The Role of NCOA4-Mediated Ferritinophagy in Health and Disease

{kind=link}
{kind=link}
Abstract
1. Introduction
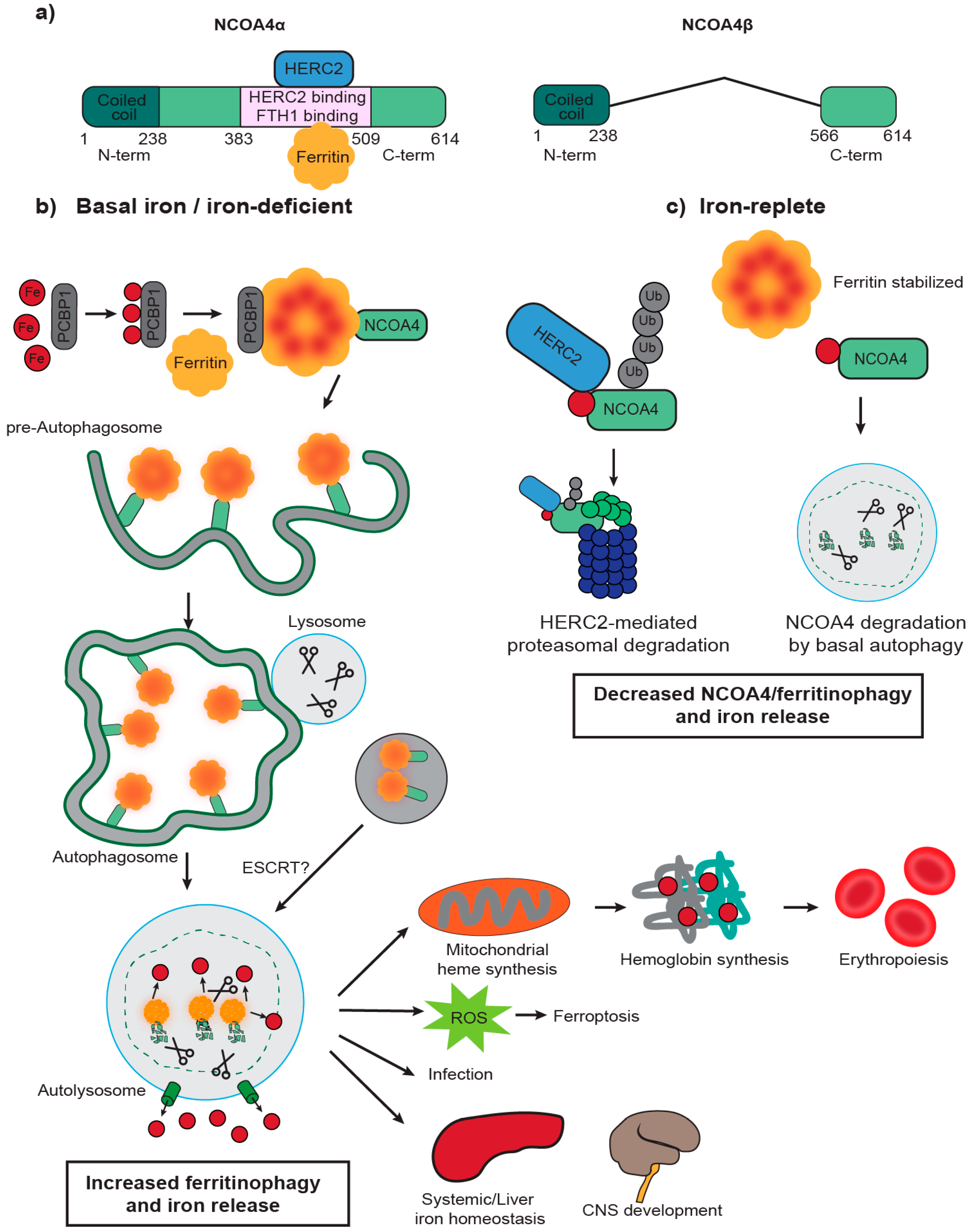
2. NCOA4 Mediates Ferritin Transport to the Lysosome for Degradation
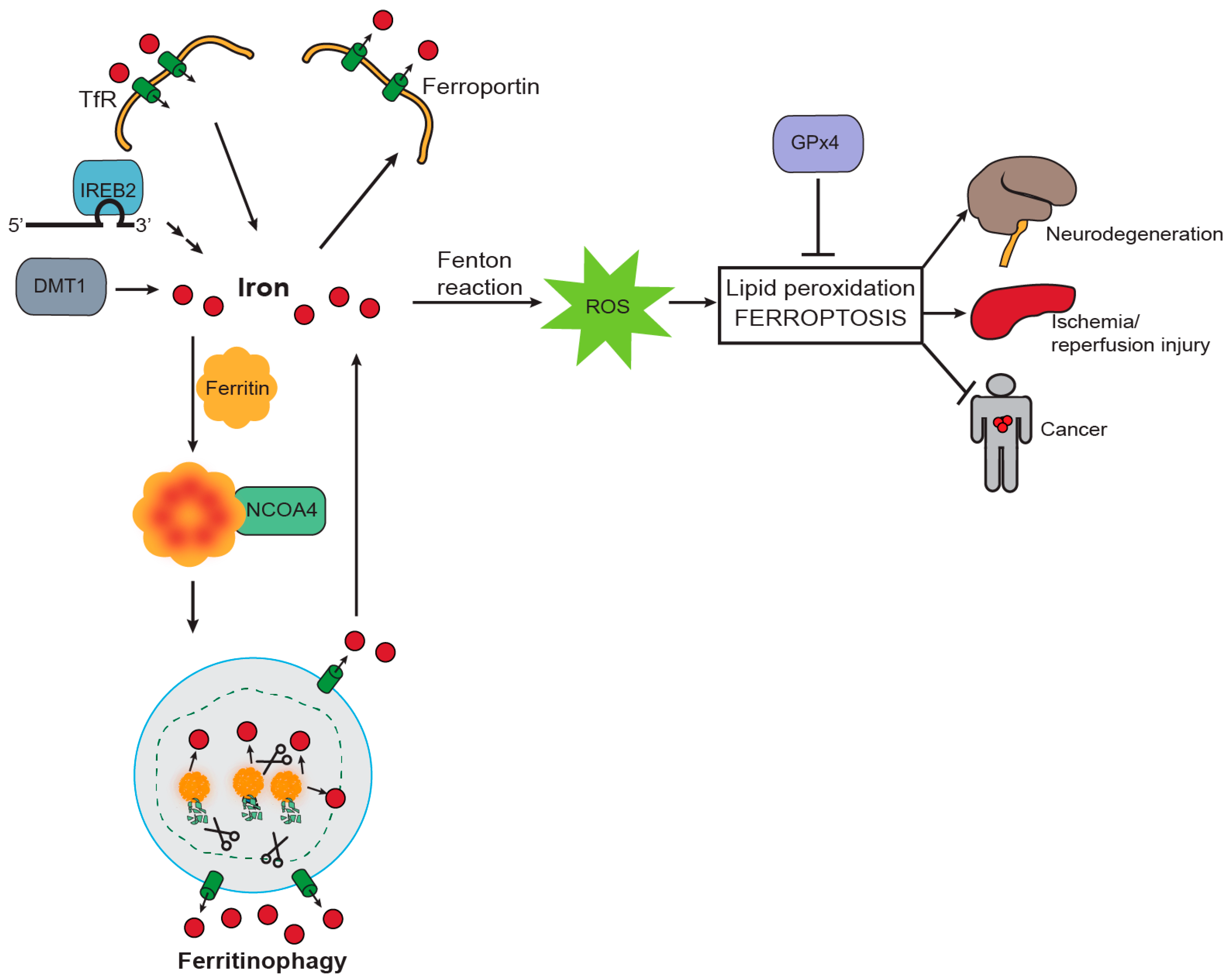
3. NCOA4 Mediates Ferritin Iron Release to Support Erythropoiesis
4. NCOA4-Mediated Ferritinophagy Modulates Ferroptosis
5. Ferritinophagy and Ferroptosis in Disease
5.1. Neurodegenerative Disease
5.2. Cancer
5.3. Infectious Disease
6. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| NCOA4 | Nuclear Receptor Co-Activator 4 |
| ATP | Adenosine Triphosphate |
| DNA | Deoxyribonucleic acid |
| ROS | Reactive Oxygen Species |
| TfR | Transferrin Receptor |
| DMT1 | Divalent Metal Transporter 1 |
| FPN | Ferroportin |
| FTH1 | Ferritin Heavy Chain 1 |
| FTL | Ferritin Light chain |
| Fe | Iron |
| PI3P | Phosphatidylinositol-3-phosphate |
| MEFs | Mouse Embryonic Fibroblasts |
| Atg7 | Autophagy-related protein 7 |
| SILAC | Stable Isotope Labeling by Amino-acids in Cell culture |
| LC-MS | Liquid Chromatography-Mass Spectrometry |
| HERC2 | HECT And RLD Domain Containing E3 Ubiquitin Protein Ligase 2 |
| NEURL4 | Neuralized E3 Ubiquitin Protein Ligase 4 |
| PCBP1 | Poly rC–binding protein 1 |
| LIR motif | LC3-interaction region |
| ESCRT | Endosomal Sorting Complex Required for Transport |
| CNS | Central Nervous System |
| AR | Androgen Receptor |
| RBC | Red Blood Cell |
| GPx4 | Glutathione Peroxidase 4 |
| RSL3 | RAS-selective lethal 3 |
| BSO | Buthionine Sulfoximine |
| DFO | Deferoxamine |
| SOD | Superoxide Dismutase |
| IREB2 | Iron Response Element Binding Protein 2 |
| ND | Neurodegenerative Disease |
| PD | Parkinson’s Disease |
| AD | Alzheimer’s Disease |
| NBIA | Neurodegeneration with Brain Iron Accumulation |
| HCMV | Human Cytomegalovirus |
References
- Bogdan, A.R.; Miyazawa, M.; Hashimoto, K.; Tsuji, Y. Regulators of Iron Homeostasis: New Players in Metabolism, Cell Death, and Disease. Trends Biochem. Sci. 2016, 41, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for Iron Metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef] [PubMed]
- Pantopoulos, K.; Porwal, S.K.; Tartakoff, A.; Devireddy, L. Mechanisms of mammalian iron homeostasis. Biochemistry 2012, 51, 5705–5724. [Google Scholar] [CrossRef] [PubMed]
- Camaschella, C. New insights into iron deficiency and iron deficiency anemia. Blood Rev. 2017, 31, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Weis, S.; Carlos, A.R.; Moita, M.R.; Singh, S.; Blankenhaus, B.; Cardoso, S.; Larsen, R.; Rebelo, S.; Schäuble, S.; Del Barrio, L.; et al. Metabolic Adaptation Establishes Disease Tolerance to Sepsis. Cell 2017, 169, 1263–1275. [Google Scholar] [CrossRef] [PubMed]
- Camaschella, C. Iron-deficiency anemia. N. Engl. J. Med. 2015, 372, 1832–1843. [Google Scholar] [CrossRef] [PubMed]
- Fenton, H.J.H. Oxidation of tartaric acid in presence of iron. J. Chem. Soc. Trans. 1894, 65, 899–910. [Google Scholar] [CrossRef]
- Sadrzadeh, S.M.; Graf, E.; Panter, S.S.; Hallaway, P.E.; Eaton, J.W. Hemoglobin. A biologic fenton reagent. J. Biol. Chem. 1984, 259, 14354–14356. [Google Scholar] [PubMed]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Mancias, J.D.; Pontano Vaites, L.; Nissim, S.; Biancur, D.E.; Kim, A.J.; Wang, X.; Liu, Y.; Goessling, W.; Kimmelman, A.C.; Harper, J.W. Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. eLife 2015, 4, e10308. [Google Scholar] [CrossRef] [PubMed]
- Arosio, P.; Ingrassia, R.; Cavadini, P. Ferritins: A family of molecules for iron storage, antioxidation and more. Biochim. Biophys. Acta 2009, 1790, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Philpott, C.C. The flux of iron through ferritin in erythrocyte development. Curr. Opin. Hematol. 2018, 3, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Kidane, T.Z.; Sauble, E.; Linder, M.C.; Theodros, Z.; Sauble, E.; Re, M.C.L. Release of iron from ferritin requires lysosomal activity. Am. J. Physiol. Cell Physiol. 2006, 291, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Radisky, D.; Kaplan, J. Iron in cytosolic ferritin can be recycled through lysosomal degradation in human fibroblasts. Biochem. J. 1998, 336, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Mikhael, M.; Xu, D.; Li, D.; Soe-Lin, S.; Ning, B.; Li, W.; Nie, G.; Zhao, Y.; Ponka, P. Lysosomal Proteolysis Is the Primary Degradation Pathway for Cytosolic Ferritin and Cytosolic Ferritin Degradation Is Necessary for Iron Exit. Antioxid. Redox Signal 2010, 13, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Mancias, J.D.; Kimmelman, A.C. Mechanisms of selective autophagy in normal physiology and cancer. J. Mol. Biol. 2016, 428, 1659–1680. [Google Scholar] [CrossRef] [PubMed]
- Asano, T.; Komatsu, M.; Yamaguchi-iwai, Y.; Ishikawa, F.; Mizushima, N.; Iwai, K. Distinct Mechanisms of Ferritin Delivery to Lysosomes in iron-depleted and iron-replete cells. Mol. Cell. Biol. 2011, 31, 2040–2052. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Debnath, J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 2015, 16, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Santana-Codina, N.; Mancias, J.D.; Kimmelman, A.C. The role of autophagy in cancer. Annu. Rev. Cancer Biol. 2017, 1, 19–39. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- Amaravadi, R.K.; Yu, D.; Lum, J.J.; Bui, T.; Christophorou, M.A.; Evan, G.I.; Thomas-Tikhonenko, A.; Thompson, C.B. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J. Clin. Investig. 2007, 117, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Dowdle, W.E.; Nyfeler, B.; Nagel, J.; Elling, R.A.; Liu, S.; Triantafellow, E.; Menon, S.; Wang, Z.; Honda, A.; Pardee, G.; et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat. Cell Biol. 2014, 16, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Van de Wijngaart, D.; van Royen, M.; Hersmus, R.; Pike, A.; Houtsmuller, A.; Jenster, G.; Trapman, J.; Dubbink, H. Novel FXXFF and FXXMF motifs in androgen receptor cofactors mediate high affinity and specific interactions with the ligand-binding domain. J. Biol. Chem. 2006, 281, 19407–19416. [Google Scholar] [CrossRef] [PubMed]
- Monaco, C.; Visconti, R.; Barone, M.; Pierantoni, G.; Berlingieri, M.; De Lorenzo, C.; Mineo, A.; Vecchio, G.; Fusco, A.; Santoro, M. The RFG oligomerization domain mediates kinase activation and re-localization of the RET/PTC3 oncoprotein to the plasma membrane. Oncogene 2001, 20, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Kollara, A.; Brown, T.J. Variable Expression of Nuclear Receptor Coactivator 4 (NcoA4) During Mouse Embryonic Development. J. Histochem. Cytochem. 2010, 58, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Alen, P.; Claessens, F.; Schoenmakers, E.; Swinnen, J.; Verhoeven, G.; Rombauts, W.; Peeters, B. Interaction of the putative androgen receptor-specific coactivator ARA70/ELE1alpha with multiple steroid receptors and identification of an internally deleted ELE1beta isoform. Mol. Endocrinol. 1999, 13, 117–128. [Google Scholar] [PubMed]
- Gryzik, M.; Srivastava, A.; Longhi, G.; Bertuzzi, M.; Gianoncelli, A.; Carmona, F.; Poli, M.; Arosio, P. Expression and characterization of the ferritin binding domain of Nuclear Receptor Coactivator-4 (NCOA4). Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 2710–2716. [Google Scholar] [CrossRef] [PubMed]
- Bauckman, K.; Mysorekar, I. Ferritinophagy drives uropathogenic Escherichia coli persistence in bladder epithelial cells. Autophagy 2016, 12, 850–863. [Google Scholar] [CrossRef] [PubMed]
- Von Muhlinen, N.; Akutsu, M.; Ravenhill, B.J.; Foeglein, Á.; Bloor, S.; Rutherford, T.J.; Freund, S.M.V.; Komander, D.; Randow, F. LC3C, bound selectively by a noncanonical LIR motif in NDP52, is required for antibacterial autophagy. Mol. Cell 2012, 48, 329–342. [Google Scholar] [CrossRef] [PubMed]
- De Domenico, I.; Vaughn, M.B.; Li, L.; Bagley, D.; Musci, G.; Ward, D.M.; Kaplan, J. Ferroportin-mediated mobilization of ferritin iron precedes ferritin degradation by the proteasome. EMBO J. 2006, 25, 5396–5404. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.M.; Dowdle, W.E.; DeJesus, R.; Wang, Z.; Bergman, P.; Kobylarz, M.; Lindeman, A.; Xavier, R.J.; McAllister, G.; Nyfeler, B.; et al. Autophagy-Independent Lysosomal Targeting Regulated by ULK1/2-FIP200 and ATG9. Cell Rep. 2017, 20, 2341–2356. [Google Scholar] [CrossRef] [PubMed]
- Mejlvang, J.; Olsvik, H.; Svenning, S.; Bruun, J.; Abudu, Y.; Larsen, K.; Brech, A.; Hansen, T.; Brenne, H.; Hansen, T.; et al. Starvation induces rapid degradation of selective autophagy receptors by endosomal microautophagy. J. Cell Biol. 2018, 217, 3640–3655. [Google Scholar] [CrossRef] [PubMed]
- Yeh, S.; Chang, C. Cloning and characterization of a specific coactivator, ARA70, for the androgen receptor in human prostate cells. Proc. Natl. Acad. Sci. USA 1996, 93, 5517–5521. [Google Scholar] [CrossRef] [PubMed]
- Heinlein, C.; Ting, H.; Yeh, S.; Chang, C. Identification of ARA70 as a ligand-enhanced coactivator for the peroxisome proliferator-activated receptor gamma. J. Biol. Chem. 1999, 274, 16147–16152. [Google Scholar] [CrossRef] [PubMed]
- Lanzino, M.; Amicis, F.; McPhaul, M.; Marsico, S.; Panno, M.; Ando, S. Endogenous coactivator ARA70 interacts with estrogen receptor alpha (ERalpha) and modulates the functional ERalpha/androgen receptor interplay in MCF-7 cells. J. Biol. Chem. 2005, 280, 20421–20430. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Brantley, K.; Bolu, E.; McPhaul, M. RFG (ARA70, ELE1) interacts with the human androgen receptor in a ligand-dependent fashion, but functions only weakly as a coactivator in cotransfection assays. Mol. Endocrinol. 1999, 13, 1645–1656. [Google Scholar] [CrossRef] [PubMed]
- Ting, H.; Bao, B.; Hsu, C.; Lee, Y. Androgen-receptor coregulators mediate the suppressive effect of androgen signals on vitamin D receptor activity. Endocrine 2005, 26, 1–9. [Google Scholar] [CrossRef]
- Kollara, A.; Brown, T. Expression and function of nuclear receptor co-activator 4: Evidence of a potential role independent of co-activator activity. Cell. Mol. Life Sci. 2012, 69, 3895–3909. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Lee, H.; Li, W.; Jeffrey, R.; Barrasa, M.I.; Ma, Q. Thyroid hormone receptor beta and NCOA4 regulate terminal erythrocyte differentiation. Proc. Natl. Acad. Sci. USA 2017, 114, 10107–10112. [Google Scholar] [CrossRef] [PubMed]
- Bellelli, R.; Castellone, M.D.; Guida, T.; Limongello, R.; Dathan, N.A.; Merolla, F.; Cirafici, A.M.; Affuso, A.; Masai, H.; Costanzo, V.; et al. NCOA4 transcriptional coactivator inhibits activation of DNA replication origins. Mol. Cell 2014, 55, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.; Prado, M.; Schmidt, P.; Sendamarai, A.; Wilson-Grady, J.; Min, M.; Campagna, D.; Tian, G.; Shi, Y.; Dederer, V.; et al. UBE2O remodels the proteome during terminal erythroid differentiation. Science 2017, 357, eaan0218. [Google Scholar] [CrossRef] [PubMed]
- Sheftel, A.D.; Zhang, A.-S.; Brown, C.; Shirihai, O.S.; Ponka, P. Direct interorganellar transfer of iron from endosome to mitochondrion. Blood 2007, 110, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Vaisman, B.; Fibach, E.; Konijn, A. Utilization of intracellular ferritin iron for hemoglobin synthesis in developing human erythroid precursors. Blood 1997, 90, 831–838. [Google Scholar] [PubMed]
- Shvartsman, M.; Cabantchik, Z.I. Intracellular iron trafficking: Role of cytosolic ligands. Biometals 2012, 25, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Ryu, M.S.; Zhang, D.; Protchenko, O.; Shakoury-Elizeh, M.; Philpott, C.C. PCBP1 and NCOA4 regulate erythroid iron storage and heme biosynthesis. J. Clin. Investig. 2017, 127, 1786–1797. [Google Scholar] [CrossRef] [PubMed]
- Weber, G.J.; Choe, S.E.; Dooley, K.A.; Paffett-Lugassy, N.N.; Zhou, Y.; Zon, L.I. Mutant-specific gene programs in the zebrafish. Blood 2005, 106, 521–530. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Schulz, V.P.; Li, J.; Wu, K.; Liu, J.; Xue, F.; Hu, J.; Mohandas, N.; Gallagher, P.G. Global transcriptome analyses of human and murine terminal erythroid differentiation. Blood 2014, 123, 3466–3477. [Google Scholar] [CrossRef] [PubMed]
- Bellelli, R.; Federico, G.; Matte, A.; Colecchia, D.; Iolascon, A.; Chiariello, M.; Santoro, M.; De Franceschi, L.; Carlomagno, F. NCOA4 Deficiency Impairs Systemic Iron Homeostasis. Cell Rep. 2016, 14, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Jia, J.; Kumar, S.; Choi, S.W.; Gu, Y.; Mudd, M.; Dupont, N.; Jiang, S.; Peters, R.; Farzam, F.; et al. Dedicated SNAREs and specialized TRIM cargo receptors mediate secretory autophagy. EMBO J. 2017, 36, 42–60. [Google Scholar] [CrossRef] [PubMed]
- Truman-Rosentsvit, M.; Berenbaum, D.; Spektor, L.; Cohen, L.A.; Belizowsky-Moshe, S.; Lifshitz, L.; Ma, J.; Li, W.; Kesselman, E.; Abutbul-Ionita, I.; et al. Ferritin is secreted via 2 distinct nonclassical vesicular pathways. Blood 2018, 131, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Doulatov, S.; Vo, L.; Macari, E.; Wahlster, L.; Kinney, M.; Taylor, A.; Barragan, J.; Gupta, M.; McGrath, K.; Lee, H.; et al. Drug discovery for Diamond-Blackfan anemia using reprogrammed hematopoietic progenitors. Sci. Transl. Med. 2017, 9, eaah5645. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ 2016, 23, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Chen, Z.; Wu, D.; Chen, L. Ferritinophagy/ferroptosis: Iron-related newcomers in human diseases. J. Cell Physiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H., III; Kang, R. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell Res. 2016, 26, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Angeli, P.F.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gasco, S.; Linkermann, A.; Murphy, M.E.; Overholtzer, M.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Stockwell, B. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Ou, Z.; Xie, M.; Kang, R.; Fan, Y.; Niu, X.; Wang, H.; Cao, L.; Tang, D. HSPB1 as a Novel Regulator of Ferroptotic Cancer Cell Death. Oncogene 2015, 34, 5617–5625. [Google Scholar] [CrossRef] [PubMed]
- Louandre, C.; Marcq, I.; Bouhlal, H.; Lachaier, E.; Godin, C.; Saidak, Z.; François, C.; Chatelain, D.; Debuysscher, V.; Barbare, J.; et al. The retinoblastoma (Rb) protein regulates ferroptosis induced by sorafenib in human hepa- tocellular carcinoma cells. Cancer Lett. 2015, 356, 971–977. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yao, Z.; Wang, L.; Ding, H.; Shao, J.; Chen, A.; Zhang, F.; Zheng, S. Activation of ferritinophagy is required for the RNA-binding protein ELAVL1/HuR to regulate ferroptosis in hepatic stellate cells. Autophagy 2018, 14, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Guiney, S.; Adlard, P.; Bush, A.; Finkelstein, D.; Ayton, S. Ferroptosis and cell death mechanisms in Parkinson’s disease. Neurochem. Int. 2017, 104, 34–48. [Google Scholar] [CrossRef] [PubMed]
- Jenner, P.; Dexter, D.; Sian, J.; Schapira, A.; Marsden, C. Oxidative stress as a cause of nigral cell death in Parkinson’s disease and incidental Lewy body disease. The Royal Kings and Queens Parkinson’s Disease Research Group. Ann. Neurol. 1992, 32 (Suppl. 1), S82–S87. [Google Scholar] [CrossRef] [PubMed]
- Salazar, J.; Mena, N.; Hunot, S.; Prigent, A.; Alvarez-Fischer, D.; Arredondo, M.; Duyckaerts, C.; Sazdovitch, V.; Zhao, L.; Garrick, L.; et al. Divalent metal transporter 1 (DMT1) contributes to neurodegeneration in animal models of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2008, 105, 18578–18583. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, S.; Buchanan, D.; Ahmed, I.; Taylor, K.; Loriot, M.; Sinsheimer, J.; Bronstein, J.; Elbaz, A.; Mellick, G.; Rotter, J.; et al. Pooled analysis of iron-related genes in Parkinson’s disease: Association with transferrin. Neurobiol. Dis. 2014, 62, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Everett, J.; Céspedes, L.; Shelford, C.; Exley, J.; Collingwood, J.; Dobson, G.; van der Laan, C.; Jenkins, E.; Arenholz, N.; Telling, D. Ferrous iron formation following the co-aggregation of ferric iron and the Alzheimer’s disease peptide β-amyloid (1–42). J. R. Soc. Interface 2014, 11, 20140165. [Google Scholar] [CrossRef] [PubMed]
- Raha, A.; Vaishnav, R.; Friedland, R.; Bomford, A.; Raha-Chowdhury, R. The systemic iron-regulatory proteins hepcidin and ferroportin are reduced in the brain in Alzheimer’s disease. Acta Neuropathol. Commun. 2013, 1, 55. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Hambright, W.S.; Na, R.; Ran, Q. Ablation of ferroptosis inhibitor glutathione peroxidase 4 in neurons results in rapid motor neuron degeneration and paralysis. J. Biol. Chem. 2015, 290, 28097–28106. [Google Scholar] [CrossRef] [PubMed]
- Do Van, B.; Gouel, F.; Jonneaux, A.; Timmerman, K.; Gelé, P.; Pétrault, M.; Bastide, M.; Laloux, C.; Moreau, C.; Bordet, R.; et al. Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol. Dis. 2016, 94, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Skouta, R.; Dixon, S.; Wang, J.; Dunn, D.; Orman, M.; Shimada, K.; Rosenberg, P.; Lo, D.C.; Weinberg, J.M.; Linkermann, A.; et al. Ferrostatins Inhibit Oxidative Lipid Damage and Cell Death in Diverse Disease Models. J. Am. Chem. Soc. 2014, 136, 4551–4556. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Saitsu, H.; Nishimura, T.; Muramatsu, K.; Kodera, H.; Kumada, S.; Sugai, K.; Kasai-Yoshida, E.; Sawaura, N.; Nishida, H.; Hoshino, A.; et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat. Genet. 2013, 45, 445. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, M.; Zheng, B.; Lu, T.; Yan, Z.; Py, B.; Ng, A.; Xavier, R.; Li, C.; Scherzer, C.; Yuan, J. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2010, 107, 14164–14169. [Google Scholar] [CrossRef] [PubMed]
- Buijs, M.; Doan, N.T.; van Rooden, S.; Versluis, M.J.; van Lew, B.; Milles, J.; van der Grond, J.; van Buchem, M.A. In vivo assessment of iron content of the cerebral cortex in healthy aging using 7-Tesla T2*-weighted phase imaging. Neurobiol. Aging 2017, 53, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Pinnix, Z.K.; Miller, L.D.; Wang, W.; D’Agostino, R.J.; Kute, T.; Willingham, M.C.; Hatcher, H.; Tesfay, L.; Sui, G.; Di, X.; et al. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci. Transl. Med. 2010, 2, 43–56. [Google Scholar] [CrossRef]
- Basuli, D.; Tesfay, L.; Deng, Z.; Paul, B.; Yamamoto, Y.; Ning, G.; Xian, W.; McKeon, F.; Lynch, M.; Crum, C.; et al. Iron addiction: A novel therapeutic target in ovarian cancer. Oncogene 2017, 36, 4089–4099. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Polack, A.; Dalla-Favera, R. Coordinated regulation of iron-controlling genes, H-ferritin and IRP2, by c-MYC. Science 1999, 283, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wang, W.; Tsuji, Y.; Torti, S.; Torti, F. Post-transcriptional modulation of iron homeostasis during p53-dependent growth arrest. J. Biol. Chem. 2008, 283, 33911–33918. [Google Scholar] [CrossRef] [PubMed]
- Kakhlon, O.; Gruenbaum, Y.; Cabantchik, Z. Repression of ferritin expression modulates cell responsiveness to H-ras-induced growth. Biochem. Soc. Trans. 2002, 30, 777–780. [Google Scholar] [CrossRef] [PubMed]
- Recalcati, S.; Locati, M.; Marini, A.; Santambrogio, P.; Zaninotto, F.; De Pizzoi, M.; Zammataro, L.; Girelli, D.; Cairo, G. Differential regulation of iron homeostasis during human macrophage polarized activation. Eur. J. Immunol. 2010, 40, 824–835. [Google Scholar] [CrossRef] [PubMed]
- Mertens, C.; Mora, J.; Ören, B.; Grein, S.; Winslow, S.; Scholich, K.; Weigert, A.; Malmström, P.; Forsare, C.; Fernö, M.; et al. Macrophage-derived lipocalin-2 transports iron in the tumor microenvironment. Oncoimmunology 2018, 7, e1408751. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.; Rittenberg, P.; Brown, T. Activation of androgen receptor-associated protein 70 (ARA70) mRNA expression in ovarian cancer. Gynecol. Oncol. 2001, 80, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Rockfield, S.; Flores, I.; Nanjundan, M. Expression and function of nuclear receptor coactivator 4 isoforms in transformed endometriotic and malignant ovarian cells. Oncotarget 2018, 9, 5344–5367. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Li, C.; Chen, F.; Wang, Z.; Ligr, M.; Melamed, J.; Wei, J.; Gerald, W.; Pagano, M.; Garabedian, M.; et al. Stimulation of prostate cancer cellular proliferation and invasion by the androgen receptor co-activator ARA70. Am. J. Pathol. 2008, 172, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Chen, F.; Sahin, A.; Albarracin, C.; Pei, Z.; Zou, X.; Singh, B.; Xu, R.; Daniels, G.; Li, Y.; et al. Distinct function of androgen receptor coactivator ARA70alpha and ARA70beta in mammary gland development, and in breast cancer. Breast Cancer Res. Treat. 2011, 128, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Lazova, R.; Camp, R.; Klump, V.; Siddiqui, S.; Amaravadi, R.; Pawelek, J. Punctate LC3B Expression Is a Common Feature of Solid Tumors and Associated with Proliferation, Metastasis, and Poor Outcome. Clin. Cancer Res. 2012, 18, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.Y.; Chen, H.Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.S.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Lock, R.; Roy, S.; Kenific, C.M.; Su, J.S.; Salas, E.; Ronen, S.M.; Debnath, J. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol. Biol. Cell 2011, 22, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Wu, S.; Guo, J.; Chen, Z.; Ge, J.; Yang, P.; Hu, B.; Chen, Z. Silencing of TKTL1 by siRNA inhibits proliferation of human gastric cancer cells in vitro and in vivo. Cancer Biol. Ther. 2010, 9, 710–716. [Google Scholar] [CrossRef] [PubMed]
- Ryschich, E.; Huszty, G.; Hartel, M.; Büchler, M.; Schmidt, J. Transferrin receptor is a marker of malignant phenotype in human pancreatic cancer and in neuroendocrine carcinoma of the pancreas. Eur. J. Cancer 2004, 40, 1418–1422. [Google Scholar] [CrossRef] [PubMed]
- Dolma, S.; Lessnick, S.L.; Hahn, W.C.; Stockwell, B.R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003, 3, 285–296. [Google Scholar] [CrossRef]
- Eling, N.; Reuter, L.; Hazin, J.; Hamacher-brady, A. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience 2015, 2, 517. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Bao, Q.; Xuan, B.; Xu, W.; Pan, D.; Li, Q.; Qian, Z. Human Cytomegalovirus Protein pUL38 Prevents Premature Cell Death by Binding to Ubiquitin-specific Protease 24 and Regulating Iron Metabolism. J. Virol. 2018, 92, e00191-18. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santana-Codina, N.; Mancias, J.D. The Role of NCOA4-Mediated Ferritinophagy in Health and Disease. Pharmaceuticals 2018, 11, 114. https://doi.org/10.3390/ph11040114
Santana-Codina N, Mancias JD. The Role of NCOA4-Mediated Ferritinophagy in Health and Disease. Pharmaceuticals. 2018; 11(4):114. https://doi.org/10.3390/ph11040114
Chicago/Turabian StyleSantana-Codina, Naiara, and Joseph D. Mancias. 2018. "The Role of NCOA4-Mediated Ferritinophagy in Health and Disease" Pharmaceuticals 11, no. 4: 114. https://doi.org/10.3390/ph11040114
APA StyleSantana-Codina, N., & Mancias, J. D. (2018). The Role of NCOA4-Mediated Ferritinophagy in Health and Disease. Pharmaceuticals, 11(4), 114. https://doi.org/10.3390/ph11040114