
Design, Synthesis, and Biological Evaluation of 4,4’-Difluorobenzhydrol Carbamates as Selective M1 Antagonists

 , ,
, ,  , ,
, ,
Abstract
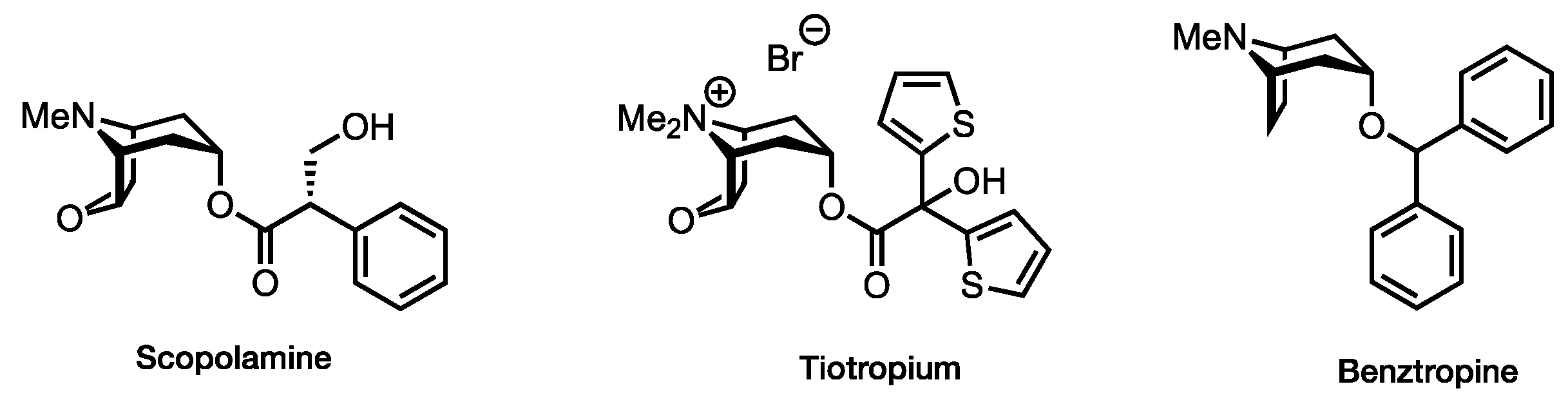
:1. Introduction
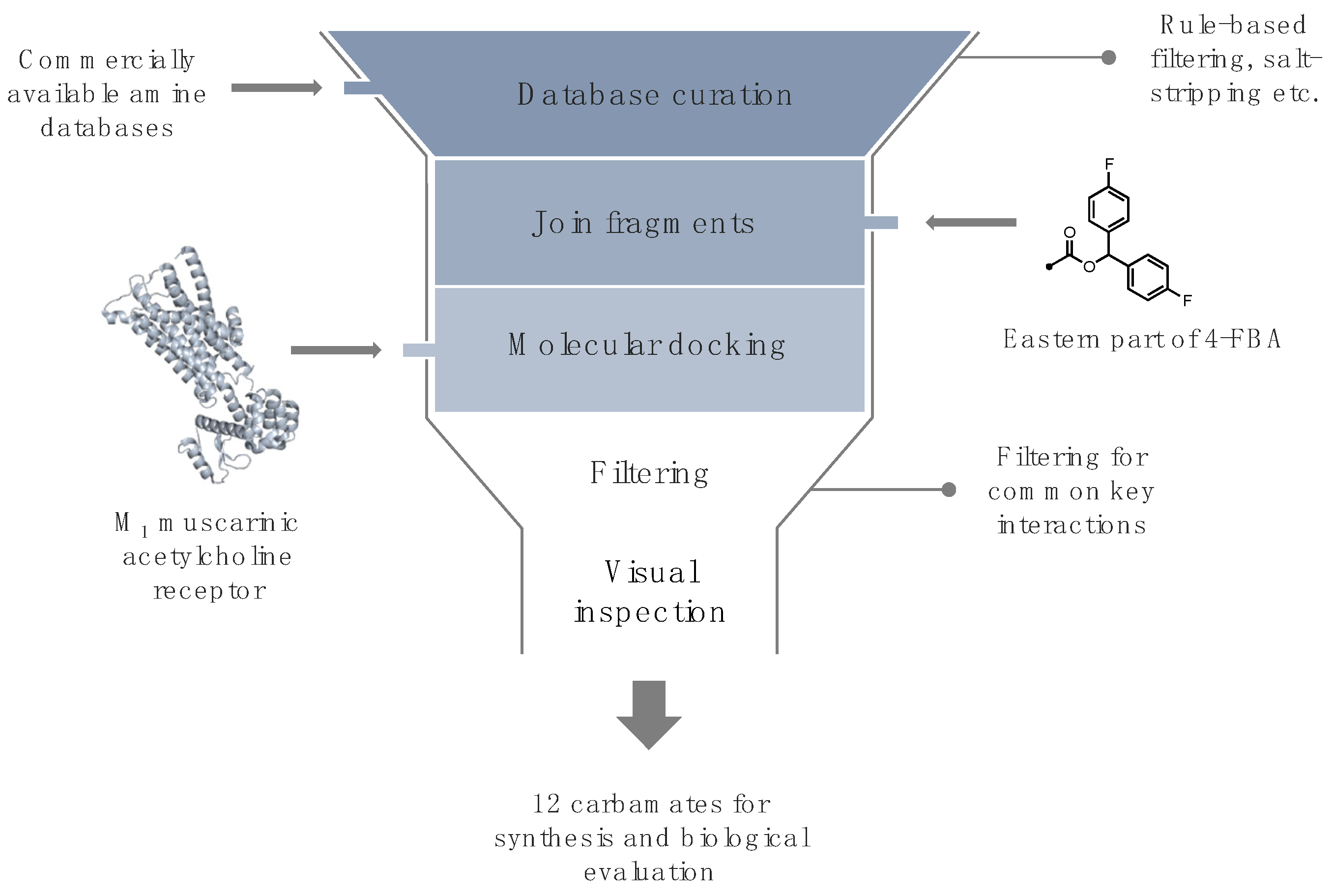
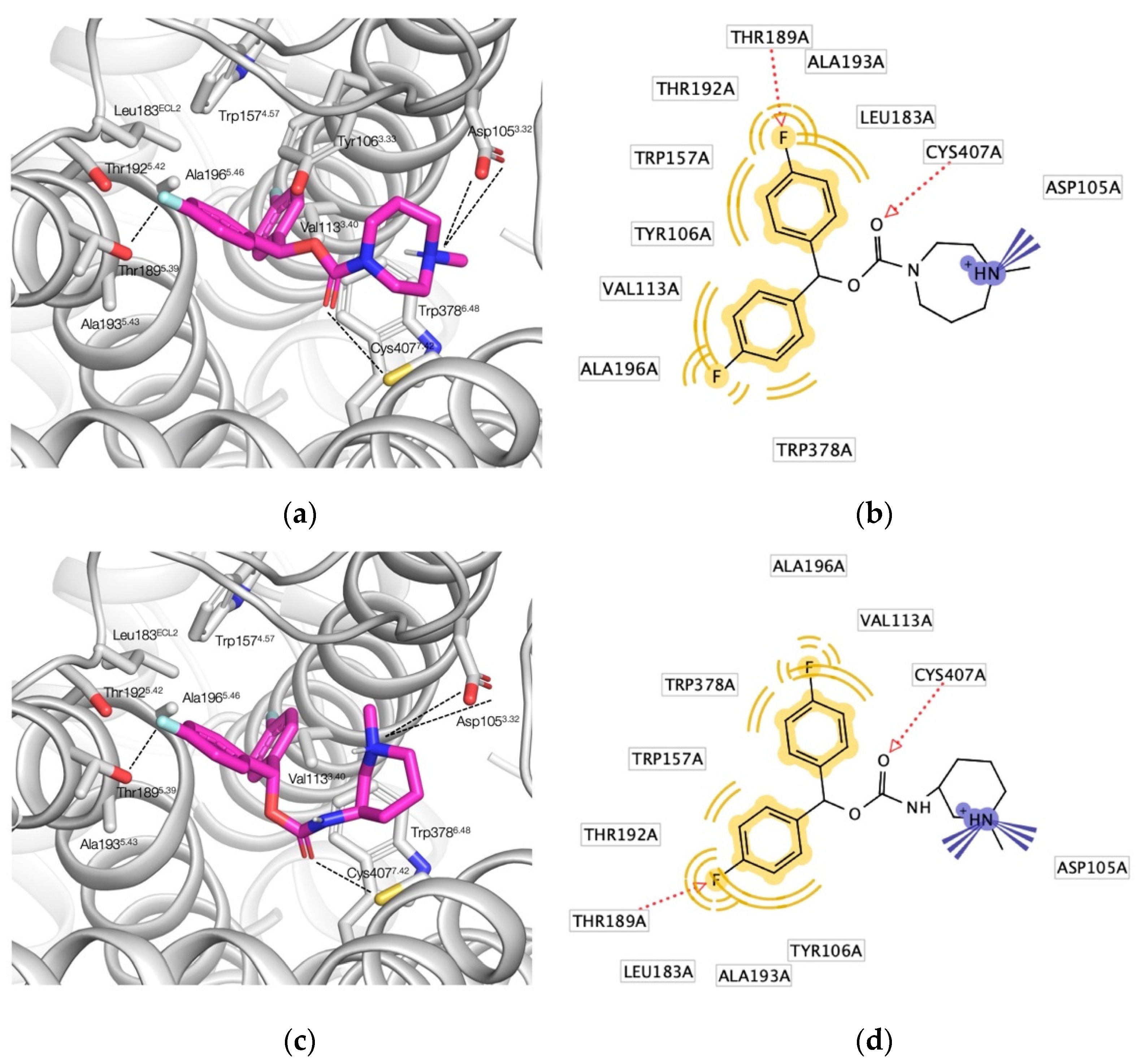
2. Results and Discussion
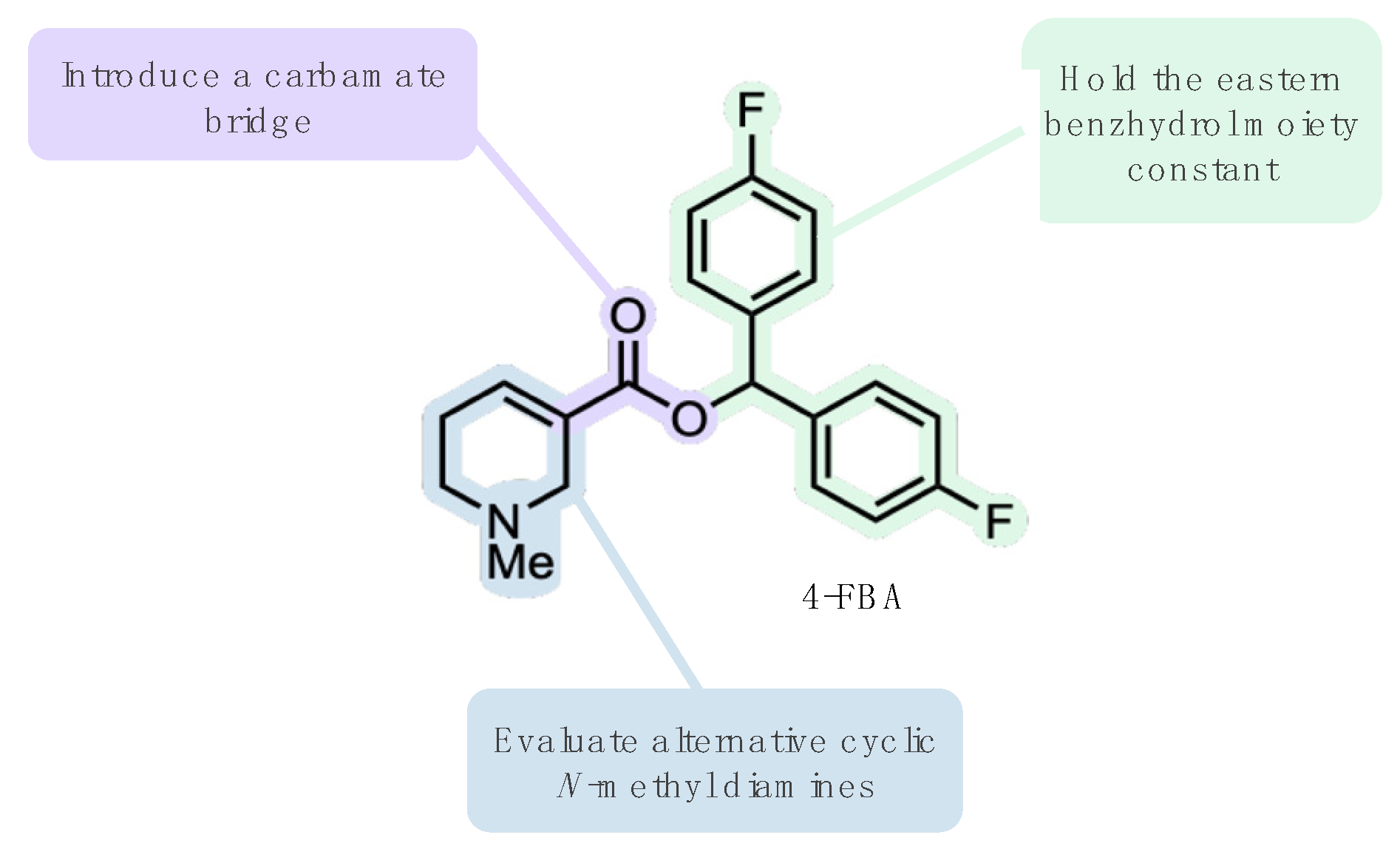
2.1. Ligand Design
2.2. Chemistry
2.3. Physico–Chemical Property Profile and Stability
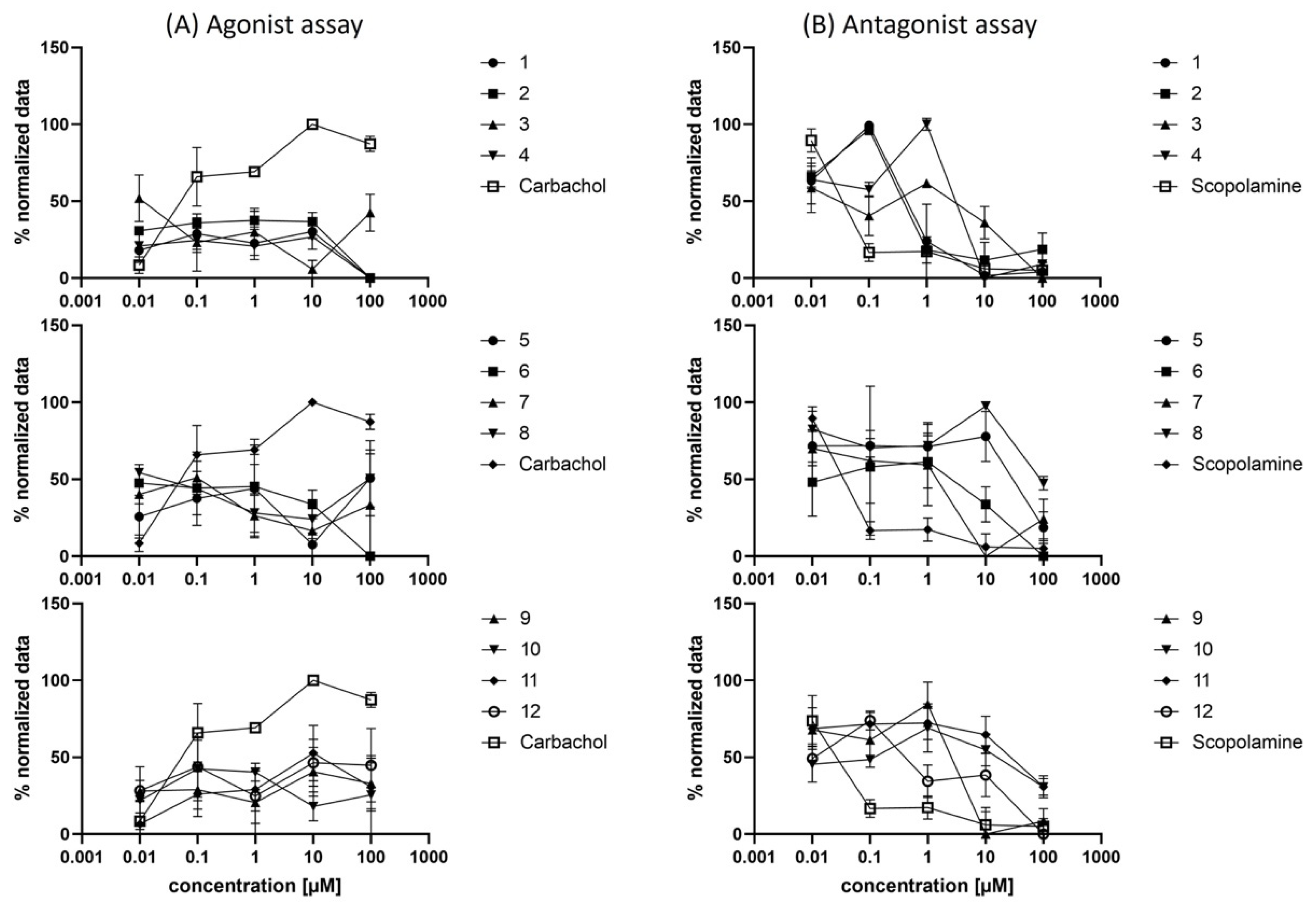
2.4. Biological Evaluation
3. Materials and Methods
3.1. Ligand Design
3.2. Chemistry
3.2.1. General Considerations
3.2.2. General Procedure for the Alkoxycarbonylation of Diamines
3.3. High Throughput HPLC-logD
3.4. Biological Evaluation
3.4.1. Materials and Methods
3.4.2. Cell Culture
3.4.3. Cell Viability (MTT Assay)
3.4.4. Stability in Cell Culture Media
3.4.5. Radioligand Binding Experiments
3.4.6. Fluo-4 Calcium Assay for Agonist-Antagonist Discrimination
3.4.7. Data Analysis and Statistics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fryer, A.D.; Christopoulos, A.; Nathanson, N.N. Muscarinic Receptors; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Kruse, A.C.; Kobilka, B.K.; Gautam, D.; Sexton, P.M.; Christopoulos, A.; Wess, J. Muscarinic acetylcholine receptors: Novel opportunities for drug development. Nat. Rev. Drug Discov. 2014, 13, 549–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moran, S.P.; Maksymetz, J.; Conn, P.J. Targeting Muscarinic Acetylcholine Receptors for the Treatment of Psychiatric and Neurological Disorders. Trends Pharmacol. Sci. 2019, 40, 1006–1020. [Google Scholar] [CrossRef] [PubMed]
- Scarr, E. Muscarinic Receptors: Their Roles in Disorders of the Central Nervous System and Potential as Therapeutic Targets. CNS Neurosci. Ther. 2012, 18, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, J.N.; Butera, J.A.; Carrick, T.; Kramer, A.; Kowal, D.; Lock, T.; Marquis, K.L.; Pausch, M.H.; Popiolek, M.; Sun, S.-C.; et al. Pharmacological comparison of muscarinic ligands: Historical versus more recent muscarinic M1-preferring receptor agonists. Eur. J. Pharmacol. 2009, 605, 53–56. [Google Scholar] [CrossRef] [PubMed]
- van der Westhuizen, E.T.; Choy, K.H.C.; Valant, C.; McKenzie-Nickson, S.; Bradley, S.J.; Tobin, A.B.; Sexton, P.M.; Christopoulos, A. Fine Tuning Muscarinic Acetylcholine Receptor Signaling Through Allostery and Bias. Front. Pharmacol. 2021, 11, 2217. [Google Scholar] [CrossRef] [PubMed]
- Felder, C.C.; Goldsmith, P.J.; Jackson, K.; Sanger, H.E.; Evans, D.A.; Mogg, A.J.; Broad, L.M. Current status of muscarinic M1 and M4 receptors as drug targets for neurodegenerative diseases. Neuropharmacology 2018, 136, 449–458. [Google Scholar] [CrossRef]
- Jones, C.K.; Byun, N.; Bubser, M. Muscarinic and Nicotinic Acetylcholine Receptor Agonists and Allosteric Modulators for the Treatment of Schizophrenia. Neuropsychopharmacology 2012, 37, 16–42. [Google Scholar] [CrossRef] [Green Version]
- Jeffrey Conn, P.; Christopoulos, A.; Lindsley, C.W. Allosteric modulators of GPCRs: A novel approach for the treatment of CNS disorders. Nat. Rev. Drug Discov. 2009, 8, 41–54. [Google Scholar] [CrossRef] [Green Version]
- Volpato, D.; Kauk, M.; Messerer, R.; Bermudez, M.; Wolber, G.; Bock, A.; Hoffmann, C.; Holzgrabe, U. The Role of Orthosteric Building Blocks of Bitopic Ligands for Muscarinic M1 Receptors. ACS Omega 2020, 5, 31706–31715. [Google Scholar] [CrossRef]
- Tong, L.; Li, W.; Lo, M.M.-C.; Gao, X.; Wai, J.M.-C.; Rudd, M.; Tellers, D.; Joshi, A.; Zeng, Z.; Miller, P.; et al. Discovery of [11C]MK-6884: A Positron Emission Tomography (PET) Imaging Agent for the Study of M4Muscarinic Receptor Positive Allosteric Modulators (PAMs) in Neurodegenerative Diseases. J. Med. Chem. 2020, 63, 2411–2425. [Google Scholar] [CrossRef] [Green Version]
- Wess, J.; Eglen, R.M.; Gautam, D. Muscarinic acetylcholine receptors: Mutant mice provide new insights for drug development. Nat. Rev. Drug Discov. 2007, 6, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Schrader, T.O.; Xiong, Y.; Lorenzana, A.O.; Broadhead, A.; Stebbins, K.J.; Poon, M.M.; Baccei, C.; Lorrain, D.S. Discovery of PIPE-359, a Brain-Penetrant, Selective M1 Receptor Antagonist with Robust Efficacy in Murine MOG-EAE. ACS Med. Chem. Lett. 2021, 12, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Toyohara, J.; Sakata, M.; Ishiwata, K. Chapter Six—Human Brain Imaging of Acetylcholine Receptors. In Imaging of the Human Brain in Health and Disease; Seeman, P., Madras, B., Eds.; Academic Press: Cambridge, MA, USA, 2014; pp. 113–160. [Google Scholar]
- Ozenil, M.; Aronow, J.; Millard, M.; Langer, T.; Wadsak, W.; Hacker, M.; Pichler, V. Update on PET Tracer Development for Muscarinic Acetylcholine Receptors. Pharmaceuticals 2021, 14, 530. [Google Scholar] [CrossRef] [PubMed]
- Ozenil, M.; Pacher, K.; Balber, T.; Vraka, C.; Roller, A.; Holzer, W.; Spreitzer, H.; Mitterhauser, M.; Wadsak, W.; Hacker, M.; et al. Enhanced arecoline derivatives as muscarinic acetylcholine receptor M1 ligands for potential application as PET radiotracers. Eur. J. Med. Chem. 2020, 204, 112623. [Google Scholar] [CrossRef]
- Gee, A.D.; Bongarzone, S.; Wilson, A.A. Small Molecules as Radiopharmaceutical Vectors. In Radiopharmaceutical Chemistry; Lewis, J.S., Windhorst, A.D., Zeglis, B.M., Eds.; Springer International Publishing: Berlin/Heidelberg, Germany, 2019; pp. 119–136. [Google Scholar]
- Thal, D.M.; Sun, B.; Feng, D.; Nawaratne, V.; Leach, K.; Felder, C.C.; Bures, M.G.; Evans, D.A.; Weis, W.I.; Bachhawat, P.; et al. Crystal structures of the M1 and M4 muscarinic acetylcholine receptors. Nature 2016, 531, 335. [Google Scholar] [CrossRef] [Green Version]
- Ballesteros, J.A.; Weinstein, H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. In Receptor Molecular Biology; Sealfon, S.C., Ed.; Academic Press: Cambridge, MA, USA, 1995; Volume 25, pp. 366–428. [Google Scholar]
- van Rhee, A.M.; Jacobson, K.A. Molecular architecture of G protein-coupled receptors. Drug Dev. Res. 1996, 37, 1–38. [Google Scholar] [CrossRef] [Green Version]
- Peng, J.Y.; Vaidehi, N.; Hall, S.E.; Goddard III, W.A. The Predicted 3D Structures of the Human M1 Muscarinic Acetylcholine Receptor with Agonist or Antagonist Bound. ChemMedChem 2006, 1, 878–890. [Google Scholar] [CrossRef]
- Wolber, G.; Langer, T. LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef]
- Bender, B.J.; Gahbauer, S.; Luttens, A.; Lyu, J.; Webb, C.M.; Stein, R.M.; Fink, E.A.; Balius, T.E.; Carlsson, J.; Irwin, J.J.; et al. A practical guide to large-scale docking. Nat. Protoc. 2021, 16, 4799–4832. [Google Scholar] [CrossRef]
- Huang, X.-P.; Nagy, P.I.; Williams, F.E.; Peseckis, S.M.; Messer, W.S., Jr. Roles of threonine 192 and asparagine 382 in agonist and antagonist interactions with M1 muscarinic receptors. Br. J. Pharmacol. 1999, 126, 735–745. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.K.; Brindisi, M. Organic Carbamates in Drug Design and Medicinal Chemistry. J. Med. Chem. 2015, 58, 2895–2940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gobbi, L.; Mercier, J.; Bang-Andersen, B.; Nicolas, J.-M.; Reilly, J.; Wagner, B.; Whitehead, D.; Briard, E.; Maguire, R.P.; Borroni, E.; et al. A Comparative Study of in vitro Assays for Predicting the Nonspecific Binding of PET Imaging Agents in vivo. ChemMedChem 2020, 15, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Vraka, C.; Nics, L.; Wagner, K.-H.; Hacker, M.; Wadsak, W.; Mitterhauser, M. LogP, a yesterday’s value? Nucl. Med. Biol. 2017, 50, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kilbourn, M.R.; Scott, P.J.H. Is logP truly dead? Nucl. Med. Biol. 2017, 54, 41–42. [Google Scholar] [CrossRef] [PubMed]
- Vraka, C.; Mitterhauser, M. Reconsider logP! Nucl. Med. Biol. 2017, 54, 42. [Google Scholar] [CrossRef] [PubMed]
- Donovan, S.F.; Pescatore, M.C. Method for measuring the logarithm of the octanol–water partition coefficient by using short octadecyl–poly(vinyl alcohol) high-performance liquid chromatography columns. J. Chromatogr. A 2002, 952, 47–61. [Google Scholar] [CrossRef]
- Pike, V.W. Considerations in the Development of Reversibly Binding PET Radioligands for Brain Imaging. Curr. Med. Chem. 2016, 23, 1818–1869. [Google Scholar] [CrossRef]
- Ghose, A.K.; Herbertz, T.; Hudkins, R.L.; Dorsey, B.D.; Mallamo, J.P. Knowledge-Based, Central Nervous System (CNS) Lead Selection and Lead Optimization for CNS Drug Discovery. ACS Chem. Neurosci. 2012, 3, 50–68. [Google Scholar] [CrossRef] [Green Version]
- Pajouhesh, H.; Lenz, G.R. Medicinal chemical properties of successful central nervous system drugs. NeuroRX 2005, 2, 541–553. [Google Scholar] [CrossRef] [Green Version]
- ACD/Percepta 2021.2.3; Advanced Chemistry Development, Inc.: Toronto, ON, Canada, 2021; Available online: www.acdlabs.com (accessed on 27 January 2022).
- Hammarlund-Udenaes, M.; Fridén, M.; Syvänen, S.; Gupta, A. On The Rate and Extent of Drug Delivery to the Brain. Pharm. Res. 2008, 25, 1737–1750. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, T.S.; Kirshner, D.A.; Lau, E.Y.; Wong, S.E.; Nilmeier, J.P.; Lightstone, F.C. A Method to Predict Blood-Brain Barrier Permeability of Drug-Like Compounds Using Molecular Dynamics Simulations. Biophys. J. 2014, 107, 630–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumaier, F.; Zlatopolskiy, B.D.; Neumaier, B. Drug Penetration into the Central Nervous System: Pharmacokinetic Concepts and In Vitro Model Systems. Pharmaceutics 2021, 13, 1542. [Google Scholar] [CrossRef] [PubMed]
- Martin, I. Prediction of blood–brain barrier penetration: Are we missing the point? Drug Discov. Today 2004, 9, 161–162. [Google Scholar] [CrossRef]
- Hammarlund-Udenaes, M. Active-Site Concentrations of Chemicals—Are They a Better Predictor of Effect than Plasma/Organ/Tissue Concentrations? Basic Clin. Pharmacol. Toxicol. 2010, 106, 215–220. [Google Scholar] [CrossRef] [PubMed]
- McCluskey, S.P.; Plisson, C.; Rabiner, E.A.; Howes, O. Advances in CNS PET: The state-of-the-art for new imaging targets for pathophysiology and drug development. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 451–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geldenhuys, W.J.; Mohammad, A.S.; Adkins, C.E.; Lockman, P.R. Molecular determinants of blood–brain barrier permeation. Ther. Deliv. 2015, 6, 961–971. [Google Scholar] [CrossRef]
- Ozenil, M.; Aronow, J.; Piljak, D.; Vraka, C.; Holzer, W.; Spreitzer, H.; Wadsak, W.; Hacker, M.; Pichler, V. Synthesis, Biological, and Computational Evaluation of Antagonistic, Chiral Hydrobenzoin Esters of Arecaidine Targeting mAChR M1. Pharmaceuticals 2020, 13, 437. [Google Scholar] [CrossRef]
- Ma, Q.; Ye, L.; Liu, H.; Shi, Y.; Zhou, N. An overview of Ca2+ mobilization assays in GPCR drug discovery. Expert Opin. Drug Discov. 2017, 12, 511–523. [Google Scholar] [CrossRef]
- Colom, M.; Vidal, B.; Zimmer, L. Is There a Role for GPCR Agonist Radiotracers in PET Neuroimaging? Front. Mol. Neurosci. 2019, 12, 255. [Google Scholar] [CrossRef]
- OMEGA 3.1.1; OpenEye Scientific Software: Santa Fe, NM, USA. Available online: https://www.eyesopen.com (accessed on 27 January 2022).
- QUACPAC 2.0.1.2; OpenEye Scientific Software: Santa Fe, NM, USA. Available online: https://www.eyesopen.com (accessed on 27 January 2022).
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Bell, E.W.; Zhang, Y. DockRMSD: An open-source tool for atom mapping and RMSD calculation of symmetric molecules through graph isomorphism. J. Cheminform. 2019, 11, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, E.J.; Lwin, C.T.; Durrant, J.D. LigGrep: A tool for filtering docked poses to improve virtual-screening hit rates. J. Cheminformatics 2020, 12, 69. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System; Version 2.3; Schrödinger, LLC: New York, NY, USA; Available online: https://www.pymol.org (accessed on 27 January 2022).
- Duspara, P.A.; Islam, M.S.; Lough, A.J.; Batey, R.A. Synthesis and Reactivity of N-Alkyl Carbamoylimidazoles: Development of N-Methyl Carbamoylimidazole as a Methyl Isocyanate Equivalent. J. Org. Chem. 2012, 77, 10362–10368. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Cmpd. |  | Yield 1 (%) | Cmpd. |  | Yield 1 (%) |
| 1 |  | 34 | 7 |  | 34 |
| 2 |  | 38 | 8 |  | 25 |
| 3 |  | 10 | 9 |  | 19 |
| 4 |  | 25 | 10 |  | 29 |
| 5 |  | 26 | 11 |  | 28 |
| 6 |  | 22 | 12 |  | 29 |
| Physico–Chemical Properties | BBB Transport Parameters | |||||
|---|---|---|---|---|---|---|
| Cmpd. | HPLC-logD | logD 1 | tPSA 2 (Å2) | pKa 3,4 | logBB 4 | logPS 4 |
| 1 | 3.16 ± 0.01 | 3.14 | 32.78 | 6.8 ± 0.1 | 0.45 | −1.2 |
| 2 | 3.20 ± 0.02 | 3.19 | 32.78 | 7.5 ± 0.1 | 0.37 | −1.2 |
| 3 | 2.69 ± 0.01 | 1.37 | 32.78 | 9.5 ± 0.2 | 0.51 | −1.6 |
| 4 | 3.28 ± 0.03 | 1.92 | 32.78 | 9.0 ± 0.2 | 0.62 | −1.5 |
| 5 | 2.69 ± 0.01 | 2.08 | 32.78 | 9.6 ± 0.2 | 0.96 | −1.4 |
| 6 | 3.25 ± 0.04 | 2.86 | 41.57 | 8.6 ± 0.1 | 0.50 | −1.5 |
| 7 | 2.2 ± 0.2 | 2.44 | 41.57 | 9.4 ± 0.1 | 0.53 | −1.5 |
| 8 | 3.21 ± 0.03 | 2.31 | 41.57 | 10.9 ± 0.4 | 0.99 | −1.5 |
| 9 | 2.8 ± 0.1 | 1.77 | 41.57 | 9.6 ± 0.4 | 0.45 | −1.7 |
| 10 | 2.69 ± 0.01 | 1.12 | 41.57 | 10.2 ± 0.4 | 0.39 | −1.7 |
| 11 | 2.5 ± 0.3 | 1.39 | 41.57 | 10.3 ± 0.4 | 0.54 | −1.6 |
| 12 | 2.82 ± 0.04 | 3.03 | 50.80 | 7.0 ± 0.4 | 0.23 | −1.4 |
| Affinity: Ki ± SD (nM) | x-Fold Selectivity for hM1 vs. hMx 1 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cmpd. | hM1 | hM2 | hM3 | hM4 | hM5 | hM2 | hM3 | hM4 | hM5 |
| 1 | 15.2 ± 3.6 | >1000 2 | 225.6 ± 85.2 | 54.8 ± 20.5 | 50.6 ± 3.9 | >66 | 14.8 | 3.6 | 3.3 |
| 2 | 1.2 ± 0.4 | 227.2 ± 85.9 | 28.4 ± 10.7 | 14.4 ± 5.5 | 4.8 ± 1.6 | 189.3 | 23.7 | 12.0 | 4.0 |
| 3 | 33.1 ± 8.1 | >1000 2 | 357.8 ± 83.0 | 115.1 ± 51.0 | 68.0 ± 22.1 | >30 | 10.8 | 3.5 | 2.1 |
| 4 | 16.5 ± 2.8 | 849.5 ± 39.8 | 141.6 ± 24.2 | 19.6 ± 5.5 | 41.8 ± 14.8 | 51.5 | 8.6 | 1.2 | 2.5 |
| 5 | 24.9 ± 6.2 | >1000 2 | 164.5 ± 37.5 | 150.3 ± 52.9 | 230.8 ± 25.7 | >40 | 6.6 | 6.0 | 9.3 |
| 7 | 1.22 ± 0.06 | 32.8 ± 11.4 | 16.1 ± 4.5 | 6.2 ± 2.1 | 3.7 ± 1.3 | 27.3 | 13.4 | 5.2 | 3.1 |
| 8 | 474.6 ± 88.5 | 623.9 ± 104.3 | >1000 2 | 562.9 ± 73.4 | 521.0 ± 172.7 | 1.3 | >2 | 1.2 | 1.1 |
| 9 | 67.8 ± 5.4 | 721.9 ± 101.19 | 181.2 ± 68.1 | 143.8 ± 37.3 | 64.5 ± 22.8 | 10.6 | 2.7 | 2.1 | 1.0 |
| 10 | 238.7 ± 67.9 | >1000 2 | 276.9 ± 45.4 | 238.2 ± 103.6 | 295.2 ± 32.8 | >4 | 1.2 | 1.0 | 1.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kilian, J.; Ozenil, M.; Millard, M.; Fürtös, D.; Maisetschläger, V.; Holzer, W.; Wadsak, W.; Hacker, M.; Langer, T.; Pichler, V. Design, Synthesis, and Biological Evaluation of 4,4’-Difluorobenzhydrol Carbamates as Selective M1 Antagonists. Pharmaceuticals 2022, 15, 248. https://doi.org/10.3390/ph15020248
Kilian J, Ozenil M, Millard M, Fürtös D, Maisetschläger V, Holzer W, Wadsak W, Hacker M, Langer T, Pichler V. Design, Synthesis, and Biological Evaluation of 4,4’-Difluorobenzhydrol Carbamates as Selective M1 Antagonists. Pharmaceuticals. 2022; 15(2):248. https://doi.org/10.3390/ph15020248
Chicago/Turabian StyleKilian, Jonas, Marius Ozenil, Marlon Millard, Dorka Fürtös, Verena Maisetschläger, Wolfgang Holzer, Wolfgang Wadsak, Marcus Hacker, Thierry Langer, and Verena Pichler. 2022. "Design, Synthesis, and Biological Evaluation of 4,4’-Difluorobenzhydrol Carbamates as Selective M1 Antagonists" Pharmaceuticals 15, no. 2: 248. https://doi.org/10.3390/ph15020248