Kinins and Their Receptors in Infectious Diseases


Centro de Pesquisa em Toxicologia e Farmacologia, Escola de Ciências da Saúde e da Vida, Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, RS 90619-900, Brazil
*
Author to whom correspondence should be addressed.
Pharmaceuticals 2020, 13(9), 215; https://doi.org/10.3390/ph13090215
Submission received: 31 July 2020
/
Revised: 23 August 2020
/
Accepted: 25 August 2020
/
Published: 27 August 2020
(This article belongs to the Special Issue Kinins and Their Receptors as Potential Therapeutic Targets —— A Tribute to Professor Domenico Regoli, A Key Figure in the Field of Kinins)
Abstract
:Kinins and their receptors have been implicated in a series of pathological alterations, representing attractive pharmacological targets for several diseases. The present review article aims to discuss the role of the kinin system in infectious diseases. Literature data provides compelling evidence about the participation of kinins in infections caused by diverse agents, including viral, bacterial, fungal, protozoan, and helminth-related ills. It is tempting to propose that modulation of kinin actions and production might be an adjuvant strategy for management of infection-related complications.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Bradykinin (BK) and related kinins are a family of small active peptides implicated in a series of biological effects. The nonapeptide BK is cleaved from the high molecular weight kininogen (HMWK), via activation of plasma kallikrein. BK generation in plasma takes part in the intrinsic coagulation pathway activation, involving the interaction of Factor XII (FXII), prekallikrein (PPK) and Factor XI (FXI) with HMWK, leading to prothrombotic and inflammatory effects [1,2]. Alternatively, kallidin (Lys-BK) is a decapeptide formed by the action of tissue kallikrein on low molecular weight kininogen (LMWK) precursor. In some circumstances, Lys-BK can be converted into BK following the cleavage of the amino-terminal lysine, by the action of plasmatic aminopeptidases. Both Lys-BK and BK are short-acting mediators that are rapidly degraded into inactive fragments by kininase 2, also called angiotensin-converting enzyme (ACE). Under certain circumstances, such as in an inflammatory milieu, there is an increased affinity of kininase 1 for kinins. In this context, this enzyme converts Lys-BK and BK into the active kinins, namely Lys-des-Arg9-BK and des-Arg9-BK, respectively [3,4,5,6]. Interestingly, ACE and its isoform ACE2 are zinc metallopeptidases involved in the metabolism of vasoactive peptides, having angiotensin- and kinin-family members as their main substrates, displaying a relevant role in a series of pathophysiological processes [7].
Predominantly, the effects of kinins are mediated by the activation of two G protein-coupled receptors, denoted as B1 and B2. These receptors have been extensively characterized in several animal species and humans, showing an identity of about 35%, with small variations across species. Both receptors share common signaling pathways, but their pattern of expression is distinctive. The B2 receptor is defined as a housekeeping molecule, being constitutively expressed by most cells at peripheral and central levels, such as vascular and non-vascular smooth muscle cells, epithelial and immune cells, besides neurons and glial cells. Conversely, despite some exceptions, the B1 receptor displays a low physiological expression, being upregulated under a variety of stressful stimuli, such as the exposure to infectious agents. Another difference refers to the affinity for the natural agonists: the B2 receptor has a high affinity for Lys-BK and BK, while the B1 receptor preferentially binds to Lys-des-Arg9-BK and des-Arg9-BK. Irrespective of these differences, both B1 and B2 receptors have been implicated in a series of responses, including the regulation of blood vessel tonus, inflammatory changes, and pain processing mechanisms [3,6,8,9,10].
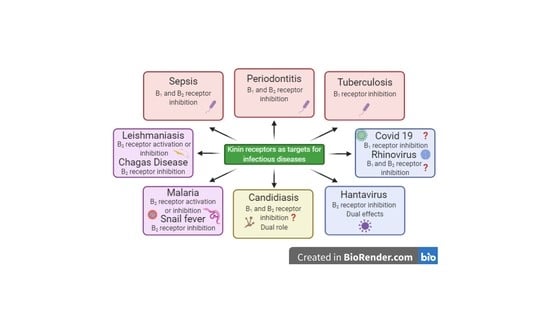
The advances in the comprehension of the pathophysiological roles of kinins and their receptors have been greatly favored by development of peptide and non-peptide selective receptor ligands by chemical synthesis [4,8,9,11,12,13]. Moreover, the modulation of kallikreins and kininases has also been explored for therapeutic purposes [14]. Interestingly, the selective pseudopeptide B2 receptor antagonist HOE-140 (Icatibant) has been approved in the Americas and Europe, for managing acute episodes of hereditary angioedema, under the commercial name Firazyr [15]. Additionally, the therapeutic benefits of ACE inhibitors, such as captopril, also rely on the modulation of kinins and their biological effects [16]. Over the last four decades, the role of either kinin receptor and other components of the kinin system has been described in a series of infectious conditions, such as sepsis, schistosomiasis, leishmaniosis, Chagas Disease, candidiasis, tuberculosis, malaria, and it has been recently correlated with Covid-19 infection [17,18,19,20,21,22]. Considering the abovementioned evidence, the present review article aims to discuss the relevance of the kinin system, mainly of kinin receptors, as pharmacological targets for management of infectious diseases, attempting to cover the implication of kinins in the progression of infection-related illnesses in humans.
2. Bacterial Infections and Kinins
According to the World Health Organization [23] communicable diseases caused by bacterial infections stand out among the top ten causes of deaths worldwide. In 2016, lower tract respiratory infections, diarrhea and tuberculosis accounted for 6 million deaths globally. The burden of bacterial infectious diseases is especially relevant in low-income countries, but it has gained more attention due to the rapid advent of antibiotic resistance in the last decades [24]. In this regard, there is a current interest in relation to the pathways implicated in bacterial infectious diseases, mainly focused on the bacteria-host interaction mechanisms. This section will discuss the most relevant data regarding the participation of the kinin family of molecules in bacterial infections. A summary of main findings discussed in this section is provided in Figure 1.
2.1. Bacterial Endotoxins and Kinin Responses
Lipopolysaccharide (LPS) is a component of the cell wall of gram-negative bacteria that elicits marked inflammatory responses. Its effects are primarily mediated by the activation of Toll-like receptors (TLR), mainly via the TLR-4-MD-2 complex, in a process dependent on LPS-binding protein (LPB) and CD14 [25]. This set of responses takes part of the host innate immune system activation triggered by pathogen-associated molecular patterns, such as LPS, aimed to suppress the infectious damage [26].
The relationship between LPS responses and the activation of the kinin system has been widely investigated in several experimental paradigms. A classical study conducted by Regoli et al. (1981) demonstrated that strips of large arteries and veins, obtained from rabbits that had been treated with LPS before euthanasia, displayed marked contractile responses to the selective B1 receptor agonists, des-Arg9-BK and Lys-des-Arg9-BK. This evidence provided the basis indicating that infectious stimuli, in addition to trauma responses, were able to promote an upregulation of B1 receptors, with variable responses regarding the B2 receptors [27]. This notion was extended by studies showing that both B1 receptor agonists, des-Arg9-BK and Lys-des-Arg9-BK, led to hypotensive responses in rabbits pre-treated with sub-lethal doses of LPS, according to assessment of mean blood pressure in comparison with untreated animals [28]. Subsequently, the induction of B1 receptors in rabbit isolated aorta preparations was also demonstrated by the in vitro incubation with bacterial LPS, which led to a time- and concentration-dependent increase of contractile responses to des-Arg9-BK [29]. The upregulation of B1 receptor-mediated contractile responses was also showed in non-vascular preparations, such as the isolated urinary bladder, after the in vitro or in vivo exposure to LPS [30].
As for inflammation models, the intravenous treatment with a low dose of LPS caused an increase of rat paw edema induced by the selective B1 receptor agonist des-Arg9-BK, whereas the edema formation produced by the selective B2 receptor agonist tyrosine8-BK was markedly reduced. The changes of kinin receptor-mediated inflammatory responses in LPS-treated animals were sensitive to glucocorticoids and cycloheximide, which suggests an implication of protein synthesis in the opposite balance of B1 and B2 receptors under inflammatory conditions [31]. Notably, the intravenous administration of LPS also produced an inversion of B2/B1 receptor-mediated febrile responses: the hyperthermia induced by the central administration of BK and tyrosine8-BK was reduced in LPS-treated rats, whereas the fever elicited by des-Arg9-BK was noticeably increased [32]. Remarkably, the effects of LPS on B1 and B2 receptors might be variable depending on the experimental approach. For instance, Passos et al. (2004) showed that local application of LPS into the rat paw resulted in a time-related increase of des-Arg9-BK-induced edema formation, preceded by an elevation of B1 receptor mRNA, without any change of the paw edema caused by the B2 receptor agonist tyrosine8-BK [33]. Additionally, it was observed that vascular hyporesponsiveness to noradrenaline, in rats that had been pre-treated with LPS, was partially blocked by the selective B2 receptor antagonist HOE-140, indicating that part of the hypotensive effects of LPS rely on the activation of B2 receptors [34]. The incubation of LPS potentiated BK-B2 receptor-mediated signaling pathways, with an increase of intracellular Ca2+ and inositolphosphates, in primary cultured tracheal smooth muscle cells. This study points out a relationship between the LPS–TLR4–BK–B2 receptor axis in the pathogenesis of bronchial hyperreactivity, by mechanisms involving the activation of Ras-Raf-Mitogen-Activated Protein Kinases (MAPK) [35]. Alternatively, the migration of neutrophils to the lungs of LPS-aerosolized mice was prevented by the ACE inhibitor enalapril, an effect that was reversed by dosing both B1 des-Arg10-HOE-140 and B2 HOE-140 receptor antagonists. In this case, the agonism of either kinin receptor was beneficial to prevent LPS-triggered lung inflammation [36]. More recently, it was demonstrated that des-Arg9-BK is a substrate for ACE2 in lungs, and the inhibition of ACE2 might favor the activation of B1 receptors by this agonist after LPS inhalation, contributing for neutrophil influx and acute lung inflammation [37]. Moreover, it was demonstrated that BK elicits a biphasic response in LPS-treated rats, with a hypotensive effect followed by a sustained increase in blood pressure, via mechanisms involving a crosstalk between kinin B2 and angiotensin-1 receptors [38]. The influence of LPS on kinin-mediated responses point out a relevant role for the kinin system in endotoxemia-related respiratory and cardiovascular complications, besides systemic inflammatory responses, with a special interest concerning septic patients, as discussed in the next section.
2.2. The Kinin System in Systemic Inflammatory Response Syndrome (SIRS)
It has been recently reviewed that pathogens have the ability to undermine contact system activation, partly by altering HMWK and BK actions, enabling bacterial dissemination via increased vascular permeability [39]. It was demonstrated that exposure of human lung fibroblasts (IMR-90 cells) to exudates of Staphylococcus aureus-treated human peripheral blood mononuclear cells (PBMC) triggered an upregulation of B1 and B2 receptor mRNA and binding sites. The same protocol of stimulation with S. aureus-primed PBMC enhanced the conversion of BK to the metabolite des-Arg9-BK. Extending somewhat this experimental evidence, biopsies from patients presenting with staphylococcal soft-tissue infection or erysipelas showed an increased expression of B1, but not B2 receptors, which had been correlated with enhanced levels of IL-1β at the same sites [40]. Thus, the increased production of des-Arg9-BK and the activation of de novo induced B1 receptors might be related to S. aureus dissemination, facilitating the development of severe conditions. In a mouse model of chronic rhinosinusitis induced by the nasal inoculation of S. aureus, there was a marked upregulation of FXII, according to the assessment of the nasal mucosa membrane, suggesting a potential role for BK release in chronification of bacterial sinusitis [41]. FXII has also been described as a plasmatic target for the metalloproteinase CpaA, which is a virulence factor of Acinetobacter baumannii, a bacterial species related to antibiotic-resistant ventilator-associated pneumonia and catheter-induced bacteremia. It has been proposed that A. baumannii CpaA inhibits the activation of FXII, reducing the host defenses and contributing for the bacterial pathogenicity [42]. The induction of a mouse model of pneumosepsis, by the intranasal administration of Klebsiella pneumoniae, triggered a reduction of PPK liver expression. Moreover, the inhibition of PPK by an antisense oligonucleotide reduced both the mortality rates and the infection burden in K. pneumoniae-infected mice, likely via activation of innate immune system in lungs [43]. The same research group demonstrated a similar protective effect in the mouse model of K. pneumoniae infection, by gene deletion of FXII [44]. However, the modulation of either HMWK or B1 and B2 kinin receptors failed to provide protection in this experimental paradigm [45,46]. The induction of experimental sepsis by Streptococcus pyogenes was associated with a reduction of FXII, kininogen-1 and PPK in mice. Extending preclinical evidence, a decline of FXII and kallikrein levels was observed in plasma of 23 septic patients, irrespective of survival outcomes. Notably, in the mouse model of sepsis elicited by S. pyogenes, the inhibition of PPK (but not FXII) markedly prevented the dissemination of S. pyogenes, according to assessment in blood and spleen. This strategy also reduced kidney damage and the serum elevation of several cytokines and chemokines, except RANTES (CCL5), which was increased after PPK knockdown. It was concluded that plasma kallikrein blockade might prevent bacterial spread, via inhibition of fibrinolysis [47]. Remarkably, S. pyogenes induces BK release from HMWK via streptokinase-activated plasmin, making possible to suggest a relevant role for the kinin system in streptococcal sepsis [48]. Furthermore, gene deletion of HMWK prevented the mortality and reduced endotoxin levels, in a rodent model of sepsis induced by a lethal dose of LPS. Nevertheless, gene ablation of FXII, PPK or B1/B2 kinin receptors did not provide a protection in this model of SIRS [49].
The participation of the kallikrein–kinin system (KKS) has also been argued in a non-human primate model of sepsis induced by Escherichia coli inoculation, indicating that inhibition of FXII, with the consequent reduction of BK release, might prevent septic shock in baboons [50]. The blockade of FXII is able to avoid respiratory complications, intravascular disseminated coagulation, and organ dysfunction, with a decrease of complement cascade activation and cytokine storm. This feasibly contributes to the extended survival rates of septic animals, likely by reducing BK levels and related vasodilation (for review, see [51]). Thus, it is tempting to propose that inhibition of the FXII–kallikrein–kininogen–kinin axis might represent an interesting strategy for preventing septic shock. In this context, a clinical study investigated the inflammatory and coagulation alterations correlating with survival rates in septic children. The authors proposed that prolonged activated partial thromboplastin time is related to poorer survival outcomes, and a reduced activation of FXII might have beneficial effects, probably due to a decline of BK production [52]. Supporting this hypothesis, studies conducted during the 1990s revealed protective effects for selective B2 receptor antagonists in preclinical and clinical sepsis. For instance, the administration of NPC17731 prevented the hypotensive responses in a porcine model of sepsis, induced by injection of living Pseudomonas aeruginosa [53]. In the clinical setting, the administration of B2 receptor antagonist CP-0127 had mild effects on septic patients, with improved survival rates in patients with gram-negative bacterial infections [18]. Compelling pre-clinical evidence also revealed a relevant role for B1 receptors in sepsis. Accordingly, the oral treatment with the selective B1 receptor antagonist SSR240612 was able to diminish LPS-induced lethality in type 1 diabetic rats by preventing sepsis-related complications such as thrombosis and multiple organ damage [54]. More recently, it was demonstrated that oral treatment with the selective B1 receptor antagonist BI113823 was able to reduce systemic inflammation, hemodynamic changes and organ failure, in a rat model of sepsis induced by cecal ligation and puncture [55]. Supporting pharmacological data, transgenic rats overexpressing B1 receptors, specifically in endothelium, displayed an increased susceptibility for endotoxic shock [56], similar to what had previously been demonstrated in mice with an overall overexpression of B1 receptors [57]. Further studies are necessary to determine how and at what level, the inhibition of the KKS might be useful for reducing mortality in patients undergoing SIRS and septic shock.
2.3. Periodontitis and Its Relationship with Kinins
Periodontitis is an inflammatory progressive disease destroying the tooth-supporting tissues, including gingiva, periodontal ligament, and alveolar bone. The interaction between periodontal pathogens and the host leads to a marked inflammatory response, finally resulting in tooth loss. Moreover, periodontal disease has serious impacts on general health, impairing the outcomes of systemic diseases, such as atherosclerosis and diabetes [58]. One of the most important pathogens leading to periodontitis is the anaerobic bacteria Porphyromonas gingivalis. Of note, P. gingivalis LPS mainly activates TLR2 receptors, differently from Escherichia and Salmonella LPS, which typically signal via TLR4 stimulation [59,60]. The high virulence of P. gingivalis is related to the production of cysteine proteinases named gingipains, which cleave LMWK and HMWK, producing Lys-BK and BK in periodontal tissues [61]. Accordingly, it was demonstrated that buccal inoculation of P. gingivalis induced both edema and gingivitis in mice, by the sequential activation of TLR2 and gingipains, leading to the release of kinins and B2 receptor stimulation, likely accounting for innate and adaptative immune responses in periodontal disease [62]. Prior evidence demonstrated that gingipain-induced inflammation in the hamster cheek pouch was reduced by the selective kinin B2 receptor antagonist NPC 17647 [63]. Furthermore, the increase of vascular permeability induced by P. gingivalis in mice was potentiated by BK or kininase inhibitors, whereas it was hindered by B2 receptor antagonism [64]. The intraplantar injection of LPS obtained from P. gingivalis led to an up-regulation of both des-Arg9-BK-induced paw edema and B1 receptor mRNA in rats, with an involvement of tumor necrosis factor and neutrophil influx [65]. This evidence suggests that kinin receptor modulation might additionally contribute for the pathogenic effects of P. gingivalis in the inflammatory milieu. Reinforcing this notion, a recent publication demonstrated that in vitro activation of TLR2 by P. gingivalis LPS, or the synthetic TLR2 agonist Pam2CSK4, increased the expression of kinin B1 and B2 receptors in human gingival fibroblasts, at both mRNA and protein levels. Interestingly, the upregulation of either kinin receptor was mimicked in vivo, after the injection of P. gingivalis LPS into the mouse gingiva [66]. It was proposed that gingipains might also play a part for the increased risk of oral cancer in patients with periodontitis—this was associated with the inactivation of SPINK6, an inhibitor of kallikrein activation in the epithelium, probably resulting in augmentation of kinins, which are recognized mitotic mediators [67]. Remarkably, it was found that formation of complex oral biofilms between P. gingivalis and Candida albicans is dependent on the action of gingipains and peptidylarginine deiminase, which in turn leads to kinin citrullination, changing the affinity for kinin receptors, what finally contributes for pathogen escaping mechanisms [68]. Altogether, literature evidence clearly highlights a relevant role for kinins and their receptors in the pathogenesis of periodontitis. Strikingly, periodontitis has negative impacts on metabolic diseases, cancer, and neurodegeneration—all of these conditions being themselves related to a shift of kinin responses. This confirms the kinin system to be a valuable target for treating chronic oral infections and the related complications.
2.4. Kinins and Tuberculosis
Tuberculosis is an infectious disease caused by bacillus Mycobacterium tuberculosis, mainly affecting the lungs. According to the Global Tuberculosis Report [69], it is the leading cause of death due to a single infectious agent, affecting 10 million people worldwide. In 2018, approximately 1.2 million TB deaths were estimated among HIV-negative people, with an additional 251,000 deaths among HIV-positive individuals. TB treatment requires the long-term use of a combined scheme of antibiotics, but the numbers of multiple drug-resistant TB cases has been dramatically increased during the last decades. The vaccination protection by M. bovis bacillus Calmette-Guérin (BCG) is partial, primarily preventing the severe forms of TB in children [69]. This indicates the current need to identify new strategies against TB infection. The relationship between Mycobaterium sp. and the kinin system has been sparsely explored. For instance, the systemic administration of BCG, as an inflammatory stimulus, triggered a long-term increase of rat paw edema induced by the selective kinin B1 receptor agonists des-Arg9-BK and Lys-des-Arg9-BK. This protocol of treatment also resulted in a synergistic inflammatory response, by the combination of des-Arg9-BK and BK, injected into the rat paw [70]. A similar treatment with BCG also potentiated the nociceptive and edematogenic responses induced by des-Arg9-BK, co-injected with formalin into the mouse paw [71]. In both cases, the effects of BCG were impeded by the treatment with the glucocorticoid dexamethasone, indicating an induction of B1 receptors. In both rats and mice, the treatment with BCG did not modify the inflammatory or painful responses caused by the selective B2 receptor agonist tyrosine8-BK, suggesting a selective modulation of B1 receptors by M. bovis. In a recent publication [72], it was proposed that kallikrein 12 (KLK12) exerts a protective role against M. bovis infection. This suggestion was based on in vivo and in vitro evidence showing that intranasal inoculation of M. bovis in mice, or its incubation with RAW264.7 macrophages, triggered an upregulation of KLK12. Additionally, KLK12 knockdown reduced the autophagy, apoptosis, and cytokine production in M. bovis-infected macrophages, favoring an increase of bacillus survival.
As for M. tuberculosis, a clinical study enrolled 13 patients that were submitted to first-line anti-TB therapy, with an induction phase of two months, including isoniazid, rifampin, ethambutol and pyrazinamide, followed by a consolidation phase with isoniazid plus rifampin. Notably, BK and des-Arg9-BK serum levels showed variations according to the phase of treatment: whereas BK contents were reduced at the induction phase, des-Arg9-BK levels were elevated. Instead, the serum contents of des-Arg9-BK were reduced during the consolidation phase of treatment. Thus, kinin levels might represent useful biological markers to predict the outcomes of TB treatment [20]. By using an in vivo model of M. tuberculosis infection, it was demonstrated that splenomegaly increased in B2 receptor knockout mice, whereas B1/B2 double knockouts presented a reduction of spleen weight [73]. Gene deletion of either B1 or B2 receptor did not alter the bacterial load in spleen or lungs. Notably, M. tuberculosis-infected wild-type mice displayed an upregulation of B1 receptors in spleen and lungs, according to assessment by immunohistochemistry [73]. Finally, the oral treatment with the selective non-peptide B1 receptor antagonist SSR240612 reduced the colony-forming units (CFU) in spleen and lungs of M. tuberculosis-infected mice. The inhibitory effects of SSR240612 on CFU counts were mirrored in vitro, in M. tuberculosis-infected RAW 264.7 macrophages [73]. Further in-depth studies are required to better understand whether the kinin system is involved in M. tuberculosis–host interactions, in either latent or active TB forms.
3. Viral Infections and Kinins
Viruses are the most common infectious agents worldwide. Although the number of viruses currently known to be pathogenic to humans is relatively small—approximately 220—epidemiological studies strongly suggest that this number is underestimated [74]. In this section, we review some key findings on how viruses modulate host cell biology to facilitate their own replication and how the kinin system impacts viral infections, with a particular focus on Covid-19, hantavirus, and rhinovirus. This emerging field of research provides insights for discovering new anti-viral and anti-inflammatory therapies for managing viral complications.
3.1. Crosstalk between SARS-CoV-2 and the Kinin System
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a highly contagious virus that was first identified in China (Wuhan city) and rapidly spread worldwide, causing the pandemic coronavirus disease 2019 (COVID-19). SARS-CoV-2 is a member of Betacoronavirus family, which includes severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV), i.e., those responsible for the outbreaks in 2002 and 2012, respectively. SARS-CoV-2 is an RNA virus that is enveloped and anchored by the spike glycoprotein (S) [75,76]. Current evidence shows that the virus invades target cells (e.g., pulmonary endothelial cells, pneumocytes type II, cardiomyocytes, blood cells) through the interaction of spike proteins with ACE2, leading to viral replication [77,78]. The common symptoms of COVID-19 are fever (98%), dry cough (76%), dyspnea (55%), myalgia and/or fatigue (44%), and only a minority of patients show gastrointestinal problems. Approximately 32% of COVID-19 patients are admitted to intensive care unit (ICU) due to respiratory problems—mainly breathing difficulty—with altered coagulation parameters (elevation of D-dimers concentration) [79].
It has been hypothesized that dysregulation of BK-related pathways could be present in Covid-19 patients, being especially related to respiratory complications. Firstly, the SARS-CoV-2 spike antigen binds to ACE2, which in turn internalizes, and thereby downregulates the expression and function of ACE2. Subsequently, there is an increase of blood pressure and pulmonary edema that might evolve to angioedema, likely through generation of BK active metabolites, such as des-Arg9-BK. In addition to the modulation of renin-angiotensin system (RAS), des-Arg9-BK binds to B1 receptors and enhance inflammation and vascular permeability, which is associated with acute lung injury [21]. The inflammatory setting evoked by the des–Arg9–BK–B1 receptor axis is related to a cytokine storm, with a marked increase in the production of several pro-inflammatory cytokines and chemokines (e.g., TNF, IL-1β, IL-6, CXCL5, CCL2 and CXCL1), finally mediating organ dysfunction [37].
Veerdonk and colleagues (2020) proposed that blocking plasma kallikrein activity (via lanadelumab), together with antiviral treatment, could prevent the acute respiratory distress syndrome in Covid-19, avoiding the ICU admission and mechanical ventilation. In particular, lanadelumab is a monoclonal antibody against the plasma kallikrein, which in turn is responsible for BK production through HMWK precursor, consequently interfering in coagulation cascade and complement system activation [80]. It is important to note that not every patient with Covid-19 will require an inhibition of the kinin signaling, whereas part of the viral load will be resolved in the lungs and thus there will be no second inflammatory wave.
In summary, different approaches could be tested to reduce SARS-CoV-2-associated symptoms and disease exacerbation, including (i) blockade of tissue and/or plasma kallikrein production, (ii) kinin degradation through recombinant ACE2, and (iii) modulation of B1 and B2 receptors and the downstream pathways.
3.2. Hantavirus Infection and BK Signaling
Hantavirus (HV) is a single-stranded, negative-sense viral RNA, belonging to the Bunyaviridae family, that causes hemorrhagic fever with renal syndrome (HFRS) and hantavirus pulmonary syndrome (HPS). The HV genus can be divided in two viral lineages: Old World and New World. The first is responsible for HFRS, which is widespread throughout Asia and Europe. The second strain is found in the Americas and is associated with the HPS. Additionally, the main pathophysiological impact in HV infection is systemic vascular leakage, i.e., the patients show impaired vascular tone and increased vascular permeability. All of these features can lead to hypotension, edema, shock, and coagulation abnormalities [81,82]. Preclinical studies have demonstrated that HV firstly invades endothelial cells for replication, and subsequently infect epithelial and vascular smooth muscle cells [83,84]. In line with this, BK promotes important outcomes on the vasculature, such as vasodilation and enhanced vascular permeability. Moreover, the BK–B2 receptor axis modulates a variety of second messengers, including intracellular Ca2+, endothelium-derived hyperpolarization factor, prostacyclin and nitric oxide, consequently leading to vascular smooth muscle cell relaxation [85]. Indeed, Taylor and coworkers (2013) demonstrated that incubation of FXII, PPK, and HMWK plasma protein, with HV-infected endothelial cells, produces enhanced secretion of BK and reduction of barrier function. In contrast, cells treated with the selective pseudopeptide B2 receptor antagonist HOE-140 (icatibant) exhibited a protective activity, with cells maintaining the endothelial integrity. In this study, the real-time cell permeability was assessed using electric cell-substrate impedance sensing [86]. Interestingly, in a case report, a 67-year-old female patient with severe HV infection was treated with HOE-140 (Icatibant, 30 mg, subcutaneously) and after 48 h, she presented a clear improvement of HFRS-related symptoms [87]. Moreover, Antonen and colleagues (2013) showed that HV caused severe capillary leakage syndrome in a 37-year-old Finnish male patient. On the other hand, when the patient received a single subcutaneous dose of the B2 receptor antagonist HOE-140 (30 mg), the condition was stabilized followed by gradual improvement. Most importantly, the patient survived. The rationale behind the clinical effects is that the drug can interfere in CD8+ T cell-promoted vascular leakage and cytokine secretion, as well as preventing complement activation [88]. It is tempting to suggest that antagonists of B2 receptors should be assessed in a broad spectrum of patients with HV infection, mainly in severe cases, i.e., those with life-threatening disease.
3.3. Interplay between Rhinovirus and Kinin Pathways
Rhinovirus (RV) is a microorganism popularly known as the “common cold virus”, which is responsible for up to half of acute upper respiratory infections in children and adults. In addition, RV infections also significantly contribute to asthma exacerbations, having a considerable economic burden due to work absenteeism. RV is a member of the family Picornaviridae and is a non-enveloped spherical virus, with a diameter of approximately 30 nm [89]. RV has been shown to potentiate the formation of pro-inflammatory mediators, especially of cytokines, via a NF-κB-dependent mechanism [90]. Indeed, mast cells, neutrophils, and airway macrophages secrete histamine, IL-8, and TNF after RV infection, leading to bronchial hyperresponsiveness [91]. Furthermore, Shelfoon and coworkers (2016) demonstrated that RV-16-inoculated human bronchial epithelial cells produced CXCL10 and CXCL8, triggering the migration of fibroblasts, with a subsequent progression of airway remodeling [92].
It was observed that severe infections by RV, mainly those affecting the upper respiratory system, are associated with the enhanced levels of kinins (i.e., BK and Lys-BK) in nasal secretion [93,94]. Alternatively, preclinical studies reported that kallikrein is also detected in lower airways in an allergic-induced asthma experimental model [95]. In 2008, Christiansen and colleagues supported these previous results by showing that tissue kallikrein is markedly activated on days 4 and 18 after RV infection, through evaluation of bronchoalveolar lavage fluid in patients with asthma. The response was accompanied by an increase of IL-8 levels, which was measured by ELISA assay [96].
dsRNA is produced by RV for replication during airway epithelium infection. As mentioned above, several observations suggest that RV infection promotes the local production of kinins. To verify whether dsRNA-derived RV enhances the expression of kinin receptors and exacerbates inflammatory response, Bengtson and coworkers used the human airway epithelial cell line BEAS-2B with dsRNA (Poly I:C) stimulation. Through RT-PCR and radioligand binding, it was possible to verify that B1 and B2 receptor levels increased in BEAS-2B cells, after Poly I:C administration, which was paralleled with p-ERK activation [97]. The key points of interference within the kinin system that might be useful for management of viral infections are depicted in Figure 2.
3.4. Viral Infections and Coagulation–KKS Axis
In the past, researchers were mainly focused on the relationship between bacteremia and the coagulation system. The current focus is on understanding its role in viral infections. Indeed, the coagulation cascade is activated during viral infections, for example by the Ebola virus, HIV, viral pneumonia (H1N1), and emerging pathogens such as Zika and Dengue virus. At first, the response is triggered as a form of protection for the organism, i.e., the immune cells together with platelets are responsible for stopping the spread of the pathogens within the body. Furthermore, fibrin (also called Factor Ia) serves as a scaffold for cell migration and adherence [98]. It is important to state that neutrophils secrete neutrophil extracellular traps (NETs) after tissue factor activation. NETs (e.g., nuclear DNA, histones plus elastase) have been shown to have a potential antiviral effect, mainly due to their negative charge [99]. In contrast, if the coagulation system is dysregulated, the viremia can initiate a disseminated intravascular coagulation and microvascular thrombosis–evoked hypoxia, promoting multiorgan failure and mortality. Hemorrhage can also occur through platelet dysregulation and consumption of coagulation factors, resulting from ongoing intravascular activation of the hemostatic system [100]. FXII can activate the coagulation cascade through FXI, or stimulate the KKS, magnifying the inflammatory responses. Enveloped viruses, such as herpes simplex virus, have been shown to increase the activation of intrinsic coagulation pathway [101]. The relevance of coagulation cascade and KKS has also been explored in COVID-19 [80]. In addition, it has been hypothesized that KKS mediates protection against H1N1 infection. On the other hand, Antoniak and colleagues (2016) demonstrated that there is no difference between HMWK-deficient and wild-type mice in terms of survival parameters, indicating that FXII-dependent KKS activation is not necessary for H1N1 protection. It seems that FXII function is independent on FXI and the KKS stimulation, which could explain the higher mortality in FXII−/− mice in H1N1 infection [102].
4. Kinins and Their Implication in Protozoan and Parasite Infections
4.1. Kinins in Leishmaniasis Pathophysiology
Leishmaniasis is caused by the protozoans of the genus Leishmania, belonging to the Trypanosomatidae family [103]. The parasites infect humans through sand-fly female bite. The extracellular forms of the parasite (promastigotes) can infect skin macrophages, transforming into the intracellular amastigote form and invading other macrophages, causing cutaneous leishmaniasis (CL). The parasite also infects mononuclear cells in the circulation and spreads to important organs such as the spleen, liver, bone marrow and lymph nodes in the intestine, leading to the potentially fatal form of visceral leishmaniasis (VL) [104]. Leishmaniasis affects tropical climate territories in Africa, Asia, Americas, and Europe. As of 2018, VL and CL were endemic in 77 and 89 countries, respectively [105]. Parasitological, immunological, and molecular approaches have been used for the diagnosis of leishmaniasis [106].
During blood-feeding, the Leishmania species vector Phlebotomy duboscq deposits saliva in the host dermis, which contains parasites and PdSP15 proteins that inhibit the activation of the contact pathway, preventing BK formation [107]. One clinical study indicated a role for kininogens in the pathophysiology of leishmaniasis, with elevated urinary levels of kininogens in 45 out of 50 kala-azar patients before treatment, suggesting that kinin precursors might be important markers of disease evolution [108]. An in vivo study reported that kinin system is activated by Leishmania donovani and L. chagasi infection. Hamster cheek pouch was infected topically by promastigotes, inducing macromolecular leakage and inflammatory edema, through the proteolytic release of kinins. The microvascular leakage was ameliorated by pre-treatment with the ACE inhibitor captopril. This response was inhibited by the peptide antagonist HOE-140 or by pre-treatment of promastigotes with K11777, an irreversible cysteine proteinase inhibitor (N-methylpiperazine-urea-Phe-homoPhe-vinylsulfone-benzene) [109].
In relation to rodent models, mice with gene deletion of kinin B2 receptors displayed a diminished resistance to visceral leishmaniasis [110]. The contribution of B2 receptor activation for development of host resistance can be explained by the modulation of the inflammatory response. Svensjo and cols. (2006) showed that captopril pretreatment was able to exacerbate the edematogenic responses in BALB/c or J129 wild-type mice in a paw edema model induced by promastigote injection, whereas HOE-140 blocked this effect. Interestingly, promastigotes were unable to induce edema in captopril-treated B2 receptor knockout mice. The in vitro step of the same study reported that B2 receptor activation modulated the uptake of promastigotes by macrophages. The treatment with BK at low (5–10 nM) and high concentrations (100–300 nM) augmented and decreased the uptake of promastigotes of L. chagasi, respectively, in captopril-treated macrophages. In both cases, this modulation relied on the activation of B2 receptors. The exposure of macrophages to K11777- or HOE-treated promastigotes led to a deficient internalization of the parasite through a mechanism dependent on protozoan-produced cysteine proteinases. Even with the reduction of parasite internalization by resident macrophages, the treatment with HOE-140 caused an increase in the rate of growth of intracellular amastigotes, making host cells susceptible to infection, likely by suppressing the protective response of macrophages [109].
It has been suggested that phagocytes are less efficient in internalizing L. major expressing inhibitors of serine peptidases (ISP), according to the evaluation of L. major WT promastigotes and L. major lineages lacking ISP2 and ISP3 (L. major Δisp2/3) [111,112]. The investigators concluded that ISP expression impaired the macrophage phagocytosis, by preventing neutrophil elastase-dependent activation of TLR4 [112,113]. More recently, based on the studies cited above, Svensjo and cols. (2014) demonstrated an interplay between the KKS and the inflammatory response evoked by exposure to L. major promastigotes in the hamster cheek pouch model. The authors demonstrated that L. major WT and L. major Δisp2/3 induced a potent microvascular leakage and leucocyte accumulation via the local kinin release. However, L. major Δisp2/3 were 20% less effective to induce transendothelial leakage of plasma, when compared with L. major WT promastigotes. The topical application of the B2 receptor antagonist HOE-140 diminished L. major WT-induced plasma leakage. In addition, the in vitro treatment with HOE-140, or with the peptide B1 receptor antagonist des-Arg9-Leu8-BK, inhibited the phagocytic uptake of L. major Δisp2/3 by macrophages and had no effects against L. major WT promastigotes. The authors also showed that L. major Δisp2/3 is capable of sequestering higher concentrations of kininogens than L. major WT, concluding that kininogens taken by L. major Δisp2/3 are opsonized and are unprotected and susceptible to proteolytic cleavage by pericellular serine proteases of macrophages. Thus, the protective role of ISPs is notable, preventing the action of phagocytes and the kinin release at the site of interaction between the parasite and the phagocytic cells [114].
4.2. Kinins Set the Tone in Trypanosoma Infections
There are two types of human trypanosomiasis that are caused by the protozoan of genus Trypanosoma [115]. Chagas disease or American trypanosomiasis is caused by Trypanosoma cruzi, and is mainly endemic in Latin America, being transmitted by the major triatomine bugs species, such as Panstrongylus megistus, Rhodnius prolixus, Triatoma (T.) brasiliensis, T. dimidiata and T. infestans. Currently, countries outside Latin America, such as the United States, have experienced a significant increase of imported cases, with an elevation in the numbers of infected individuals. At least eleven species of triatomine bugs have been reported to transmit Chagas in North America (T. gerstaeckeri, T. incrassata, T. indictiva, T. lecticularia, T. neotomae, T. protracta, T. recurva, T. rubida, T. rubrofasciata, T. sanguisuga, and Paratriatoma hirsute) [116]. Sleeping sickness, or African trypanosomiasis, is caused by T. brucei (b.) gambiense or T. b. rhodesiense, affecting western sub-Saharan Africa and eastern sub-Saharan Africa, respectively. In this form, the protozoan is transmitted by the tsetse fly bite [117]. The trypomastigote form arrives the bloodstream after contact with a wound or a mucous membrane of the host. The amastigote form is found in muscle and nerve cells [118]. P. chinai, T. dimidiata, T. infestans and T. rubrofasciata salivary triafestin inhibits both hemostatic and inflammatory pathways in the host skin, preventing KKS activation [119,120,121,122].
A very few studies describe the association between the KKS and the T. brucei infection. Kinin levels increased in the blood of volunteers infected with T. b. rhodesiense after 2 days of infection, and just after the maximum parasitaemia was reached (12th day). The kininogen levels were inversely proportional to the kinin levels in these individuals [123]. Kinin concentrations were augmented in urine, plasma, ears, skin and feet of mice, rats, rabbits or cattle infected with trypanosomes [123,124,125]. The tropolysin oligopeptidase was described in African trypanosomes, being able to degrade kinins, consequently abolishing the activation of kinin B2 receptors [126].
Regarding Chagas disease, a group of Brazilian researchers reported the involvement of the KKS in T. cruzi infection, in a sequence of scientific publications [17,127,128,129,130,131]. Much has been studied about cruzipain, the most abundant cysteinyl proteinase in T. cruzi, as a potential target for development of new therapies for treating T. cruz infection. Firstly, it was demonstrated that cruzipain might be inhibited by rat T-kininogen, depending on the enzyme isoform [132]. In addition, cruzipain is able to release Lys-BK by proteolysis of LMWK, and it also activates PPK [133]. The kinin release, after invasion of non-phagocytic cells (human primary umbilical vein endothelial cells (HUVECs) or Chinese hamster ovary (CHO) cells overexpressing the B2 receptor), by T. cruzi trypomastigotes, was potentiated by captopril treatment and inhibited by HOE-140. T. cruzi invasion was also hindered by treatment with the membrane-permeable cysteine proteinase inhibitor Z-(SBz)Cys-Phe-CHN2, demonstrating that cruzipain action is directly associated with kinin release [134]. Moreover, the kinin release by cruzipain activation was modulated through heparan sulfate treatment [135]. Scharfstein and cols. also proposed that B2 receptor-mediated intracellular Ca2+ elevation takes part of the strategies of protozoan invasion [136]. Alterations in intracellular Ca2+ were previously detected in endothelial cells infected by T. cruzi [137]. More recently, Mijares and cols. (2020) described that BK treatment elevates intracellular Ca2+ concentrations in cardiomyocytes from Chagasic subjects [138].
An in vitro study identified that treatment with DL-2-mercaptomethyl-3-guanidino-ethylthiopropanoic acid (MGTA), an inhibitor of kininase I (carboxypeptidase M/N), reduced the infection by T. cruzi in B1 receptor-expressing cells. In the same study, the mouse paw edema induced by trypomastigotes was mediated by the activation of constitutive B2 receptors and induced B1 receptors, in the early (3-h) and late-phase (24-h), respectively. It is important to highlight that edema formation was enhanced by captopril treatment [139]. Considering the immune cell infection, Monteiro and cols. demonstrated that dendritic cells (DCs) are sensors to kinins released by T. cruzi, with an involvement of B2 receptors [140]. Firstly, the authors described a cooperative activation of TLR2 and B2 receptors for induction of type 1 immunity after T. cruzi subcutaneous injection [141]. Additionally, the interplay between TLR2 and B2 receptors was linked with CXCR2 activation. Parasitemia and mortality rates were higher in mice lacking B2 receptors that had been infected with T. cruzi. Of note, the parasite mRNA expression was augmented in the heart of B2 receptor knockout mice. Additionally, effector T cells (CD4+ and CD8+) isolated from hearts of mice lacking B2 receptors produced diminished IFN-γ levels. The type-1 response impairment was also observed in CD11c+ DCs isolated from spleen of B2 receptor knockout mice that had been infected by trypomastigotes, with reduced IL-12 production. Collectively, these data suggest that B2 receptor activation in DCs has a putative role in T. cruzi infection resistance [142]. Corroborating this notion, an in vitro study reported that captopril enhanced the extent of parasite uptake by monocytes, increasing IL-17 expression by CD4+ T cells, via activation of kinin receptors [143].
The interaction between TLR2, CXCR2 and kinin B2 receptors was assessed in the hamster cheek pouch and mouse paw edema models induced by trypomastigotes. The investigators showed that trypomastigote infection stimulated the production of CXC chemokines via TLR2 activation by macrophages. TLR2 recognition was related to the activation of both B2 and CXCR2 receptors, as the inflammatory response induced by trypomastigotes was blocked by HOE-140 and repertaxin (a CXCR2 antagonist) [144]. The endothelial transmigration by T. cruzi was facilitated by BK and increased by the chemokine CCL2, substantiating the relationship between BK and chemokines in T. cruzi pathogenesis [145].
A link between T. cruzi infection and functional changes of endothelin-1 and kinin activity has been described [146]. Initially, Camargos et al. (2002) showed that ETA receptor activation is correlated with the immune response against T. cruzi infection [147]. Afterward, Andrade and cols. (2012) demonstrated that T. cruzi infection is mediated by an interplay between endothelin (ETAR and ETBR) and kinin B2 receptors. In this work, the antagonists of B2 receptor (HOE-140), ETAR (BQ-123) and ETBR (BQ-788) decreased parasite internalization in smooth muscle cells (HSMCs) infected by T. cruzi, and prevented the leucocyte accumulation in the hamster cheek pouch model, while the exogenous endothelin-1 enhanced T. cruzi uptake by HSMCs [148]. Based on this evidence, D’Orléans-Juste and cols. (2012) raised the importance of investigations about the role of de novo induction of B1 receptors by cytokines, in Chagas vasculopathies [149].
Schmitz and cols. (2014) suggested a crosstalk between the complement cascade and the kallikrein–kinin system in T. cruzi infection. The inhibition of C5a receptor (C5aR) or B2 receptor reduced the plasma leakage in the hamster cheek pouch model induced by trypomastigotes, attenuating the trypomastigote-induced IL-12p40/70 responses in CD11c+ DCs [150]. More recently, this research group demonstrated that T. cruzi-induced edema is dependent on mast cell degranulation (upstream) and FXII-mediated generation of BK (downstream). Sodium cromoglycate (mast cell stabilizer), infestin-4 (a specific inhibitor of FXIIa), HOE-140 and bosentan (a non-selective antagonist of ETAR/ETBR) treatments decreased the intracardiac parasite load in mice inoculated with trypomastigotes [151].
The kinin system involvement in chronic Chagas disease (CCHD) associated with systemic arterial hypertension (SAH) was also analyzed. The authors demonstrated an elevation of kinins and nitric oxide in patients with CCHD-SAH, based on increased plasma and tissue kallikrein and plasma kininase II activities, in addition to decreased kininogen concentrations (both HMWK and LMWK) in these subjects [152].
4.3. The Kinin System in Malaria
Malaria is caused by Plasmodium parasites, encompassing Plasmodium falciparum, P. vivax, P. malariae, P. ovale and P. knowlesi. The disease has a high prevalence, reaching 228 million cases in 2018, mainly affecting Africa; the P. falciparum species is responsible for most cases [153]. The transmission occurs through the bites of infected female Anopheles mosquitoes. The symptoms include fever, headache and chills, and if not treated can lead to death [153]. Remarkably, salivary hamadarin and anophensin proteins from A. stephensi inhibit BK release and the subsequent host inflammatory reactions [154,155].
In the 1970s and 1980s, researchers held some pieces of evidence about the relevance of kinins in Plasmodium infections. HMWK and LMWK levels decreased, while serum kallikrein concentrations were elevated in monkeys infected by P. knowlesi. These results were accompanied by increased parasitemia after 3 days of infection and were correlated with inflammatory responses in malaria disease (for review see: [156,157]). In guinea-pig injected intradermally and subdurally with active kallikrein fractions from P. knowlesi-infected monkeys, there was an increase of leucocyte infiltrates and endothelial permeability in skin and brain, respectively [158,159]. There was also an elevation of serum kininase activity in infected monkeys, demonstrating the importance of balance among the different components of the kinin system, minimizing the endothelial damage [160]. Ohtomo and Katori (1972) reproduced these data in mice infected by P. berghei. The authors observed that severe cases of malaria infection results in parasitemia elevation, diminished plasmatic kininogen and increased kinin formation, with development of hypotension [161]. The increase of kinin levels and kininase activity in the acute stages of malarial infection were correlated with circulatory disturbances [162]. More recently, the release of kinins by P. chabaudi and P. falciparum parasites was analyzed. In this study, the parasites internalized HMWK and liberated vasoactive kinins (Lys-BK, BK, and des-Arg9-BK) through the activation of cysteine proteases falcipain-2 and falcipain-3. The B1 and B2 receptor activation triggered intracellular Ca2+ increase in HUVEC, while the B1 and B2 receptor antagonists Des-Arg9[Leu8]-BK and HOE-140 restored this effect [19]. Additionally, Cotrin and cols. (2013) described kininogenase activity for falcipain-2 and falcipain-3 [163].
An in vitro assay reported that BK and its P2 and RI-BbKI analogs presented anti-plasmodial activity against P. gallinaceum sporozoites, but not to P. falciparum [164]. Another study demonstrated a crosstalk between B2 and Mas receptors in invasion of human erythrocytes by P. falciparum. BK and Ang-(1–7) impaired the erythrocytic cycle of P. falciparum, via PKA activity inhibition, an effect that was inhibited by the selective B2 and Mas receptor antagonists, HOE-140 and A779, respectively [165]. Interestingly, kinins have been correlated with placental malaria pathogenesis—P. berghei-infected erythrocytes display an upregulation of B2 receptors, triggering BK binding and receptor internalization, with erytrocyte engulfment by the trophoblast. In this case, alterations of the BK–B2 receptor axis are likely related to placental dysfunction in malaria [166]. Moreover, the activation of either B1 or B2 receptor by kinins released from P. falciparum-infected erythrocytes modulates the adhesion of infected erythrocytes to endothelial cells, disrupting the blood-brain barrier integrity [167]. In an in vivo study, P. chabaudi infection was related to an upregulation of B1 receptors in endothelial cells of sinusoids and other blood vessels of mouse liver. Of note, the anti-malarial drug chloroquine modulated the expression of B1 receptors in this tissue [168]. A clinical study showed the activation of coagulation cascade in severe malaria caused by P. falciparum. The patients presented a reduction of plasma antithrombin III concentrations, an elevation of thrombin-AT 111 complexes and neutrophil elastase, accompanied by a decrease of FXII and PPK activities. The authors suggested that neutrophil elastase and BK influence the pathophysiology of malaria [169]. The role of FXII-PPK was confirmed by Isawa et al. (2007). The investigators showed that anophensin, a KKS inhibitor identified in the salivary glands of vector Anopheles stephensi, inhibited kinin production, hindering the activation of both FXII and PPK [155].
4.4. Kinins and Snail Fever
Schistosomiasis is a parasitic disease caused by blood flukes (trematode worms) of Schistosoma genus; in 2018, it was endemic in 78 countries [170]. Schistosomiasis life cycle occurs by asexual reproduction in snails and sexual reproduction in mammals [171]. Snails release larval forms in freshwater, and these can penetrate skin, infecting individuals. The larvae turn into adult worms that live in the blood vessels inside of body. Eggs released by females, if not expelled, provoke immune reactions and organ damage. Four main species (S. intercalatum, S. japonicum, S. mansoni and S. mekongi) and S. haematobium are trematodes that infect humans and provoke intestinal and urogenital schistosomiasis forms, respectively [170]. Schistosomiasis is a public health problem that severely affects children and pregnant women, and disease complications can lead to increased morbidity and mortality [172].
Clinical data suggest the participation of kinins in schistosomiasis pathophysiology. PPK and HMWK concentrations were lower in hepatosplenic schistosomiasis patients, when compared with the control group [173,174]. A therapeutic approach employing a short human kininogen insertion into S. japonicum glutathione S-transferases, ameliorated their biological activity, and inhibited human umbilical vein endothelial cell proliferation [175].
A study identified a serine protease from S. mansoni cercariae genomic DNA: a kallikrein-like protease [176]. The same research group isolated the serine protease SmSP1 from S. mansoni, which presents homology with mouse plasma kallikrein and human factor I light chain. This protease likely plays a pivotal role in evasion to the host immune response. Surprisingly, S. mansoni infection did not produce specific antibodies to recombinant SmSP1 [177]. In another work, the investigators isolated the sK1 enzyme from S. mansoni adult worm homogenate supernatant, which presents kallikrein-like activity. sK1 is present on the surface of adult male worms causing hypotension in a kinin-dependent manner and facilitating the worm movement into the host visceral vasculature. The reduction of arterial blood pressure was demonstrated by intravenous injection of 3 µg of sK1 in rats [178]. As well, BK and its fragment 1–5 were associated with S. mansoni schistosomula navigation within the host tissues [179].
Ranasinghe and cols. (2015) identified a gene encoding a single domain Kunitz type protease inhibitor, SjKI-1, a coagulation inhibitor from S. japonicum that can be used for treatment of hematological disorders, being able to inhibit plasma kallikrein. These authors described SmKI-1 expression in S. mansoni, referring to anti-inflammatory and anti-coagulant properties. Kunitz proteins also protect the schistosomes from the host defense [180]. More recently, the ability of adult schistosomes to cause changes in the mouse plasma proteome was analyzed. In this work, the schistosomes generated carboxyl-truncated forms of HK in comparison with control (murine plasma without parasites), which produces the full-length protein. Schistosomula or adult male worms cleaves HMWK through the action of two cysteine proteases belonging to the calpain family (SmCalp1 and SmCalp2), independent on the activation of kallikreins or BK formation [181]. An anti-hemostatic function was observed to SmSP2 serine protease from S. mansoni. The SmSP2 releases BK from kininogen and modulates the host vasodilatation and fibrinolysis, through plasmin activation [182].
In an animal study, an intradermal injection of S. mansoni cercariae extracts into guinea pig skin induced edema formation, dependent on local BK production and leukocyte accumulation, while HOE-140 decreased the edema response. The authors also showed that BK did not induce significant leukocyte accumulation [183]. BK administration, in the presence or absence of PGE2, before cercarial extracts infection, significantly enhanced edema responses in mouse skin [184]. The therapeutic properties of BK on schistosomal hepatic fibrosis were analyzed in schistosoma cercariae-inoculated mice. The authors demonstrated a reduction of hepatic fibrosis in BK-treated groups, via RAS blockage, with decreased collagen deposition and TGFβ1 protein expression [185]. The role of kinin system in parasite infections is illustrated in Figure 3.
5. Kinins and Fungal Infections
Candida albicans is the most common opportunistic fungal pathogen in humans and is responsible for up to 50% of invasive candidiasis, albeit it is commensally present in skin and mucosal surfaces (i.e., oral cavity, genitourinary system, and gastrointestinal tract). Additionally, non-albicans species have been recognized as serious infection agents in humans, such as C. glabrata, C. parapsilosis, C. tropicalis, C. krusei, C. kefyr, C. lusitanie, and C. dubliniensis [186]. In general, immunocompetent individuals do not show Candida-related infections. In contrast, subjects with a compromised immunity can display annoying and painful infections, including oral thrush, cutaneous candidiasis, and/or vulvovaginitis. In severe cases, i.e., immunosuppressed patients (HIV, cancer therapy, organ transplantation, or diabetes), the development of systemic candidiasis can occur, being associated with high morbidity and mortality rates [187]. It is important to highlight that the conditions mentioned above are dependent on geographical location, population susceptibility, as well as the use of antifungal drugs.
In this context, kinin production is enhanced in infection as a host defense against invading pathogens, including the recruitment of neutrophils or monocytes to the infection site, or promotion of the pro-inflammatory cytokine secretion through other immune cells. Yet, kinins can exert a dual role, i.e., can be beneficial to pathogens by promoting the increase of vascular permeability, which is necessary for the microorganism nutrition or to colonize the host tissue (Figure 4) [22]. Therefore, tight control over the system is essential to define its activity. Candidadepsin is an extracellular aspartic proteinase that is present in Candida spp. and is the major virulence factor of yeasts. Candidadepsin has been proposed to activate kinin production through two routes: (i) the modulation of FXII, and (ii) the enzymatic cleavage of kininogen. Indeed, Kozik and colleagues (2015) showed that LMWK, rather than HMWK, is a substrate for C. albicans Sap3 (secreted aspartic protease 3) at sites of infection to produce an amount of kinins [188]. These results were also observed for C. parapsilosis Sap1 and Sap2, and all recombinant C. albicans Saps, except for Sap7 [189,190]. An approach using proteomics has been used to survey the HMWK-binding proteins, related to C. albicans cell wall [191]. Seweryn and coworkers (2015) extended the studies to FXII and PPK, showing that candidal cell wall interacts with these components, mainly five proteins (Als3, Eno1, Eft2, Tpi1 and Gpm1) [192]. The affinity of FXII, PPK and HMWK to fungal protein is evidenced by the works cited above and could be used as potential targets for candidiasis management. More recently, it was demonstrated that BK or kallikrein 1 overexpression protected IL-17 receptor A-knockout mice from candidiasis, by improving kidney function and survival rates. The authors also demonstrated that combination of BK with the antifungal drug fluconazol, resulted in increased survival rates of mice infected with C. albicans [193]. Thus, kinin-based strategies might be useful in patients with systemic yeast infections.
6. Conclusions
From the pioneer studies showing the relevance of the kinin system in Plasmodium infections in the 1970s, passing through the elegant contributions by Dr. Regoli about the effects of bacterial LPS on de novo induction of kinin B1 receptors in the 1980s, we arrived in the 1990s with a massive development of kinin antagonists, reaching the 2000s with the approval of icatibant for the treatment of hereditary angioedema, finally meeting the year of 2020 with startling evidence about the relevance of ACE2 and kinins for Covid-19 burden. For every infectious agent—bacteria, virus, fungus, protozoan or helminth—the mechanisms of invasion and escaping from the immune system involve one or more components of the kinin system. A huge number of preclinical studies and some pieces of clinical evidence clearly indicate that pharmacological modulation of kinin precursors, active peptides, kinin receptors and/or related enzymes might provide benefits for managing infectious diseases, including life-threatening situations. Despite this, the number of approved drugs targeting the kinin system is still low. Definitely, there is an urgent need for new options to treat infectious diseases, and the reasons involve the emerging resistance of pathogens and the high toxicity of most antimicrobial drugs, besides unmet clinical needs, such as complex cases of Covid-19. It is about time to reassess the available molecules and to develop novel tools targeting the kinin system as part of the endeavors against infectious diseases.
Funding
This work was supported by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES; Finance Code 001), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), and PUCRS. A.P.A.D. and R.B.M.S. are post-doc researchers receiving grants from CAPES and CNPq, respectively. MMC is a researcher career awardee of CNPq (304042/2018-8).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kaplan, A.P.; Joseph, K.; Silverberg, M. Pathways for bradykinin formation and inflammatory disease. J. Allergy Clin. Immunol. 2002, 109, 195–209. [Google Scholar] [CrossRef]
- Wu, Y. Contact pathway of coagulation and inflammation. Thromb. J. 2015, 13, 17. [Google Scholar] [CrossRef] [Green Version]
- Couture, R.; Harrisson, M.; Vianna, R.M.J.; Cloutier, F. Kinin receptors in pain and inflammation. Eur. J. Pharmacol. 2001, 429, 161–176. [Google Scholar] [CrossRef]
- Marceau, F.; Regoli, D. Bradykinin receptor ligands: Therapeutic perspectives. Nat. Rev. Drug Discov. 2004, 3, 845–852. [Google Scholar] [CrossRef]
- Leeb-Lundberg, L.M.F.; Marceau, F.; Müller-Esterl, W.; Pettibone, D.J.; Zuraw, B.L.; Leeb-Lundberg, L.M.F. International Union of Pharmacology. XLV. Classification of the Kinin Receptor Family: From Molecular Mechanisms to Pathophysiological Consequences. Pharmacol. Rev. 2005, 57, 27–77. [Google Scholar] [CrossRef] [Green Version]
- Costa-Neto, C.M.; Dillenburg-Pilla, P.; Heinrich, T.A.; Parreiras-E-Silva, L.T.; Pereira, M.G.; Reis, R.I.; Souza, P.P.C. Participation of kallikrein–kinin system in different pathologies. Int. Immunopharmacol. 2008, 8, 135–142. [Google Scholar] [CrossRef]
- Turner, A.J.; Hooper, N.M. The angiotensin–converting enzyme gene family: Genomics and pharmacology. Trends Pharmacol. Sci. 2002, 23, 177–183. [Google Scholar] [CrossRef]
- Calixto, J.B.; Cabrini, D.A.; Ferreira, J.; Campos, M.M. Kinins in pain and inflammation. Pain 2000, 87, 1–5. [Google Scholar] [CrossRef]
- Campos, M.M.; Leal, P.C.; Yunes, R.A.; Calixto, J.B. Non-peptide antagonists for kinin B1 receptors: New insights into their therapeutic potential for the management of inflammation and pain. Trends Pharmacol. Sci. 2006, 27, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Asraf, K.; Torika, N.; Danon, A.; Fleisher-Berkovich, S. Involvement of the Bradykinin B1 Receptor in Microglial Activation: In vitro and in vivo Studies. Front. Endocrinol. 2017, 8, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreau, M.E.; Garbacki, N.; Molinaro, G.; Brown, N.J.; Marceau, F.; Adam, A. The Kallikrein-Kinin System: Current and Future Pharmacological Targets. J. Pharmacol. Sci. 2005, 99, 6–38. [Google Scholar] [CrossRef] [Green Version]
- Okada, Y.; Takasawa, R.; Kubo, D.; Iwanaga, N.; Fujita, S.; Suzuki, K.; Suzuki, H.; Kamiya, H.; Chiba, K. Improved Tag-Assisted Liquid-Phase Peptide Synthesis: Application to the Synthesis of the Bradykinin Receptor Antagonist Icatibant Acetate. Org. Process. Res. Dev. 2019, 23, 2576–2581. [Google Scholar] [CrossRef]
- Lesage, A.; Gibson, C.; Marceau, F.; Ambrosi, H.-D.; Saupe, J.; Katzer, W.; Loenders, B.; Charest-Morin, X.; Knolle, J. In vitro Pharmacological Profile of a New Small Molecule Bradykinin B2 Receptor Antagonist. Front. Pharmacol. 2020, 11, 916. [Google Scholar] [CrossRef] [PubMed]
- Bhatwadekar, A.D.; Kansara, V.S.; Ciulla, T.A. Investigational plasma kallikrein inhibitors for the treatment of diabetic macular edema: An expert assessment. Expert Opin. Investig. Drugs 2020, 29, 237–244. [Google Scholar] [CrossRef]
- Levi, M.; Cohn, D.M.; Zeerleder, S. Hereditary angioedema: Linking complement regulation to the coagulation system. Res. Pract. Thromb. Haemost. 2019, 3, 38–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, J.N. Activation of the Bradykinin System by Angiotensin-Converting Enzyme Inhibitors. Eur. J. Inflamm. 2010, 8, 55–61. [Google Scholar] [CrossRef] [Green Version]
- Scharfstein, J.; Ramos, P.I.P.; Barral-Netto, M. G Protein-Coupled Kinin Receptors and Immunity against Pathogens. In Advances in Immunology; Elsevier BV: Amsterdam, The Netherlands, 2017; pp. 29–84. [Google Scholar]
- Fein, A.M. Treatment of severe systemic inflammatory response syndrome and sepsis with a novel bradykinin antagonist, deltibant (CP-0127). Results of a randomized, double-blind, placebo-controlled trial. CP-0127 SIRS and Sepsis Study Group. JAMA J. Am. Med. Assoc. 1997, 277, 482–487. [Google Scholar] [CrossRef]
- Bagnaresi, P.; de Barros, N.M.; Assis, D.M.; Melo, P.M.S.; Fonseca, R.G.; Juliano, M.A.; Pesquero, J.B.; Juliano, L.; Rosenthal, P.J.; Carmona, A.K.; et al. Intracellular proteolysis of kininogen by malaria parasites promotes release of active kinins. Malar. J. 2012, 11, 156. [Google Scholar] [CrossRef] [Green Version]
- Qian, X.; Nguyen, D.T.M.; Li, Y.; Lyu, J.; Graviss, E.A.; Hu, T.Y. Predictive value of serum bradykinin and desArg9-bradykinin levels for chemotherapeutic responses in active tuberculosis patients: A retrospective case series. Tuberculosis 2016, 101, S109–S118. [Google Scholar] [CrossRef] [Green Version]
- Sriram, K.; Insel, P.A. A hypothesis for pathobiology and treatment of COVID-19: The centrality of ACE1/ACE2 imbalance. Br. J. Pharmacol. 2020, in press. [Google Scholar] [CrossRef]
- Frick, I.-M.; Björck, L.; Herwald, H. The dual role of the contact system in bacterial infectious disease. Thromb. Haemost. 2007, 98, 497–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. Available online: https://apps.who.int/neglected_diseases/ntddata/sch/sch.html (accessed on 21 July 2020).
- Cassini, A.; Högberg, L.D.; Plachouras, D.; Quattrocchi, A.; Hoxha, A.; Simonsen, G.S.; Colomb-Cotinat, M.; Kretzschmar, M.E.; Devleesschauwer, B.; Cecchini, M.; et al. Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European Economic Area in 2015: A population-level modelling analysis. Lancet Infect. Dis. 2019, 19, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Park, B.S.; Lee, J.-O. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp. Mol. Med. 2013, 45, e66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front. Immunol. 2018, 9, 2379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regoli, D.C.; Marceau, F.; Lavigne, J. Induction of B1-receptors for kinins in the rabbit by a bacterial lipopolysaccharide. Eur. J. Pharmacol. 1981, 71, 105–115. [Google Scholar] [CrossRef]
- Marceau, F.; Lussier, A.; St-Pierre, S. Selective Induction of Cardiovascular Responses to des-Arg9-Bradykinin by Bacterial Endotoxin. Pharmacology 1984, 29, 70–74. [Google Scholar] [CrossRef]
- Bouthillier, J.; Deblois, D.; Marceau, F. Studies on the induction of pharmacological responses to des-Arg9-bradykinin in vitro and in vitro. Br. J. Pharmacol. 1987, 92, 257–264. [Google Scholar] [CrossRef] [Green Version]
- Saban, R. Lipopolysaccharide upregulates bradykinin 1 receptors in the isolated mouse bladder. J. Urol. 1998, 160, 2267–2273. [Google Scholar]
- Campos, M.M.; Souza, G.E.P.; Calixto, J.B. Upregulation of B1 receptor mediating des-Arg9-BK-induced rat paw oedema by systemic treatment with bacterial endotoxin. Br. J. Pharmacol. 1996, 117, 793–798. [Google Scholar] [CrossRef] [Green Version]
- Coelho, M.M.; Oliveira, C.R.; Pajolla, G.P.; Calixto, J.B.; Pelá, I.R. Central involvement of kinin B1 and B2 receptors in the febrile response induced by endotoxin in rats. Br. J. Pharmacol. 1997, 121, 296–302. [Google Scholar] [CrossRef] [Green Version]
- Passos, G.F.; Fernandes, E.S.; Campos, M.M.; Araújo, J.G.V.C.; Pesquero, J.L.; Souza, G.E.P.; Avellar, M.C.W.; Teixeira, M.M.; Calixto, J.B. Kinin B1 Receptor Up-Regulation after Lipopolysaccharide Administration: Role of Proinflammatory Cytokines and Neutrophil Influx. J. Immunol. 2004, 172, 1839–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paya, D.; Stoclet, J.C. Involvement of bradykinin and nitric oxide in the early hemodynamic effects of lipopolysaccharide in rats. Shock 1995, 3, 376–379. [Google Scholar] [PubMed]
- Luo, S.-F.; Wang, C.-C.; Chiu, C.-T.; Chien, C.-S.; Hsiao, L.-D.; Lin, C.-H.; Yang, C.-M. Lipopolysaccharide enhances bradykinin-induced signal transduction via activation of Ras/Raf/MEK/MAPK in canine tracheal smooth muscle cells. Br. J. Pharmacol. 2000, 130, 1799–1808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arndt, P.G.; Young, S.K.; Poch, K.R.; Nick, J.A.; Falk, S.; Schrier, R.W.; Worthen, G.S. Systemic Inhibition of the Angiotensin-Converting Enzyme Limits Lipopolysaccharide-Induced Lung Neutrophil Recruitment through Both Bradykinin and Angiotensin II-Regulated Pathways. J. Immunol. 2006, 177, 7233–7241. [Google Scholar] [CrossRef]
- Sodhi, C.P.; Wohlford-Lenane, C.; Yamaguchi, Y.; Prindle, T.; Fulton, W.B.; Wang, S.; McCray, P.B.; Chappell, M.; Hackam, D.J.; Jia, H. Attenuation of pulmonary ACE2 activity impairs inactivation of des-Arg 9 bradykinin/BKB1R axis and facilitates LPS-induced neutrophil infiltration. Am. J. Physiol. Cell. Mol. Physiol. 2018, 314, L17–L31. [Google Scholar] [CrossRef]
- Anton, E.L.; Fernandes, D.; Assreuy, J.; Silva-Santos, J.E. Bradykinin increases BP in endotoxemic rat: Functional and biochemical evidence of angiotensin II AT1/bradykinin B2 receptor heterodimerization. Br. J. Pharmacol. 2019, 176, 2608–2626. [Google Scholar] [CrossRef]
- Oehmcke-Hecht, S.; Köhler, J. Interaction of the Human Contact System with Pathogens—An Update. Front. Immunol. 2018, 9, 312. [Google Scholar] [CrossRef]
- Bengtson, S.H. Kinin receptor expression during Staphylococcus aureus infection. Blood 2006, 108, 2055–2063. [Google Scholar] [CrossRef]
- Geng, L.; Wang, S.; Zhao, Y.; Hu, H. Gene expression profile in mouse bacterial chronic rhinosinusitis. Exp. Ther. Med. 2019, 17, 3451–3458. [Google Scholar] [CrossRef] [Green Version]
- Waack, U.; Warnock, M.; Yee, A.; Huttinger, Z.; Smith, S.; Kumar, A.; Deroux, A.; Ginsburg, D.; Mobley, H.L.T.; Lawrence, D.A.; et al. CpaA Is a Glycan-Specific Adamalysin-like Protease Secreted by Acinetobacter baumannii That Inactivates Coagulation Factor XII. mBio 2018, 9, e01606-18. [Google Scholar] [CrossRef] [Green Version]
- Ding, C.; Scicluna, B.P.; Stroo, I.; Yang, J.; Roelofs, J.J.T.H.; Boer, O.J.; Vos, A.F.; Nürnberg, P.; Revenko, A.S.; Crosby, J.; et al. Prekallikrein inhibits innate immune signaling in the lung and impairs host defense during pneumosepsis in mice. J. Pathol. 2020, 250, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Stroo, I.; Zeerleder, S.; Ding, C.; Luken, B.; Roelofs, J.; de Boer, O.; Meijers, J.; Castellino, F.; van’t Veer, C.; van der Poll, T. Coagulation factor XI improves host defence during murine pneumonia-derived sepsis independent of factor XII activation. Thromb. Haemost. 2017, 117, 1601–1614. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; van’t Veer, C.; Roelofs, J.J.T.H.; Shukla, M.; McCrae, K.R.; Revenko, A.S.; Crosby, J.; van der Poll, T. Limited role of kininogen in the host response during gram-negative pneumonia-derived sepsis. Am. J. Physiol. Cell. Mol. Physiol. 2018, 314, L397–L405. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Yang, J.; van’t Veer, C.; van der Poll, T. Bradykinin receptor deficiency or antagonism do not impact the host response during gram-negative pneumonia-derived sepsis. Intensive Care Med. Exp. 2019, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Köhler, J.; Maletzki, C.; Koczan, D.; Frank, M.; Trepesch, C.; Revenko, A.S.; Crosby, J.R.; MacLeod, A.R.; Mikkat, S.; Oehmcke-Hecht, S. The contact system proteases play disparate roles in streptococcal sepsis. Haematologica 2020, 105, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Nitzsche, R.; Rosenheinrich, M.; Kreikemeyer, B.; Oehmcke-Hecht, S. Streptococcus pyogenes Triggers Activation of the Human Contact System by Streptokinase. Infect. Immun. 2015, 83, 3035–3042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, A.; Xie, Z.; Wang, B.; Colman, R.W.; Dai, J.; Wu, Y. An essential role of high-molecular-weight kininogen in endotoxemia. J. Exp. Med. 2017, 214, 2649–2670. [Google Scholar] [CrossRef] [PubMed]
- Pixley, R.A.; De La Cadena, R.; Page, J.D.; Kaufman, N.; Wyshock, E.G.; Chang, A.; Taylor, F.B.; Colman, R.W. The contact system contributes to hypotension but not disseminated intravascular coagulation in lethal bacteremia. In vitro use of a monoclonal anti-factor XII antibody to block contact activation in baboons. J. Clin. Investig. 1993, 91, 61–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghunathan, V.; Zilberman-Rudenko, J.; Olson, S.R.; Lupu, F.; McCarty, O.J.T.; Shatzel, J.J. The contact pathway and sepsis. Res. Pract. Thromb. Haemost. 2019, 3, 331–339. [Google Scholar] [CrossRef]
- Niederwanger, C.; Bachler, M.; Hell, T.; Linhart, C.; Entenmann, A.; Balog, A.; Auer, K.; Innerhofer, P. Inflammatory and coagulatory parameters linked to survival in critically ill children with sepsis. Ann. Intensive Care 2018, 8, 111. [Google Scholar] [CrossRef]
- Ridings, P.C.; Sugerman, H.J.; Blocher, C.R.; Fisher, B.J.; Fowler, A.A. Hemodynamic Effects of Bradykinin Antagonism in Porcine Gram-Negative Sepsis. J. Investig. Surg. 1995, 8, 115–122. [Google Scholar] [CrossRef]
- Tidjane, N.; Hachem, A.; Zaid, Y.; Merhi, Y.; Gaboury, L.; Girolami, J.-P.; Couture, R. A primary role for kinin B1 receptor in inflammation, organ damage, and lethal thrombosis in a rat model of septic shock in diabetes. Eur. J. Inflamm. 2015, 13, 40–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murugesan, P.; Jung, B.; Lee, D.; Khang, G.; Doods, H.; Wu, D. Kinin B1 Receptor Inhibition with BI113823 Reduces Inflammatory Response, Mitigates Organ Injury, and Improves Survival among Rats with Severe Sepsis. J. Infect. Dis. 2016, 213, 532–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merino, V.F.; Todiras, M.; Campos, L.A.; Saul, V.; Popova, E.; Baltatu, O.C.; Pesquero, J.B.; Bader, M. Increased susceptibility to endotoxic shock in transgenic rats with endothelial overexpression of kinin B1 receptors. J. Mol. Med. 2008, 86, 791–798. [Google Scholar] [CrossRef]
- Ni, A.; Yin, H.; Agata, J.; Yang, Z.; Chao, L.; Chao, J. Overexpression of kinin B1 receptors induces hypertensive response to Des-Arg9-bradykinin and susceptibility to inflammation. J. Biol. Chem. 2003, 278, 219–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bui, F.Q.; Almeida-da-Silva, C.L.C.; Huynh, B.; Trinh, A.; Liu, J.; Woodward, J.; Asadi, H.; Ojcius, D.M. Association between periodontal pathogens and systemic disease. Biomed. J. 2019, 42, 27–35. [Google Scholar] [CrossRef]
- Tapping, R.I.; Akashi, S.; Miyake, K.; Godowski, P.J.; Tobias, P.S. Toll-Like Receptor 4, But Not Toll-Like Receptor 2, Is a Signaling Receptor for Escherichia and Salmonella Lipopolysaccharides. J. Immunol. 2000, 165, 5780–5787. [Google Scholar] [CrossRef] [Green Version]
- Burns, E.; Bachrach, G.; Shapira, L.; Nussbaum, G. Cutting Edge: TLR2 Is Required for the Innate Response to Porphyromonas gingivalis: Activation Leads to Bacterial Persistence and TLR2 Deficiency Attenuates Induced Alveolar Bone Resorption. J. Immunol. 2006, 177, 8296–8300. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Collyer, C.A. Gingipains from Porphyromonas gingivalis—Complex domain structures confer diverse functions. Eur. J. Microbiol. Immunol. 2011, 1, 41–58. [Google Scholar] [CrossRef] [Green Version]
- Monteiro, A.C.; Scovino, A.; Raposo, S.; Gaze, V.M.; Cruz, C.; Svensjö, E.; Narciso, M.S.; Colombo, A.P.; Pesquero, J.B.; Feres-Filho, E.; et al. Kinin Danger Signals Proteolytically Released by Gingipain Induce Fimbriae-Specific IFN-γ-and IL-17-Producing T Cells in Mice Infected Intramucosally with Porphyromonas gingivalis. J. Immunol. 2009, 183, 3700–3711. [Google Scholar] [CrossRef] [Green Version]
- Rubinstein, I.; Potempa, J.; Travis, J.; Gao, X.-P. Mechanisms Mediating Porphyromonas gingivalis Gingipain RgpA-Induced Oral Mucosa Inflammation in vitro. Infect. Immun. 2001, 69, 1199–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.-W.; Huang, C.-H.; Huang, H.-C.; Lai, Y.-Y.; Lin, Y.-Y. Transvascular dissemination of Porphyromonas gingivalis from a sequestered site is dependent upon activation of the kallikrein/kinin pathway. J. Periodontal Res. 2006, 41, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Dornelles, F.N.; Santos, D.S.; Van Dyke, T.E.; Calixto, J.B.; Batista, E.L.; Campos, M.M. In vitro Up-Regulation of Kinin B1 Receptors after Treatment with Porphyromonas gingivalis Lipopolysaccharide in Rat Paw. J. Pharmacol. Exp. Ther. 2009, 330, 756–763. [Google Scholar] [CrossRef] [PubMed]
- Souza, P.P.C.; Lundberg, P.; Lundgren, I.; Magalhães, F.A.C.; Costa-Neto, C.M.; Lerner, U.H. Activation of Toll-like receptor 2 induces B1 and B2 kinin receptors in human gingival fibroblasts and in mouse gingiva. Sci. Rep. 2019, 9, 2973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plaza, K.; Kalinska, M.; Bochenska, O.; Meyer-Hoffert, U.; Wu, Z.; Fischer, J.; Falkowski, K.; Sasiadek, L.; Bielecka, E.; Potempa, B.; et al. Gingipains of Porphyromonas gingivalis Affect the Stability and Function of Serine Protease Inhibitor of Kazal-type 6 (SPINK6), a Tissue Inhibitor of Human Kallikreins. J. Biol. Chem. 2016, 291, 18753–18764. [Google Scholar] [CrossRef] [Green Version]
- Karkowska-Kuleta, J.; Surowiec, M.; Gogol, M.; Koziel, J.; Potempa, B.; Potempa, J.; Kozik, A.; Rapala-Kozik, M. Peptidylarginine Deiminase of Porphyromonas gingivalis Modulates the Interactions between Candida albicans Biofilm and Human Plasminogen and High-Molecular-Mass Kininogen. Int. J. Mol. Sci. 2020, 21, 2495. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Global Tuberculosis Report 2019. Available online: https://www.who.int/tb/publications/global_report/en/ (accessed on 25 July 2020).
- Campos, M.M.; Henriques, M.G.M.O.; Calixto, J.B. The role of B1 and B2 kinin receptors in oedema formation after long-term treatment with Mycobacterium bovis bacillus Calmette-Guérin (BCG). Br. J. Pharmacol. 1997, 120, 502–508. [Google Scholar] [CrossRef] [PubMed]
- de Campos, R.O.; Henriques, M.G.M.; Calixto, J. Systemic treatment with Mycobacterium bovis bacillus calmette-guerin (BCG) potentiates kinin B1 receptor agonist-induced nociception and oedema formation in the formalin test in mice. Neuropeptides 1998, 32, 393–403. [Google Scholar] [CrossRef]
- Sabir, N.; Hussain, T.; Liao, Y.; Wang, J.; Song, Y.; Shahid, M.; Cheng, G.; Mangi, M.H.; Yao, J.; Yang, L.; et al. Kallikrein 12 Regulates Innate Resistance of Murine Macrophages against Mycobacterium bovis Infection by Modulating Autophagy and Apoptosis. Cells 2019, 8, 415. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues-Junior, V.S.; Pail, P.B.; Villela, A.D.; Falcão, V.C.A.; Dadda, A.S.; Abbadi, B.L.; Pesquero, J.B.; Santos, D.S.; Basso, L.A.; Campos, M.M. Effect of the bradykinin 1 receptor antagonist SSR240612 after oral administration in Mycobacterium tuberculosis-infected mice. Tuberculosis 2018, 109, 1–7. [Google Scholar] [CrossRef]
- McArthur, D.B. Emerging Infectious Diseases. Nurs. Clin. North. Am. 2019, 54, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.; Ni, Z.; Hu, Y.; Liang, W.; Ou, C.; He, J.; Liu, L.; Shan, H.; Lei, C.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Chen, P.; Wang, J.; Feng, J.; Zhou, H.; Li, X.; Zhong, W.; Hao, P. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci. China Life Sci. 2020, 63, 457–460. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- van de Veerdonk, F.L.; Netea, M.G.; van Deuren, M.; van der Meer, J.W.M.; de Mast, Q.; Brüggemann, R.J.; van der Hoeven, H. Kallikrein-kinin blockade in patients with COVID-19 to prevent acute respiratory distress syndrome. eLife 2020, 9, e57555. [Google Scholar] [CrossRef]
- Khaiboullina, S.; Morzunov, S.; St. Jeor, S. Hantaviruses: Molecular Biology, Evolution and Pathogenesis. Curr. Mol. Med. 2005, 5, 773–790. [Google Scholar] [CrossRef]
- Simpson, S.Q.; Spikes, L.; Patel, S.; Faruqi, I. Hantavirus Pulmonary Syndrome. Infect. Dis. Clin. N. Am. 2010, 24, 159–173. [Google Scholar] [CrossRef]
- Zaki, S.R.; Greer, P.W.; Coffield, L.M.; Goldsmith, C.S.; Nolte, K.B.; Foucar, K.; Feddersen, R.M.; Zumwalt, R.E.; Miller, G.L.; Khan, A.S.; et al. Hantavirus pulmonary syndrome: Pathogenesis of an emerging infectious disease. Am. J. Pathol. 1995, 146, 552–579. [Google Scholar]
- Krüger, D.H.; Schönrich, G.; Klempa, B. Human pathogenic hantaviruses and prevention of infection. Hum. Vaccines 2011, 7, 685–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giles, T.D.; Sander, G.E.; Nossaman, B.D.; Kadowitz, P.J. Impaired Vasodilation in the Pathogenesis of Hypertension: Focus on Nitric Oxide, Endothelial-Derived Hyperpolarizing Factors, and Prostaglandins. J. Clin. Hypertens. 2012, 14, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.L.; Wahl-Jensen, V.; Copeland, A.M.; Jahrling, P.B.; Schmaljohn, C.S. Endothelial Cell Permeability during Hantavirus Infection Involves Factor XII-Dependent Increased Activation of the Kallikrein-Kinin System. PLoS Pathog. 2013, 9, e1003470. [Google Scholar] [CrossRef] [PubMed]
- Laine, O.; Leppänen, I.; Koskela, S.; Antonen, J.; Mäkelä, S.; Sinisalo, M.; Vaheri, A.; Mustonen, J. Severe Puumala virus infection in a patient with a lymphoproliferative disease treated with icatibant. Infect. Dis. 2015, 47, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Antonen, J.; Leppänen, I.; Tenhunen, J.; Arvola, P.; Mäkelä, S.; Vaheri, A.; Mustonen, J. A severe case of Puumala hantavirus infection successfully treated with bradykinin receptor antagonist icatibant. Scand. J. Infect. Dis. 2013, 45, 494–496. [Google Scholar] [CrossRef]
- To, K.K.W.; Yip, C.C.Y.; Yuen, K.-Y. Rhinovirus—From bench to bedside. J. Formos. Med. Assoc. 2017, 116, 496–504. [Google Scholar] [CrossRef] [Green Version]
- Edwards, M.R.; Hewson, C.A.; Laza-Stanca, V.; Lau, H.-T.H.; Mukaida, N.; Hershenson, M.B.; Johnston, S.L. Protein kinase R, IκB kinase-β and NF-κB are required for human rhinovirus induced pro-inflammatory cytokine production in bronchial epithelial cells. Mol. Immunol. 2007, 44, 1587–1597. [Google Scholar] [CrossRef]
- Custovic, A.; Belgrave, D.; Lin, L.; Bakhsoliani, E.; Telcian, A.G.; Solari, R.; Murray, C.S.; Walton, R.P.; Curtin, J.; Edwards, M.R.; et al. Cytokine Responses to Rhinovirus and Development of Asthma, Allergic Sensitization, and Respiratory Infections during Childhood. Am. J. Respir. Crit. Care Med. 2018, 197, 1265–1274. [Google Scholar] [CrossRef]
- Shelfoon, C.; Shariff, S.; Traves, S.L.; Kooi, C.; Leigh, R.; Proud, D. Chemokine release from human rhinovirus–infected airway epithelial cells promotes fibroblast migration. J. Allergy Clin. Immunol. 2016, 138, 114–122.e4. [Google Scholar] [CrossRef] [Green Version]
- Sheahan, P.; McConn-Walsh, R.; Walsh, M.; Costello, R.W. Subjects with non-allergic non-infectious perennial rhinitis do not show nasal hyper-responsiveness to bradykinin. Eur. Arch. Oto-Rhino-Laryngol. 2007, 264, 33–37. [Google Scholar] [CrossRef]
- Naclerio, R.M.; Proud, D.; Lichtenstein, L.M.; Kagey-Sobotka, A.; Hendley, J.O.; Sorrentino, J.; Gwaltney, J.M. Kinins are Generated During Experimental Rhinovirus Colds. J. Infect. Dis. 1988, 157, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, S.C.; Proud, D.; Sarnoff, R.B.; Juergens, U.; Cochrane, C.G.; Zuraw, B.L. Elevation of Tissue Kallikrein and Kinin in the Airways of Asthmatic Subjects after Endobronchial Allergen Challenge. Am. Rev. Respir. Dis. 1992, 145, 900–905. [Google Scholar] [CrossRef]
- Christiansen, S.C.; Eddleston, J.; Bengtson, S.H.; Jenkins, G.R.; Sarnoff, R.B.; Turner, R.B.; Gwaltney, J.M.; Zuraw, B.L. Experimental rhinovirus infection increases human tissue kallikrein activation in allergic subjects. Int. Arch. Allergy Immunol. 2008, 147, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Bengtson, S.H.; Eddleston, J.; Christiansen, S.C.; Zuraw, B.L. Double-stranded RNA increases kinin B1 receptor expression and function in human airway epithelial cells. Int. Immunopharmacol. 2007, 7, 1880–1887. [Google Scholar] [CrossRef] [PubMed]
- Gaertner, F.; Massberg, S. Blood coagulation in immunothrombosis—At the frontline of intravascular immunity. Semin. Immunol. 2016, 28, 561–569. [Google Scholar] [CrossRef]
- Agraz-Cibrian, J.M.; Giraldo, D.M.; Mary, F.-M.; Urcuqui-Inchima, S. Understanding the molecular mechanisms of NETs and their role in antiviral innate immunity. Virus Res. 2017, 228, 124–133. [Google Scholar] [CrossRef]
- Goeijenbier, M.; van Wissen, M.; van de Weg, C.; Jong, E.; Gerdes, V.E.A.; Meijers, J.C.M.; Brandjes, D.P.M.; van Gorp, E.C.M. Review: Viral infections and mechanisms of thrombosis and bleeding. J. Med. Virol. 2012, 84, 1680–1696. [Google Scholar] [CrossRef]
- Gershom, E.S.; Sutherland, M.R.; Lollar, P.; Pryzdial, E.L.G. Involvement of the contact phase and intrinsic pathway in herpes simplex virus-initiated plasma coagulation. J. Thromb. Haemost. 2010, 8, 1037–1043. [Google Scholar] [CrossRef]
- Antoniak, S.; Tatsumi, K.; Hisada, Y.; Milner, J.J.; Neidich, S.D.; Shaver, C.M.; Pawlinski, R.; Beck, M.A.; Bastarache, J.A.; Mackman, N. Tissue factor deficiency increases alveolar hemorrhage and death in influenza A virus-infected mice. J. Thromb. Haemost. 2016, 14, 1238–1248. [Google Scholar] [CrossRef]
- Claborn, D. The biology and control of leishmaniasis vectors. J. Glob. Infect. Dis. 2010, 2, 127. [Google Scholar] [CrossRef]
- Steverding, D. The history of leishmaniasis. Parasit. Vectors 2017, 10, 82. [Google Scholar] [CrossRef] [Green Version]
- WHO. Available online: https://apps.who.int/neglected_diseases/ntddata/leishmaniasis/leishmaniasis.html (accessed on 14 July 2020).
- Thakur, S.; Joshi, J.; Kaur, S. Leishmaniasis diagnosis: An update on the use of parasitological, immunological and molecular methods. J. Parasit. Dis. 2020, 44, 253–272. [Google Scholar] [CrossRef] [PubMed]
- Alvarenga, P.H.; Xu, X.; Oliveira, F.; Chagas, A.C.; Nascimento, C.R.; Francischetti, I.M.B.; Juliano, M.A.; Juliano, L.; Scharfstein, J.; Valenzuela, J.G.; et al. Novel Family of Insect Salivary Inhibitors Blocks Contact Pathway Activation by Binding to Polyphosphate, Heparin, and Dextran Sulfate. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2759–2770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Mishra, M.; Rajput, S.K.; Bajpai, S.; Singh, R.K. Detection and diagnostic applicability of human urinary kininogen in kala-azar patients. Parasitol. Res. 2012, 111, 1851–1855. [Google Scholar] [CrossRef] [PubMed]
- Svensjö, E.; Batista, P.R.; Brodskyn, C.I.; Silva, R.; Lima, A.P.C.A.; Schmitz, V.; Saraiva, E.; Pesquero, J.B.; Mori, M.A.S.; Müller-Esterl, W.; et al. Interplay between parasite cysteine proteases and the host kinin system modulates microvascular leakage and macrophage infection by promastigotes of the Leishmania donovani complex. Microbes Infect. 2006, 8, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Nico, D.; Feijó, D.; Maran, N.; Morrot, A.; Scharfstein, J.; Palatnik, M.; Palatnik-de-Sousa, C. Resistance to visceral leishmaniasis is severely compromised in mice deficient of bradykinin B2-receptors. Parasit. Vectors 2012, 5, 261. [Google Scholar] [CrossRef] [Green Version]
- Eschenlauer, S.C.P.; Faria, M.S.; Morrison, L.S.; Bland, N.; Ribeiro-Gomes, F.L.; DosReis, G.A.; Coombs, G.H.; Lima, A.P.C.A.; Mottram, J.C. Influence of parasite encoded inhibitors of serine peptidases in early infection of macrophages with Leishmania major. Cell. Microbiol. 2009, 11, 106–120. [Google Scholar] [CrossRef] [Green Version]
- Faria, M.S.; Reis, F.C.G.; Azevedo-Pereira, R.L.; Morrison, L.S.; Mottram, J.C.; Lima, A.P.C.A. Leishmania Inhibitor of Serine Peptidase 2 Prevents TLR4 Activation by Neutrophil Elastase Promoting Parasite Survival in Murine Macrophages. J. Immunol. 2011, 186, 411–422. [Google Scholar] [CrossRef] [Green Version]
- Faria, M.S.; Calegari-Silva, T.C.; Vivarini, A.D.C.; Mottram, J.C.; Lopes, U.G.; Lima, A.P.C.A. Role of protein kinase R in the killing of Leishmania major by macrophages in response to neutrophil elastase and TLR4 via TNFα and IFNβ. FASEB J. 2014, 28, 3050–3063. [Google Scholar] [CrossRef]
- Svensjö, E.; Nogueira de Almeida, L.; Vellasco, L.; Juliano, L.; Scharfstein, J. Ecotin-Like ISP of L. major Promastigotes Fine-Tunes Macrophage Phagocytosis by Limiting the Pericellular Release of Bradykinin from Surface-Bound Kininogens: A Survival Strategy Based on the Silencing of Proinflammatory G-Protein Coupled Kinin B2 and B1. Mediat. Inflamm. 2014, 2014, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Maxfield, L.; Bermudez, R. Trypanosomiasis (Trypansomiasis); StatPearls: Treasure Island, FL, USA, 2019. [Google Scholar]
- Bern, C.; Kjos, S.; Yabsley, M.J.; Montgomery, S.P. Trypanosoma cruzi and Chagas’ Disease in the United States. Clin. Microbiol. Rev. 2011, 24, 655–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.; Waseem, M. Chagas Disease (American Trypanosomiasis); StatPearls: Treasure Island, FL, USA, 2020. [Google Scholar]
- Dunn, N.; Wang, S.; Adigun, R. African Trypanosomiasis (Sleeping Sickness); StatPearls: Treasure Island, FL, USA, 2020. [Google Scholar]
- Kato, H.; Jochim, R.C.; Gomez, E.A.; Sakoda, R.; Iwata, H.; Valenzuela, J.G.; Hashiguchi, Y. A repertoire of the dominant transcripts from the salivary glands of the blood-sucking bug, Triatoma dimidiata, a vector of Chagas disease. Infect. Genet. Evol. 2010, 10, 184–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isawa, H.; Orito, Y.; Jingushi, N.; Iwanaga, S.; Morita, A.; Chinzei, Y.; Yuda, M. Identification and characterization of plasma kallikrein-kinin system inhibitors from salivary glands of the blood-sucking insect Triatoma infestans. FEBS J. 2007, 274, 4271–4286. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, D.; Tabbabi, A.; Yamamoto, D.S.; Kien, L.T.; Kato, H. Salivary gland transcriptome of the Asiatic Triatoma rubrofasciata. Acta Trop. 2020, 210, 105473. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Jochim, R.C.; Gomez, E.A.; Tsunekawa, S.; Valenzuela, J.G.; Hashiguchi, Y. Salivary gland transcripts of the kissing bug, Panstrongylus chinai, a vector of Chagas disease. Acta Trop. 2017, 174, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Boreham, P.F.L. Kinin release and the immune reaction in human trypanosomiasis caused by Trypanosoma rhodesiense. Trans. R. Soc. Trop. Med. Hyg. 1970, 64, 394–400. [Google Scholar] [CrossRef]
- Richards, W.H.G. Pharmacologically active substances in the blood, tissues and urine of mice infected with Trypanosoma brucei. Br. J. Pharmacol. Chemother. 1965, 24, 124–131. [Google Scholar] [CrossRef] [Green Version]
- Boreham, P.F.L. Pharmacologically Active Peptides produced in the Tissues of the Host during Chronic Trypanosome Infections. Nature 1966, 212, 190–191. [Google Scholar] [CrossRef]
- Morty, R.E.; Vadász, I.; Bulau, P.; Dive, V.; Oliveira, V.; Seeger, W.; Juliano, L. Tropolysin, a New Oligopeptidase from African Trypanosomes. Biochemistry 2005, 44, 14658–14669. [Google Scholar] [CrossRef]
- Scharfstein, J. Subverting bradykinin-evoked inflammation by co-opting the contact system. Curr. Opin. Hematol. 2018, 25, 347–357. [Google Scholar] [CrossRef]
- Scharfstein, J.; Gomes, J.D.A.S.; Correa-Oliveira, R. Back to the future in Chagas disease: From animal models to patient cohort studies, progress in immunopathogenesis research. Mem. Inst. Oswaldo Cruz 2009, 104, 187–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scharfstein, J. Parasite cysteine proteinase interactions with α2-macroglobulin or kininogens: Differential pathways modulating inflammation and innate immunity in infection by pathogenic trypanosomatids. Immunobiology 2006, 211, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Scharfstein, J.; Andrade, D. Infection-Associated Vasculopathy in Experimental Chagas Disease. In Advances in Parasitology Volume 57; Elsevier BV: Amsterdam, The Netherlands, 2011; pp. 101–127. [Google Scholar]
- Scharfstein, J.; Monteiro, A.C.; Schmitz, V.; Svensjö, E. Angiotensin-converting enzyme limits inflammation elicited by Trypanosoma cruzi cysteine proteases: A peripheral mechanism regulating adaptive immunity via the innate kinin pathway. Biol. Chem. 2008, 389, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Serveau, C.; Lalmanach, G.; Juliano, M.A.; Scharfstein, J.; Juliano, L.; Gauthier, F. Investigation of the substrate specificity of cruzipain, the major cysteine proteinase of Trypanosoma cruzi, through the use of cystatin-derived substrates and inhibitors. Biochem. J. 1996, 313, 951–956. [Google Scholar] [CrossRef]
- Del Nery, E.; Juliano, M.A.; Lima, A.P.C.A.; Scharfstein, J.; Juliano, L. Kininogenase Activity by the Major Cysteinyl Proteinase (Cruzipain) from Trypanosoma cruzi. J. Biol. Chem. 1997, 272, 25713–25718. [Google Scholar] [CrossRef] [Green Version]
- Scharfstein, J.; Morrot, A. A role for extracellular amastigotes in the immunopathology of Chagas disease. Mem. Inst. Oswaldo Cruz 1999, 94, 51–63. [Google Scholar] [CrossRef] [Green Version]
- Lima, A.P.C.A.; Almeida, P.C.; Tersariol, I.L.S.; Schmitz, V.; Schmaier, A.H.; Juliano, L.; Hirata, I.Y.; Müller-Esterl, W.; Chagas, J.R.; Scharfstein, J. Heparan Sulfate Modulates Kinin Release by Trypanosoma cruzi through the Activity of Cruzipain. J. Biol. Chem. 2002, 277, 5875–5881. [Google Scholar] [CrossRef] [Green Version]
- Scharfstein, J.; Schmitz, V.; Morandi, V.; Capella, M.M.A.; Lima, A.P.C.A.; Morrot, A.; Juliano, L.; Müller-Esterl, W. Host Cell Invasion by Trypanosoma cruzi is Potentiated by Activation of Bradykinin B2 Receptors. J. Exp. Med. 2000, 192, 1289–1300. [Google Scholar] [CrossRef]
- Morris, S.; Tanowitz, H.; Hatcher, V.; Bilezikian, J.; Wittner, M. Alterations in intracellular calcium following infection of human endothelial cells with Trypanosoma cruzi. Mol. Biochem. Parasitol. 1988, 29, 213–221. [Google Scholar] [CrossRef]
- Mijares, A.; Espinosa, R.; Adams, J.; Lopez, J.R. Increases in [IP3]i aggravates diastolic [Ca2+] and contractile dysfunction in Chagas’ human cardiomyocytes. PLoS Negl. Trop. Dis. 2020, 14, e0008162. [Google Scholar] [CrossRef]
- Todorov, A.G.; Andrade, D.; Pesquero, J.B.; Carvalho Araujo, R.; Bader, M.; Stewart, J.; Gera, L.; Müller-Esterl, W.; Morandi, V.; Goldenberg, R.C.S.; et al. Trypanosoma cruzi induces edematogenic responses in mice and invades cardiomyocytes and endothelial cells in vitro by activating distinct kinin receptor subtypes (B1/B2). FASEB J. 2003, 17, 73–75. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, A.C.; Schmitz, V.; Morrot, A.; de Arruda, L.B.; Nagajyothi, F.; Granato, A.; Pesquero, J.B.; Müller-Esterl, W.; Tanowitz, H.B.; Scharfstein, J. Bradykinin B2 Receptors of Dendritic Cells, Acting as Sensors of Kinins Proteolytically Released by Trypanosoma cruzi, Are Critical for the Development of Protective Type-1 Responses. PLoS Pathog. 2007, 3, e185. [Google Scholar] [CrossRef]
- Monteiro, A.C.; Schmitz, V.; Svensjo, E.; Gazzinelli, R.T.; Almeida, I.C.; Todorov, A.; de Arruda, L.B.; Torrecilhas, A.C.T.; Pesquero, J.B.; Morrot, A.; et al. Cooperative Activation of TLR2 and Bradykinin B2 Receptor Is Required for Induction of Type 1 Immunity in a Mouse Model of Subcutaneous Infection by Trypanosoma cruzi. J. Immunol. 2006, 177, 6325–6335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scharfstein, J.; Schmitz, V.; Svensjö, E.; Granato, A.; Monteiro, A.C. Kininogens Coordinate Adaptive Immunity through the Proteolytic Release of Bradykinin, an Endogenous Danger Signal Driving Dendritic Cell Maturation. Scand. J. Immunol. 2007, 66, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Coelho dos Santos, J.S.; Menezes, C.A.S.; Villani, F.N.A.; Magalhães, L.M.D.; Scharfstein, J.; Gollob, K.J.; Dutra, W.O. Captopril increases the intensity of monocyte infection by Trypanosoma cruzi and induces human T helper type 17 cells. Clin. Exp. Immunol. 2010, 162, 528–536. [Google Scholar] [CrossRef]
- Schmitz, V.; Svensjö, E.; Serra, R.R.; Teixeira, M.M.; Scharfstein, J. Proteolytic generation of kinins in tissues infected by Trypanosoma cruzi depends on CXC chemokine secretion by macrophages activated via Toll-like 2 receptors. J. Leukoc. Biol. 2009, 85, 1005–1014. [Google Scholar] [CrossRef]
- Coates, B.M.; Sullivan, D.P.; Makanji, M.Y.; Du, N.Y.; Olson, C.L.; Muller, W.A.; Engman, D.M.; Epting, C.L. Endothelial Transmigration by Trypanosoma cruzi. PLoS ONE 2013, 8, e81187. [Google Scholar] [CrossRef] [Green Version]
- Tanowitz, H.B. Role of vasoactive mediators in the pathogenesis of Chagas disease. Front. Biosci. 2003, 8, 1103. [Google Scholar] [CrossRef] [Green Version]
- Camargos, E.R.S.; Machado, C.R.S.; Teixeira, A.L.; Rocha, L.L.V.; Ferreira, A.J.; Almeida, A.P.; Barton, M.; Teixeira, M.M. Role of endothelin during experimental Trypanosoma cruzi infection in rats. Clin. Sci. 2002, 103, 64S–67S. [Google Scholar] [CrossRef]
- Andrade, D.; Serra, R.; Svensjö, E.; Lima, A.P.C.; Ramos Junior, E.S.; Fortes, F.S.; Morandini, A.C.F.; Morandi, V.; Soeiro, M.D.N.; Tanowitz, H.B.; et al. Trypanosoma cruzi invades host cells through the activation of endothelin and bradykinin receptors: A converging pathway leading to chagasic vasculopathy. Br. J. Pharmacol. 2012, 165, 1333–1347. [Google Scholar] [CrossRef] [Green Version]
- D’Orléans-Juste, P.; Bkaily, G.; Rae, G.A. Endothelin and bradykinin: ‘brothers-in-arms’ in Chagas vasculopathies? Br. J. Pharmacol. 2012, 165, 1330–1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, V.; Almeida, L.N.; Svensjö, E.; Monteiro, A.C.; Köhl, J.; Scharfstein, J. C5a and Bradykinin Receptor Cross-Talk Regulates Innate and Adaptive Immunity in Trypanosoma cruzi Infection. J. Immunol. 2014, 193, 3613–3623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nascimento, C.R.; Andrade, D.; Carvalho-Pinto, C.E.; Serra, R.R.; Vellasco, L.; Brasil, G.; Ramos-Junior, E.S.; da Mota, J.B.; Almeida, L.N.; Andrade, M.V.; et al. Mast Cell Coupling to the Kallikrein–Kinin System Fuels Intracardiac Parasitism and Worsens Heart Pathology in Experimental Chagas Disease. Front. Immunol. 2017, 8, 840. [Google Scholar] [CrossRef] [PubMed]
- Dellalibera-Joviliano, R.; Bestetti, R.B.; Lopes, G.S.; Furlan-Daniel, R.; Lopes, K.C.; Faria-Junior, M.; Junior, N.I. Kinins and nitric oxide in patients with chronic chagas disease and systemic arterial hypertension. Cardiovasc. Pathol. 2020, 49, 107257. [Google Scholar] [CrossRef]
- World Health Organization. World Malaria Report 2019. Available online: https://www.who.int/publications/i/item/world-malaria-report-2019 (accessed on 20 July 2020).
- Isawa, H.; Yuda, M.; Orito, Y.; Chinzei, Y. A Mosquito Salivary Protein Inhibits Activation of the Plasma Contact System by Binding to Factor XII and High Molecular Weight Kininogen. J. Biol. Chem. 2002, 277, 27651–27658. [Google Scholar] [CrossRef] [Green Version]
- Isawa, H.; Orito, Y.; Iwanaga, S.; Jingushi, N.; Morita, A.; Chinzei, Y.; Yuda, M. Identification and characterization of a new kallikrein-kinin system inhibitor from the salivary glands of the malaria vector mosquito Anopheles stephensi. Insect Biochem. Mol. Biol. 2007, 37, 466–477. [Google Scholar] [CrossRef]
- Maegraith, B.; Fletcher, A. The Pathogenesis of Mammalian Malaria. Adv. Parasitol. 1972, 10, 49–75. [Google Scholar]
- Maegraith, B. Other pathological processes in malaria. Bull. World Health Organ. 1974, 50, 187–193. [Google Scholar]
- Onabanjo, A.O.; Maegraith, B.G. Inflammatory changes in small blood vessels induced by kallikrein (kininogenase) in the blood of Macaca mulatta, infected with Plasmodium knowlesi. Ann. Trop. Med. Parasitol. 1970, 64, 227–236. [Google Scholar] [CrossRef]
- Onabanjo, A.O.; Maegraith, B.G. Pathological lesions produced in the brain by kallikrein (kininogenase) in Macaca mulatta, infected with Plasmodium knowlesi. Ann. Trop. Med. Parasitol. 1970, 64, 237–242. [Google Scholar] [CrossRef]
- Onabanjo, A.O.; Bhabani, A.R.; Maegraith, B.G. The significance of kinin-destroying enzymes activity in Plasmodium knowlesi malarial infection. Br. J. Exp. Pathol. 1970, 51, 534–540. [Google Scholar] [PubMed]
- Ohtomo, H.; Katori, M. Realible evidence of involvement of the kinin system in mouse malaria. Jpn. J. Pharmacol. 1972, 22, 493–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, I.G. The kallikrein-kinin system and its role in the hypotensive shock syndrome of animals infected with the haemoprotozoan parasites Babesia, Plasmodium and Trypanosoma. Gen. Pharmacol. Vasc. Syst. 1979, 10, 319–325. [Google Scholar] [CrossRef]
- Cotrin, S.S.; Gouvêa, I.E.; Melo, P.M.S.; Bagnaresi, P.; Assis, D.M.; Araújo, M.S.; Juliano, M.A.; Gazarini, M.L.; Rosenthal, P.J.; Juliano, L.; et al. Substrate specificity studies of the cysteine peptidases falcipain-2 and falcipain-3 from Plasmodium falciparum and demonstration of their kininogenase activity. Mol. Biochem. Parasitol. 2013, 187, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.F.; Alves, F.L.; Pedron, C.N.; Torres, M.D.T.; Silva, L.S.; Pinheiro, A.A.S.; Miranda, A.; Oliveira, V.X. Anti-plasmodial activity of bradykinin and analogs. Bioorg. Med. Chem. Lett. 2015, 25, 3311–3313. [Google Scholar] [CrossRef]
- Silva, L.D.S.; Peruchetti, D.D.B.; Silva, C.T.F.-D.; Ferreira-DaSilva, A.T.; Perales, J.; Caruso-Neves, C.; Pinheiro, A.A.S. Interaction between bradykinin B2 and Ang-(1–7) Mas receptors regulates erythrocyte invasion by Plasmodium falciparum. Biochim. Biophys. Acta Gen. Subj. 2016, 1860, 2438–2444. [Google Scholar] [CrossRef]
- de Moraes, L.V.; Barateiro, A.; Sousa, P.M.; Penha-Gonçalves, C. Bradykinin Sequestration by Plasmodium berghei Infected Erythrocytes Conditions B2R Signaling and Parasite Uptake by Fetal Trophoblasts. Front. Microbiol. 2018, 9, 3106. [Google Scholar] [CrossRef] [Green Version]
- Silva, L.S.; Pinheiro, A.S.; Teixeira, D.E.; Silva-Aguiar, R.P.; Peruchetti, D.B.; Scharfstein, J.; Caruso-Neves, C.; Pinheiro, A.A.S. Kinins Released by Erythrocytic Stages of Plasmodium falciparum Enhance Adhesion of Infected Erythrocytes to Endothelial Cells and Increase Blood Brain Barrier Permeability via Activation of Bradykinin Receptors. Front. Med. 2019, 6, 1–9. [Google Scholar] [CrossRef]
- Ventura, P.D.S.; Carvalho, C.P.F.; Barros, N.M.T.; Martins-Silva, L.; Dantas, E.O.; Martinez, C.; Melo, P.M.S.; Pesquero, J.B.; Carmona, A.K.; Nagaoka, M.R.; et al. Malaria infection promotes a selective expression of kinin receptors in murine liver. Malar. J. 2019, 18, 213. [Google Scholar] [CrossRef]
- Clemens, R.; Pramoolsinsap, C.; Lorenz, R.; Pukrittayakamee, S.; Bock, H.L.; White, N.J. Activation of the coagulation cascade in severe falciparum malaria through the intrinsic pathway. Br. J. Haematol. 1994, 87, 100–105. [Google Scholar] [CrossRef]
- WHO. Available online: https://www.who.int/news-room/fact-sheets/detail/schistosomiasis (accessed on 28 July 2020).
- Nelwan, M.L. Schistosomiasis: Life Cycle, Diagnosis, and Control. Curr. Ther. Res. 2019, 91, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Verjee, M.A. Schistosomiasis: Still a Cause of Significant Morbidity and Mortality. Res. Rep. Trop. Med. 2020, 10, 153–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manoukian, N.; Borges, D.R. Prealbumina, precalicreína e protrombina na forma hepatesplênica da esquistossomose: Catabolismo aumentado de proteínas da coagulação? Rev. Inst. Med. Trop. Sao Paulo 1984, 26, 237–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omran, S.A.; Amer, A.M.; El-Kaliouby, A.H.; Eldin, A.A.S. Study of contact activation in endemic hepatosplenomegaly. Blood Coagul. Fibrinolysis 1991, 2, 659–662. [Google Scholar] [CrossRef] [PubMed]
- Bentley, A.A.; Merkulov, S.M.; Peng, Y.; Rozmarynowycz, R.; Qi, X.; Pusztai-Carey, M.; Merrick, W.C.; Yee, V.C.; McCrae, K.R.; Komar, A.A. Chimeric Glutathione S-Transferases Containing Inserts of Kininogen Peptides. J. Biol. Chem. 2012, 287, 22142–22150. [Google Scholar] [CrossRef] [Green Version]
- Cocude, C.; Pierrot, C.; Cetre, C.; Godin, C.; Capron, A.; Khalife, J. Molecular characterization of a partial sequence encoding a novel Schistosoma mansoni serine protease. Parasitology 1997, 115, 395–402. [Google Scholar] [CrossRef]
- Cocude, C.; Pierrot, C.; Cetre, C.; Fontaine, J.; Godin, C.; Capron, A.; Khalife, J. Identification of a developmentally regulated Schistosoma mansoni serine protease homologous to mouse plasma kallikrein and human factor I. Parasitology 1999, 118, 389–396. [Google Scholar] [CrossRef]
- Da’dara, A.; Skelly, P.J. Manipulation of vascular function by blood flukes? Blood Rev. 2011, 25, 175–179. [Google Scholar] [CrossRef] [Green Version]
- Grabe, K.; Haas, W. Navigation within host tissues: Schistosoma mansoni and Trichobilharzia ocellata schistosomula respond to chemical gradients. Int. J. Parasitol. 2004, 34, 927–934. [Google Scholar] [CrossRef]
- Ranasinghe, S.L.; Fischer, K.; Gobert, G.N.; McManus, D.P. Functional expression of a novel Kunitz type protease inhibitor from the human blood fluke Schistosoma mansoni. Parasit. Vectors 2015, 8, 408. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Da’dara, A.A.; Skelly, P.J. The blood fluke Schistosoma mansoni cleaves the coagulation protein high molecular weight kininogen (HK) but does not generate the vasodilator bradykinin. Parasit. Vectors 2018, 11, 182. [Google Scholar] [CrossRef] [Green Version]
- Leontovyč, A.; Ulrychová, L.; O’Donoghue, A.J.; Vondrášek, J.; Marešová, L.; Hubálek, M.; Fajtová, P.; Chanová, M.; Jiang, Z.; Craik, C.S.; et al. SmSP2: A serine protease secreted by the blood fluke pathogen Schistosoma mansoni with anti-hemostatic properties. PLoS Negl. Trop. Dis. 2018, 12, e0006446. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, M.M.; Doenhoff, M.J.; McNeice, C.; Williams, T.J.; Hellewell, P.G. Mechanisms of the inflammatory response induced by extracts of Schistosoma mansoni larvae in guinea pig skin. J. Immunol. 1993, 151, 5525–5534. [Google Scholar] [PubMed]
- Fallon, P.G.; Teixeira, M.M.; Neice, C.M.; Williams, T.J.; Hellewell, P.G.; Doenhoff, M.J. Enhancement of Schistosoma mansoni Infectivity by Intradermal Injections of Larval Extracts: A Putative Role for Larval Proteases. J. Infect. Dis. 1996, 173, 1460–1466. [Google Scholar] [CrossRef] [PubMed]
- Parreira, N.A.; Ramalho, F.S.; Augusto, M.J.; Silva, D.M.; Prado, C.M.; Elias Júnior, J.; Rodrigues, V.; Ramalho, L.N.Z. The comparative efficacy of renin-angiotensin system blockers in schistosomal hepatic fibrosis. Exp. Parasitol. 2018, 191, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Silva, S.; Negri, M.; Henriques, M.; Oliveira, R.; Williams, D.W.; Azeredo, J. Candida glabrata, Candida parapsilosis and Candida tropicalis: Biology, epidemiology, pathogenicity and antifungal resistance. FEMS Microbiol. Rev. 2012, 36, 288–305. [Google Scholar] [CrossRef] [Green Version]
- Kullberg, B.J.; Arendrup, M.C. Invasive Candidiasis. N. Engl. J. Med. 2015, 373, 1445–1456. [Google Scholar] [CrossRef] [Green Version]
- Kozik, A.; Gogol, M.; Bochenska, O.; Karkowska-Kuleta, J.; Wolak, N.; Kamysz, W.; Aoki, W.; Ueda, M.; Faussner, A.; Rapala-Kozik, M. Kinin release from human kininogen by 10 aspartic proteases produced by pathogenic yeast Candida albicans. BMC Microbiol. 2015, 15, 60. [Google Scholar] [CrossRef] [Green Version]
- Bras, G.; Bochenska, O.; Rapala-Kozik, M.; Guevara-Lora, I.; Faussner, A.; Kamysz, W.; Kozik, A. Release of biologically active kinin peptides, Met-Lys-bradykinin and Leu-Met-Lys-bradykinin from human kininogens by two major secreted aspartic proteases of Candida parapsilosis. Peptides 2013, 48, 114–123. [Google Scholar] [CrossRef]
- Bras, G.; Bochenska, O.; Rapala-Kozik, M.; Guevara-Lora, I.; Faussner, A.; Kozik, A. Extracellular aspartic protease SAP2 of Candida albicans yeast cleaves human kininogens and releases proinflammatory peptides, Met-Lys-bradykinin and des-Arg9-Met-Lys-bradykinin. Biol. Chem. 2012, 393, 829–839. [Google Scholar] [CrossRef] [Green Version]
- Karkowska-Kuleta, J.; Kedracka-Krok, S.; Rapala-Kozik, M.; Kamysz, W.; Bielinska, S.; Karafova, A.; Kozik, A. Molecular determinants of the interaction between human high molecular weight kininogen and Candida albicans cell wall: Identification of kininogen-binding proteins on fungal cell wall and mapping the cell wall-binding regions on kininogen molecule. Peptides 2011, 32, 2488–2496. [Google Scholar] [CrossRef] [PubMed]
- Seweryn, K.; Karkowska-Kuleta, J.; Wolak, N.; Bochenska, O.; Kedracka-Krok, S.; Kozik, A.; Rapala-Kozik, M. Kinetic and thermodynamic characterization of the interactions between the components of human plasma kinin-forming system and isolated and purified cell wall proteins of Candida albicans. Acta Biochim. Pol. 2015, 62, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Ramani, K.; Garg, A.V.; Jawale, C.V.; Conti, H.R.; Whibley, N.; Jackson, E.K.; Shiva, S.S.; Horne, W.; Kolls, J.K.; Gaffen, S.L.; et al. The Kallikrein-Kinin System: A Novel Mediator of IL-17-Driven Anti-Candida Immunity in the Kidney. PLoS Pathog. 2016, 12, e1005952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Relevance of components of the kinin system in bacterial infections. (A) E. coli LPS is a classical stimulus for kinin B1 receptor induction in vascular tissues, likely contributing to hypotension in systemic inflammation. E. coli LPS-induced B1 receptor upregulation rely on TLR4 activation and de novo protein synthesis, mediated by the transcriptional factor NF-κB. Part of the hypotensive effects of LPS are related to an increased production of BK and B2 receptor activation. (B) Streptococcal respiratory infections caused by S. pyogenes trigger the activation of contact system, via modulation of FXII, PPK and kallikrein. Plasmin activation by S. pyogenes streptokinase increases BK levels. Upon the bacteria spread throughout the blood stream due to increased vascular permeability, the patient develops systemic inflammatory response syndrome (SIRS) with coagulopathy and hypotension, causing multiple organ failure and shock. The modulation of one or more components of kinin formation pathways might be useful for management of septic patients. (C) P. gingivalis-produced gingipains cleave kininogen precursors, leading to BK and Lys-BK formation in periodontal tissues. Part of tissue destruction is mediated by the activation of constitutive B2 receptors. P. gingivalis LPS activates TLR2 and induces an upregulation of B1 receptors, which might sustain the chronic inflammation in periodontal disease. (D) M. tuberculosis infection is able to upregulate kinin B1 receptors in lungs, with an increased formation of the selective B1 receptor agonist, des-Arg9-BK from BK. Pharmacological inhibitors of B1 receptors might be therapeutic adjuvants for reducing TB infection burden.
Figure 1.
Relevance of components of the kinin system in bacterial infections. (A) E. coli LPS is a classical stimulus for kinin B1 receptor induction in vascular tissues, likely contributing to hypotension in systemic inflammation. E. coli LPS-induced B1 receptor upregulation rely on TLR4 activation and de novo protein synthesis, mediated by the transcriptional factor NF-κB. Part of the hypotensive effects of LPS are related to an increased production of BK and B2 receptor activation. (B) Streptococcal respiratory infections caused by S. pyogenes trigger the activation of contact system, via modulation of FXII, PPK and kallikrein. Plasmin activation by S. pyogenes streptokinase increases BK levels. Upon the bacteria spread throughout the blood stream due to increased vascular permeability, the patient develops systemic inflammatory response syndrome (SIRS) with coagulopathy and hypotension, causing multiple organ failure and shock. The modulation of one or more components of kinin formation pathways might be useful for management of septic patients. (C) P. gingivalis-produced gingipains cleave kininogen precursors, leading to BK and Lys-BK formation in periodontal tissues. Part of tissue destruction is mediated by the activation of constitutive B2 receptors. P. gingivalis LPS activates TLR2 and induces an upregulation of B1 receptors, which might sustain the chronic inflammation in periodontal disease. (D) M. tuberculosis infection is able to upregulate kinin B1 receptors in lungs, with an increased formation of the selective B1 receptor agonist, des-Arg9-BK from BK. Pharmacological inhibitors of B1 receptors might be therapeutic adjuvants for reducing TB infection burden.

Figure 2.
The role of kinin system in viral infections. (A) The kallikrein–kinin pathways and their intermediates, including enzymes, active metabolites, and receptors. In addition, the current pharmacological targets to modulate the system (e.g., lanadelumab and icatibant). Dashed lines designate the ligands and their respective receptors. (B) Hypothetical mechanism of crosstalk between SARS-CoV-2 and the kinin system, in which it could be one of the responsible routes for the worsening of Covid-19 clinical evolution. (C) Hantavirus (HV) modulates the bradykinin (BK)–B2 receptor axis promoting the release of Ca2+, endothelium-derived hyperpolarization factor (EDHF), prostacyclin and, nitric oxide (NO). Consequently, increasing the vascular permeability. (D) The “common cold virus”, called Rhinovirus (RV), has been suggested to enhance the expression of BK receptors (B1 and B2) by increasing the kinin agonists (i.e., BK, kallidin, and des-Arg9-BK) in nasal secretion of patients with respiratory syndromes. LMWK = low molecular weight kininogen; HMWK = high molecular weight kininogen; ACE2 = angiotensin-converting enzyme 2.
Figure 2.
The role of kinin system in viral infections. (A) The kallikrein–kinin pathways and their intermediates, including enzymes, active metabolites, and receptors. In addition, the current pharmacological targets to modulate the system (e.g., lanadelumab and icatibant). Dashed lines designate the ligands and their respective receptors. (B) Hypothetical mechanism of crosstalk between SARS-CoV-2 and the kinin system, in which it could be one of the responsible routes for the worsening of Covid-19 clinical evolution. (C) Hantavirus (HV) modulates the bradykinin (BK)–B2 receptor axis promoting the release of Ca2+, endothelium-derived hyperpolarization factor (EDHF), prostacyclin and, nitric oxide (NO). Consequently, increasing the vascular permeability. (D) The “common cold virus”, called Rhinovirus (RV), has been suggested to enhance the expression of BK receptors (B1 and B2) by increasing the kinin agonists (i.e., BK, kallidin, and des-Arg9-BK) in nasal secretion of patients with respiratory syndromes. LMWK = low molecular weight kininogen; HMWK = high molecular weight kininogen; ACE2 = angiotensin-converting enzyme 2.
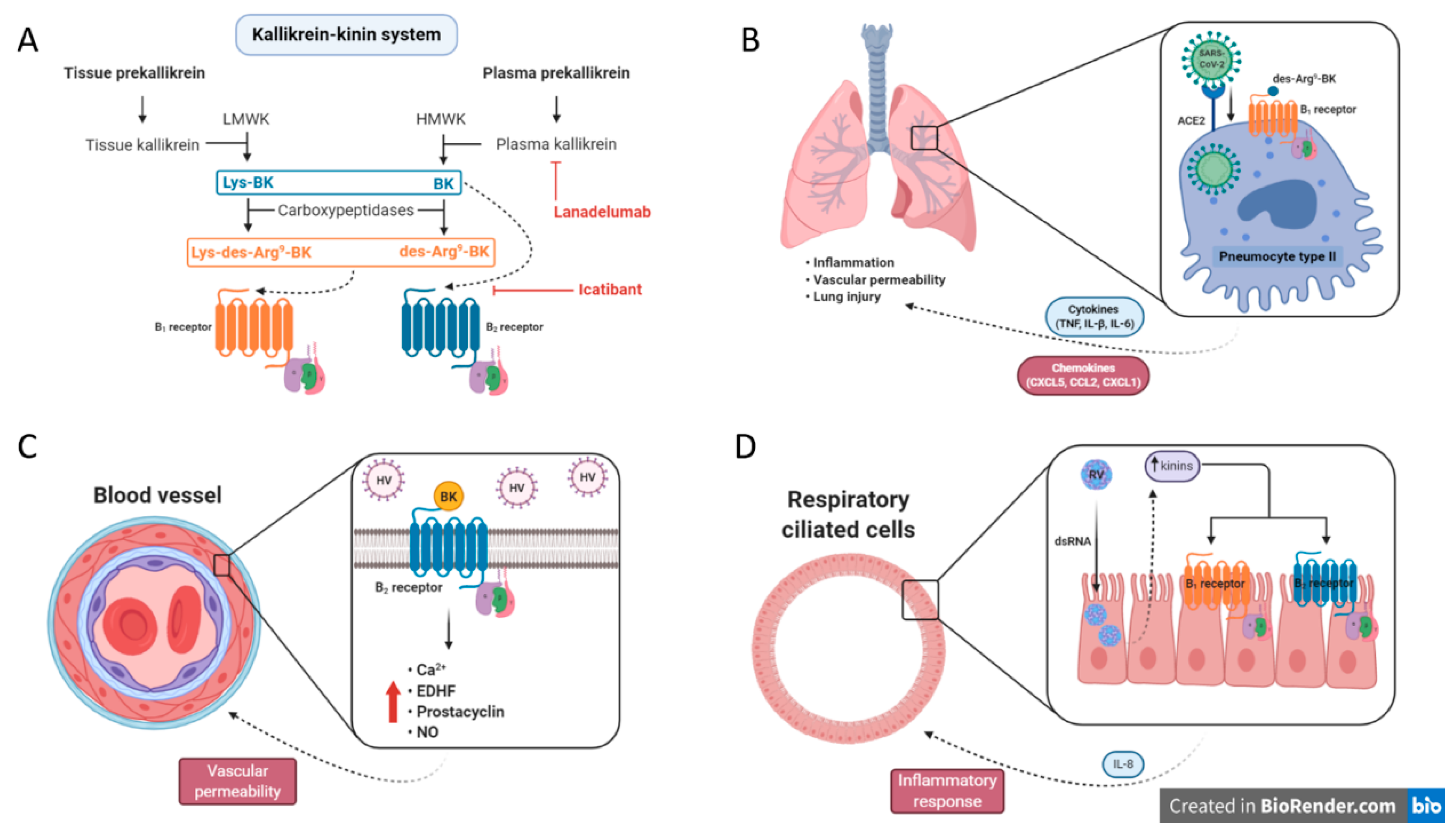
Figure 3.
Animal models of parasite infection and the role of kinins. (A) Leishmania promastigotes induced macrophage effector responses through B2 receptor activation by BK in the paw edema model. The treatment with BK increased the uptake of promastigotes by macrophages, whereas HOE-140 blocked this effect. Even with the reduction of parasite internalization, the treatment with HOE-140 caused an increase in the rate of growth of intracellular amastigotes, making host cells susceptible to infection. (B) Trypomastigotes release kinins and sensitize dendritic cells (DCs) via B2 receptor activation. A cooperative activation of TLR2, CXCR2 and B2 receptors induces type 1 immunity. An impairment of type-1 responses was observed in CD11c+ DCs isolated from spleen of B2 receptor knockout mice, that had been infected by trypomastigotes, with reduced IL-12 production. (C) Monkeys or mice infected by Plasmodium presented decreased HMWK and LMWK levels, while serum kallikrein concentrations and kinin formation were elevated. These results were accompanied by increased parasitemia and kininase activity. Parasites internalized plasmatic kininogen and liberated vasoactive kinins (Lys-BK, BK, and des-Arg9-BK) through the activation of cysteine proteases falcipain-2 and falcipain-3. B1 and B2 receptor activation triggered intracellular Ca2+ increase in endothelial cells causing circulatory disturbances, while the selective kinin antagonists des-Arg9[Leu8]-BK and HOE-140 restored this effect. (D) Intradermal injection of Schistosoma cercariae into the guinea pig skin induced edema formation, BK release and leukocyte accumulation, while HOE-140 decreased the edema response. Adult male worms cleave HMWK through protease activation. Proteases trigger BK production from kininogen, stimulating the release of tissue plasminogen activator (tPA) from vascular endothelial cells, which would promote fibrinolysis and anticoagulant effects.
Figure 3.
Animal models of parasite infection and the role of kinins. (A) Leishmania promastigotes induced macrophage effector responses through B2 receptor activation by BK in the paw edema model. The treatment with BK increased the uptake of promastigotes by macrophages, whereas HOE-140 blocked this effect. Even with the reduction of parasite internalization, the treatment with HOE-140 caused an increase in the rate of growth of intracellular amastigotes, making host cells susceptible to infection. (B) Trypomastigotes release kinins and sensitize dendritic cells (DCs) via B2 receptor activation. A cooperative activation of TLR2, CXCR2 and B2 receptors induces type 1 immunity. An impairment of type-1 responses was observed in CD11c+ DCs isolated from spleen of B2 receptor knockout mice, that had been infected by trypomastigotes, with reduced IL-12 production. (C) Monkeys or mice infected by Plasmodium presented decreased HMWK and LMWK levels, while serum kallikrein concentrations and kinin formation were elevated. These results were accompanied by increased parasitemia and kininase activity. Parasites internalized plasmatic kininogen and liberated vasoactive kinins (Lys-BK, BK, and des-Arg9-BK) through the activation of cysteine proteases falcipain-2 and falcipain-3. B1 and B2 receptor activation triggered intracellular Ca2+ increase in endothelial cells causing circulatory disturbances, while the selective kinin antagonists des-Arg9[Leu8]-BK and HOE-140 restored this effect. (D) Intradermal injection of Schistosoma cercariae into the guinea pig skin induced edema formation, BK release and leukocyte accumulation, while HOE-140 decreased the edema response. Adult male worms cleave HMWK through protease activation. Proteases trigger BK production from kininogen, stimulating the release of tissue plasminogen activator (tPA) from vascular endothelial cells, which would promote fibrinolysis and anticoagulant effects.


Figure 4.
Involvement of kinin system in candidiasis. The pathogenicity of Candida spp. in sequential stages of interaction (Sap -> LMWK -> kinins -> effects) with kinin signaling in human host infection. Sap = secreted aspartic protease; LMWK = low molecular weight kininogen.
Figure 4.
Involvement of kinin system in candidiasis. The pathogenicity of Candida spp. in sequential stages of interaction (Sap -> LMWK -> kinins -> effects) with kinin signaling in human host infection. Sap = secreted aspartic protease; LMWK = low molecular weight kininogen.
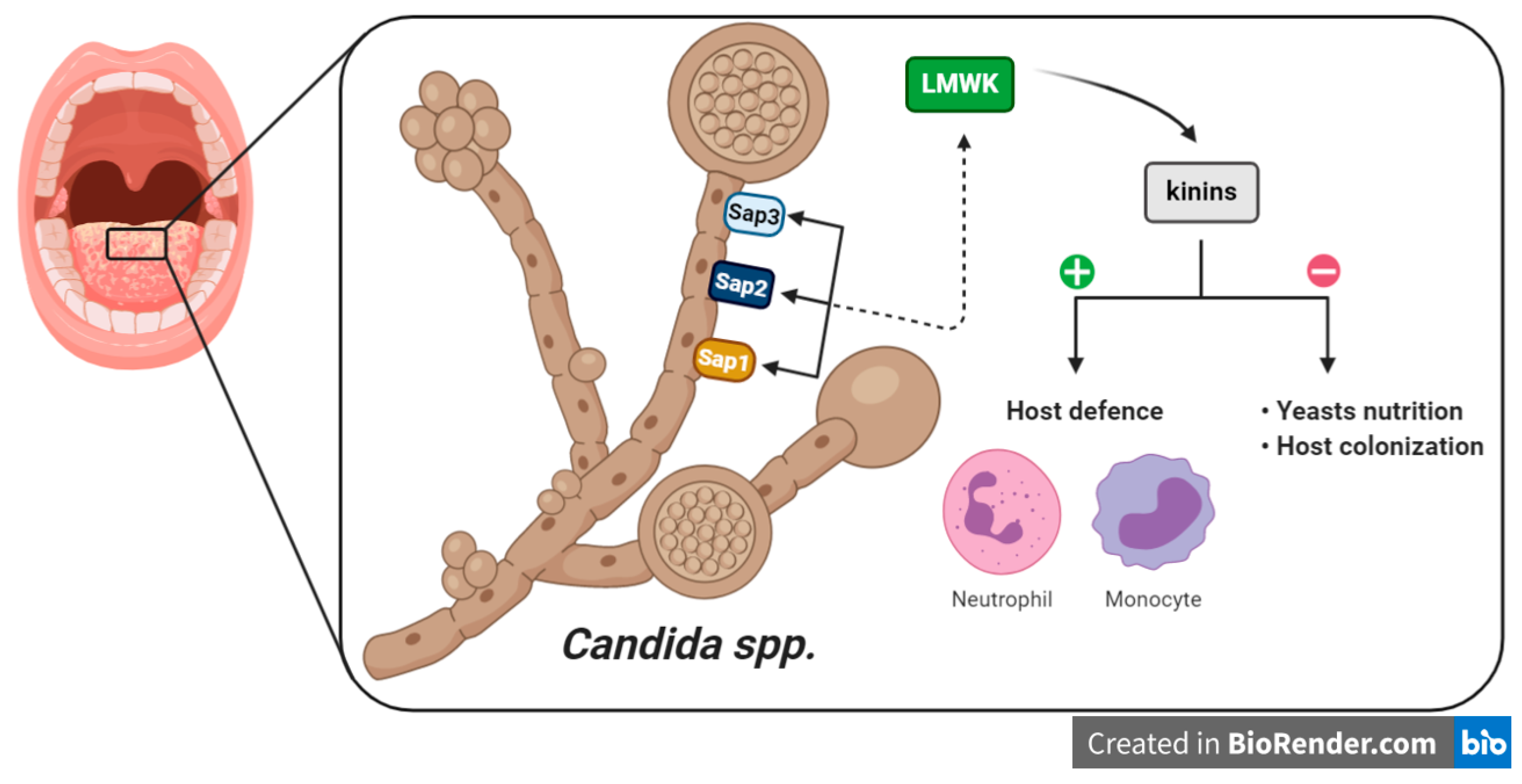
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Dagnino, A.P.A.; Campos, M.M.; Silva, R.B.M. Kinins and Their Receptors in Infectious Diseases. Pharmaceuticals 2020, 13, 215. https://doi.org/10.3390/ph13090215
AMA Style
Dagnino APA, Campos MM, Silva RBM. Kinins and Their Receptors in Infectious Diseases. Pharmaceuticals. 2020; 13(9):215. https://doi.org/10.3390/ph13090215
Chicago/Turabian StyleDagnino, Ana Paula A., Maria M. Campos, and Rodrigo B. M. Silva. 2020. "Kinins and Their Receptors in Infectious Diseases" Pharmaceuticals 13, no. 9: 215. https://doi.org/10.3390/ph13090215
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.