heme scavenger Hx causes defective myelination of axons; the impairment of intracellular heme export by FLVCR1a is associated with increased oxidative stress. On the other hand, heme deficiency, due to defective synthesis, leads to mitochondrial decay and the blocking of neurite growth. These events all result in neuronal cell death. In the figure, neurons are represented as the main target for heme-mediated effects; however, other cell types of the nervous system could be affected by the same phenomena.
Figure 1.
Implication of heme in neurodegeneration. Both heme excess and heme deficiency contribute to neurodegeneration. Heme released during hemorrhages leads to inflammation, lipid peroxidation and oxidative stress; the loss of the heme scavenger Hx causes defective myelination of axons; the impairment of intracellular heme export by FLVCR1a is associated with increased oxidative stress. On the other hand, heme deficiency, due to defective synthesis, leads to mitochondrial decay and the blocking of neurite growth. These events all result in neuronal cell death. In the figure, neurons are represented as the main target for heme-mediated effects; however, other cell types of the nervous system could be affected by the same phenomena.

Figure 2.
Mitochondrial dependent mechanisms in neurodegeneration. Mitochondria contribute to neurodegeneration by several mechanisms, including alterations in calcium homeostasis, mitochondrial biogenesis (mitobiogenesis), mitochondrial dynamics, metabolism and mitophagy. Moreover, mutations in mitochondrial DNA (mtDNA) and inappropriate activation of apoptosis can be alternative mechanisms. Finally, additional systems include mutations in nuclear DNA (nDNA) at the level of genes encoding for mitochondrial proteins, the compromised exchange of mitochondria-derived vesicles (MDVs) among mitochondria and peroxisomes and the inefficient interaction among mitochondria and the endoplasmic reticulum at the level of mitochondrial associated membranes (MAMs).
Figure 2.
Mitochondrial dependent mechanisms in neurodegeneration. Mitochondria contribute to neurodegeneration by several mechanisms, including alterations in calcium homeostasis, mitochondrial biogenesis (mitobiogenesis), mitochondrial dynamics, metabolism and mitophagy. Moreover, mutations in mitochondrial DNA (mtDNA) and inappropriate activation of apoptosis can be alternative mechanisms. Finally, additional systems include mutations in nuclear DNA (nDNA) at the level of genes encoding for mitochondrial proteins, the compromised exchange of mitochondria-derived vesicles (MDVs) among mitochondria and peroxisomes and the inefficient interaction among mitochondria and the endoplasmic reticulum at the level of mitochondrial associated membranes (MAMs).

Figure 3.
The “heme-mitochondria” relationship and the putative heme-related targets for the therapy of neurodegenerative disorders. Heme and mitochondria share a strong relationship based on several elements: heme synthesis occurs partly in the mitochondrion and acts as a cataplerotic pathway for the Kreb’s cycle; heme is a cofactor for cytochromes c and cytochromes in complexes II-III-IV of the mitochondrial ETC [137]; heme influences the ATP translocation between mitochondria and cytosol mediated by adenine nucleotide translocases (ANTs); heme export influences calcium (Ca2+) flux in mitochondria. Therefore, modulation of heme metabolism can lead to modification of mitochondrial functions. The control of intracellular heme levels is achieved by a balance among synthesis, catabolism and proper trafficking of heme. Thus, all these processes (highlighted with red boxes in the figure) represent putative good targets for the therapy of neurodegenerative disorder.
Figure 3.
The “heme-mitochondria” relationship and the putative heme-related targets for the therapy of neurodegenerative disorders. Heme and mitochondria share a strong relationship based on several elements: heme synthesis occurs partly in the mitochondrion and acts as a cataplerotic pathway for the Kreb’s cycle; heme is a cofactor for cytochromes c and cytochromes in complexes II-III-IV of the mitochondrial ETC [137]; heme influences the ATP translocation between mitochondria and cytosol mediated by adenine nucleotide translocases (ANTs); heme export influences calcium (Ca2+) flux in mitochondria. Therefore, modulation of heme metabolism can lead to modification of mitochondrial functions. The control of intracellular heme levels is achieved by a balance among synthesis, catabolism and proper trafficking of heme. Thus, all these processes (highlighted with red boxes in the figure) represent putative good targets for the therapy of neurodegenerative disorder.


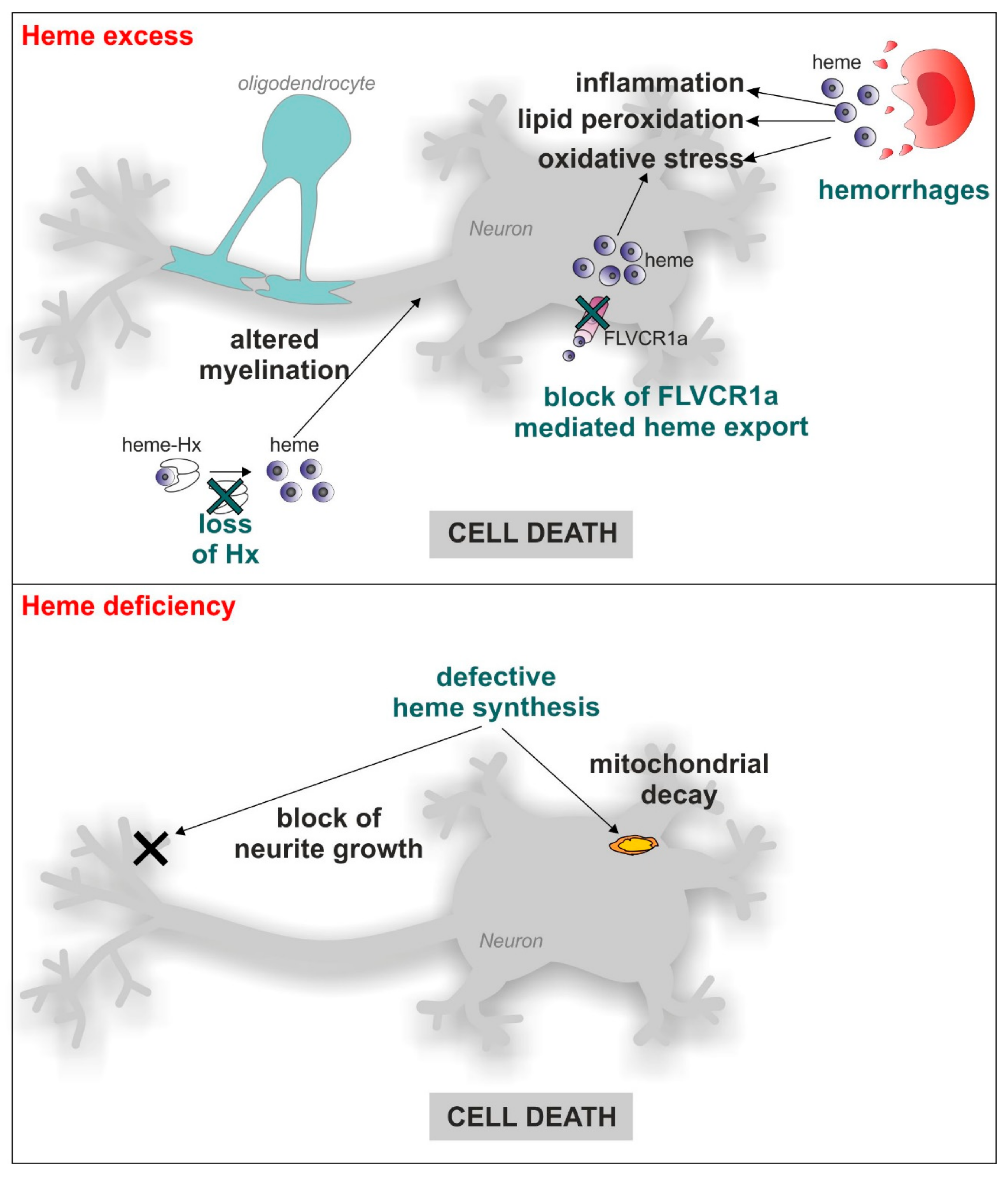{kind=link}
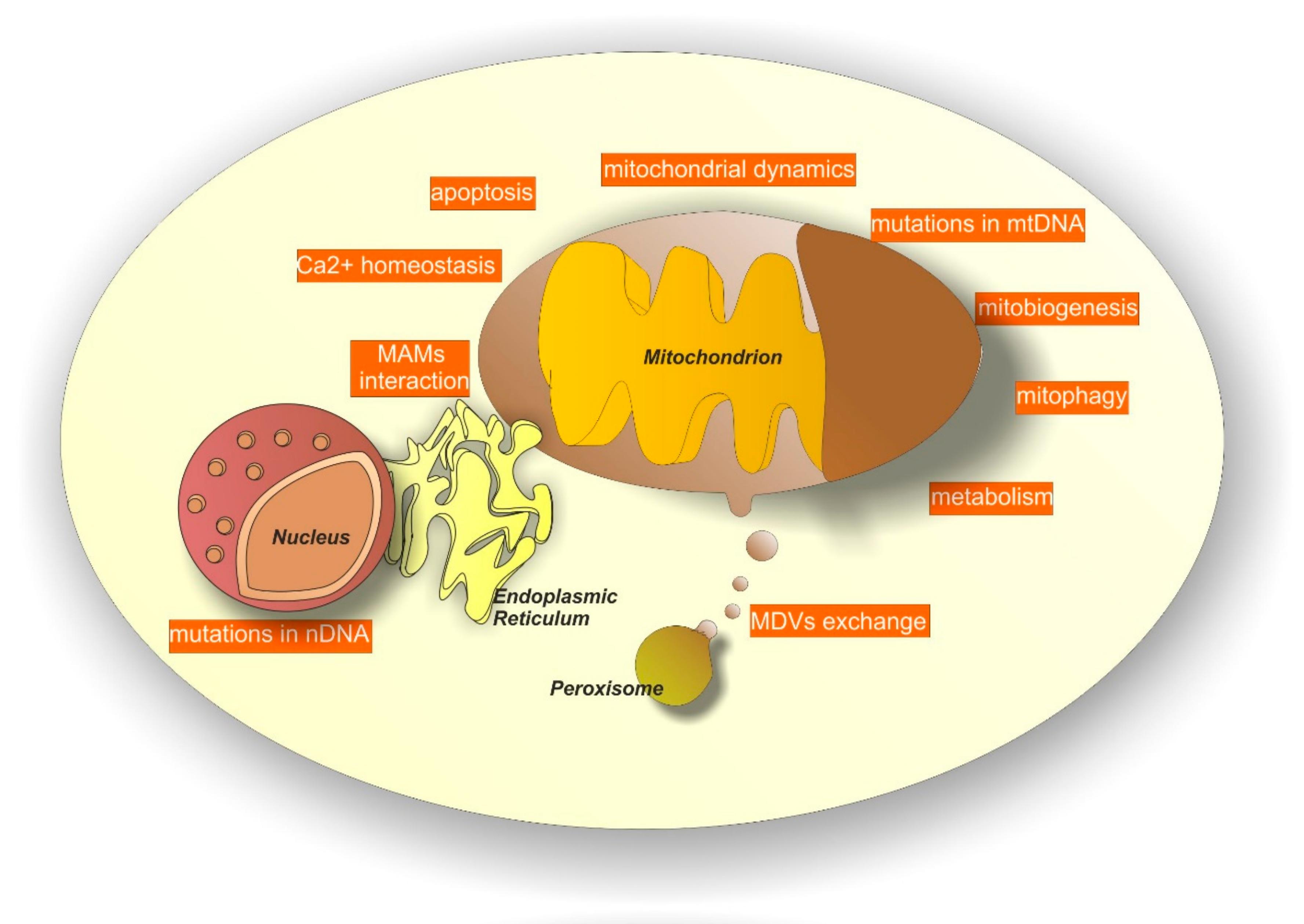{kind=link}
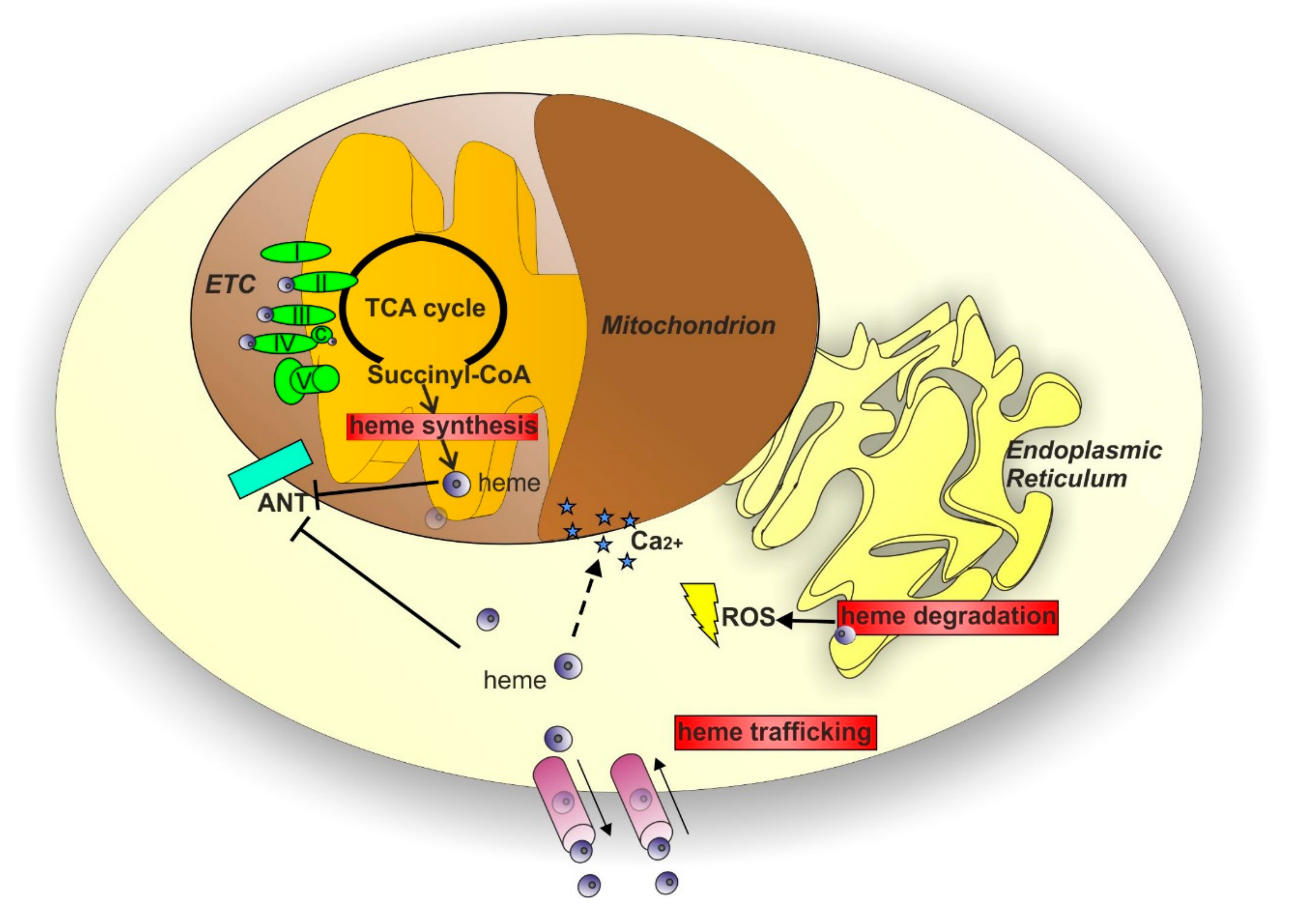{kind=link}
Table 1.
Rare neurodegenerative disorders linked to defective heme metabolism.
| Disease | Gene | Inheritance | Clinical Features | OMIM |
|---|---|---|---|---|
| ALAD deficiency | 5-aminolevulinate dehydratase (ALAD) | autosomal recessive | Neuropathic Porphyria: acute neurovisceral attacks involving severe abdominal pain, peripheral neuropathies and psychiatric disturbances | 612740 |
| Acute intermittent porphyria (AIP) | Hydroxymethylbilane synthase (HMBS) | autosomal dominant | 176000 | |
| Hereditary coproporphyria (HCP) | Coproporphyrinogen oxidase (CPOX) | autosomal dominant | 121300 | |
| Variegate porphyria (VP) | Protoporphyrinogen oxidase (PPOX) | autosomal dominant | 176200 | |
| Friederich Ataxia (FRDA) | Frataxin (FXN) | autosomal recessive | Progressive gait and limb ataxia associated with cardiomyopathy and diabetes | 229300 |
| Posterior Column Ataxia and Retinitis Pigmentosa (PCARP) | Feline Leukemia Virus Subgroup C Receptor 1 (FLVCR1) | autosomal recessive | Sensory ataxia and retinitis pigmentosa | 609033 |
| Non syndromic Retinitis pigmentosa (RP) | Feline Leukemia Virus Subgroup C Receptor 1 (FLVCR1) | autosomal recessive | Retinitis pigmentosa | 268000 |
| Hereditary Sensory and Autonomic Neuropathy (HSAN) | Feline Leukemia Virus Subgroup C Receptor 1 (FLVCR1) | autosomal recessive | Loss of pain perception | 201300 |
| Fowler syndrome (PVHH) | Feline Leukemia Virus Subgroup C Receptor 2 (FLVCR2) | autosomal recessive | Proliferative glomerular vasculopathy in the central nervous system associated with severe hydrocephaly, ventriculomegaly, cortical thinning and hypoplastic cerebellum. | 225790 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Fiorito, V.; Chiabrando, D.; Tolosano, E. Mitochondrial Targeting in Neurodegeneration: A Heme Perspective. Pharmaceuticals 2018, 11, 87. https://doi.org/10.3390/ph11030087
AMA Style
Fiorito V, Chiabrando D, Tolosano E. Mitochondrial Targeting in Neurodegeneration: A Heme Perspective. Pharmaceuticals. 2018; 11(3):87. https://doi.org/10.3390/ph11030087
Chicago/Turabian StyleFiorito, Veronica, Deborah Chiabrando, and Emanuela Tolosano. 2018. "Mitochondrial Targeting in Neurodegeneration: A Heme Perspective" Pharmaceuticals 11, no. 3: 87. https://doi.org/10.3390/ph11030087
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.