
A3 Adenosine Receptor Antagonists with Nucleoside Structures and Their Anticancer Activity

 , , , , ,
, , , , ,  and
and
Abstract
:
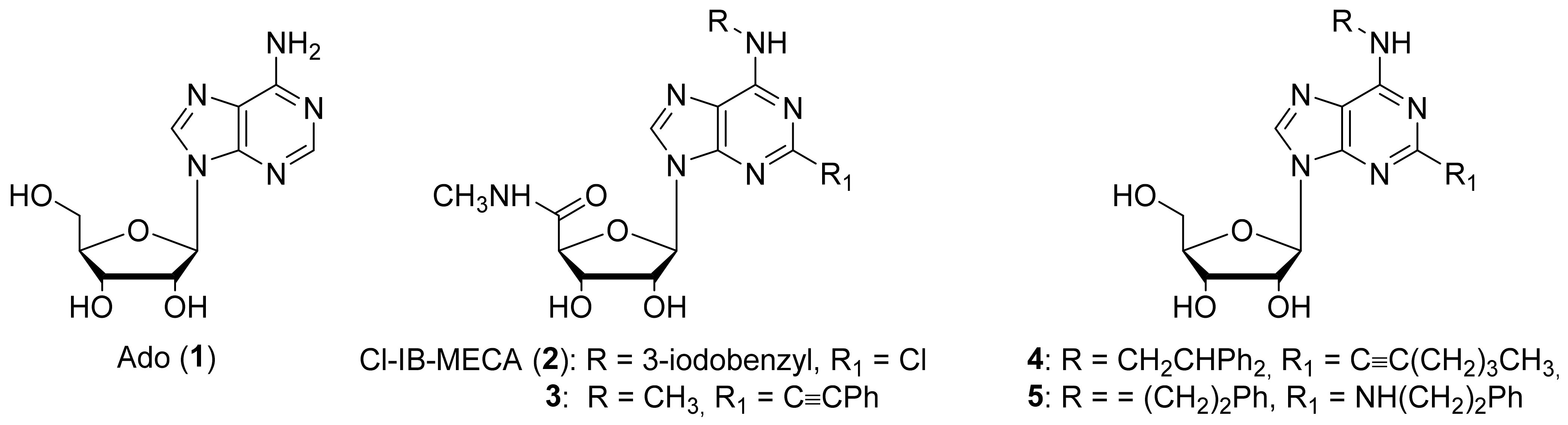
1. Introduction
2. Results and Discussion
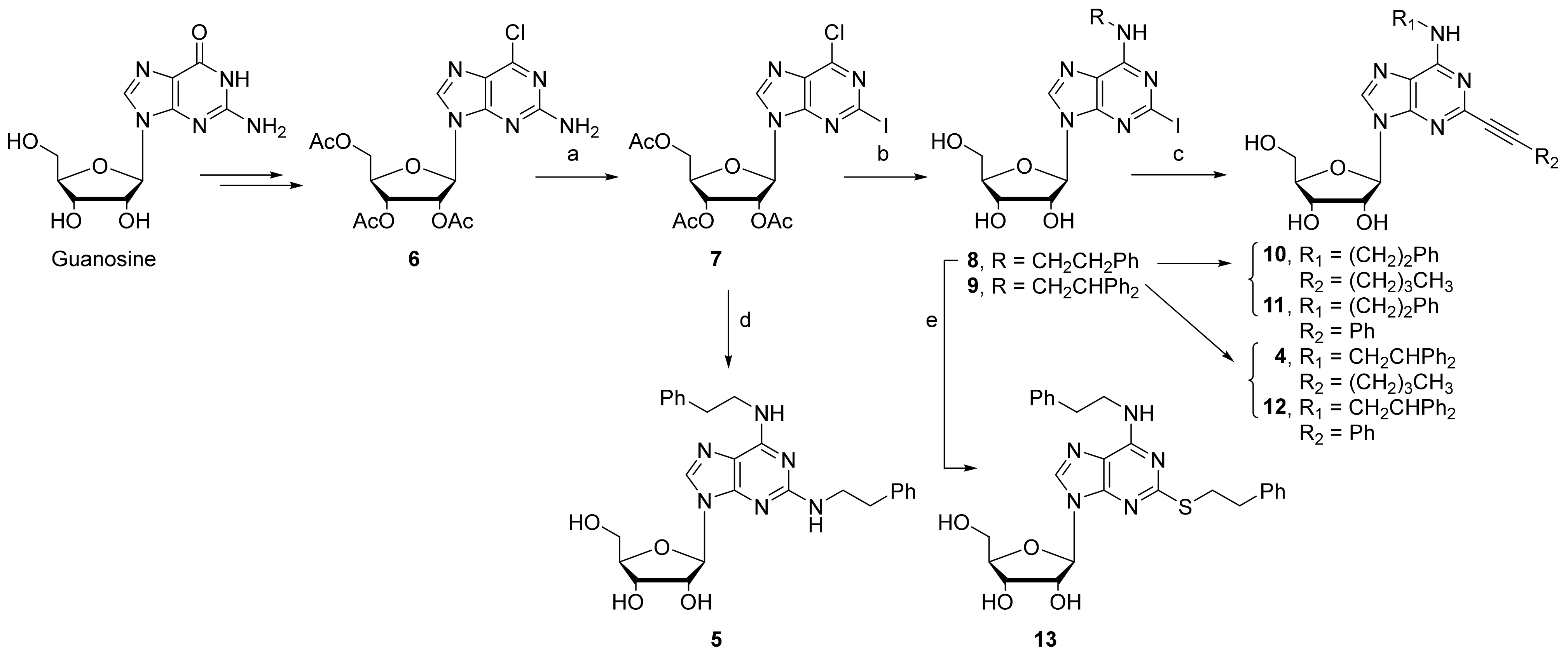
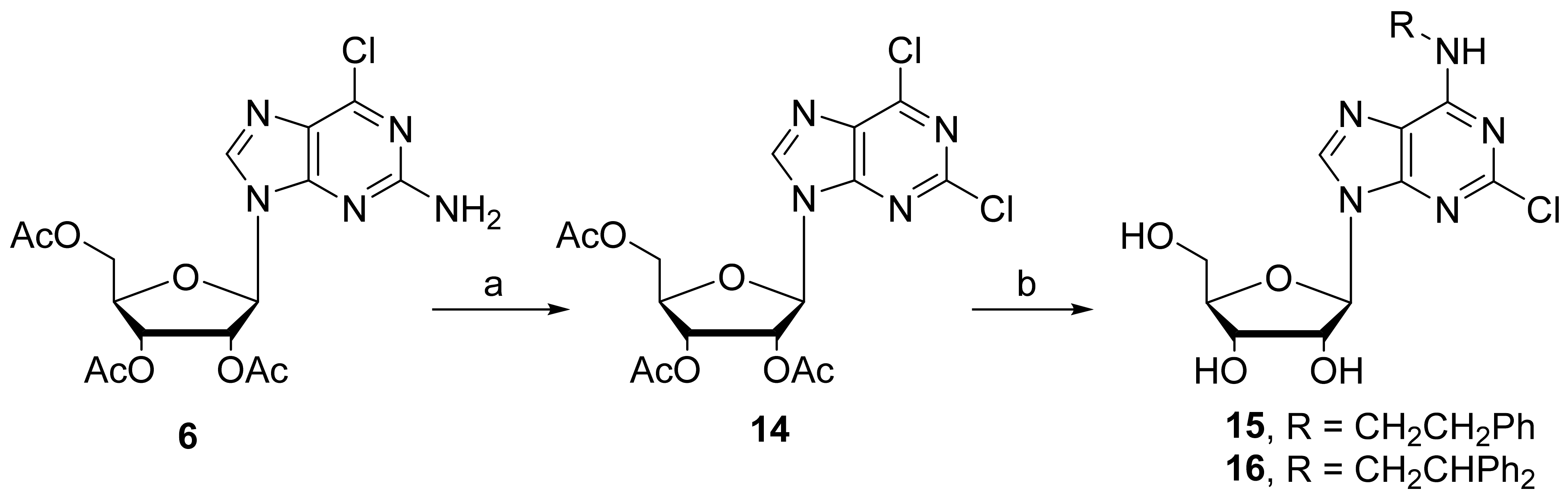
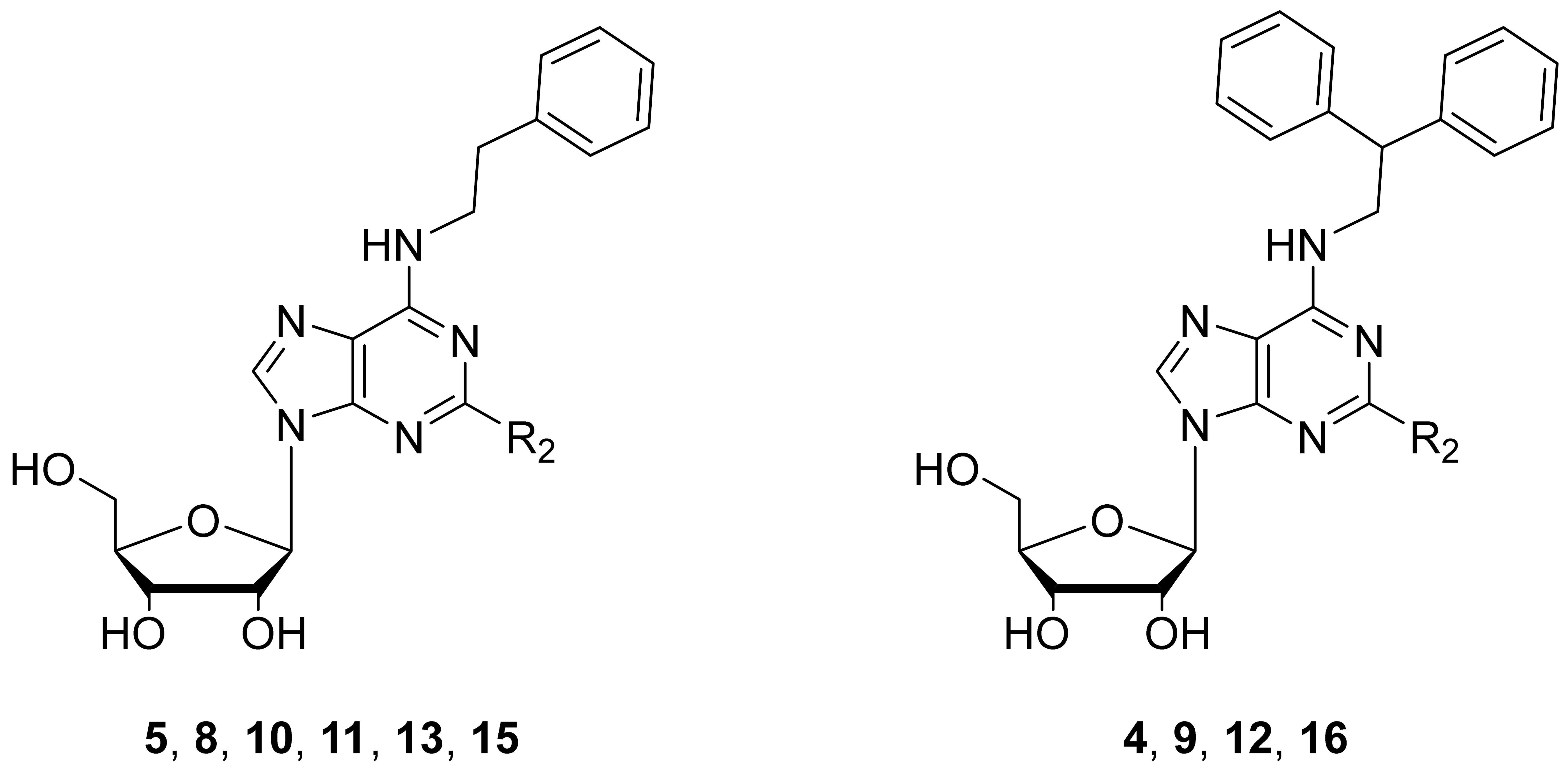
2.1. Chemistry
2.2. Binding Assay at A1, A2A, and A3 ARs and Functional Studies at A2BARs
2.3. Antiproliferative and Cytotoxic Assays
2.4. Functional Activity at Human A3AR
3. Materials and Methods
3.1. Chemical Synthesis
3.1.1. General Methods
3.1.2. Synthesis of 6-Chloro-2-iodo-2′,3′,5′-tri-O-acetyladenosine (7)
3.1.3. Synthesis of 2-Iodo-N6-(2-phenylethyl)adenosine (8)
3.1.4. Synthesis of 2-Iodo-N6-(2,2-diphenylethyl)adenosine (9)
3.1.5. 2-Phenylethylamino-N6-(2-phenylethyl)adenosine (5)
3.1.6. Synthesis of N6-(2-Phenylethyl)-2-phenylethylthioadenosine (13)
3.1.7. General Procedure for the Synthesis of 2-Alkynyl-N6-substituted Adenosines 10, 11, 4, and 12
2-Hexynyl-N6-(2-phenylethyl)adenosine (10)
2-Phenylethynyl-N6-(2-phenethyl)adenosine (11)
2-Hexynyl-N6-(2,2-diphenylethyl)adenosine (4)
N6-(2,2-Diphenylethyl)-2-phenylethynyladenosine (12)
3.1.8. Synthesis of 2,6-Dichloro-2′,3′,5′-tri-O-acetyl-(β-D-ribofuranosyl)purine (14)
3.1.9. General Procedure for the Synthesis of 2-Chloro-N6-substituted adenosines 15 and 16
3.1.10. 2-Chloro-N6-(2-phenylethyl)adenosine (15)
3.1.11. 2-Chloro-N6-(2,2-diphenylethyl)-adenosine (16)
3.2. Biological Assays at Human Adenosine Receptors
3.2.1. Cell Culture
3.2.2. Membrane Preparation
3.2.3. Binding Assay
3.2.4. Functional Agonism or Antagonism at A2B or A3 ARs in GloSensor cAMP Assay
3.3. Biological Studies on Cancer Cell Line
3.3.1. Cell Lines
3.3.2. Reagents
3.3.3. Cell Growth Inhibition Assay
3.3.4. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Di Virgilio, F.; Adinolfi, E. Extracellular purines, purinergic receptors and tumor growth. Oncogene 2017, 36, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Kazemi, M.H.; Raoofi Mohseni, S.; Hojjat-Farsangi, M.; Anvari, E.; Ghalamfarsa, G.; Mohammadi, H.; Jadidi-Niaragh, F. Adenosine and adenosine receptors in the immunopathogenesis and treatment of cancer. J. Cell. Physiol. 2018, 233, 2032–2057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobson, K.A.; Merighi, S.; Varani, K.; Borea, P.A.; Baraldi, S.; Aghazadeh Tabrizi, M.; Romagnoli, R.; Baraldi, P.G.; Ciancetta, A.; Tosh, D.K.; et al. A3 Adenosine Receptors as Modulators of Inflammation: From Medicinal Chemistry to Therapy. Med. Res. Rev. 2018, 38, 1031–1072. [Google Scholar] [CrossRef] [PubMed]
- Contini, C.; Rotondo, J.C.; Magagnoli, F.; Maritati, M.; Seraceni, S.; Graziano, A.; Poggi, A.; Capucci, R.; Vesce, F.; Tognon, M.; et al. Investigation on silent bacterial infections in specimens from pregnant women affected by spontaneous miscarriage. J. Cell. Physiol. 2018, 234, 100–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gessi, S.; Merighi, S.; Borea, P.A.; Cohen, S.; Fishman, P. Adenosine receptors and current opportunities to treat cancer. Receptors 2018, 34, 543–555. [Google Scholar]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of Adenosine Receptors: The State of the Art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef]
- Varani, K.; Maniero, S.; Vincenzi, F.; Targa, M.; Stefanelli, A.; Maniscalco, P.; Martini, F.; Tognon, M.; Borea, P.A. A(3) receptors are overexpressed in pleura from patients with mesothelioma and reduce cell growth via Akt/nuclear factor-kappaB pathway. Am. J. Respir. Crit. Care Med. 2011, 183, 522–530. [Google Scholar] [CrossRef]
- Mlejnek, P.; Dolezel, P.; Frydrych, I. Effects of synthetic A3 adenosine receptor agonists on cell proliferation and viability are receptor independent at micromolar concentrations. J. Physiol. Biochem. 2013, 69, 405–417. [Google Scholar] [CrossRef]
- Gorain, B.; Choudhury, H.; Yee, G.S.; Bhattamisra, S.K. Adenosine Receptors as Novel Targets for the Treatment of Various Cancers. Curr. Pharm. Des. 2019, 25, 2828–2841. [Google Scholar] [CrossRef]
- Mazziotta, C.; Rotondo, J.C.; Lanzillotti, C.; Campione, G.; Martini, F.; Tognon, M. Cancer biology and molecular genetics of A3 adenosine receptor. Oncogene 2021, 41, 301–308. [Google Scholar] [CrossRef]
- Jacobson, K.A.; Reitman, M.L. Adenosine-Related Mechanisms in Non-Adenosine Receptor Drugs. Cells 2020, 9, 956. [Google Scholar] [CrossRef]
- Man, S.; Lu, Y.; Yin, L.; Cheng, X.; Ma, L. Potential and promising anticancer drugs from adenosine and its analogs. Drug Discov Today 2021, 26, 1490–1500. [Google Scholar] [CrossRef] [PubMed]
- Bednarska-Szczepaniak, K.; Mieczkowski, A.; Kierozalska, A.; Pavlovic Saftic, D.; Glabala, K.; Przygodzki, T.; Stanczyk, L.; Karolczak, K.; Watala, C.; Rao, H.; et al. Synthesis and evaluation of adenosine derivatives as A1, A2A, A2B and A3 adenosine receptor ligands containing boron clusters as phenyl isosteres and selective A3 agonists. Eur. J. Med. Chem. 2021, 223, 113607. [Google Scholar] [CrossRef] [PubMed]
- Bednarska-Szczepaniak, K.; Przelazly, E.; Kania, K.D.; Szwed, M.; Litecka, M.; Gruner, B.; Lesnikowski, Z.J. Interaction of Adenosine, Modified Using Carborane Clusters, with Ovarian Cancer Cells: A New Anticancer Approach against Chemoresistance. Cancers 2021, 13, 3855. [Google Scholar] [CrossRef] [PubMed]
- Rotondo, J.C.; Giari, L.; Guerranti, C.; Tognon, M.; Castaldelli, G.; Fano, E.A.; Martini, F. Environmental doses of perfluorooctanoic acid change the expression of genes in target tissues of common carp. Environ. Toxicol. Chem. 2018, 37, 942–948. [Google Scholar] [CrossRef] [PubMed]
- Fishman, P.; Bar-Yehuda, S.; Barer, F.; Madi, L.; Multani, A.S.; Pathak, S. The A3 adenosine receptor as a new target for cancer therapy and chemoprotection. Exp. Cell. Res. 2001, 269, 230–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stemmer, S.M.; Manojlovic, N.S.; Marinca, M.V.; Petrov, P.; Cherciu, N.; Ganea, D.; Ciuleanu, T.E.; Pusca, I.A.; Beg, M.S.; Purcell, W.T.; et al. Namodenoson in Advanced Hepatocellular Carcinoma and Child-Pugh B Cirrhosis: Randomized Placebo-Controlled Clinical Trial. Cancers 2021, 13, 187. [Google Scholar] [CrossRef]
- Marucci, G.; Santinelli, C.; Buccioni, M.; Navia, A.M.; Lambertucci, C.; Zhurina, A.; Yli-Harja, O.; Volpini, R.; Kandhavelu, M. Anticancer activity study of A3 adenosine receptor agonists. Life Sci. 2018, 205, 155–163. [Google Scholar] [CrossRef]
- Volpini, R.; Buccioni, M.; Dal Ben, D.; Lambertucci, C.; Lammi, C.; Marucci, G.; Ramadori, A.T.; Klotz, K.N.; Cristalli, G. Synthesis and biological evaluation of 2-alkynyl-N6-methyl-5’-N-methylcarboxamidoadenosine derivatives as potent and highly selective agonists for the human adenosine A3 receptor. J. Med. Chem. 2009, 52, 7897–7900. [Google Scholar] [CrossRef]
- Cristalli, G.; Eleuteri, A.; Vittori, S.; Volpini, R.; Lohse, M.J.; Klotz, K.N. 2-Alkynyl derivatives of adenosine and adenosine-5’-N-ethyluronamide as selective agonists at A2 adenosine receptors. J. Med. Chem. 1992, 35, 2363–2368. [Google Scholar] [CrossRef]
- Grunewald, C.; Kwon, T.; Piton, N.; Forster, U.; Wachtveitl, J.; Engels, J.W. RNA as scaffold for pyrene excited complexes. Bioorg. Med. Chem. 2008, 16, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Klotz, K.N.; Hessling, J.; Hegler, J.; Owman, C.; Kull, B.; Fredholm, B.B.; Lohse, M.J. Comparative pharmacology of human adenosine receptor subtypes—characterization of stably transfected receptors in CHO cells. Naunyn-Schmiedeberg’s Arch. Pharm. 1998, 357, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Klotz, K.N.; Falgner, N.; Kachler, S.; Lambertucci, C.; Vittori, S.; Volpini, R.; Cristalli, G. [3H]HEMADO—A novel tritiated agonist selective for the human adenosine A3 receptor. Eur. J. Pharm. 2007, 556, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.G.; Blaustein, J.B.; Gross, A.S.; Melman, N.; Jacobson, K.A. N6-Substituted adenosine derivatives: Selectivity, efficacy, and species differences at A3 adenosine receptors. Biochem. Pharmacol. 2003, 65, 1675–1684. [Google Scholar] [CrossRef] [Green Version]
- Voigt, W. Sulforhodamine B assay and chemosensitivity. Methods Mol. Med. 2005, 110, 39–48. [Google Scholar]
- Volpini, R.; Dal Ben, D.; Lambertucci, C.; Taffi, S.; Vittori, S.; Klotz, K.N.; Cristalli, G. N6-methoxy-2-alkynyladenosine derivatives as highly potent and selective ligands at the human A3 adenosine receptor. J. Med. Chem. 2007, 50, 1222–1230. [Google Scholar] [CrossRef]
- Gao, Z.G.; Kim, S.K.; Biadatti, T.; Chen, W.; Lee, K.; Barak, D.; Kim, S.G.; Johnson, C.R.; Jacobson, K.A. Structural determinants of A(3) adenosine receptor activation: Nucleoside ligands at the agonist/antagonist boundary. J. Med. Chem. 2002, 45, 4471–4484. [Google Scholar] [CrossRef]
- Gessi, S.; Merighi, S.; Varani, K.; Leung, E.; Mac Lennan, S.; Borea, P.A. The A(3) adenosine receptor: An enigmatic player in cell biology. Pharmacol. Ther. 2008, 117, 123–140. [Google Scholar] [CrossRef]
- Kim, H.; Kang, J.W.; Lee, S.; Choi, W.J.; Jeong, L.S.; Yang, Y.; Hong, J.T.; Yoon, D.Y. A3 adenosine receptor antagonist, truncated Thio-Cl-IB-MECA, induces apoptosis in T24 human bladder cancer cells. Anticancer. Res. 2010, 30, 2823–2830. [Google Scholar]
- Thomas, A.; Buccioni, M.; Dal Ben, D.; Lambertucci, C.; Marucci, G.; Santinelli, C.; Spinaci, A.; Kachler, S.; Klotz, K.N.; Volpini, R. The Length and Flexibility of the 2-Substituent of 9-Ethyladenine Derivatives Modulate Affinity and Selectivity for the Human A2A Adenosine Receptor. ChemMedChem 2016, 11, 1829–1839. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cmp | R2 | hA1R a (Ki nM) | hA2AR b (Ki nM) | hA3R c (Ki nM) | A1/A3 | A2A/A3 |
|---|---|---|---|---|---|---|
| Cl-IB-MECA [24] | 1240 | 5360 | 1.4 | 886 | 3829 | |
| 5 [18] | NHCH2CH2Ph | 357 | 1368 | 0.33 | 1082 | 4145 |
| 8 | I | 144 ± 32 | 4119 ± 998 | 5 ± 1.2 | 29 | 824 |
| 15 | Cl | 1.64 ± 0.37 | 660 ± 129 | 0.024 ± 0.005 | 68 | 27,500 |
| 13 | SCH2CH2Ph | 263 ± 19 | 3359 ± 752 | 30 ± 7.1 | 9 | 112 |
| 10 | C≡C(CH2)3CH3 | 129 ± 8.2 | 146 ± 48 | 1.5 ± 0.3 | 86 | 97 |
| 11 | C≡CPh | 809 ± 117 | 2983 ± 271 | 3.8 ± 0.6 | 213 | 785 |
| 9 | I | 182 ± 25.4 | 1243 ± 253.1 | 11 ± 1.8 | 17 | 113 |
| 16 | Cl | 0.76 ± 0.2 | 266 ± 35 | 0.13 ± 0.01 | 6 | 2046 |
| 4 [18] | C≡C(CH2)3CH3 | 984 | 153 | 27 | 36 | 6 |
| 12 | C≡CPh | 211 ± 53 | 450 ± 84 | 106 ± 25 | 2 | 4 |

| Cpd | R2 | GI50 (μM) | TGI (μM) | LC50 (μM) | A3AR (nM) |
|---|---|---|---|---|---|
| Cl-IB-MECA | 18 ± 0.4 | 44 ± 3.3 | 110 ± 8.4 | 1.4 | |
| 5 | NHCH2CH2Ph | 237 ± 9.4 | >500 | >500 | 0.33 |
| 8 | I | 42 ± 2.7 | 113 ± 6.7 | 301 ± 13.4 | 5.0 |
| 15 | Cl | 51 ± 5.2 | 262 ± 8.3 | >500 | 0.024 |
| 13 | SCH2CH2Ph | 41 ± 4.2 | >500 | >500 | 30 |
| 10 | C≡C(CH2)3CH3 | 13 ± 1.6 | 77 ± 2.9 | 452 ± 10.5 | 1.5 |
| 11 | C≡CPh | 2.5 ± 0.8 | 19 ± 6.1 | 151 ± 8.9 | 3.8 |
| 9 | I | 24 ± 1.2 | 48 ± 3.5 | 94 ± 3.5 | 11 |
| 16 | Cl | 35 ± 3.1 | 94 ± 4.1 | 253 ± 11.5 | 0.13 |
| 4 | C≡C(CH2)3CH3 | 16 ± 0.5 | 35 ± 1.5 | 80 ± 9.8 | 27 |
| 12 | C≡CPh | 14 ± 0.9 | 29 ± 4.9 | 59 ± 10.1 | 106 |
| Cpd | A3AR CHO Cells | |
|---|---|---|
| EC50, nM | IC50, nM | |
| Cl-IB-MECA | 2.8 ± 1.4 | |
| 15 | 14 ± 3.4 | |
| 10 | 31 ± 6 | |
| 11 | 79 ± 15 | |
| 4 | 380 ± 80 | |
| 12 | 153 ± 12 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spinaci, A.; Buccioni, M.; Dal Ben, D.; Maggi, F.; Marucci, G.; Francucci, B.; Santoni, G.; Lambertucci, C.; Volpini, R. A3 Adenosine Receptor Antagonists with Nucleoside Structures and Their Anticancer Activity. Pharmaceuticals 2022, 15, 164. https://doi.org/10.3390/ph15020164
Spinaci A, Buccioni M, Dal Ben D, Maggi F, Marucci G, Francucci B, Santoni G, Lambertucci C, Volpini R. A3 Adenosine Receptor Antagonists with Nucleoside Structures and Their Anticancer Activity. Pharmaceuticals. 2022; 15(2):164. https://doi.org/10.3390/ph15020164
Chicago/Turabian StyleSpinaci, Andrea, Michela Buccioni, Diego Dal Ben, Federica Maggi, Gabriella Marucci, Beatrice Francucci, Giorgio Santoni, Catia Lambertucci, and Rosaria Volpini. 2022. "A3 Adenosine Receptor Antagonists with Nucleoside Structures and Their Anticancer Activity" Pharmaceuticals 15, no. 2: 164. https://doi.org/10.3390/ph15020164