
Levetiracetam Mechanisms of Action: From Molecules to Systems


 ,
,  , , and
, , and
Abstract
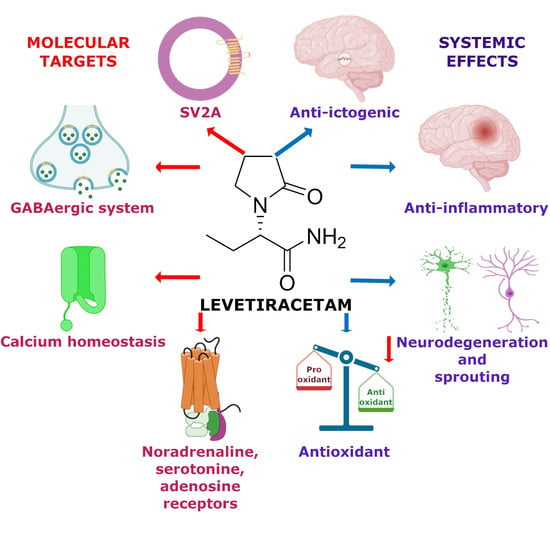
:
1. Introduction
2. Methods
2.1. Inclusion Criteria
2.2. Exclusion Criteria
3. Clinical Indications in Epilepsy
4. Other Clinical Applications
5. Generalities, Chemical Structure, and Analogous
6. Levetiracetam Binding Site (LBS)
7. Molecular Mechanism
7.1. Synaptic Vesicle Protein 2A (SV2A)
7.2. Calcium Homeostasis
7.3. GABAergic System
7.4. SV2A and GABAergic System
7.5. AMPA Receptors
7.6. Noradrenaline, Adenosine and Serotonin Receptors
7.7. Intracellular pH Regulation
7.8. Single or Integrated LEV Molecular Mechanism of Action?
8. Genetic Mechanism
Effect of Gene Polymorphisms in LEV Treatment in Clinical Studies
9. Anti-Ictogenic Mechanism
10. Antiepileptogenic Mechanism
11. Neuroprotective Mechanism
12. Anti-Inflammatory and Antioxidant Mechanisms
13. Rebound Effect and Aggressiveness Behavior
14. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- World Health Organization (WHO). Epilepsy. Available online: https://www.who.int/news-room/fact-sheets/detail/epilepsy (accessed on 6 October 2021).
- Fisher, R.S.; Acevedo, C.; Arzimanoglou, A.; Bogacz, A.; Cross, J.H.; Elger, C.E.; Engel, J.J.; Forsgren, L.; French, J.A.; Glynn, M.; et al. ILAE official report: A practical clinical definition of epilepsy. Epilepsia 2014, 55, 475–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, R.S.; Cross, J.H.; French, J.A.; Higurashi, N.; Hirsch, E.; Jansen, F.E.; Lagae, L.; Moshé, S.L.; Peltola, J.; Roulet Perez, E.; et al. Operational classification of seizure types by the International League Against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 522–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Pérez, D.; Frías-Soria, C.L.; Rocha, L. Drug-resistant epilepsy: From multiple hypotheses to an integral explanation using preclinical resources. Epilepsy Behav. 2021, 121, 106430. [Google Scholar] [CrossRef] [PubMed]
- Pitkänen, A.; Sutula, T.P. Is epilepsy a progressive disorder? Prospects for new therapeutic approaches in temporal-lobe epilepsy. Lancet Neurol. 2002, 1, 173–181. [Google Scholar] [CrossRef]
- Klein, P.; Friedman, A.; Hameed, M.Q.; Kaminski, R.M.; Bar-Klein, G.; Klitgaard, H.; Koepp, M.; Jozwiak, S.; Prince, D.A.; Rotenberg, A.; et al. Repurposed molecules for antiepileptogenesis: Missing an opportunity to prevent epilepsy? Epilepsia 2020, 61, 359–386. [Google Scholar] [CrossRef] [PubMed]
- Alrabiah, H. Levetiracetam. Profiles Drug Subst. Excip. Relat. Methodol. 2019, 44, 167–204. [Google Scholar] [CrossRef]
- Löscher, W.; Gillard, M.; Sands, Z.A.; Kaminski, R.M.; Klitgaard, H. Synaptic Vesicle Glycoprotein 2A Ligands in the Treatment of Epilepsy and Beyond. CNS Drugs 2016, 30, 1055–1077. [Google Scholar] [CrossRef] [Green Version]
- Lynch, B.A.; Lambeng, N.; Nocka, K.; Kensel-Hammes, P.; Bajjalieh, S.M.; Matagne, A.; Fuks, B. The synaptic vesicle is the protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proc. Natl. Acad. Sci. USA 2004, 101, 9861–9866. [Google Scholar] [CrossRef] [Green Version]
- Crepeau, A.Z.; Treiman, D.M. Levetiracetam: A comprehensive review. Expert Rev. Neurother. 2010, 10, 159–171. [Google Scholar] [CrossRef]
- Cortes-Altamirano, J.L.; Olmos-Hernández, A.; Bonilla-Jaime, H.; Bandala, C.; González-Maciel, A.; Alfaro-Rodríguez, A. Levetiracetam as an antiepileptic, neuroprotective, and hyperalgesic drug. Neurol. India 2016, 64, 1266–1275. [Google Scholar] [CrossRef]
- Wong, L.C.; Freeburg, J.D.; Montouris, G.D.; Hohler, A.D. Two patients with Hashimoto’s encephalopathy and uncontrolled diabetes successfully treated with levetiracetam. J. Neurol. Sci. 2015, 348, 251–252. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.; Mataluni, G.; Codecà, C.; Fiore, S.; Buttari, F.; Musella, A.; Castelli, M.; Bernardi, G.; Centonze, D. Effects of levetiracetam on chronic pain in multiple sclerosis: Results of a pilot, randomized, placebo-controlled study. Eur. J. Neurol. 2009, 16, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Falah, M.; Madsen, C.; Holbech, J.V.; Sindrup, S.H. A randomized, placebo-controlled trial of levetiracetam in central pain in multiple sclerosis. Eur. J. Pain 2012, 16, 860–869. [Google Scholar] [CrossRef]
- Steinhoff, B.J.; Staack, A.M. Levetiracetam and brivaracetam: A review of evidence from clinical trials and clinical experience. Ther. Adv. Neurol. Disord. 2019, 12, 3518. [Google Scholar] [CrossRef] [Green Version]
- Kenda, B.M.; Matagne, A.C.; Talaga, P.E.; Pasau, P.M.; Differding, E.; Lallemand, B.I.; Frycia, A.M.; Moureau, F.G.; Klitgaard, H.V.; Gillard, M.R.; et al. Discovery of 4-substituted pyrrolidone butanamides as new agents with significant antiepileptic activity. J. Med. Chem. 2004, 47, 530–549. [Google Scholar] [CrossRef]
- Leclercq, K.; Matagne, A.; Provins, L.; Klitgaard, H.; Kaminski, R.M. Pharmacological Profile of the Novel Antiepileptic Drug Candidate Padsevonil: Characterization in Rodent Seizure and Epilepsy Models. J. Pharmacol. Exp. Ther. 2020, 372, 11–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surges, R.; Volynski, K.E.; Walker, M.C. Is levetiracetam different from other antiepileptic drugs? Levetiracetam and its cellular mechanism of action in epilepsy revisited. Ther. Adv. Neurol. Disord. 2008, 1, 13–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogl, C.; Mochida, S.; Wolff, C.; Whalley, B.J.; Stephens, G.J. The synaptic vesicle glycoprotein 2A ligand levetiracetam inhibits presynaptic Ca2+ channels through an intracellular pathway. Mol. Pharmacol. 2012, 82, 199–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnet, U.; Bingmann, D.; Speckmann, E.-J.; Wiemann, M. Levetiracetam mediates subtle pH-shifts in adult human neocortical pyramidal cells via an inhibition of the bicarbonate-driven neuronal pH-regulation—Implications for excitability and plasticity modulation. Brain Res. 2019, 1710, 146–156. [Google Scholar] [CrossRef]
- Lévesque, M.; Behr, C.; Avoli, M. The anti-ictogenic effects of levetiracetam are mirrored by interictal spiking and high-frequency oscillation changes in a model of temporal lobe epilepsy. Seizure 2015, 25, 18–25. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Inamine, M.; Oshima, W.; Kotani, M.; Chiba, Y.; Ueno, M.; Ishihara, Y. Prevention of status epilepticus-induced brain edema and neuronal cell loss by repeated treatment with high-dose levetiracetam. Brain Res. 2015, 1608, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Taniguchi, R.; Matsuo, T.; Oguro, A.; Vogel, C.F.A.; Yamazaki, T.; Ishihara, Y. Suppressive effects of levetiracetam on neuroinflammation and phagocytic microglia: A comparative study of levetiracetam, valproate and carbamazepine. Neurosci. Lett. 2019, 708, 134363. [Google Scholar] [CrossRef] [PubMed]
- Sarangi, S.C.; Pattnaik, S.S.; Katyal, J.; Kaleekal, T.; Dinda, A.K. An interaction study of Ocimum sanctum L. and levetiracetam in pentylenetetrazole kindling model of epilepsy. J. Ethnopharmacol. 2020, 249, 112389. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W. The holy grail of epilepsy prevention: Preclinical approaches to antiepileptogenic treatments. Neuropharmacology 2020, 167, 107605. [Google Scholar] [CrossRef]
- Hakami, T. Neuropharmacology of Antiseizure Drugs. Neuropsychopharmacol. Rep. 2021, 41, 336–351. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration (FDA). FDA-Approved Drugs. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=021035 (accessed on 22 November 2021).
- U.S. Food and Drug Administration (FDA). FDA-Approved Drugs. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=021872 (accessed on 22 November 2021).
- European Medicines Agency (EMA). Keppra. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/keppra#authorisation-details-section (accessed on 22 November 2021).
- Italiano, D.; Ferlazzo, E.; Gasparini, S.; Spina, E.; Mondello, S.; Labate, A.; Gambardella, A.; Aguglia, U. Generalized versus partial reflex seizures: A review. Seizure 2014, 23, 512–520. [Google Scholar] [CrossRef] [Green Version]
- Auvin, S. Treatment of juvenile myoclonic epilepsy. CNS Neurosci. Ther. 2008, 14, 227–233. [Google Scholar] [CrossRef]
- Cereghino, J.J.; Biton, V.; Abou-Khalil, B.; Dreifuss, F.; Gauer, L.J.; Leppik, I. Levetiracetam for partial seizures: Results of a double-blind, randomized clinical trial. Neurology 2000, 55, 236–242. [Google Scholar] [CrossRef]
- Abou-Khalil, B. Levetiracetam in the treatment of epilepsy. Neuropsychiatr. Dis. Treat. 2008, 4, 507–523. [Google Scholar] [CrossRef] [Green Version]
- Shorvon, S.D.; Löwenthal, A.; Janz, D.; Bielen, E.; Loiseau, P. Multicenter double-blind, randomized, placebo-controlled trial of levetiracetam as add-on therapy in patients with refractory partial seizures. European Levetiracetam Study Group. Epilepsia 2000, 41, 1179–1186. [Google Scholar] [CrossRef]
- Ben-Menachem, E.; Falter, U. Efficacy and tolerability of levetiracetam 3000 mg/d in patients with refractory partial seizures: A multicenter, double-blind, responder-selected study evaluating monotherapy. European Levetiracetam Study Group. Epilepsia 2000, 41, 1276–1283. [Google Scholar] [CrossRef] [PubMed]
- Abdelmesih, S.K.; Elkhateeb, N.; Zakaria, M.; Girgis, M.Y. Initial levetiracetam versus valproate monotherapy in antiseizure medicine (ASM)-naïve pediatric patients with idiopathic generalized epilepsy with tonic-clonic seizures. Seizure 2021, 91, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Falsaperla, R.; Scalia, B.; Giugno, A.; Pavone, P.; Motta, M.; Caccamo, M.; Ruggieri, M. Treating the symptom or treating the disease in neonatal seizures: A systematic review of the literature. Ital. J. Pediatrics 2021, 47, 85. [Google Scholar] [CrossRef] [PubMed]
- Özalkaya, E.; Topcuoglu, S.; Karatepe, H.; Tüten, A.; Gokmen, T.; Karatekin, G. Efficacy of levetiracetam in premature infants: Our experience and review of the literature. J. Matern.-Fetal Neonatal Med. 2019, 32, 4093–4096. [Google Scholar] [CrossRef]
- Hughes, K.; Garrity, L.; Nelson, A.S.; Lane, A.; Teusink-Cross, A. Comparison of levetiracetam versus phenytoin/fosphenytoin for busulfan seizure prophylaxis at a pediatric institution. Pediatric Transplant. 2021, 25, e14026. [Google Scholar] [CrossRef]
- Glauser, T.A.; Pellock, J.M.; Bebin, E.M.; Fountain, N.B.; Ritter, F.J.; Jensen, C.M.; Shields, W.D. Efficacy and safety of levetiracetam in children with partial seizures: An open-label trial. Epilepsia 2002, 43, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Piña-Garza, J.E.; Nordli, D.R.J.; Rating, D.; Yang, H.; Schiemann-Delgado, J.; Duncan, B. Adjunctive levetiracetam in infants and young children with refractory partial-onset seizures. Epilepsia 2009, 50, 1141–1149. [Google Scholar] [CrossRef]
- Fayyazi, A.; Ebrahimi, M.H.; Roshanaei, G.; Bazmamoun, H. Evaluation of the Levetiracetam treatment on reduction of epileptic discharges in electroencephalogram in children with epilepsy. Iran. J. Child Neurol. 2021, 15, 67–73. [Google Scholar] [CrossRef]
- Trinka, E.; Cock, H.; Hesdorffer, D.; Rossetti, A.O.; Scheffer, I.E.; Shinnar, S.; Shorvon, S.; Lowenstein, D.H. A definition and classification of status epilepticus--Report of the ILAE Task Force on Classification of Status Epilepticus. Epilepsia 2015, 56, 1515–1523. [Google Scholar] [CrossRef]
- Chu, S.-S.; Wang, H.-J.; Zhu, L.-N.; Xu, D.; Wang, X.-P.; Liu, L. Therapeutic effect of intravenous levetiracetam in status epilepticus: A meta-analysis and systematic review. Seizure 2020, 74, 49–55. [Google Scholar] [CrossRef]
- Yang, L.; Dong, X.-Z.; Cui, X.-H.; Liu, J.-M.; Liu, W.-N.; Zhang, L. Comparison of the efficacy and safety of levetiracetam and phenytoin in the treatment of established status epilepticus: A systematic review and meta-analysis. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2021, 89, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Chen, Y.; Jia, Y.; Wang, Z.; Wang, X.; Jiang, L.; Ai, C.; Li, W.; Liu, Y. Efficacy and safety of levetiracetam versus (fos)phenytoin for second-line treatment of epilepticus: A meta-analysis of latest randomized controlled trials. Seizure 2021, 91, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Haller, J.T.; Bonnin, S.; Radosevich, J. Rapid administration of undiluted intravenous levetiracetam. Epilepsia 2021, 62, 1865–1870. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Bian, H.; Zhang, L. A meta-analysis of levetiracetam for randomized placebo-controlled trials in patients with refractory epilepsy. Neuropsychiatr. Dis. Treat. 2019, 15, 905–917. [Google Scholar] [CrossRef] [Green Version]
- Qiao, M.-Y.; Cui, H.-T.; Zhao, L.-Z.; Miao, J.-K.; Chen, Q.-X. Efficacy and Safety of Levetiracetam vs. Phenobarbital for Neonatal Seizures: A Systematic Review and Meta-Analysis. Front. Neurol. 2021, 12, 747745. [Google Scholar] [CrossRef]
- Ruangritkul, P.; Tiamkao, S.; Chainirun, N.; Pranboon, S.; Tiamkao, S.; Sawanyawisuth, K.; Khamsai, S. The Efficacy and Safety Profile of Generic Intravenous Levetiracetam in a Real-World Setting. Curr. Ther. Res. Clin. Exp. 2021, 95, 100648. [Google Scholar] [CrossRef]
- Wright, C.; Downing, J.; Mungall, D.; Khan, O.; Williams, A.; Fonkem, E.; Garrett, D.; Aceves, J.; Kirmani, B. Clinical pharmacology and pharmacokinetics of levetiracetam. Front. Neurol. 2013, 4, 192. [Google Scholar] [CrossRef] [Green Version]
- Hnaini, M.; Darwich, M.; Koleilat, N.; Jaafar, F.; Hanneyan, S.; Rahal, S.; El Mikati, I.; Shbarou, R.M.; Nabout, R.; Maalouf, F.I.; et al. High-Dose Levetiracetam for Neonatal Seizures: A Retrospective Review. Seizure 2020, 82, 7–11. [Google Scholar] [CrossRef]
- Besli, G.E.; Yuksel Karatoprak, E.; Yilmaz, S. Efficacy and safety profile of intravenous levetiracetam versus phenytoin in convulsive status epilepticus and acute repetitive seizures in children. Epilepsy Behav. 2020, 111, 107289. [Google Scholar] [CrossRef]
- Yi, Z.-M.; Wen, C.; Cai, T.; Xu, L.; Zhong, X.-L.; Zhan, S.-Y.; Zhai, S.-D. Levetiracetam for epilepsy: An evidence map of efficacy, safety and economic profiles. Neuropsychiatr. Dis. Treat. 2019, 15, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Verrotti, A.; Prezioso, G.; Di Sabatino, F.; Franco, V.; Chiarelli, F.; Zaccara, G. The adverse event profile of levetiracetam: A meta-analysis on children and adults. Seizure 2015, 31, 49–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Choi, H.; Hirsch, L.J.; Katz, A.; Legge, A.; Buchsbaum, R.; Detyniecki, K. Psychiatric and behavioral side effects of antiepileptic drugs in adults with epilepsy. Epilepsy Behav. 2017, 76, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Cortes, C.; Manterola, C. Behavioral alterations associated with levetiracetam in pediatric epilepsy. Epilepsy Behav. 2020, 112, 107472. [Google Scholar] [CrossRef] [PubMed]
- Kawai, M.; Goji, H.; Kanemoto, K. Differences in aggression as psychiatric side effect of levetiracetam and perampanel in patients with epilepsy. Epilepsy Behav. 2022, 126, 108493. [Google Scholar] [CrossRef]
- Moon, J.U.; Han, J.Y. Comparative Efficacy of Levetiracetam for Epilepsy in School-Aged Children with Intellectual Disability and Normal Intelligence. Brain Sci. 2021, 11, 1452. [Google Scholar] [CrossRef]
- Liu, B.-K.; Jiang, L.; Li, X.-J.; Hong, S.-Q.; Chen, W.; Hu, Y. Efficacy and safety of levetiracetam in the off-label treatment of neonatal seizures. Int. J. Neurosci. 2020, 130, 336–342. [Google Scholar] [CrossRef]
- Bangash, O.; Simonin, A.; Tsimiklis, C.; Ramakonar, H.; Honeybul, S. Prophylactic levetiracetam-induced pancytopenia with traumatic extra-dural hematoma: Case report. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2020, 80, 264–266. [Google Scholar] [CrossRef]
- Fagan, A.; Fuld, J.; Soon, E. Levetiracetam-induced eosinophilic pneumonia. BMJ Case Rep. 2017, 2017, 9121. [Google Scholar] [CrossRef]
- Gayatri, P.; Selvam, M.M.; Sreeharsha, S.V. Levetiracetam-Induced Hepatic Dysfunction. Neurol. India 2020, 68, 910–912. [Google Scholar] [CrossRef]
- Moinuddin, I.A. Suspected Levetiracetam-Induced Rhabdomyolysis: A Case Report and Literature Review. Am. J. Case Rep. 2020, 21, e926064. [Google Scholar] [CrossRef]
- Spencer, D. Levetiracetam in Men With Epilepsy: Testosterone Is Left Alone But Sperm Count Is Paramount. Epilepsy Curr. 2017, 17, 99–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brighina, F.; Palermo, A.; Aloisio, A.; Francolini, M.; Giglia, G.; Fierro, B. Levetiracetam in the prophylaxis of migraine with aura: A 6-month open-label study. Clin. Neuropharmacol. 2006, 29, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Hamza, M.S.; Anderson, D.G.; Snyder, J.W.; Deschner, S.; Cifu, D.X. Effectiveness of levetiracetam in the treatment of lumbar radiculopathy: An open-label prospective cohort study. PMR 2009, 1, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Awaad, Y.; Michon, A.M.; Minarik, S. Use of levetiracetam to treat tics in children and adolescents with Tourette syndrome. Mov. Disord. 2005, 20, 714–718. [Google Scholar] [CrossRef] [PubMed]
- Solaro, C.; de Sire, A.; Messmer Uccelli, M.; Mueller, M.; Bergamaschi, R.; Gasperini, C.; Restivo, D.A.; Stabile, M.R.; Patti, F. Efficacy of levetiracetam on upper limb movement in multiple sclerosis patients with cerebellar signs: A multicenter double-blind, placebo-controlled, crossover study. Eur. J. Neurol. 2020, 27, 2209–2216. [Google Scholar] [CrossRef]
- Solaro, C.; Brichetto, G.; Capello, E.; Abuarqub, S.; Sanguineti, V. Activity, tolerability and efficacy of levetiracetam on cerebellar symptoms in multiple sclerosis patients: A pilot kinematic study. Eur. J. Neurol. 2008, 15, 619–626. [Google Scholar] [CrossRef]
- D’Amelio, M.; Callari, G.; Gammino, M.; Saia, V.; Lupo, I.; Salemi, G.; Ragonese, P.; Savettieri, G. Levetiracetam in the treatment of vascular chorea: A case report. Eur. J. Clin. Pharmacol. 2005, 60, 835–836. [Google Scholar] [CrossRef]
- Direk, M.; Epcacan, S.; Epcacan, Z.; Yildirim, D.D.; Okuyaz, C. Efficacy of levetiracetam in the treatment of Sydenham chorea. Pediatrics Int. 2020, 62, 1264–1268. [Google Scholar] [CrossRef]
- Şahin, S.; Cansu, A. A New Alternative Drug With Fewer Adverse Effects in the Treatment of Sydenham Chorea: Levetiracetam Efficacy in a Child. Clin. Neuropharmacol. 2015, 38, 144–146. [Google Scholar] [CrossRef]
- Wang, M.; Jiang, L.; Tang, X. Levetiracetam is associated with decrease in subclinical epileptiform discharges and improved cognitive functions in pediatric patients with autism spectrum disorder. Neuropsychiatr. Dis. Treat. 2017, 13, 2321–2326. [Google Scholar] [CrossRef] [Green Version]
- Deriaz, N.; Willi, J.P.; Orihuela-Flores, M.; Galli Carminati, G.; Ratib, O. Treatment with levetiracetam in a patient with pervasive developmental disorders, severe intellectual disability, self-injurious behavior, and seizures: A case report. Neurocase 2012, 18, 386–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farooq, M.U.; Bhatt, A.; Majid, A.; Gupta, R.; Khasnis, A.; Kassab, M.Y. Levetiracetam for managing neurologic and psychiatric disorders. Am. J. Health Pharm. AJHP Off. J. Am. Soc. Health Pharm. 2009, 66, 541–561. [Google Scholar] [CrossRef] [PubMed]
- Mattes, J.A. Levetiracetam in patients with impulsive aggression: A double-blind, placebo-controlled trial. J. Clin. Psychiatry 2008, 69, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Çökmüş, F.P.; Aşçıbaşı, K.; Öztekin, S.; Demet, M.M. Relationship of levetiracetam and obsessive-compulsive disorder: A case report. Psychiatry Clin. Psychopharmacol. 2017, 27, 319–321. [Google Scholar] [CrossRef] [Green Version]
- Esang, M.; Santos, M.G.; Ahmed, S. Levetiracetam and Suicidality: A Case Report and Literature Review. Prim. Care Compan. CNS Disord. 2020, 22, 19nr02502. [Google Scholar] [CrossRef]
- Müller, C.A.; Schäfer, M.; Schneider, S.; Heimann, H.M.; Hinzpeter, A.; Volkmar, K.; Förg, A.; Heinz, A.; Hein, J. Efficacy and safety of levetiracetam for outpatient alcohol detoxification. Pharmacopsychiatry 2010, 43, 184–189. [Google Scholar] [CrossRef]
- Jabbarli, R.; Ahmadipour, Y.; Rauschenbach, L.; Santos, A.N.; Darkwah Oppong, M.; Pierscianek, D.; Quesada, C.M.; Kebir, S.; Dammann, P.; Guberina, N.; et al. How about Levetiracetam in Glioblastoma? An Institutional Experience and Meta-Analysis. Cancers 2021, 13, 3370. [Google Scholar] [CrossRef]
- Woods, S.W.; Saksa, J.R.; Baker, C.B.; Cohen, S.J.; Tek, C. Effects of levetiracetam on tardive dyskinesia: A randomized, double-blind, placebo-controlled study. J. Clin. Psychiatry 2008, 69, 546–554. [Google Scholar] [CrossRef]
- Kakisaka, Y.; Jin, K.; Fujikawa, M.; Kitazawa, Y.; Kato, K.; Nakasato, N. Levetiracetam improves symptoms of multiple chemical sensitivity: Case report. J. Med. Investig. 2017, 64, 296–298. [Google Scholar] [CrossRef] [Green Version]
- Meena, N.; Satia, M.P.S. A study of the obstetric and perinatal outcomes of eclampsia and the use of levetiracetam in its management. Int. J. Reprod. Contracept. Obs. Gynecol. 2016, 5, 4266–4270. [Google Scholar]
- Grüter, T.; Ayzenberg, I.; Gold, R.; Börnke, C. Charles Bonnet syndrome successfully treated with levetiracetam. J. Neurol. 2016, 263, 1872–1875. [Google Scholar] [CrossRef] [PubMed]
- Hejazi, R.A.; Reddymasu, S.C.; Namin, F.; Lavenbarg, T.; Foran, P.; McCallum, R.W. Efficacy of tricyclic antidepressant therapy in adults with cyclic vomiting syndrome: A two-year follow-up study. J. Clin. Gastroenterol. 2010, 44, 18–21. [Google Scholar] [CrossRef] [PubMed]
- Clouse, R.E.; Sayuk, G.S.; Lustman, P.J.; Prakash, C. Zonisamide or levetiracetam for adults with cyclic vomiting syndrome: A case series. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2007, 5, 44–48. [Google Scholar] [CrossRef]
- Gower, A.J.; Noyer, M.; Verloes, R.; Gobert, J.; Wülfert, E. ucb L059, a novel anti-convulsant drug: Pharmacological profile in animals. Eur. J. Pharmacol. 1992, 222, 193–203. [Google Scholar] [CrossRef]
- Gualtieri, F.; Manetti, D.; Romanelli, M.N.; Ghelardini, C. Design and study of piracetam-like nootropics, controversial members of the problematic class of cognition-enhancing drugs. Curr. Pharm. Des. 2002, 8, 125–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frycia, A.; Starck, J.P.; Jadot, S.; Lallemand, B.; Leclercq, K.; Brutto, P.L.; Matagne, A.; Verbois, V.; Mercier, J.; Kenda, B. Discovery of indolone acetamides as novel SV2A ligands with improved potency toward seizure suppression. ChemMedChem 2010, 5, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Daniels, V.; Wood, M.; Leclercq, K.; Kaminski, R.M.; Gillard, M. Modulation of the conformational state of the SV2A protein by an allosteric mechanism as evidenced by ligand binding assays. Br. J. Pharmacol. 2013, 169, 1091–1101. [Google Scholar] [CrossRef] [Green Version]
- Bennett, B.; Matagne, A.; Michel, P.; Leonard, M.; Cornet, M.; Meeus, M.-A.; Toublanc, N. Seletracetam (UCB 44212). Neurotherapeutics 2007, 4, 117–122. [Google Scholar] [CrossRef]
- Pollard, J.R. Seletracetam, a small molecule SV2A modulator for the treatment of epilepsy. Curr. Opin. Investig. Drugs 2008, 9, 101–107. [Google Scholar]
- Malykh, A.G.; Sadaie, M.R. Piracetam and piracetam-like drugs: From basic science to novel clinical applications to CNS disorders. Drugs 2010, 70, 287–312. [Google Scholar] [CrossRef]
- Abram, M.; Rapacz, A.; Mogilski, S.; Latacz, G.; Lubelska, A.; Kamiński, R.M.; Kamiński, K. Multitargeted Compounds Derived from (2,5-Dioxopyrrolidin-1-yl)(phenyl)-Acetamides as Candidates for Effective Anticonvulsant and Antinociceptive Agents. ACS Chem. Neurosci. 2020, 11, 1996–2008. [Google Scholar] [CrossRef]
- Niespodziany, I.; Ghisdal, P.; Mullier, B.; Wood, M.; Provins, L.; Kaminski, R.M.; Wolff, C. Functional characterization of the antiepileptic drug candidate, padsevonil, on GABA(A) receptors. Epilepsia 2020, 61, 914–923. [Google Scholar] [CrossRef] [Green Version]
- Wood, M.; Daniels, V.; Provins, L.; Wolff, C.; Kaminski, R.M.; Gillard, M. Pharmacological Profile of the Novel Antiepileptic Drug Candidate Padsevonil: Interactions with Synaptic Vesicle 2 Proteins and the GABA(A) Receptor. J. Pharmacol. Exp. Ther. 2020, 372, 1–10. [Google Scholar] [CrossRef] [Green Version]
- UCB. Available online: Chrome-extension://efaidnbmnnnibpcajpcglclefindmkaj/viewer.html?pdfurl=https%3A%2F%2Fwww.ucb.com%2Fsites%2Fdefault%2Ffiles%2Fpress_files%2Fa1e5de61474c0bbf.pdf&clen=513698&chunk=true (accessed on 10 January 2022).
- Noyer, M.; Gillard, M.; Matagne, A.; Hénichart, J.P.; Wülfert, E. The novel antiepileptic drug levetiracetam (ucb L059) appears to act via a specific binding site in CNS membranes. Eur. J. Pharmacol. 1995, 286, 137–146. [Google Scholar] [CrossRef]
- Gillard, M.; Fuks, B.; Michel, P.; Vertongen, P.; Massingham, R.; Chatelain, P. Binding characteristics of [3H]ucb 30889 to levetiracetam binding sites in rat brain. Eur. J. Pharmacol. 2003, 478, 1–9. [Google Scholar] [CrossRef]
- Rogawski, M.A. Brivaracetam: A rational drug discovery success story. Br. J. Pharmacol. 2008, 154, 1555–1557. [Google Scholar] [CrossRef] [Green Version]
- Bajjalieh, S.M.; Peterson, K.; Shinghal, R.; Scheller, R.H. SV2, a brain synaptic vesicle protein homologous to bacterial transporters. Science 1992, 257, 1271–1273. [Google Scholar] [CrossRef]
- Gillard, M.; Fuks, B.; Leclercq, K.; Matagne, A. Binding characteristics of brivaracetam, a selective, high affinity SV2A ligand in rat, mouse and human brain: Relationship to anti-convulsant properties. Eur. J. Pharmacol. 2011, 664, 36–44. [Google Scholar] [CrossRef]
- Shi, J.; Anderson, D.; Lynch, B.A.; Castaigne, J.-G.; Foerch, P.; Lebon, F. Combining modelling and mutagenesis studies of synaptic vesicle protein 2A to identify a series of residues involved in racetam binding. Biochem. Soc. Trans. 2011, 39, 1341–1347. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Daniels, V.; Sands, Z.A.; Lebon, F.; Shi, J.; Biggin, P.C. Exploring the interaction of SV2A with racetams using homology modelling, molecular dynamics and site-directed mutagenesis. PLoS ONE 2015, 10, e0116589. [Google Scholar] [CrossRef]
- Correa-Basurto, J.; Cuevas-Hernández, R.I.; Phillips-Farfán, B.V.; Martínez-Archundia, M.; Romo-Mancillas, A.; Ramírez-Salinas, G.L.; Pérez-González, Ó.A.; Trujillo-Ferrara, J.; Mendoza-Torreblanca, J.G. Identification of the antiepileptic racetam binding site in the synaptic vesicle protein 2A by molecular dynamics and docking simulations. Front. Cell. Neurosci. 2015, 9, 125. [Google Scholar] [CrossRef] [Green Version]
- Wood, M.D.; Gillard, M. Evidence for a differential interaction of brivaracetam and levetiracetam with the synaptic vesicle 2A protein. Epilepsia 2017, 58, 255–262. [Google Scholar] [CrossRef]
- Wood, M.D.; Sands, Z.A.; Vandenplas, C.; Gillard, M. Further evidence for a differential interaction of brivaracetam and levetiracetam with the synaptic vesicle 2A protein. Epilepsia 2018, 59, e147–e151. [Google Scholar] [CrossRef]
- Janz, R.; Goda, Y.; Geppert, M.; Missler, M.; Südhof, T.C. SV2A and SV2B function as redundant Ca2+ regulators in neurotransmitter release. Neuron 1999, 24, 1003–1016. [Google Scholar] [CrossRef] [Green Version]
- Chang, W.-P.; Südhof, T.C. SV2 renders primed synaptic vesicles competent for Ca2+ -induced exocytosis. J. Neurosci. 2009, 29, 883–897. [Google Scholar] [CrossRef] [Green Version]
- Pichardo, L.A.; Contreras, I.J.; Zamudio, S.R.; Mixcoha, E.; Mendoza, J.G. Synaptic Vesicle Protein 2A as a novel pharmacological target with broad potential for new antiepileptic drugs. In Antiepileptic Drug Discovery: Novel Approaches, Methods in Pharmacology and Toxicology, 1st ed.; Talevi, A., Rocha, L., Eds.; Springer: Berlin, Germany, 2016; pp. 53–65. ISBN 13:978-1493963539. [Google Scholar]
- Crowder, K.M.; Gunther, J.M.; Jones, T.A.; Hale, B.D.; Zhang, H.Z.; Peterson, M.R.; Scheller, R.H.; Chavkin, C.; Bajjalieh, S.M. Abnormal neurotransmission in mice lacking synaptic vesicle protein 2A (SV2A). Proc. Natl. Acad. Sci. USA 1999, 96, 15268–15273. [Google Scholar] [CrossRef] [Green Version]
- Custer, K.L.; Austin, N.S.; Sullivan, J.M.; Bajjalieh, S.M. Synaptic vesicle protein 2 enhances release probability at quiescent synapses. J. Neurosci. 2006, 26, 1303–1313. [Google Scholar] [CrossRef]
- Vogl, C.; Tanifuji, S.; Danis, B.; Daniels, V.; Foerch, P.; Wolff, C.; Whalley, B.J.; Mochida, S.; Stephens, G.J. Synaptic vesicle glycoprotein 2A modulates vesicular release and calcium channel function at peripheral sympathetic synapses. Eur. J. Neurosci. 2015, 41, 398–409. [Google Scholar] [CrossRef]
- Xu, T.; Bajjalieh, S.M. SV2 modulates the size of the readily releasable pool of secretory vesicles. Nat. Cell Biol. 2001, 3, 691–698. [Google Scholar] [CrossRef]
- Venkatesan, K.; Alix, P.; Marquet, A.; Doupagne, M.; Niespodziany, I.; Rogister, B.; Seutin, V. Altered balance between excitatory and inhibitory inputs onto CA1 pyramidal neurons from SV2A-deficient but not SV2B-deficient mice. J. Neurosci. Res. 2012, 90, 2317–2327. [Google Scholar] [CrossRef]
- Schivell, A.E.; Mochida, S.; Kensel-Hammes, P.; Custer, K.L.; Bajjalieh, S.M. SV2A and SV2C contain a unique synaptotagmin-binding site. Mol. Cell. Neurosci. 2005, 29, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Nowack, A.; Kensel-Hammes, P.; Gardner, R.G.; Bajjalieh, S.M. Cotrafficking of SV2 and synaptotagmin at the synapse. J. Neurosci. 2010, 30, 5569–5578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stout, K.A.; Dunn, A.R.; Hoffman, C.; Miller, G.W. The Synaptic Vesicle Glycoprotein 2: Structure, Function, and Disease Relevance. ACS Chem. Neurosci. 2019, 10, 3927–3938. [Google Scholar] [CrossRef] [PubMed]
- Ciruelas, K.; Marcotulli, D.; Bajjalieh, S.M. Synaptic vesicle protein 2: A multi-faceted regulator of secretion. Semin. Cell Dev. Biol. 2019, 95, 130–141. [Google Scholar] [CrossRef]
- Nowack, A.; Malarkey, E.B.; Yao, J.; Bleckert, A.; Hill, J.; Bajjalieh, S.M. Levetiracetam reverses synaptic deficits produced by overexpression of SV2A. PLoS ONE 2011, 6, e29560. [Google Scholar] [CrossRef]
- Meehan, A.L.; Yang, X.; McAdams, B.D.; Yuan, L.L.; Rothman, S.M. A new mechanism for antiepileptic drug action: Vesicular entry may mediate the effects of levetiracetam. J. Neurophysiol. 2011, 106, 1227–1239. [Google Scholar] [CrossRef]
- Mendoza-Torreblanca, J.G.; Vanoye-Carlo, A.; Phillips-Farfán, B.V.; Carmona-Aparicio, L.; Gómez-Lira, G. Synaptic vesicle protein 2A: Basic facts and role in synaptic function. Eur. J. Neurosci. 2013, 38, 3529–3539. [Google Scholar] [CrossRef]
- Nicolas, J.-M.; Hannestad, J.; Holden, D.; Kervyn, S.; Nabulsi, N.; Tytgat, D.; Huang, Y.; Chanteux, H.; Staelens, L.; Matagne, A.; et al. Brivaracetam, a selective high-affinity synaptic vesicle protein 2A (SV2A) ligand with preclinical evidence of high brain permeability and fast onset of action. Epilepsia 2016, 57, 201–209. [Google Scholar] [CrossRef] [Green Version]
- Doheny, H.C.; Ratnaraj, N.; Whittington, M.A.; Jefferys, J.G.; Patsalos, P.N. Blood and cerebrospinal fluid pharmacokinetics of the novel anticonvulsant levetiracetam (ucb L059) in the rat. Epilepsy Res. 1999, 34, 161–168. [Google Scholar] [CrossRef]
- Patsalos, P.N. Pharmacokinetic profile of levetiracetam: Toward ideal characteristics. Pharmacol. Ther. 2000, 85, 77–85. [Google Scholar] [CrossRef]
- Tong, X.; Patsalos, P.N. A microdialysis study of the novel antiepileptic drug levetiracetam: Extracellular pharmacokinetics and effect on taurine in rat brain. Br. J. Pharmacol. 2001, 133, 867–874. [Google Scholar] [CrossRef] [Green Version]
- Kaminski, R.M.; Gillard, M.; Leclercq, K.; Hanon, E.; Lorent, G.; Dassesse, D.; Matagne, A.; Klitgaard, H. Proepileptic phenotype of SV2A-deficient mice is associated with reduced anticonvulsant efficacy of levetiracetam. Epilepsia 2009, 50, 1729–1740. [Google Scholar] [CrossRef]
- De Groot, M.; Aronica, E.; Heimans, J.J.; Reijneveld, J.C. Synaptic vesicle protein 2A predicts response to levetiracetam in patients with glioma. Neurology 2011, 77, 532–539. [Google Scholar] [CrossRef]
- Ohno, Y.; Ishihara, S.; Terada, R.; Kikuta, M.; Sofue, N.; Kawai, Y.; Serikawa, T.; Sasa, M. Preferential increase in the hippocampal synaptic vesicle protein 2A (SV2A) by pentylenetetrazole kindling. Biochem. Biophys. Res. Commun. 2009. [Google Scholar] [CrossRef]
- Matveeva, E.A.; Vanaman, T.C.; Whiteheart, S.W.; Slevin, J.T. Levetiracetam prevents kindling-induced asymmetric accumulation of hippocampal 7S SNARE complexes. Epilepsia 2008, 49, 1749–1758. [Google Scholar] [CrossRef] [Green Version]
- Inaba, T.; Miyamoto, N.; Hira, K.; Ueno, Y.; Yamashiro, K.; Watanabe, M.; Shimada, Y.; Hattori, N.; Urabe, T. Protective Role of Levetiracetam Against Cognitive Impairment And Brain White Matter Damage in Mouse prolonged Cerebral Hypoperfusion. Neuroscience 2019, 414, 255–264. [Google Scholar] [CrossRef]
- Contreras-García, I.J.; Gómez-Lira, G.; Phillips-Farfán, B.V.; Pichardo-Macías, L.A.; García-Cruz, M.E.; Chávez-Pacheco, J.L.; Mendoza-Torreblanca, J.G. Synaptic Vesicle Protein 2A Expression in Glutamatergic Terminals Is Associated with the Response to Levetiracetam Treatment. Brain Sci. 2021, 11, 531. [Google Scholar] [CrossRef]
- Marcotulli, D.; Fattorini, G.; Bragina, L.; Perugini, J.; Conti, F. Levetiracetam Affects Differentially Presynaptic Proteins in Rat Cerebral Cortex. Front. Cell. Neurosci. 2017, 11, 389. [Google Scholar] [CrossRef]
- Niespodziany, I.; Klitgaard, H.; Margineanu, D.G. Levetiracetam inhibits the high-voltage-activated Ca(2+) current in pyramidal neurones of rat hippocampal slices. Neurosci. Lett. 2001, 306, 5–8. [Google Scholar] [CrossRef]
- Costa, C.; Martella, G.; Picconi, B.; Prosperetti, C.; Pisani, A.; Di Filippo, M.; Pisani, F.; Bernardi, G.; Calabresi, P. Multiple mechanisms underlying the neuroprotective effects of antiepileptic drugs against in vitro ischemia. Stroke 2006, 37, 1319–1326. [Google Scholar] [CrossRef]
- Pisani, A.; Bonsi, P.; Martella, G.; De Persis, C.; Costa, C.; Pisani, F.; Bernardi, G.; Calabresi, P. Intracellular calcium increase in epileptiform activity: Modulation by levetiracetam and lamotrigine. Epilepsia 2004, 45, 719–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukyanetz, E.A.; Shkryl, V.M.; Kostyuk, P.G. Selective blockade of N-type calcium channels by levetiracetam. Epilepsia 2002, 43, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.-D.; Ishihara, K.; Seki, T.; Hanaya, R.; Kurisu, K.; Arita, K.; Serikawa, T.; Sasa, M. Inhibitory effects of levetiracetam on the high-voltage-activated L-type Ca2+ channels in hippocampal CA3 neurons of spontaneously epileptic rat (SER). Brain Res. Bull. 2013, 90, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, L.S.; Delorenzo, R.J. Mechanisms of levetiracetam in the control of status epilepticus and epilepsy. Front. Neurol. 2014, 5, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angehagen, M.; Margineanu, D.G.; Ben-Menachem, E.; Rönnbäck, L.; Hansson, E.; Klitgaard, H. Levetiracetam reduces caffeine-induced Ca2+ transients and epileptiform potentials in hippocampal neurons. Neuroreport 2003, 14, 471–475. [Google Scholar] [CrossRef]
- Nagarkatti, N.; Deshpande, L.S.; DeLorenzo, R.J. Levetiracetam inhibits both ryanodine and IP3 receptor activated calcium induced calcium release in hippocampal neurons in culture. Neurosci. Lett. 2008, 436, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Cataldi, M.; Lariccia, V.; Secondo, A.; di Renzo, G.; Annunziato, L. The antiepileptic drug levetiracetam decreases the inositol 1,4,5-trisphosphate-dependent [Ca2+]I increase induced by ATP and bradykinin in PC12 cells. J. Pharmacol. Exp. Ther. 2005, 313, 720–730. [Google Scholar] [CrossRef] [Green Version]
- Navidhamidi, M.; Ghasemi, M.; Mehranfard, N. Epilepsy-associated alterations in hippocampal excitability. Rev. Neurosci. 2017, 28, 307–334. [Google Scholar] [CrossRef]
- Rigo, J.-M.; Hans, G.; Nguyen, L.; Rocher, V.; Belachew, S.; Malgrange, B.; Leprince, P.; Moonen, G.; Selak, I.; Matagne, A.; et al. The anti-epileptic drug levetiracetam reverses the inhibition by negative allosteric modulators of neuronal GABA- and glycine-gated currents. Br. J. Pharmacol. 2002, 136, 659–672. [Google Scholar] [CrossRef]
- Doelken, M.T.; Hammen, T.; Bogner, W.; Mennecke, A.; Stadlbauer, A.; Boettcher, U.; Doerfler, A.; Stefan, H. Alterations of intracerebral γ-aminobutyric acid (GABA) levels by titration with levetiracetam in patients with focal epilepsies. Epilepsia 2010, 51, 1477–1482. [Google Scholar] [CrossRef]
- Li, Q.; Chen, C.; Gong, T. High-field MRS study of GABA+ in patients with migraine: Response to levetiracetam treatment. Neuroreport 2018, 29, 1007–1010. [Google Scholar] [CrossRef] [PubMed]
- Klitgaard, H.; Matagne, A.; Grimee, R.; Vanneste-Goemaere, J.; Margineanu, D.G. Electrophysiological, neurochemical and regional effects of levetiracetam in the rat pilocarpine model of temporal lobe epilepsy. Seizure 2003, 12, 92–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuyama, K.; Tanahashi, S.; Nakagawa, M.; Yamamura, S.; Motomura, E.; Shiroyama, T.; Tanii, H.; Okada, M. Levetiracetam inhibits neurotransmitter release associated with CICR. Neurosci. Lett. 2012, 518, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Pichardo Macías, L.A.; Ramírez Mendiola, B.A.; Contreras García, I.J.; Zamudio Hernández, S.R.; Chávez Pacheco, J.L.; Sánchez Huerta, K.B.; Mendoza Torreblanca, J.G. Effect of levetiracetam on extracellular amino acid levels in the dorsal hippocampus of rats with temporal lobe epilepsy. Epilepsy Res. 2018, 140, 111–119. [Google Scholar] [CrossRef]
- Löscher, W.; Hönack, D.; Bloms-Funke, P. The novel antiepileptic drug levetiracetam (ucb L059) induces alterations in GABA metabolism and turnover in discrete areas of rat brain and reduces neuronal activity in substantia nigra pars reticulata. Brain Res. 1996, 735, 208–216. [Google Scholar] [CrossRef]
- Mazzuferi, M.; Palma, E.; Martinello, K.; Maiolino, F.; Roseti, C.; Fucile, S.; Fabene, P.F.; Schio, F.; Pellitteri, M.; Sperk, G.; et al. Enhancement of GABA(A)-current run-down in the hippocampus occurs at the first spontaneous seizure in a model of temporal lobe epilepsy. Proc. Natl. Acad. Sci. USA 2010, 107, 3180–3185. [Google Scholar] [CrossRef] [Green Version]
- Cifelli, P.; Palma, E.; Roseti, C.; Verlengia, G.; Simonato, M. Changes in the sensitivity of GABAA current rundown to drug treatments in a model of temporal lobe epilepsy. Front. Cell. Neurosci. 2013, 7, 108. [Google Scholar] [CrossRef] [Green Version]
- Palma, E.; Ragozzino, D.; Di Angelantonio, S.; Mascia, A.; Maiolino, F.; Manfredi, M.; Cantore, G.; Esposito, V.; Di Gennaro, G.; Quarato, P.; et al. The antiepileptic drug levetiracetam stabilizes the human epileptic GABAA receptors upon repetitive activation. Epilepsia 2007, 48, 1842–1849. [Google Scholar] [CrossRef]
- Malatynska, E.; Knapp, R.; Ikeda, M.; Yamamura, H.I. Beta-carboline interactions at the BZ-GABA receptor chloride-ionophore complex in the rat cerebral cortex. Brain Res. Bull. 1989, 22, 845–848. [Google Scholar] [CrossRef]
- Evans, A.K.; Lowry, C.A. Pharmacology of the beta-carboline FG-7,142, a partial inverse agonist at the benzodiazepine allosteric site of the GABA A receptor: Neurochemical, neurophysiological, and behavioral effects. CNS Drug Rev. 2007, 13, 475–501. [Google Scholar] [CrossRef]
- Kulick, C.V.; Gutherz, S.B.; Beck, V.C.; Medvedeva, N.; Soper, C.; Forcelli, P.A. Profile of anticonvulsant action of levetiracetam, tiagabine and phenobarbital against seizures evoked by DMCM (methyl-6,7-dimethoxy-4-ethyl-β-carboline-3-carboxylate) in neonatal rats. Eur. J. Pharmacol. 2014, 743, 63–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakita, M.; Kotani, N.; Kogure, K.; Akaike, N. Inhibition of excitatory synaptic transmission in hippocampal neurons by levetiracetam involves Zn2+-dependent GABA type A receptor-mediated presynaptic modulation. J. Pharmacol. Exp. Ther. 2014, 348, 246–259. [Google Scholar] [CrossRef] [PubMed]
- Buckley, K.; Kelly, R.B. Identification of a transmembrane glycoprotein specific for secretory vesicles of neural and endocrine cells. J. Cell Biol. 1985, 100, 1284–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajjalieh, S.M.; Frantz, G.D.; Weimann, J.M.; McConnell, S.K.; Scheller, R.H. Differential expression of synaptic vesicle protein 2 (SV2) isoforms. J. Neurosci. 1994, 14, 5223–5235. [Google Scholar] [CrossRef] [Green Version]
- Bragina, L.; Fattorini, G.; Giovedí, S.; Melone, M.; Bosco, F.; Benfenati, F.; Conti, F. Analysis of Synaptotagmin, SV2, and Rab3 Expression in Cortical Glutamatergic and GABAergic Axon Terminals. Front. Cell. Neurosci. 2011, 5, 32. [Google Scholar] [CrossRef] [Green Version]
- Grønborg, M.; Pavlos, N.J.; Brunk, I.; Chua, J.J.E.; Münster-Wandowski, A.; Riedel, D.; Ahnert-Hilger, G.; Urlaub, H.; Jahn, R. Quantitative comparison of glutamatergic and GABAergic synaptic vesicles unveils selectivity for few proteins including MAL2, a novel synaptic vesicle protein. J. Neurosci. 2010, 351, 981–984. [Google Scholar] [CrossRef] [Green Version]
- Tokudome, K.; Okumura, T.; Terada, R.; Shimizu, S.; Kunisawa, N.; Mashimo, T.; Serikawa, T.; Sasa, M.; Ohno, Y. A Missense Mutation of the Gene Encoding Synaptic Vesicle Glycoprotein 2A (SV2A) Confers Seizure Susceptibility by Disrupting Amygdalar Synaptic GABA Release. Front. Pharmacol. 2016, 7, 210. [Google Scholar] [CrossRef] [Green Version]
- Ohno, Y.; Tokudome, K. Therapeutic Role of Synaptic Vesicle Glycoprotein 2A (SV2A) in Modulating Epileptogenesis. CNS Neurol. Disord. Drug Targets 2017, 16, 463–471. [Google Scholar] [CrossRef]
- Contreras-García, I.J.; Pichardo-Macías, L.A.; Santana-Gómez, C.E.; Sánchez-Huerta, K.; Ramírez-Hernández, R.; Gómez-González, B.; Rocha, L.; Mendoza Torreblanca, J.G. Differential expression of synaptic vesicle protein 2A after status epilepticus and during epilepsy in a lithium-pilocarpine model. Epilepsy Behav. 2018, 88, 283–294. [Google Scholar] [CrossRef]
- Tokudome, K.; Okumura, T.; Shimizu, S.; Mashimo, T.; Takizawa, A.; Serikawa, T.; Terada, R.; Ishihara, S.; Kunisawa, N.; Sasa, M.; et al. Synaptic vesicle glycoprotein 2A (SV2A) regulates kindling epileptogenesis via GABAergic neurotransmission. Sci. Rep. 2016, 6, 27420. [Google Scholar] [CrossRef]
- Mendoza-Torreblanca, J.G.; García-Cruz, M.E.; Sánchez-Cruz, I.; Gomez-Gonzalez, B.; Juárez-Méndez, S.; Gómez-Lira, G. Analysis of Differential Expression of Synaptic Vesicle Protein 2A in the Adult Rat Brain. Neuroscience 2019, 419, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Micov, A.; Tomić, M.; Popović, B.; Stepanović-Petrović, R. The antihyperalgesic effect of levetiracetam in an inflammatory model of pain in rats: Mechanism of action. Br. J. Pharmacol. 2010, 161, 384–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stepanović-Petrović, R.M.; Micov, A.M.; Tomić, M.A.; Ugrešić, N.D. The local peripheral antihyperalgesic effect of levetiracetam and its mechanism of action in an inflammatory pain model. Anesth. Analg. 2012, 115, 1457–1466. [Google Scholar] [CrossRef] [PubMed]
- Carunchio, I.; Pieri, M.; Ciotti, M.T.; Albo, F.; Zona, C. Modulation of AMPA receptors in cultured cortical neurons induced by the antiepileptic drug levetiracetam. Epilepsia 2007, 48, 654–662. [Google Scholar] [CrossRef]
- Hentschke, M.; Wiemann, M.; Hentschke, S.; Kurth, I.; Hermans-Borgmeyer, I.; Seidenbecher, T.; Jentsch, T.J.; Gal, A.; Hübner, C.A. Mice with a targeted disruption of the Cl-/HCO3- exchanger AE3 display a reduced seizure threshold. Mol. Cell. Biol. 2006, 26, 182–191. [Google Scholar] [CrossRef] [Green Version]
- Svichar, N.; Esquenazi, S.; Chen, H.-Y.; Chesler, M. Preemptive regulation of intracellular pH in hippocampal neurons by a dual mechanism of depolarization-induced alkalinization. J. Neurosci. 2011, 31, 6997–7004. [Google Scholar] [CrossRef]
- Sander, T.; Toliat, M.R.; Heils, A.; Leschik, G.; Becker, C.; Rüschendorf, F.; Rohde, K.; Mundlos, S.; Nürnberg, P. Association of the 867Asp variant of the human anion exchanger 3 gene with common subtypes of idiopathic generalized epilepsy. Epilepsy Res. 2002, 51, 249–255. [Google Scholar] [CrossRef]
- Leniger, T.; Thöne, J.; Bonnet, U.; Hufnagel, A.; Bingmann, D.; Wiemann, M. Levetiracetam inhibits Na+-dependent Cl-/HCO3- exchange of adult hippocampal CA3 neurons from guinea-pigs. Br. J. Pharmacol. 2004, 142, 1073–1080. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.; Lynch, B.A.; Anderson, D.; Klitgaard, H.; Lu, S.; Elashoff, M.; Ebert, U.; Potschka, H.; Löscher, W. The antiepileptic drug levetiracetam selectively modifies kindling-induced alterations in gene expression in the temporal lobe of rats. Eur. J. Neurosci. 2004, 19, 334–345. [Google Scholar] [CrossRef]
- Husum, H.; Bolwig, T.G.; Sánchez, C.; Mathé, A.A.; Hansen, S.L. Levetiracetam prevents changes in levels of brain-derived neurotrophic factor and neuropeptide Y mRNA and of Y1- and Y5-like receptors in the hippocampus of rats undergoing amygdala kindling: Implications for antiepileptogenic and mood-stabilizing proper. Epilepsy Behav. 2004, 5, 204–215. [Google Scholar] [CrossRef]
- Christensen, K.V.; Leffers, H.; Watson, W.P.; Sánchez, C.; Kallunki, P.; Egebjerg, J. Levetiracetam attenuates hippocampal expression of synaptic plasticity-related immediate early and late response genes in amygdala-kindled rats. BMC Neurosci. 2010, 11, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.-E.; Choi, H.-C.; Song, H.-K.; Jo, S.-M.; Kim, D.-S.; Choi, S.-Y.; Kim, Y.-I.; Kang, T.-C. Levetiracetam inhibits interleukin-1 beta inflammatory responses in the hippocampus and piriform cortex of epileptic rats. Neurosci. Lett. 2010, 471, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Rassu, M.; Biosa, A.; Galioto, M.; Fais, M.; Sini, P.; Greggio, E.; Piccoli, G.; Crosio, C.; Iaccarino, C. Levetiracetam treatment ameliorates LRRK2 pathological mutant phenotype. J. Cell. Mol. Med. 2019, 23, 8505–8510. [Google Scholar] [CrossRef]
- Kovacevic, J.; Maroteaux, G.; Schut, D.; Loos, M.; Dubey, M.; Pitsch, J.; Remmelink, E.; Koopmans, B.; Crowley, J.; Cornelisse, L.N.; et al. Protein instability, haploinsufficiency, and cortical hyper-excitability underlie STXBP1 encephalopathy. Brain 2018, 141, 1350–1374. [Google Scholar] [CrossRef] [PubMed]
- Dilena, R.; Striano, P.; Traverso, M.; Viri, M.; Cristofori, G.; Tadini, L.; Barbieri, S.; Romeo, A.; Zara, F. Dramatic effect of levetiracetam in early-onset epileptic encephalopathy due to STXBP1 mutation. Brain Dev. 2016, 38, 128–131. [Google Scholar] [CrossRef]
- Parveen, B.; Tripathi, M.; Vohora, D. A Cross-Sectional Study to Assess the Modulation of Wnt Inhibitors following Anti-Epileptic Drug Therapy and their Correlation with Vitamin D and Receptor Activator of Nuclear Factor κ B Ligand in Indian Women with Epilepsy. Basic Clin. Pharmacol. Toxicol. 2018, 123, 271–276. [Google Scholar] [CrossRef] [Green Version]
- Lange, F.; Weßlau, K.; Porath, K.; Hörnschemeyer, J.; Bergner, C.; Krause, B.J.; Mullins, C.S.; Linnebacher, M.; Köhling, R.; Kirschstein, T. AMPA receptor antagonist perampanel affects glioblastoma cell growth and glutamate release in vitro. PLoS ONE 2019, 14, e0211644. [Google Scholar] [CrossRef] [Green Version]
- Niidome, K.; Taniguchi, R.; Yamazaki, T.; Tsuji, M.; Itoh, K.; Ishihara, Y. FosL1 Is a Novel Target of Levetiracetam for Suppressing the Microglial Inflammatory Reaction. Int. J. Mol. Sci. 2021, 22, 10962. [Google Scholar] [CrossRef]
- Hassel, B.; Taubøll, E.; Shaw, R.; Gjerstad, L.; Dingledine, R. Region-specific changes in gene expression in rat brain after chronic treatment with levetiracetam or phenytoin. Epilepsia 2010, 51, 1714–1720. [Google Scholar] [CrossRef] [Green Version]
- Sills, G.J.; Rogawski, M.A. Mechanisms of action of currently used antiseizure drugs. Neuropharmacology 2020, 168, 107966. [Google Scholar] [CrossRef] [PubMed]
- Belcastro, V.; Pierguidi, L.; Tambasco, N. Levetiracetam in brain ischemia: Clinical implications in neuroprotection and prevention of post-stroke epilepsy. Brain Dev. 2011, 33, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Rakhade, S.N.; Shah, A.K.; Agarwal, R.; Yao, B.; Asano, E.; Loeb, J.A. Activity-dependent gene expression correlates with interictal spiking in human neocortical epilepsy. Epilepsia 2007, 48 (Suppl. 5), 86–95. [Google Scholar] [CrossRef] [PubMed]
- Arion, D.; Sabatini, M.; Unger, T.; Pastor, J.; Alonso-Nanclares, L.; Ballesteros-Yáñez, I.; García Sola, R.; Muñoz, A.; Mirnics, K.; DeFelipe, J. Correlation of transcriptome profile with electrical activity in temporal lobe epilepsy. Neurobiol. Dis. 2006, 22, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Margineanu, D.G.; Matagne, A.; Kaminski, R.M.; Klitgaard, H. Effects of chronic treatment with levetiracetam on hippocampal field responses after pilocarpine-induced status epilepticus in rats. Brain Res. Bull. 2008, 77, 282–285. [Google Scholar] [CrossRef]
- Zhao, T.; Yu, J.; Wang, T.-T.; Feng, J.; Zhao, W.-B.; Sun, L.; Yu, L.-H.; Li, H.-J.; Sun, Y. Impact of ABCB1 Polymorphism on Levetiracetam Serum Concentrations in Epileptic Uygur Children in China. Ther. Drug Monit. 2020, 42, 886–892. [Google Scholar] [CrossRef]
- Calame, D.G.; Herman, I.; Riviello, J.J. A de novo heterozygous rare variant in SV2A causes epilepsy and levetiracetam-induced drug-resistant status epilepticus. Epilepsy Behav. Rep. 2021, 15, 100425. [Google Scholar] [CrossRef]
- Wolking, S.; Moreau, C.; Nies, A.T.; Schaeffeler, E.; McCormack, M.; Auce, P.; Avbersek, A.; Becker, F.; Krenn, M.; Møller, R.S.; et al. Testing association of rare genetic variants with resistance to three common antiseizure medications. Epilepsia 2020, 61, 657–666. [Google Scholar] [CrossRef]
- Grimminger, T.; Pernhorst, K.; Surges, R.; Niehusmann, P.; Priebe, L.; von Lehe, M.; Hoffmann, P.; Cichon, S.; Schoch, S.; Becker, A.J. Levetiracetam resistance: Synaptic signatures & corresponding promoter SNPs in epileptic hippocampi. Neurobiol. Dis. 2013, 60, 115–125. [Google Scholar] [CrossRef]
- Helmstaedter, C.; Mihov, Y.; Toliat, M.R.; Thiele, H.; Nuernberg, P.; Schoch, S.; Surges, R.; Elger, C.E.; Kunz, W.S.; Hurlemann, R. Genetic variation in dopaminergic activity is associated with the risk for psychiatric side effects of levetiracetam. Epilepsia 2013, 54, 36–44. [Google Scholar] [CrossRef]
- Ulloa, C.M.; Towfigh, A.; Safdieh, J. Review of levetiracetam, with a focus on the extended release formulation, as adjuvant therapy in controlling partial-onset seizures. Neuropsychiatr. Dis. Treat. 2009, 5, 467–476. [Google Scholar] [CrossRef] [Green Version]
- Löscher, W.; Hönack, D. Differences in anticonvulsant potency and adverse effects between dextromethorphan and dextrorphan in amygdala-kindled and non-kindled rats. Eur. J. Pharmacol. 1993, 238, 191–200. [Google Scholar] [CrossRef]
- Klitgaard, H.; Matagne, A.; Gobert, J.; Wülfert, E. Evidence for a unique profile of levetiracetam in rodent models of seizures and epilepsy. Eur. J. Pharmacol. 1998, 353, 191–206. [Google Scholar] [CrossRef]
- Kupferberg, H. Animal models used in the screening of antiepileptic drugs. Epilepsia 2001, 42 (Suppl. 4), 7–12. [Google Scholar] [CrossRef]
- Klitgaard, H. Levetiracetam: The preclinical profile of a new class of antiepileptic drugs? Epilepsia 2001, 42 (Suppl. 4), 13–18. [Google Scholar] [CrossRef]
- Birnstiel, S.; Wülfert, E.; Beck, S.G. Levetiracetam (ucb LO59) affects in vitro models of epilepsy in CA3 pyramidal neurons without altering normal synaptic transmission. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1997, 356, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Morgan, O.; Medenwald, B. Safety and Tolerability of Rapid Administration Undiluted Levetiracetam. Neurocrit. Care 2020, 32, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Glien, M.; Brandt, C.; Potschka, H.; Löscher, W. Effects of the novel antiepileptic drug levetiracetam on spontaneous recurrent seizures in the rat pilocarpine model of temporal lobe epilepsy. Epilepsia 2002, 43, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Ji-Qun, C.; Ishihara, K.; Nagayama, T.; Serikawa, T.; Sasa, M. Long-lasting antiepileptic effects of levetiracetam against epileptic seizures in the spontaneously epileptic rat (SER): Differentiation of levetiracetam from conventional antiepileptic drugs. Epilepsia 2005, 46, 1362–1370. [Google Scholar] [CrossRef]
- Oliveira, A.A.; Nogueira, C.R.A.; Nascimento, V.S.; Aguiar, L.M.V.; Freitas, R.M.; Sousa, F.C.F.; Viana, G.S.B.; Fonteles, M.M.F. Evaluation of levetiracetam effects on pilocarpine-induced seizures: Cholinergic muscarinic system involvement. Neurosci. Lett. 2005, 385, 184–188. [Google Scholar] [CrossRef]
- Löscher, W.; Hönack, D. Development of tolerance during chronic treatment of kindled rats with the novel antiepileptic drug levetiracetam. Epilepsia 2000, 41, 1499–1506. [Google Scholar] [CrossRef]
- Song, H.; Tufa, U.; Chow, J.; Sivanenthiran, N.; Cheng, C.; Lim, S.; Wu, C.; Feng, J.; Eubanks, J.H.; Zhang, L. Effects of Antiepileptic Drugs on Spontaneous Recurrent Seizures in a Novel Model of Extended Hippocampal Kindling in Mice. Front. Pharmacol. 2018, 9, 451. [Google Scholar] [CrossRef] [PubMed]
- Doheny, H.C.; Whittington, M.A.; Jefferys, J.G.R.; Patsalos, P.N. A comparison of the efficacy of carbamazepine and the novel anti-epileptic drug levetiracetam in the tetanus toxin model of focal complex partial epilepsy. Br. J. Pharmacol. 2002, 135, 1425–1434. [Google Scholar] [CrossRef]
- Gower, A.J.; Hirsch, E.; Boehrer, A.; Noyer, M.; Marescaux, C. Effects of levetiracetam, a novel antiepileptic drug, on convulsant activity in two genetic rat models of epilepsy. Epilepsy Res. 1995, 22, 207–213. [Google Scholar] [CrossRef]
- Bouwman, B.M.; van Rijn, C.M. Effects of levetiracetam on spike and wave discharges in WAG/Rij rats. Seizure 2004, 13, 591–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talos, D.M.; Chang, M.; Kosaras, B.; Fitzgerald, E.; Murphy, A.; Folkerth, R.D.; Jensen, F.E. Antiepileptic effects of levetiracetam in a rodent neonatal seizure model. Pediatric Res. 2013, 73, 24–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Vliet, E.A.; van Schaik, R.; Edelbroek, P.M.; da Silva, F.H.L.; Wadman, W.J.; Gorter, J.A. Development of tolerance to levetiracetam in rats with chronic epilepsy. Epilepsia 2008, 49, 1151–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Moussally, J.; Cash, S.S.; Karnam, H.B.; Cole, A.J. Intravenous levetiracetam in the rat pilocarpine-induced status epilepticus model: Behavioral, physiological and histological studies. Neuropharmacology 2010, 58, 793–798. [Google Scholar] [CrossRef] [Green Version]
- ClinicalTrials.gov U.S. National Library of Medicine. Available online: www.clinicaltrials.gov (accessed on 3 November 2021).
- Stodieck, S.; Steinhoff, B.J.; Kolmsee, S.; van Rijckevorsel, K. Effect of levetiracetam in patients with epilepsy and interictal epileptiform discharges. Seizure 2001, 10, 583–587. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.F.; Weisenfeld, A.; Rothman, S.M. Prolonged exposure to levetiracetam reveals a presynaptic effect on neurotransmission. Epilepsia 2007, 48, 1861–1869. [Google Scholar] [CrossRef]
- Lee, C.-Y.; Chen, C.-C.; Liou, H.-H. Levetiracetam inhibits glutamate transmission through presynaptic P/Q-type calcium channels on the granule cells of the dentate gyrus. Br. J. Pharmacol. 2009, 158, 1753–1762. [Google Scholar] [CrossRef] [Green Version]
- Löscher, W.; Hönack, D.; Rundfeldt, C. Antiepileptogenic effects of the novel anticonvulsant levetiracetam (ucb L059) in the kindling model of temporal lobe epilepsy. J. Pharmacol. Exp. Ther. 1998, 284, 474–479. [Google Scholar] [PubMed]
- Stratton, S.C.; Large, C.H.; Cox, B.; Davies, G.; Hagan, R.M. Effects of lamotrigine and levetiracetam on seizure development in a rat amygdala kindling model. Epilepsy Res. 2003, 53, 95–106. [Google Scholar] [CrossRef]
- Vinogradova, L.V.; van Rijn, C.M. Anticonvulsive and antiepileptogenic effects of levetiracetam in the audiogenic kindling model. Epilepsia 2008, 49, 1160–1168. [Google Scholar] [CrossRef]
- Yan, H.-D.; Ji-qun, C.; Ishihara, K.; Nagayama, T.; Serikawa, T.; Sasa, M. Separation of antiepileptogenic and antiseizure effects of levetiracetam in the spontaneously epileptic rat (SER). Epilepsia 2005, 46, 1170–1177. [Google Scholar] [CrossRef] [PubMed]
- Sugaya, Y.; Jinde, S.; Kato, N.; Maru, E. Levetiracetam inhibits kindling-induced synaptic potentiation in the dentate gyrus of freely moving rats. Neurosci. Res. 2010, 66, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Leo, A.; De Caro, C.; Nesci, V.; Palma, E.; Tallarico, M.; Iannone, M.; Constanti, A.; De Sarro, G.; Russo, E.; Citraro, R. Antiepileptogenic effects of Ethosuximide and Levetiracetam in WAG/Rij rats are only temporary. Pharmacol. Rep. 2019, 71, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Sugaya, Y.; Maru, E.; Kudo, K.; Shibasaki, T.; Kato, N. Levetiracetam suppresses development of spontaneous EEG seizures and aberrant neurogenesis following kainate-induced status epilepticus. Brain Res. 2010, 1352, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Brandt, C.; Glien, M.; Gastens, A.M.; Fedrowitz, M.; Bethmann, K.; Volk, H.A.; Potschka, H.; Löscher, W. Prophylactic treatment with levetiracetam after status epilepticus: Lack of effect on epileptogenesis, neuronal damage, and behavioral alterations in rats. Neuropharmacology 2007, 53, 207–221. [Google Scholar] [CrossRef]
- Christensen, J.; Pedersen, M.G.; Pedersen, C.B.; Sidenius, P.; Olsen, J.; Vestergaard, M. Long-term risk of epilepsy after traumatic brain injury in children and young adults: A population-based cohort study. Lancet 2009, 373, 1105–1110. [Google Scholar] [CrossRef]
- Fiani, B.; Andraos, C.; Mabry, I.; Siddiqi, J. A Comparison of Seizure Prophylaxis: Phenytoin Versus Levetiracetam. Cureus 2021, 13, e14956. [Google Scholar] [CrossRef]
- Fang, T.; Valdes, E.; Frontera, J.A. Levetiracetam for Seizure Prophylaxis in Neurocritical Care: A Systematic Review and Meta-analysis. Neurocrit. Care 2022, 36, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W.; Brandt, C. Prevention or modification of epileptogenesis after brain insults: Experimental approaches and translational research. Pharmacol. Rev. 2010, 62, 668–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milligan, T.A.; Hurwitz, S.; Bromfield, E.B. Efficacy and tolerability of levetiracetam versus phenytoin after supratentorial neurosurgery. Neurology 2008, 71, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Iuchi, T.; Kuwabara, K.; Matsumoto, M.; Kawasaki, K.; Hasegawa, Y.; Sakaida, T. Levetiracetam versus phenytoin for seizure prophylaxis during and early after craniotomy for brain tumours: A phase II prospective, randomised study. J. Neurol. Neurosurg. Psychiatry 2015, 86, 1158–1162. [Google Scholar] [CrossRef]
- Klein, P.; Herr, D.; Pearl, P.L.; Natale, J.; Levine, Z.; Nogay, C.; Sandoval, F.; Trzcinski, S.; Atabaki, S.M.; Tsuchida, T.; et al. Results of phase 2 safety and feasibility study of treatment with levetiracetam for prevention of posttraumatic epilepsy. Arch. Neurol. 2012, 69, 1290–1295. [Google Scholar] [CrossRef]
- Kruer, R.M.; Harris, L.H.; Goodwin, H.; Kornbluth, J.; Thomas, K.P.; Slater, L.A.; Haut, E.R. Changing trends in the use of seizure prophylaxis after traumatic brain injury: A shift from phenytoin to levetiracetam. J. Crit. Care 2013, 28, 883.e9–883.e13. [Google Scholar] [CrossRef]
- Radic, J.A.E.; Chou, S.H.-Y.; Du, R.; Lee, J.W. Levetiracetam versus phenytoin: A comparison of efficacy of seizure prophylaxis and adverse event risk following acute or subacute subdural hematoma diagnosis. Neurocrit. Care 2014, 21, 228–237. [Google Scholar] [CrossRef]
- Falsaperla, R.; Mauceri, L.; Pavone, P.; Barbagallo, M.; Vitaliti, G.; Ruggieri, M.; Pisani, F.; Corsello, G. Short-Term Neurodevelopmental Outcome in Term Neonates Treated with Phenobarbital versus Levetiracetam: A Single-Center Experience. Behav. Neurol. 2019, 2019, 3683548. [Google Scholar] [CrossRef] [Green Version]
- Fuller, K.L.; Wang, Y.Y.; Cook, M.J.; Murphy, M.A.; D’Souza, W.J. Tolerability, safety, and side effects of levetiracetam versus phenytoin in intravenous and total prophylactic regimen among craniotomy patients: A prospective randomized study. Epilepsia 2013, 54, 45–57. [Google Scholar] [CrossRef]
- Taylor, S.; Heinrichs, R.J.; Janzen, J.M.; Ehtisham, A. Levetiracetam is associated with improved cognitive outcome for patients with intracranial hemorrhage. Neurocrit. Care 2011, 15, 80–84. [Google Scholar] [CrossRef]
- Pang, X.-M.; Liang, X.-L.; Zhou, X.; Liu, J.-P.; Zhang, Z.; Zheng, J.-O. Alterations in intra- and internetwork functional connectivity associated with levetiracetam treatment in temporal lobe epilepsy. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2020, 41, 2165–2174. [Google Scholar] [CrossRef] [PubMed]
- Wandschneider, B.; Stretton, J.; Sidhu, M.; Centeno, M.; Kozák, L.R.; Symms, M.; Thompson, P.J.; Duncan, J.S.; Koepp, M.J. Levetiracetam reduces abnormal network activations in temporal lobe epilepsy. Neurology 2014, 83, 1508–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavarsan, C.F.; Malheiros, J.; Hamani, C.; Najm, I.; Covolan, L. Is mossy fiber sprouting a potential therapeutic target for epilepsy? Front. Neurol. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutlu, G.; Gomceli, Y.B.; Unal, Y.; Inan, L.E. Levetiracetam monotherapy for late poststroke seizures in the elderly. Epilepsy Behav. 2008, 13, 542–544. [Google Scholar] [CrossRef]
- Marini, H.; Altavilla, D.; Bellomo, M.; Adamo, E.B.; Marini, R.; Laureanti, F.; Bonaccorso, M.C.; Seminara, P.; Passaniti, M.; Minutoli, L.; et al. Modulation of IL-1 beta gene expression by lipid peroxidation inhibition after kainic acid-induced rat brain injury. Exp. Neurol. 2004, 188, 178–186. [Google Scholar] [CrossRef]
- Lee, D.-S.; Ryu, H.J.; Kim, J.-E.; Choi, H.-C.; Kim, Y.-I.; Song, H.-K.; Kang, T.-C. The effect of levetiracetam on status epilepticus-induced neuronal death in the rat hippocampus. Seizure 2013, 22, 368–377. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Ishihara, Y.; Komori, R.; Nochi, H.; Taniguchi, R.; Chiba, Y.; Ueno, M.; Takata-Tsuji, F.; Dohgu, S.; Kataoka, Y. Levetiracetam treatment influences blood-brain barrier failure associated with angiogenesis and inflammatory responses in the acute phase of epileptogenesis in post-status epilepticus mice. Brain Res. 2016, 1652, 1–13. [Google Scholar] [CrossRef]
- Shetty, A.K. Prospects of levetiracetam as a neuroprotective drug against status epilepticus, traumatic brain injury, and stroke. Front. Neurol. 2013, 4, 172. [Google Scholar] [CrossRef] [Green Version]
- Gibbs, J.E.; Walker, M.C.; Cock, H.R. Levetiracetam: Antiepileptic properties and protective effects on mitochondrial dysfunction in experimental status epilepticus. Epilepsia 2006, 47, 469–478. [Google Scholar] [CrossRef]
- Santana-Gómez, C.E.; Valle-Dorado, M.G.; Domínguez-Valentín, A.E.; Hernández-Moreno, A.; Orozco-Suárez, S.; Rocha, L. Neuroprotective effects of levetiracetam, both alone and combined with propylparaben, in the long-term consequences induced by lithium-pilocarpine status epilepticus. Neurochem. Int. 2018, 120, 224–232. [Google Scholar] [CrossRef]
- Hanon, E.; Klitgaard, H. Neuroprotective properties of the novel antiepileptic drug levetiracetam in the rat middle cerebral artery occlusion model of focal cerebral ischemia. Seizure 2001, 10, 287–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilicdag, H.; Daglıoglu, K.; Erdogan, S.; Guzel, A.; Sencar, L.; Polat, S.; Zorludemir, S. The effect of levetiracetam on neuronal apoptosis in neonatal rat model of hypoxic ischemic brain injury. Early Hum. Dev. 2013, 89, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Yang, W.; Ren, Z.; Zhang, H.; Shi, D.; Li, Y.; Yu, Z.; Guo, Q.; Yang, G.; Gu, Y.; et al. Neuroprotective and Angiogenesis Effects of Levetiracetam Following Ischemic Stroke in Rats. Front. Pharmacol. 2021, 12, 638209. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Gao, J.; Lassiter, T.F.; McDonagh, D.L.; Sheng, H.; Warner, D.S.; Lynch, J.R.; Laskowitz, D.T. Levetiracetam is neuroprotective in murine models of closed head injury and subarachnoid hemorrhage. Neurocrit. Care 2006, 5, 71–78. [Google Scholar] [CrossRef]
- Xiong, J.; Zhou, H.; Lu, D.; Wang, Z.; Liu, H.; Sun, Y.; Xu, J.; Feng, Y.; Xing, A. Levetiracetam Reduces Early Inflammatory Response After Experimental Intracerebral Hemorrhage by Regulating the Janus Kinase 2 (JAK2)-Signal Transducer and Activator of Transcription 3 (STAT3) Signaling Pathway. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2020, 26, e922741. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, H.M.F.; Sami, M.M.; Makary, S.; Toraih, E.A.; Mohamed, A.O.; El-Ghaiesh, S.H. Neuroprotective effect of levetiracetam in mouse diabetic retinopathy: Effect on glucose transporter-1 and GAP43 expression. Life Sci. 2019, 232, 116588. [Google Scholar] [CrossRef] [PubMed]
- Lima, R.; Gomes, E.D.; Cibrão, J.R.; Rocha, L.A.; Assunção-Silva, R.C.; Rodrigues, C.S.; Neves-Carvalho, A.; Monteiro, S.; Salgado, A.J.; Silva, N.A. Levetiracetam treatment leads to functional recovery after thoracic or cervical injuries of the spinal cord. NPJ Regen. Med. 2021, 6, 11. [Google Scholar] [CrossRef]
- Bonifacio, S.L.; Gonzalez, F.; Ferriero, D.M. Chapter 61—Central Nervous System Injury and Neuroprotection. In Avery’s Diseases of the Newborn (Ninth Edition); Christine, A.G., Devaskar, S.U., Eds.; W.B. Saunders: Philadelpia, PA, USA, 2012; pp. 869–891. ISBN 978-1-4377-0134-0. [Google Scholar]
- Klitgaard, H.; Pitkänen, A. Antiepileptogenesis, neuroprotection, and disease modification in the treatment of epilepsy: Focus on levetiracetam. Epileptic Disord. 2003, 5 (Suppl. 1), S9–S16. [Google Scholar]
- Calabresi, P.; Cupini, L.M.; Centonze, D.; Pisani, F.; Bernardi, G. Antiepileptic drugs as a possible neuroprotective strategy in brain ischemia. Ann. Neurol. 2003, 53, 693–702. [Google Scholar] [CrossRef]
- Vezzani, A.; Fujinami, R.S.; White, H.S.; Preux, P.-M.; Blümcke, I.; Sander, J.W.; Löscher, W. Infections, inflammation and epilepsy. Acta Neuropathol. 2016, 131, 211–234. [Google Scholar] [CrossRef]
- Ambrogini, P.; Torquato, P.; Bartolini, D.; Albertini, M.C.; Lattanzi, D.; Di Palma, M.; Marinelli, R.; Betti, M.; Minelli, A.; Cuppini, R.; et al. Excitotoxicity, neuroinflammation and oxidant stress as molecular bases of epileptogenesis and epilepsy-derived neurodegeneration: The role of vitamin E. Biochim. Biophys. Acta. Mol. Basis Dis. 2019, 1865, 1098–1112. [Google Scholar] [CrossRef]
- Sanz, P.; Garcia-Gimeno, M.A. Reactive Glia Inflammatory Signaling Pathways and Epilepsy. Int. J. Mol. Sci. 2020, 21, 4096. [Google Scholar] [CrossRef] [PubMed]
- Vrinda, M.; Arun, S.; Srikumar, B.N.; Kutty, B.M.; Shankaranarayana Rao, B.S. Temporal lobe epilepsy-induced neurodegeneration and cognitive deficits: Implications for aging. J. Chem. Neuroanat. 2019, 95, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; French, J.; Bartfai, T.; Baram, T.Z. The role of inflammation in epilepsy. Nat. Rev. Neurol. 2011, 7, 31–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pracucci, E.; Pillai, V.; Lamers, D.; Parra, R.; Landi, S. Neuroinflammation: A Signature or a Cause of Epilepsy? Int. J. Mol. Sci. 2021, 22, 6981. [Google Scholar] [CrossRef]
- Devinsky, O.; Vezzani, A.; Najjar, S.; De Lanerolle, N.C.; Rogawski, M.A. Glia and epilepsy: Excitability and inflammation. Trends Neurosci. 2013, 36, 174–184. [Google Scholar] [CrossRef]
- Behl, T.; Makkar, R.; Sehgal, A.; Singh, S.; Sharma, N.; Zengin, G.; Bungau, S.; Andronie-Cioara, F.L.; Munteanu, M.A.; Brisc, M.C.; et al. Current Trends in Neurodegeneration: Cross Talks between Oxidative Stress, Cell Death, and Inflammation. Int. J. Mol. Sci. 2021, 22, 7432. [Google Scholar] [CrossRef]
- Farrell, J.S.; Wolff, M.D.; Teskey, G.C. Neurodegeneration and Pathology in Epilepsy: Clinical and Basic Perspectives. Adv. Neurobiol. 2017, 15, 317–334. [Google Scholar] [CrossRef]
- Martinc, B.; Grabnar, I.; Vovk, T. Antioxidants as a preventive treatment for epileptic process: A review of the current status. Curr. Neuropharmacol. 2014, 12, 527–550. [Google Scholar] [CrossRef] [Green Version]
- Beltrán-Sarmiento, E.; Arregoitia-Sarabia, C.K.; Floriano-Sánchez, E.; Sandoval-Pacheco, R.; Galván-Hernández, D.E.; Coballase-Urrutia, E.; Carmona-Aparicio, L.; Ramos-Reyna, E.; Rodríguez-Silverio, J.; Cárdenas-Rodríguez, N. Effects of Valproate Monotherapy on the Oxidant-Antioxidant Status in Mexican Epileptic Children: A Longitudinal Study. Oxid. Med. Cell. Longev. 2018, 2018, 7954371. [Google Scholar] [CrossRef] [Green Version]
- Nazıroğlu, M.; Yürekli, V.A. Effects of antiepileptic drugs on antioxidant and oxidant molecular pathways: Focus on trace elements. Cell. Mol. Neurobiol. 2013, 33, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Hansson, E.; Björklund, U.; Skiöldebrand, E.; Rönnbäck, L. Anti-inflammatory effects induced by pharmaceutical substances on inflammatory active brain astrocytes-promising treatment of neuroinflammation. J. Neuroinflamm. 2018, 15, 321. [Google Scholar] [CrossRef] [PubMed]
- Osuntokun, O.S.; Abdulwahab, U.F.; Akanji, N.O.; Adedokun, K.I.; Adekomi, A.D.; Olayiwola, G. Anticonvulsant and neuroprotective effects of carbamazepine-levetiracetam adjunctive treatment in convulsive status epilepticus rat model: Inhibition of cholinergic transmission. Neurosci. Lett. 2021, 762, 136167. [Google Scholar] [CrossRef]
- Bayhan, I.; Turtay, M.G.; Ciftci, O.; Cetin, A.; Basak, N.; Namık Oztanır, M.; Oguzturk, H.; Gurbuz, S.; Guven, T. Comparison of immunological, histological and oxidative effects of felbamate and levetiracetam in traumatic brain injury. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 7083–7091. [Google Scholar] [CrossRef] [PubMed]
- de Souza, A.G.; Chaves Filho, A.J.M.; Souza Oliveira, J.V.; de Souza, D.A.A.; Lopes, I.S.; de Carvalho, M.A.J.; de Lima, K.A.; Florenço Sousa, F.C.; Mendes Vasconcelos, S.M.; Macedo, D.; et al. Prevention of pentylenetetrazole-induced kindling and behavioral comorbidities in mice by levetiracetam combined with the GLP-1 agonist liraglutide: Involvement of brain antioxidant and BDNF upregulating properties. Biomed. Pharmacother. 2019, 109, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, B.A.; Shaikh, I.A.; Khateeb, M.M.; Habeeb, S.M. Omega 3 polyunsaturated fatty acids enhance the protective effect of levetiracetam against seizures, cognitive impairment and hippocampal oxidative DNA damage in young kindled rats. Pharmacol. Biochem. Behav. 2015, 135, 105–113. [Google Scholar] [CrossRef]
- Mazhar, F.; Malhi, S.M.; Simjee, S.U. Comparative studies on the effects of clinically used anticonvulsants on the oxidative stress biomarkers in pentylenetetrazole-induced kindling model of epileptogenesis in mice. J. Basic Clin. Physiol. Pharmacol. 2017, 28, 31–42. [Google Scholar] [CrossRef]
- Imran, I.; Koch, K.; Schöfer, H.; Lau, H.; Klein, J. Effects of Three Anti-Seizure Drugs on Cholinergic and Metabolic Activity in Experimental Status Epilepticus. J. Pharm. Pharm. Sci. Publ. Can. Soc. Pharm. Sci. Soc. Can. Sci. Pharm. 2019, 22, 340–351. [Google Scholar] [CrossRef]
- Oliveira, A.A.; Almeida, J.P.C.; Freitas, R.M.; Nascimento, V.S.; Aguiar, L.M.V.; Júnior, H.V.N.; Fonseca, F.N.; Viana, G.S.B.; Sousa, F.C.F.; Fonteles, M.M.F. Effects of levetiracetam in lipid peroxidation level, nitrite-nitrate formation and antioxidant enzymatic activity in mice brain after pilocarpine-induced seizures. Cell. Mol. Neurobiol. 2007, 27, 395–406. [Google Scholar] [CrossRef]
- Dircio-Bautista, M.; Colín-González, A.L.; Aguilera, G.; Maya-López, M.; Villeda-Hernández, J.; Galván-Arzate, S.; García, E.; Túnez, I.; Santamaría, A. The Antiepileptic Drug Levetiracetam Protects Against Quinolinic Acid-Induced Toxicity in the Rat Striatum. Neurotox. Res. 2018, 33, 837–845. [Google Scholar] [CrossRef]
- Erbaş, O.; Yılmaz, M.; Taşkıran, D. Levetiracetam attenuates rotenone-induced toxicity: A rat model of Parkinson’s disease. Environ. Toxicol. Pharmacol. 2016, 42, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Erbaş, O.; Oltulu, F.; Yılmaz, M.; Yavaşoğlu, A.; Taşkıran, D. Neuroprotective effects of chronic administration of levetiracetam in a rat model of diabetic neuropathy. Diabetes Res. Clin. Pract. 2016, 114, 106–116. [Google Scholar] [CrossRef]
- Akman, L.; Erbas, O.; Akdemir, A.; Yavasoglu, A.; Taskiran, D.; Kazandi, M. Levetiracetam ameliorates ovarian function in streptozotocin-induced diabetic rats. Gynecol. Endocrinol. Off. J. Int. Soc. Gynecol. Endocrinol. 2015, 31, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Sarangi, S.C.; Kakkar, A.K.; Kumar, R.; Gupta, Y.K. Effect of lamotrigine, levetiracetam & topiramate on neurobehavioural parameters & oxidative stress in comparison with valproate in rats. Indian J. Med. Res. 2016, 144, 104–111. [Google Scholar] [CrossRef]
- Baysal, M.; Ilgin, S.; Kilic, G.; Kilic, V.; Ucarcan, S.; Atli, O. Reproductive toxicity after levetiracetam administration in male rats: Evidence for role of hormonal status and oxidative stress. PLoS ONE 2017, 12, e0175990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ersan, S.; Cigdem, B.; Bakir, D.; Dogan, H.O. Determination of levels of oxidative stress and nitrosative stress in patients with epilepsy. Epilepsy Res. 2020, 164, 106352. [Google Scholar] [CrossRef] [PubMed]
- Mahdavi, A.; Naeini, A.A.; Najafi, M.; Maracy, M.; Ghazvini, M.A. Effect of levetiracetam drug on antioxidant and liver enzymes in epileptic patients: Case-control study. Afr. Health Sci. 2020, 20, 984–990. [Google Scholar] [CrossRef]
- Haznedar, P.; Doğan, Ö.; Albayrak, P.; Öz Tunçer, G.; Teber, S.; Deda, G.; Eminoglu, F.T. Effects of levetiracetam and valproic acid treatment on liver function tests, plasma free carnitine and lipid peroxidation in childhood epilepsies. Epilepsy Res. 2019, 153, 7–13. [Google Scholar] [CrossRef]
- Ozden, H.; Kabay, S.C.; Toker, A.; Ustüner, M.C.; Ozbayer, C.; Ustüner, D.; Günes, H.V. The effects of levetiracetam on urinary 15f-2t-isoprostane levels in epileptic patients. Seizure 2010, 19, 514–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varoglu, A.O.; Yildirim, A.; Aygul, R.; Gundogdu, O.L.; Sahin, Y.N. Effects of valproate, carbamazepine, and levetiracetam on the antioxidant and oxidant systems in epileptic patients and their clinical importance. Clin. Neuropharmacol. 2010, 33, 155–157. [Google Scholar] [CrossRef]
- Morimoto, M.; Hashimoto, T.; Kitaoka, T.; Kyotani, S. Impact of Oxidative Stress and Newer Antiepileptic Drugs on the Albumin and Cortisol Value in Severe Motor and Intellectual Disabilities With Epilepsy. J. Clin. Med. Res. 2018, 10, 137–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morimoto, M.; Satomura, S.; Hashimoto, T.; Kyotani, S. A study of oxidative stress and the newer antiepileptic drugs in epilepsy associated with severe motor and intellectual disabilities. J. Chin. Med. Assoc. 2017, 80, 19–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Tan, Y.; Ge, Y.; Chen, Y.; Liu, X. The Effects of Levetiracetam on Cerebrospinal Fluid and Plasma NPY and GAL, and on the Components of Stress Response System, hs-CRP, and S100B Protein in Serum of Patients with Refractory Epilepsy. Cell Biochem. Biophys. 2015, 73, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Stienen, M.N.; Haghikia, A.; Dambach, H.; Thöne, J.; Wiemann, M.; Gold, R.; Chan, A.; Dermietzel, R.; Faustmann, P.M.; Hinkerohe, D.; et al. Anti-inflammatory effects of the anticonvulsant drug levetiracetam on electrophysiological properties of astroglia are mediated via TGFβ1 regulation. Br. J. Pharmacol. 2011, 162, 491–507. [Google Scholar] [CrossRef] [Green Version]
- Haghikia, A.; Ladage, K.; Hinkerohe, D.; Vollmar, P.; Heupel, K.; Dermietzel, R.; Faustmann, P.M. Implications of antiinflammatory properties of the anticonvulsant drug levetiracetam in astrocytes. J. Neurosci. Res. 2008, 86, 1781–1788. [Google Scholar] [CrossRef]
- Thöne, J.; Ellrichmann, G.; Faustmann, P.M.; Gold, R.; Haghikia, A. Anti-inflammatory effects of levetiracetam in experimental autoimmune encephalomyelitis. Int. Immunopharmacol. 2012, 14, 9–12. [Google Scholar] [CrossRef]
- Guenther, S.; Bauer, S.; Hagge, M.; Knake, S.; Olmes, D.G.; Tackenberg, B.; Rosenow, F.; Hamer, H.M. Chronic valproate or levetiracetam treatment does not influence cytokine levels in humans. Seizure 2014, 23, 666–669. [Google Scholar] [CrossRef] [Green Version]
- Labh, R.; Gupta, R.; Narang, M.; Halder, S.; Kar, R. Effect of valproate and add-on levetiracetam on inflammatory biomarkers in children with epilepsy. Epilepsy Behav. 2021, 125, 108358. [Google Scholar] [CrossRef]
- Gulcebi, M.I.; Kendirli, T.; Turgan, Z.A.; Patsalos, P.N.; Onat Yilmaz, F. The effect of serum levetiracetam concentrations on therapeutic response and IL1-beta concentration in patients with epilepsy. Epilepsy Res. 2018, 148, 17–22. [Google Scholar] [CrossRef]
- Stettner, M.; Dehmel, T.; Mausberg, A.K.; Köhne, A.; Rose, C.R.; Kieseier, B.C. Levetiracetam exhibits protective properties on rat Schwann cells in vitro. J. Peripher. Nerv. Syst. 2011, 16, 250–260. [Google Scholar] [CrossRef]
- Irwin, M.H.; Moos, W.H.; Faller, D.V.; Steliou, K.; Pinkert, C.A. Epigenetic Treatment of Neurodegenerative Disorders: Alzheimer and Parkinson Diseases. Drug Dev. Res. 2016, 77, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Kann, O.; Ohlemeyer, C.; Hanisch, U.-K.; Kettenmann, H. Elevation of basal intracellular calcium as a central element in the activation of brain macrophages (microglia): Suppression of receptor-evoked calcium signaling and control of release function. J. Neurosci. 2003, 23, 4410–4419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marciani, M.G.; Gotman, J.; Andermann, F.; Olivier, A. Patterns of Seizure Activation after Withdrawal of Antiepileptic Medication. Neurology 1985, 35, 1537–1543. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.-M.; Lucchi, C.; Malkoç, A.; Rustichelli, C.; Biagini, G. Relationship between Delta Rhythm, Seizure Occurrence and Allopregnanolone Hippocampal Levels in Epileptic Rats Exposed to the Rebound Effect. Pharmaceuticals 2021, 14, 127. [Google Scholar] [CrossRef] [PubMed]
- Boon, P.; Chauvel, P.; Pohlmann-Eden, B.; Otoul, C.; Wroe, S. Dose-Response Effect of Levetiracetam 1000 and 2000 Mg/Day in Partial Epilepsy. Epilepsy Res. 2002, 48, 77–89. [Google Scholar] [CrossRef]
- Hansen, C.C.; Ljung, H.; Brodtkorb, E.; Reimers, A. Mechanisms Underlying Aggressive Behavior Induced by Antiepileptic Drugs: Focus on Topiramate, Levetiracetam, and Perampanel. Behav. Neurol. 2018, 2018, 2064027. [Google Scholar] [CrossRef] [Green Version]
- Brodie, M.J.; Besag, F.; Ettinger, A.B.; Mula, M.; Gobbi, G.; Comai, S.; Aldenkamp, A.P.; Steinhoff, B.J. Epilepsy, Antiepileptic Drugs, and Aggression: An Evidence-Based Review. Pharmacol. Rev. 2016, 68, 563–602. [Google Scholar] [CrossRef]
- Steinhoff, B.J.; Klein, P.; Klitgaard, H.; Laloyaux, C.; Moseley, B.D.; Ricchetti-Masterson, K.; Rosenow, F.; Sirven, J.I.; Smith, B.; Stern, J.M.; et al. Behavioral Adverse Events with Brivaracetam, Levetiracetam, Perampanel, and Topiramate: A Systematic Review. Epilepsy Behav. 2021, 118, 107939. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
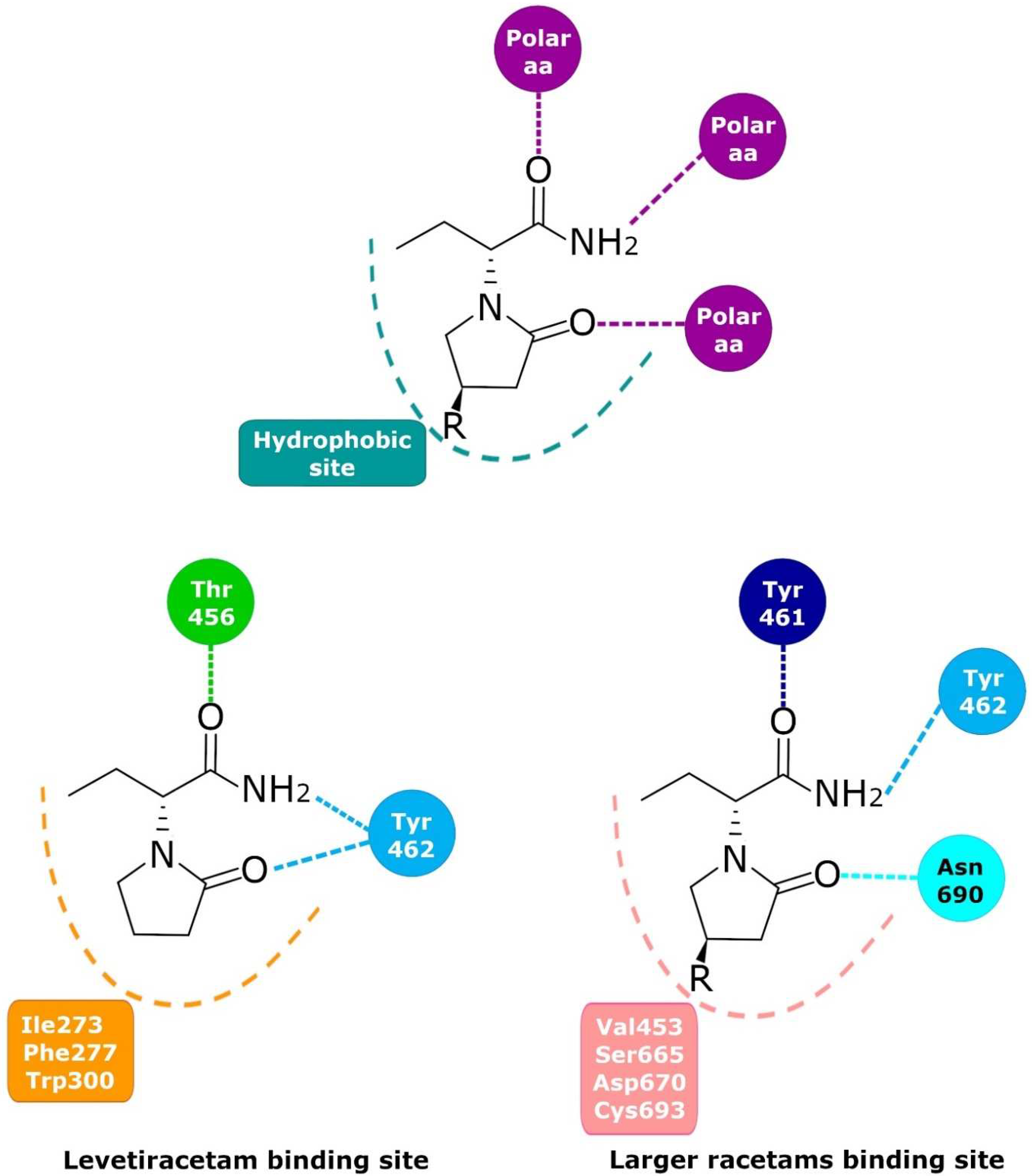{kind=link}
{kind=link}
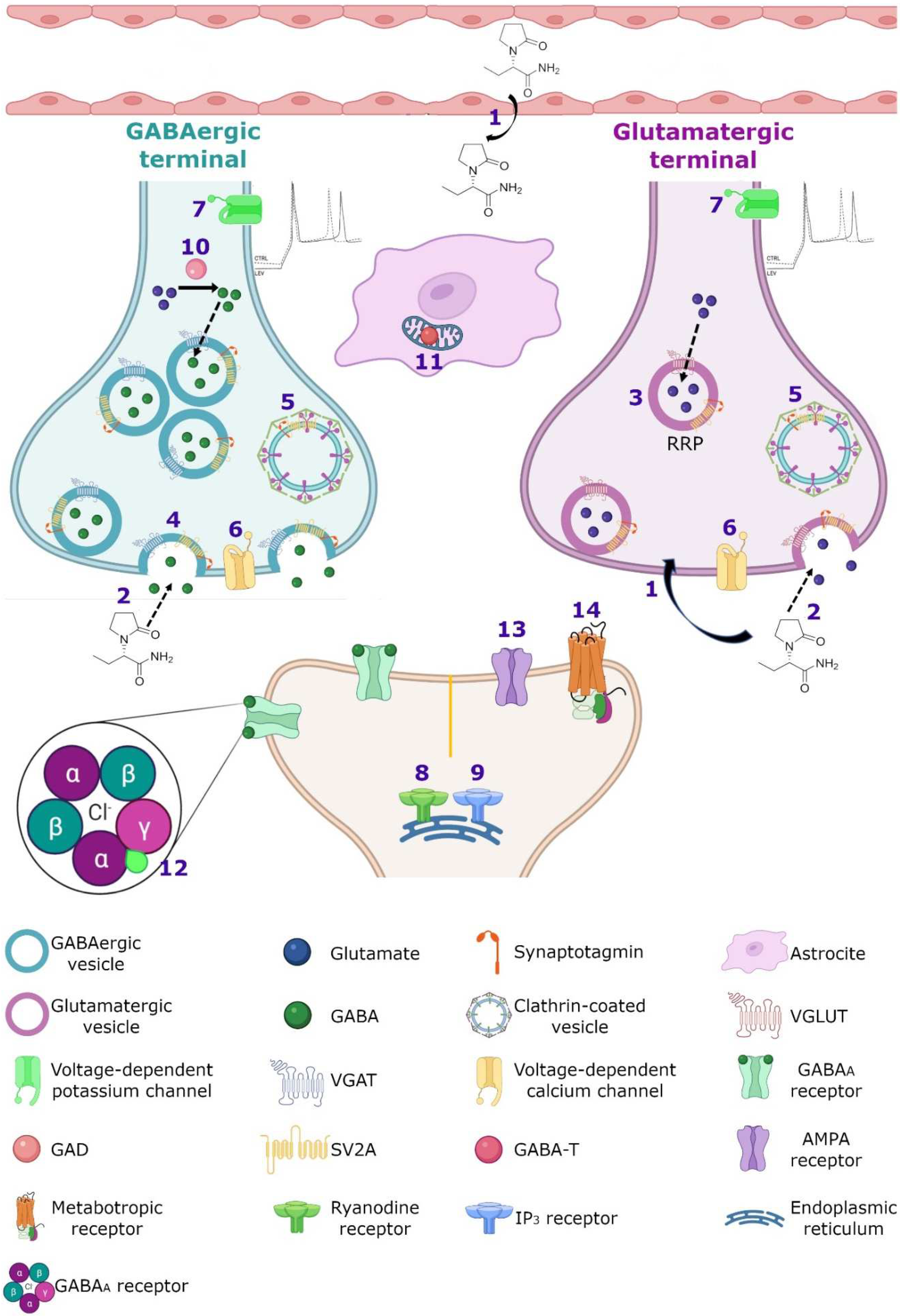{kind=link}
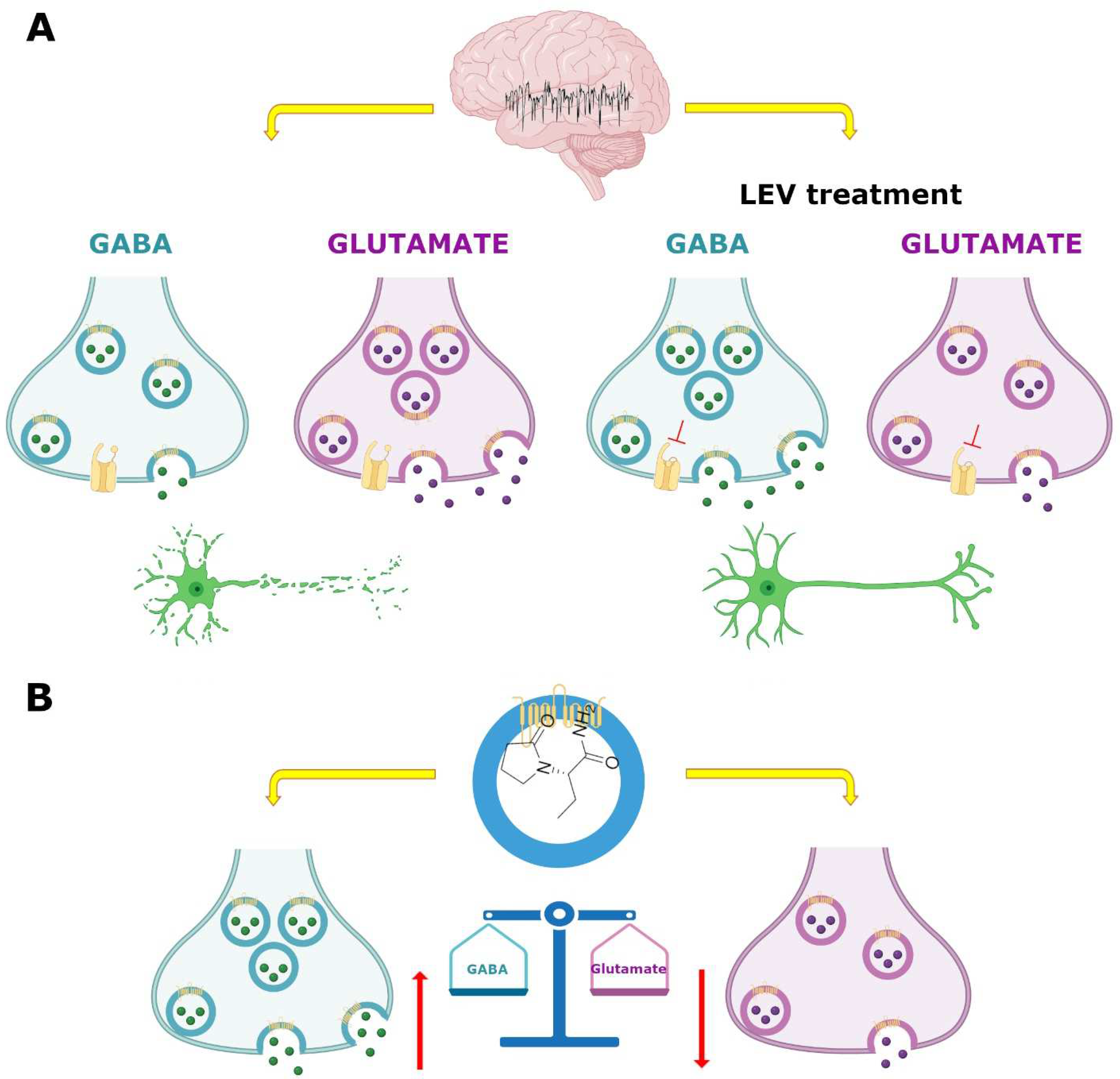{kind=link}
{kind=link}
{kind=link}
| Autor | Model | LEV Treatment | Findings |
|---|---|---|---|
| Löscher et al. [218] | Rat amygdala kindling model | 13, 27 or 54 mg/kg i.p. (16 days) | After daily LEV (54 mg/kg), behavioral seizure and amygdala after discharge duration remained shorter. |
| Stratton et al. [219] | Rat amygdala kindling model | 50 mg/kg i.p. (16 days) | LEV blocked seizure development when administered 1 h prior to electrical stimulation during the kindling phase. This effect continued after washout, suggesting that LEV blocked the underlying kindling mechanism and not simply masked seizure expression by its anticonvulsant action. |
| Vinogradova and van Rijn [220] | Rat audiogenic kindling model | 50 mg/kg i.p. (one) | A single LEV injection significantly suppressed kindling progression. This is evidence of a long-lasting antiepileptogenic activity. |
| Yan et al. [221] | Spontaneously epileptic rat | 80 mg/kg/day i.p. (from postnatal weeks 5 to 8) | Long-term LEV treatment inhibited development of seizure activity and this effect was still evident 5 weeks after cessation of treatment. |
| Sugaya et al. [222] | Rat perforant path kindling model | 100 mg/kg, i.p. (21 days) | LEV inhibited both the development of kindling-induced potentiation in perforant path-granule cell synapses and development of perforant path kindling. |
| Leo et al. [223] | WAG/Rij rats Rat model of absence epilepsy | 80 mg/kg/day i.p. (17 weeks) | LEV showed antiepileptogenic effects 1 month after discontinuation. However, it did not maintain its antiepileptogenic effect 5 months after suspension, and worsened depressive-like behavior. |
| Itoh et al. [22] | Mice, SE induced with pilocarpine | 500 mg/kg v.o. (28 days; twice a day) | LEV treatment prevented the development of spontaneous recurrent seizures for at least 28 days. |
| Sugaya et al. [224] | Rat kainate-induced SE model | 100 mg/kg, i.p. (21 days) | LEV treatment decreased the duration of spontaneous electrographic seizures 58 days after SE. |
| Brandt et al. [225] | Rat, SE induced by sustained electrical stimulation of the basal amygdala | 1000 mg/kg Via osmotic minipumps (5 weeks) | LEV treatment did not prevent seizure development when administrated 4 after SE onset. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Contreras-García, I.J.; Cárdenas-Rodríguez, N.; Romo-Mancillas, A.; Bandala, C.; Zamudio, S.R.; Gómez-Manzo, S.; Hernández-Ochoa, B.; Mendoza-Torreblanca, J.G.; Pichardo-Macías, L.A. Levetiracetam Mechanisms of Action: From Molecules to Systems. Pharmaceuticals 2022, 15, 475. https://doi.org/10.3390/ph15040475
Contreras-García IJ, Cárdenas-Rodríguez N, Romo-Mancillas A, Bandala C, Zamudio SR, Gómez-Manzo S, Hernández-Ochoa B, Mendoza-Torreblanca JG, Pichardo-Macías LA. Levetiracetam Mechanisms of Action: From Molecules to Systems. Pharmaceuticals. 2022; 15(4):475. https://doi.org/10.3390/ph15040475
Chicago/Turabian StyleContreras-García, Itzel Jatziri, Noemí Cárdenas-Rodríguez, Antonio Romo-Mancillas, Cindy Bandala, Sergio R. Zamudio, Saúl Gómez-Manzo, Beatriz Hernández-Ochoa, Julieta Griselda Mendoza-Torreblanca, and Luz Adriana Pichardo-Macías. 2022. "Levetiracetam Mechanisms of Action: From Molecules to Systems" Pharmaceuticals 15, no. 4: 475. https://doi.org/10.3390/ph15040475