
Emergence of Nanotechnology as a Powerful Cavalry against Triple-Negative Breast Cancer (TNBC)

 , , ,
, , ,  , and
, and
Abstract
:
1. Introduction
1.1. Epidemiology
1.2. TNBC—Metastasis Driven Complexity
1.3. Heterogeneity of TNBC
2. Subtypes of TNBC
2.1. Subtypes Based on Gene-Expression Profiling

2.1.1. BL Subtypes TNBC
2.1.2. IM Subtype TNBC
2.1.3. M Subtype TNBC
2.1.4. MSL Subtype TNBC
2.1.5. LAR Subtype
2.1.6. BLIS Subtype TNBC
2.2. Subtypes Based on the Histology of TNBC Cells
2.3. Subtypes Based on the Tumor Microenvironment (TME)
3. Potential Therapeutic Targets for TNBC Therapy
3.1. Notch Signaling Pathways
3.2. Hedgehog (Hh) Signaling Pathway
3.3. Wnt/β-Catenin Pathway
3.4. TGF-β Signaling Pathway
3.5. PI3K/AKT/mTOR Signaling Pathway
3.6. EGFR
3.7. IGF1R
3.8. PARP1
3.9. Src Kinases
3.10. Immune-System Targeting
3.10.1. PD-L1
3.10.2. CTLA-4
3.11. CSPG4 Proteins
3.12. Androgen Receptor (AR)
4. Available Drugs Used in TNBC Treatment
4.1. Chemotherapy
4.1.1. Taxanes
4.1.2. Anthracyclines
4.1.3. Cyclophosphamide
4.1.4. Antimetabolites
4.1.5. Platinum Compounds
4.2. Targeted Therapy
4.2.1. γ-Secretase Inhibitors (GSIs)
4.2.2. PARP Inhibitors
4.2.3. PI3K/AKT/mTOR Inhibitors
4.2.4. Growth Factor Inhibitors
4.2.5. Src Inhibitors
4.2.6. Immune Checkpoint Inhibitors
4.2.7. Antiandrogens
5. Nanotechnology: Cavalry for TNBC Therapy
5.1. Liposomes
5.2. Dendrimers
5.3. Polymeric Micelles
5.4. Polymeric Nanoparticles
5.5. Carbon Nanotubes
5.6. Metallic Nanoparticles
5.7. Nanoemulsion/Self-Nanoemulsifying Drug Delivery System (SNEDDS)
5.8. Solid Lipid Nanoparticles (SLN)
5.9. Nanostructured Lipid Carriers (NLC)
6. Nanomedicine: From Pre-Clinical Design to Clinical Practice
7. Toxicity of Nanoparticles
8. Conclusions
9. Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Makki, J. Diversity of breast carcinoma: Histological subtypes and clinical relevance. Clin. Med. Insights Pathol. 2015, 8, 23–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Carlson, R.W.; Allred, D.C.; Anderson, B.O.; Burstein, H.J.; Carter, W.B.; Edge, S.B.; Erban, J.K.; Farrar, W.B.; Forero, A.; Giordano, S.H.; et al. Invasive breast cancer: Clinical practice guidelines in oncology. JNCCN J. Natl. Compr. Cancer Netw. 2011, 9, 136–222. [Google Scholar] [CrossRef] [PubMed]
- Wolff, A.C.; Hammond, M.E.H.; Hicks, D.G.; Dowsett, M.; McShane, L.M.; Allison, K.H.; Allred, D.C.; Bartlett, J.M.S.; Bilous, M.; Fitzgibbons, P.; et al. Recommendations for Human Epidermal Growth Factor Receptor 2 Testing in Breast Cancer: American Society of Clinical Oncology/College ofAmerican Pathologists Clinical Practice Guideline Update. J. Clin. Oncol. 2013, 31, 3997–4013. [Google Scholar] [CrossRef] [PubMed]
- Anders, C.K.; Carey, L.A. Biology, metastatic patterns, and treatment of patients with triple-negative breast cancer. Clin. Breast Cancer 2009, 9, S73–S81. [Google Scholar] [CrossRef]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef] [Green Version]
- Lin, N.U.; Claus, E.; Sohl, J.; Razzak, A.R.; Arnaout, A.; Winer, E.P. Sites of distant recurrence and clinical outcomes in patients with metastatic triple-negative breast cancer: High incidence of central nervous system metastases. Cancer 2008, 113, 2638–2645. [Google Scholar] [CrossRef] [Green Version]
- Almansour, N.M. Triple-Negative Breast Cancer: A Brief Review About Epidemiology, Risk Factors, Signaling Pathways, Treatment and Role of Artificial Intelligence. Front. Mol. Biosci. 2022, 9, 836417. [Google Scholar] [CrossRef]
- Morris, G.J.; Naidu, S.; Topham, A.K.; Guiles, F.; Xu, Y.; McCue, P.; Schwartz, G.F.; Park, P.K.; Rosenberg, A.L.; Brill, K.; et al. Differences in breast carcinoma characteristics in newly diagnosed African-American and Caucasian patients: A single-institution compilation compared with the national cancer institute’s surveillance, epidemiology, and end results database. Cancer 2007, 110, 876–884. [Google Scholar] [CrossRef]
- Howlader, N.; Altekruse, S.F.; Li, C.I.; Chen, V.W.; Clarke, C.A.; Ries, L.A.G.; Cronin, K.A. US incidence of breast cancer subtypes defined by joint hormone receptor and HER2 status. J. Natl. Cancer Inst. 2014, 106, dju055. [Google Scholar] [CrossRef] [Green Version]
- Plasilova, M.L.; Hayse, B.; Killelea, B.K.; Horowitz, N.R.; Chagpar, A.B.; Lannin, D.R. Features of triple-negative breast cancer-Analysis of 38,813 cases from the national cancer database. Medicine 2016, 95, e4614. [Google Scholar] [CrossRef] [PubMed]
- Boyle, P. Triple-negative breast cancer: Epidemiological considerations and recommendations. Ann. Oncol. 2012, 23, vi7–vi12. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, G.S.; Erqou, S.; Patterson, H.; Mathew, A. Prevalence of Triple-Negative Breast Cancer in India: Systematic Review and Meta-Analysis. J. Glob. Oncol. 2016, 2, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Thakur, K.K.; Bordoloi, D.; Kunnumakkara, A.B. Alarming Burden of Triple-Negative Breast Cancer in India. Clin. Breast Cancer 2018, 18, e393–e399. [Google Scholar] [CrossRef]
- Al-Mahmood, S.; Sapiezynski, J.; Garbuzenko, O.B.; Minko, T. Metastatic and triple-negative breast cancer: Challenges and treatment options. Drug Deliv. Transl. Res. 2018, 8, 1483–1507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craene, B.D.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef]
- Ferrari-Amorotti, G.; Chiodoni, C.; Shen, F.; Cattelani, S.; Soliera, A.R.; Manzotti, G.; Grisendi, G.; Dominici, M.; Rivasi, F.; Colombo, M.P.; et al. Suppression of Invasion and Metastasis of Triple-Negative Breast Cancer Lines by Pharmacological or Genetic Inhibition of Slug Activity. Neoplasia 2014, 16, 1047–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, H.D.; Luitel, K.; Kim, M.; Zhang, K.; Longmore, G.D.; Tran, D.D. Transient SNAIL1 expression is necessary for metastatic competence in breast cancer. Cancer Res. 2014, 74, 6330–6340. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.K.; Kim, H.S.; Jin, T.; Moon, W.K. LOXL4 knockdown enhances tumor growth and lung metastasis through collagen-dependent extracellular matrix changes in triple-negative breast cancer. Oncotarget 2017, 8, 11977–11989. [Google Scholar] [CrossRef] [Green Version]
- Neophytou, C.; Boutsikos, P.; Papageorgis, P. Molecular mechanisms and emerging therapeutic targets of triple-negative breast cancer metastasis. Front. Oncol. 2018, 8, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, Y.F.; Tseng, L.M.; Hsu, C.Y.; Yang, M.H.; Chiu, J.H.; Shyr, Y.M. Brain-derived neurotrophic factor (BDNF)-TrKB signaling modulates cancer-endothelial cells interaction and affects the outcomes of triple negative breast cancer. PLoS ONE 2017, 12, e0178173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimbung, S.; Loman, N.; Hedenfalk, I. Clinical and molecular complexity of breast cancer metastases. Semin. Cancer Biol. 2015, 35, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Jhan, J.R.; Andrechek, E.R. Triple-negative breast cancer and the potential for targeted therapy. Pharmacogenomics 2017, 18, 1595–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; Ding, L.; Mardis, E.R.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Abramson, V.G.; Mayer, I.A. Molecular heterogeneity of triple-negative breast cancer. Curr. Breast Cancer Rep. 2014, 6, 154–158. [Google Scholar] [CrossRef] [Green Version]
- Darvishi, B.; Farahmand, L.; Majidzadeh-A, K. Stimuli-responsive Mesoporous Silica NPs as Non-viral Dual siRNA/Chemotherapy Carriers for Triple Negative Breast Cancer. Mol. Ther.—Nucleic Acids 2017, 7, 164–180. [Google Scholar] [CrossRef] [Green Version]
- Hirshfield, K.M.; Ganesan, S. Triple-negative breast cancer: Molecular subtypes and targeted therapy. Curr. Opin. Obstet. Gynecol. 2014, 26, 34–40. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [Green Version]
- Juul, N.; Szallasi, Z.; Eklund, A.C.; Li, Q.; Burrell, R.A.; Gerlinger, M.; Valero, V.; Andreopoulou, E.; Esteva, F.J.; Symmans, W.F.; et al. Assessment of an RNA interference screen-derived mitotic and ceramide pathway metagene as a predictor of response to neoadjuvant paclitaxel for primary triple-negative breast cancer: A retrospective analysis of five clinical trials. Lancet Oncol. 2010, 11, 358–365. [Google Scholar] [CrossRef]
- Perou, C.M.; Sørile, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; Ress, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Hubalek, M.; Czech, T.; Müller, H. Biological Subtypes of Triple-Negative Breast Cancer. Breast Care 2017, 12, 8–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertucci, F.; Finetti, P.; Cervera, N.; Charafe-Jauffret, E.; Mamessier, E.; Adélaïde, J.; Debono, S.; Houvenaeghel, G.; Maraninchi, D.; Viens, P.; et al. Gene expression profiling shows medullary breast cancer is a subgroup of basal breast cancers. Cancer Res. 2006, 66, 4636–4644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, B.D.; Jovanović, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of triple-negative breast cancer molecular subtypes: Implications for neoadjuvant chemotherapy selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef]
- Jiang, Y.Z.; Ma, D.; Suo, C.; Shi, J.; Xue, M.; Hu, X.; Xiao, Y.; Yu, K.D.; Liu, Y.R.; Yu, Y.; et al. Genomic and Transcriptomic Landscape of Triple-Negative Breast Cancers: Subtypes and Treatment Strategies. Cancer Cell 2019, 35, 428–440.e5. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, B.D.; Pietenpol, J.A. Identification and use of biomarkers in treatment strategies for triple-negative breast cancer subtypes. J. Pathol. 2014, 232, 142–150. [Google Scholar] [CrossRef]
- Stefansson, O.A.; Jonasson, J.G.; Johannsson, O.T.; Olafsdottir, K.; Steinarsdottir, M.; Valgeirsdottir, S.; Eyfjord, J.E. Genomic profiling of breast tumours in relation to BRCA abnormalities and phenotypes. Breast Cancer Res. 2009, 11, R47. [Google Scholar] [CrossRef] [Green Version]
- Adams, S.; Gray, R.J.; Demaria, S.; Goldstein, L.; Perez, E.A.; Shulman, L.N.; Martino, S.; Wang, M.; Jones, V.E.; Saphner, T.J.; et al. Prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancers from two phase III randomized adjuvant breast cancer trials: ECOG 2197 and ECOG 1199. J. Clin. Oncol. 2014, 32, 2959–2966. [Google Scholar] [CrossRef]
- Desmedt, C.; Haibe-Kains, B.; Wirapati, P.; Buyse, M.; Larsimont, D.; Bontempi, G.; Delorenzi, M.; Piccart, M.; Sotiriou, C. Biological processes associated with breast cancer clinical outcome depend on the molecular subtypes. Clin. Cancer Res. 2008, 14, 5158–5165. [Google Scholar] [CrossRef] [Green Version]
- Denkert, C.; Loibl, S.; Noske, A.; Roller, M.; Müller, B.M.; Komor, M.; Budczies, J.; Darb-Esfahani, S.; Kronenwett, R.; Hanusch, C.; et al. Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J. Clin. Oncol. 2010, 28, 105–113. [Google Scholar] [CrossRef]
- Denkert, C.; Von Minckwitz, G.; Brase, J.C.; Sinn, B.V.; Gade, S.; Kronenwett, R.; Pfitzner, B.M.; Salat, C.; Loi, S.; Schmitt, W.D.; et al. Tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy with or without carboplatin in human epidermal growth factor receptor 2-positive and triple-negative primary breast cancers. J. Clin. Oncol. 2015, 33, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Burstein, M.D.; Tsimelzon, A.; Poage, G.M.; Covington, K.R.; Contreras, A.; Fuqua, S.A.W.; Savage, M.I.; Osborne, C.K.; Hilsenbeck, S.G.; Chang, J.C.; et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin. Cancer Res. 2015, 21, 1688–1698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, G.R.; Qian, D.; Ku, J.K.; Lai, L.L. Metaplastic breast cancer: Clinical features and outcomes. Am. Surg. 2005, 71, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Hayes, M.J.; Thomas, D.; Emmons, A.; Giordano, T.J.; Kleer, C.G. Genetic changes of Wnt pathway genes are common events in metaplastic carcinomas of the breast. Clin. Cancer Res. 2008, 14, 4038–4044. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.R.; Jiang, Y.Z.; Xu, X.E.; Yu, K.D.; Jin, X.; Hu, X.; Zuo, W.J.; Hao, S.; Wu, J.; Liu, G.Y.; et al. Comprehensive transcriptome analysis identifies novel molecular subtypes and subtype-specific RNAs of triple-negative breast cancer. Breast Cancer Res. 2016, 18, 33. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Loi, S.; Drubay, D.; Adams, S.; Pruneri, G.; Francis, P.A.; Lacroix-Triki, M.; Joensuu, H.; Dieci, M.V.; Badve, S.; Demaria, S.; et al. Tumor-infiltrating lymphocytes and prognosis: A pooled individual patient analysis of early-stage triple-negative breast cancers. J. Clin. Oncol. 2019, 37, 559–569. [Google Scholar] [CrossRef]
- Park, J.H.; Jonas, S.F.; Bataillon, G.; Criscitiello, C.; Salgado, R.; Loi, S.; Viale, G.; Lee, H.J.; Dieci, M.V.; Kim, S.B.; et al. Prognostic value of tumor-infiltrating lymphocytes in patients with early-stage triple-negative breast cancers (TNBC) who did not receive adjuvant chemotherapy. Ann. Oncol. 2019, 30, 1941–1949. [Google Scholar] [CrossRef]
- Stanton, S.E.; Adams, S.; Disis, M.L. Variation in the Incidence and Magnitude of Tumor-Infiltrating Lymphocytes in Breast Cancer Subtypes: A Systematic Review. JAMA Oncol. 2016, 2, 1354–1360. [Google Scholar] [CrossRef]
- Xiao, Y.; Ma, D.; Zhao, S.; Suo, C.; Shi, J.; Xue, M.Z.; Ruan, M.; Wang, H.; Zhao, J.; Li, Q.; et al. Multi-omics profiling reveals distinct microenvironment characterization and suggests immune escape mechanisms of triple-negative breast cancer. Clin. Cancer Res. 2019, 25, 5002–5014. [Google Scholar] [CrossRef] [Green Version]
- Jamdade, V.S.; Sethi, N.; Mundhe, N.A.; Kumar, P.; Lahkar, M.; Sinha, N. Therapeutic targets of triple-negative breast cancer: A review. Br. J. Pharmacol. 2015, 172, 4228–4237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dontu, G.; Jackson, K.W.; McNicholas, E.; Kawamura, M.J.; Abdallah, W.M.; Wicha, M.S. Role of Notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Res. 2004, 6, R605–R615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, R.B.; Spence, K.; Anderson, E.; Howell, A.; Okano, H.; Potten, C.S. A putative human breast stem cell population is enriched for steroid receptor-positive cells. Dev. Biol. 2005, 277, 443–456. [Google Scholar] [CrossRef] [PubMed]
- Shih, I.M.; Wang, T.L. Notch signaling, γ-secretase inhibitors, and cancer therapy. Cancer Res. 2007, 67, 1879–1882. [Google Scholar] [CrossRef] [Green Version]
- Brou, C.; Logeat, F.; Gupta, N.; Bessia, C.; LeBail, O.; Doedens, J.R.; Cumano, A.; Roux, P.; Black, R.A.; Israël, A. A novel proteolytic cleavage involved in Notch signaling: The role of the disintegrin-metalloprotease TACE. Mol. Cell 2000, 5, 207–216. [Google Scholar] [CrossRef]
- Mumm, J.S.; Schroeter, E.H.; Saxena, M.T.; Griesemer, A.; Tian, X.; Pan, D.J.; Ray, W.J.; Kopan, R. A ligand-induced extracellular cleavage regulates γ-secretase-like proteolytic activation of Notch1. Mol. Cell 2000, 5, 197–206. [Google Scholar] [CrossRef]
- Shah, S.; Lee, S.F.; Tabuchi, K.; Hao, Y.H.; Yu, C.; LaPlant, Q.; Ball, H.; Dann, C.E.; Südhof, T.; Yu, G. Nicastrin functions as a γ-secretase-substrate receptor. Cell 2005, 122, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Gordon, W.R.; Arnett, K.L.; Blacklow, S.C. The molecular logic of Notch signaling—A structural and biochemical perspective. J. Cell Sci. 2008, 121, 3109–3119. [Google Scholar] [CrossRef] [Green Version]
- Kovall, R.A. More complicated than it looks: Assembly of Notch pathway transcription complexes. Oncogene 2008, 27, 5099–5109. [Google Scholar] [CrossRef] [Green Version]
- O’Toole, S.A.; Beith, J.M.; Millar, E.K.A.; West, R.; McLean, A.; Cazet, A.; Swarbrick, A.; Oakes, S.R. Therapeutic targets in triple negative breast cancer. J. Clin. Pathol. 2013, 66, 530–542. [Google Scholar] [CrossRef]
- Pece, S.; Serresi, M.; Santolini, E.; Capra, M.; Hulleman, E.; Galimberti, V.; Zurrida, S.; Maisonneuve, P.; Viale, G.; Di Fiore, P.P. Loss of negative regulation by Numb over Notch is relevant to human breast carcinogenesis. J. Cell Biol. 2004, 167, 215–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Dontu, G.; Wicha, M.S. Mammary stem cells, self-renewal pathways, and carcinogenesis. Breast Cancer Res. 2005, 7, 86–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speiser, J.; Foreman, K.; Drinka, E.; Godellas, C.; Perez, C.; Salhadar, A.; Erşahin, Ç.; Rajan, P. Notch-1 and notch-4 biomarker expression in triple-negative breast cancer. Int. J. Surg. Pathol. 2012, 20, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Liu, M.; Gonzalez-Perez, R.R. Role of Notch and its oncogenic signaling crosstalk in breast cancer. Biochim. Biophys. Acta—Rev. Cancer 2011, 1815, 197–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, H.; Sylvestre, J.R.; Jolicoeur, P. Notch1-induced mammary tumor development is cyclin D1-dependent and correlates with expansion of pre-malignant multipotent duct-limited progenitors. Oncogene 2010, 29, 4543–4554. [Google Scholar] [CrossRef] [Green Version]
- Efstratiadis, A.; Szabolcs, M.; Klinakis, A. Notch, Myc and breast cancer. Cell Cycle 2007, 6, 418–429. [Google Scholar] [CrossRef]
- Graziani, I.; Eliasz, S.; De Marco, M.A.; Chen, Y.; Pass, H.I.; De May, R.M.; Strack, P.R.; Miele, L.; Bocchetta, M. Opposite effects of Notch-1 and Notch-2 on mesothelioma cell survival under hypoxia are exerted through the Akt pathway. Cancer Res. 2008, 68, 9678–9685. [Google Scholar] [CrossRef] [Green Version]
- Fultang, N.; Chakraborty, M.; Peethambaran, B. Regulation of cancer stem cells in triple negative breast cancer. Cancer Drug Resist. 2021, 4, 321–342. [Google Scholar] [CrossRef]
- Rennstam, K.; McMichael, N.; Berglund, P.; Honeth, G.; Hegardt, C.; Rydén, L.; Luts, L.; Bendahl, P.O.; Hedenfalk, I. Numb protein expression correlates with a basal-like phenotype and cancer stem cell markers in primary breast cancer. Breast Cancer Res. Treat. 2010, 122, 315–324. [Google Scholar] [CrossRef] [Green Version]
- Gallahan, D.; Callahan, R. Mammary tumorigenesis in feral mice: Identification of a new int locus in mouse mammary tumor virus (Czech II)-induced mammary tumors. J. Virol. 1987, 61, 66–74. [Google Scholar] [CrossRef] [Green Version]
- Reipas, K.M.; Law, J.H.; Couto, N.; Islam, S.; Li, Y.; Li, H.; Cherkasov, A.; Jung, K.; Cheema, A.S.; Jones, S.J.M.; et al. Luteolin is a novel p90 ribosomal s6 kinase (RSK) inhibitor that suppresses notch4 signaling by blocking the activation of Y-box binding protein-1 (YB-1). Oncotarget 2013, 4, 329–345. [Google Scholar] [CrossRef] [PubMed]
- Lewis, M.T.; Veltmaat, J.M. Next stop, the twilight zone: Hedgehog network regulation of mammary gland development. J. Mammary Gland Biol. Neoplasia 2004, 9, 165–181. [Google Scholar] [CrossRef] [PubMed]
- Habib, J.G.; O’Shaughnessy, J.A. The hedgehog pathway in triple-negative breast cancer. Cancer Med. 2016, 5, 2989–3006. [Google Scholar] [CrossRef] [PubMed]
- Katoh, Y.; Katoh, M. Hedgehog signaling, epithelial-to-mesenchymal transition and miRNA (Review). Int. J. Mol. Med. 2008, 22, 271–275. [Google Scholar] [CrossRef] [Green Version]
- Merchant, A.A.; Matsui, W. Targeting Hedgehog—A cancer stem cell pathway. Clin. Cancer Res. 2010, 16, 3130–3140. [Google Scholar] [CrossRef] [Green Version]
- Polkinghorn, W.R.; Tarbell, N.J. Medulloblastoma: Tumorigenesis, current clinical paradigm, and efforts to improve risk stratification. Nat. Clin. Pract. Oncol. 2007, 4, 295–304. [Google Scholar] [CrossRef]
- Mukherjee, S.; Frolova, N.; Sadlonova, A.; Novak, Z.; Steg, A.; Page, G.P.; Welch, D.R.; Lobo-Ruppert, S.M.; Michael Ruppert, J.; Johnson, M.R.; et al. Hedgehog signaling and response to cyclopamine differ in epithelial and stromal cells in benign breast and breast cancer. Cancer Biol. Ther. 2006, 5, 674–683. [Google Scholar] [CrossRef] [Green Version]
- King, T.D.; Suto, M.J.; Li, Y. The wnt/β-catenin signaling pathway: A potential therapeutic target in the treatment of triple negative breast cancer. J. Cell. Biochem. 2012, 113, 13–18. [Google Scholar] [CrossRef]
- Izrailit, J.; Reedijk, M. Developmental pathways in breast cancer and breast tumor-initiating cells: Therapeutic implications. Cancer Lett. 2012, 317, 115–126. [Google Scholar] [CrossRef]
- Lu, W.; Lin, C.; Roberts, M.J.; Waud, W.R.; Piazza, G.A.; Li, Y. Niclosamide suppresses cancer cell growth by inducing Wnt co-receptor LRP6 degradation and inhibiting the Wnt/β-catenin pathway. PLoS ONE 2011, 6, e29290. [Google Scholar] [CrossRef]
- Geyer, F.C.; Lacroix-Triki, M.; Savage, K.; Arnedos, M.; Lambros, M.B.; MacKay, A.; Natrajan, R.; Reis-Filho, J.S. B-Catenin pathway activation in breast cancer is associated with triple-negative phenotype but not with CTNNB1 mutation. Mod. Pathol. 2011, 24, 209–231. [Google Scholar] [CrossRef] [PubMed]
- Ghadami, M.; Makita, Y.; Yoshida, K.; Nishimura, G.; Fukushima, Y.; Wakui, K.; Ikegawa, S.; Yamada, K.; Kondo, S.; Niikawa, N.; et al. Genetic mapping of the Camurati-Engelmann disease locus to chromosome 19q13.1-q13.3. Am. J. Hum. Genet. 2000, 66, 143–147. [Google Scholar] [CrossRef] [Green Version]
- Vaughn, S.P.; Broussard, S.; Hall, C.R.; Scott, A.; Blanton, S.H.; Milunsky, J.M.; Hecht, J.T. Confirmation of the mapping of the Camurati-Englemann locus to 19q13.2 and refinement to a 3.2-cM region. Genomics 2000, 66, 119–121. [Google Scholar] [CrossRef] [PubMed]
- Bhola, N.E.; Balko, J.M.; Dugger, T.C.; Kuba, M.G.; Sánchez, V.; Sanders, M.; Stanford, J.; Cook, R.S.; Arteaga, C.L. TGF-β inhibition enhances chemotherapy action against triple-negative breast cancer. J. Clin. Investig. 2013, 123, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, F.; Sun, H.; Deng, C.X. Potential therapeutic targets of triple-negative breast cancer based on its intrinsic subtype. Oncotarget 2017, 8, 73329–73344. [Google Scholar] [CrossRef]
- LoRusso, P.M. Inhibition of the PI3K/AKT/mTOR pathway in solid tumors. J. Clin. Oncol. 2016, 34, 3803–3815. [Google Scholar] [CrossRef]
- Costa, R.L.B.; Han, H.S.; Gradishar, W.J. Targeting the PI3K/AKT/mTOR pathway in triple-negative breast cancer: A review. Breast Cancer Res. Treat. 2018, 169, 397–406. [Google Scholar] [CrossRef]
- Hay, N.; Sonenberg, N. Upstream and downstream of mTOR. Genes. Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef] [Green Version]
- Beevers, C.S.; Li, F.; Liu, L.; Huang, S. Curcumin inhibits the mammalian target of rapamycin-mediated signaling pathways in cancer cells. Int. J. Cancer 2006, 119, 757–764. [Google Scholar] [CrossRef]
- Liu, T.; Yacoub, R.; Taliaferro-Smith, L.T.D.; Sun, S.Y.; Graham, T.R.; Dolan, R.; Lobo, C.; Tighiouart, M.; Yang, L.; Adams, A.; et al. Combinatorial effects of lapatinib and rapamycin in triple-negative breast cancer cells. Mol. Cancer Ther. 2011, 10, 1460–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cossu-Rocca, P.; Orrù, S.; Muroni, M.R.; Sanges, F.; Sotgiu, G.; Ena, S.; Pira, G.; Murgia, L.; Manca, A.; Uras, M.G.; et al. Analysis of PIK3CA mutations and activation pathways in triple negative breast cancer. PLoS ONE 2015, 10, e0141763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ooms, L.M.; Binge, L.C.; Davies, E.M.; Rahman, P.; Conway, J.R.W.; Gurung, R.; Ferguson, D.T.; Papa, A.; Fedele, C.G.; Vieusseux, J.L.; et al. The Inositol Polyphosphate 5-Phosphatase PIPP Regulates AKT1-Dependent Breast Cancer Growth and Metastasis. Cancer Cell 2015, 28, 155–169. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Murphy, C.J.; Karreth, F.A.; Emdal, K.B.; Yang, K.; White, F.M.; Elemento, O.; Toker, A.; Wulf, G.M.; Cantley, L.C. Identifying and targeting sporadic oncogenic genetic aberrations in mouse models of triple-negative breast cancer. Cancer Discov. 2018, 8, 354–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.J.X.; Loh, K.; Yap, Y.S. PI3K/Akt/mTOR inhibitors in breast cancer. Cancer Biol. Med. 2015, 12, 342–354. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S. Review of epidermal growth factor receptor biology. Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, S21–S26. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Berezov, A.; Wang, Q.; Zhang, G.; Drebin, J.; Murali, R.; Greene, M.I. ErbB receptors: From oncogenes to targeted cancer therapies. J. Clin. Investig. 2007, 117, 2051–2058. [Google Scholar] [CrossRef] [Green Version]
- Gauthier, M.L.; Torretto, C.; Ly, J.; Francescutti, V.; O’Day, D.H. Protein kinase Cα negatively regulates cell spreading and motility in MDA-MB-231 human breast cancer cells downstream of epidermal growth factor receptor. Biochem. Biophys. Res. Commun. 2003, 307, 839–846. [Google Scholar] [CrossRef]
- Tabach, Y.; Milyavsky, M.; Shats, I.; Brosh, R.; Zuk, O.; Yitzhaky, A.; Mantovani, R.; Domany, E.; Rotter, V.; Pilpel, Y. The promoters of human cell cycle genes integrate signals from two tumor suppressive pathways during cellular transformation. Mol. Syst. Biol. 2005, 1, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Park, H.S.; Jang, M.H.; Kim, E.J.; Kim, H.J.; Lee, H.J.; Kim, Y.J.; Kim, J.H.; Kang, E.; Kim, S.W.; Kim, I.A.; et al. High EGFR gene copy number predicts poor outcome in triple-negative breast cancer. Mod. Pathol. 2014, 27, 1212–1222. [Google Scholar] [CrossRef] [Green Version]
- Kumaraswamy, E.; Wendt, K.L.; Augustine, L.A.; Stecklein, S.R.; Sibala, E.C.; Li, D.; Gunewardena, S.; Jensen, R.A. BRCA1 regulation of epidermal growth factor receptor (EGFR) expression in human breast cancer cells involves microRNA-146a and is critical for its tumor suppressor function. Oncogene 2015, 34, 4333–4346. [Google Scholar] [CrossRef] [Green Version]
- Nakai, K.; Hung, M.C.; Yamaguchi, H. A perspective on anti-EGFR therapies targeting triple-negative breast cancer. Am. J. Cancer Res. 2016, 6, 1609–1623. [Google Scholar] [PubMed]
- Changavi, A.A.; Shashikala, A.; Ramji, A.S. Epidermal growth factor receptor expression in triple negative and nontriple negative breast Ccarcinomas. J. Lab. Physicians 2015, 7, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.; Sabnis, N.; McConathy, W.J.; Lacko, A.G. The potential role of nanotechnology in therapeutic approaches for triple negative breast cancer. Pharmaceutics 2013, 5, 353–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tentori, L.; Graziani, G. Chemopotentiation by PARP inhibitors in cancer therapy. Pharmacol. Res. 2005, 52, 25–33. [Google Scholar] [CrossRef]
- Veuger, S.J.; Curtin, N.J.; Smith, G.C.M.; Durkacz, B.W. Effects of novel inhibitors of poly(ADP-ribose) polymerase-1 and the DNA-dependent protein kinase on enzyme activities and DNA repair. Oncogene 2004, 23, 7322–7329. [Google Scholar] [CrossRef] [Green Version]
- De Vos, M.; Schreiber, V.; Dantzer, F. The diverse roles and clinical relevance of PARPs in DNA damage repair: Current state of the art. Biochem. Pharmacol. 2012, 84, 137–146. [Google Scholar] [CrossRef]
- Geenen, J.J.J.; Linn, S.C.; Beijnen, J.H.; Schellens, J.H.M. PARP Inhibitors in the Treatment of Triple-Negative Breast Cancer. Clin. Pharmacokinet. 2018, 57, 427–437. [Google Scholar] [CrossRef]
- Hwang, S.Y.; Park, S.; Kwon, Y. Recent therapeutic trends and promising targets in triple negative breast cancer. Pharmacol. Ther. 2019, 199, 30–57. [Google Scholar] [CrossRef]
- Finn, R.S. Targeting Src in breast cancer. Ann. Oncol. 2008, 19, 1379–1386. [Google Scholar] [CrossRef]
- Tryfonopoulos, D.; Walsh, S.; Collins, D.M.; Flanagan, L.; Quinn, C.; Corkery, B.; McDermott, E.W.; Evoy, D.; Pierce, A.; O’Donovan, N.; et al. Src: A potential target for the treatment of triple-negative breast cancer. Ann. Oncol. 2011, 22, 2234–2240. [Google Scholar] [CrossRef]
- Sánchez-Bailón, M.P.; Calcabrini, A.; Gómez-Domínguez, D.; Morte, B.; Martín-Forero, E.; Gómez-López, G.; Molinari, A.; Wagner, K.U.; Martín-Pérez, J. Src kinases catalytic activity regulates proliferation, migration and invasiveness of MDA-MB-231 breast cancer cells. Cell. Signal. 2012, 24, 1276–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harde, H.; Siddhapura, K.; Agrawal, A.K.; Jain, S. Development of dual toxoid-loaded layersomes for complete immunostimulatory response following peroral administration. Nanomedicine 2015, 10, 1077–1091. [Google Scholar] [CrossRef] [PubMed]
- Wojtukiewicz, M.Z.; Rek, M.M.; Karpowicz, K.; Górska, M.; Polityńska, B.; Wojtukiewicz, A.M.; Moniuszko, M.; Radziwon, P.; Tucker, S.C.; Honn, K.V. Inhibitors of immune checkpoints—PD-1, PD-L1, CTLA-4—new opportunities for cancer patients and a new challenge for internists and general practitioners. Cancer Metastasis Rev. 2021, 40, 949–982. [Google Scholar] [CrossRef] [PubMed]
- Collignon, J.; Lousberg, L.; Schroeder, H.; Jerusalem, G. Triple-negative breast cancer: Treatment challenges and solutions. Breast Cancer Targets Ther. 2016, 8, 93–107. [Google Scholar] [CrossRef] [Green Version]
- Jia, H.; Truica, C.I.; Wang, B.; Wang, Y.; Ren, X.; Harvey, H.A.; Song, J.; Yang, J.M. Immunotherapy for triple-negative breast cancer: Existing challenges and exciting prospects. Drug Resist. Updates 2017, 32, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cimino-Mathews, A.; Thompson, E.; Taube, J.M.; Ye, X.; Lu, Y.; Meeker, A.; Xu, H.; Sharma, R.; Lecksell, K.; Cornish, T.C.; et al. PD-L1 (B7-H1) expression and the immune tumor microenvironment in primary and metastatic breast carcinomas. Hum. Pathol. 2016, 47, 52–63. [Google Scholar] [CrossRef] [Green Version]
- Khosravi-Shahi, P.; Cabezón-Gutiérrez, L.; Custodio-Cabello, S. Metastatic triple negative breast cancer: Optimizing treatment options, new and emerging targeted therapies. Asia-Pac. J. Clin. Oncol. 2018, 14, 32–39. [Google Scholar] [CrossRef] [Green Version]
- Gatalica, Z.; Snyder, C.; Maney, T.; Ghazalpour, A.; Holterman, D.A.; Xiao, N.; Overberg, P.; Rose, I.; Basu, G.D.; Vranic, S.; et al. Programmed cell death 1 (PD-1) and its ligand (PD-L1) in common cancers and their correlation with molecular cancer type. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2965–2970. [Google Scholar] [CrossRef] [Green Version]
- Akiki, M.; Haddad, F.G.; Kourie, H.R.; Khaddage, A.; Smayra, V.T. PD-L1: An unavoidable biomarker in advanced triple-negative breast cancer. Biomark. Med. 2019, 13, 1539–1541. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.-A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Mittendorf, E.A.A.V.P.; Meric-Bernstam, F.; Qiao, N.; Wu, Y.; Harrington, S.; Su, X.; Wang, Y.; Gonzalez-Angulo, A.M.; Akcakanat, A.; Chawla, A.; et al. PD-L1 Expression in Triple Negative Breast Cancer. Cancer Immunol. Res. 2014, 2, 361–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, H.; Zhang, L.; Yang, Y.; Zuo, W.; Bi, Y.; Gao, W.; Deng, B.; Sun, J.; Shao, Q.; Qu, X. New Insights of CTLA-4 into Its Biological Function in Breast Cancer. Curr. Cancer Drug Targets 2010, 10, 728–736. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Y.; Yu, L.; Sakakura, K.; Visus, C.; Schwab, J.H.; Ferrone, C.R.; Favoino, E.; Koya, Y.; Campoli, M.R.; et al. CSPG4 in Cancer: Multiple Roles. Curr. Mol. Med. 2010, 10, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Katayama, A.; Wang, Y.; Yu, L.; Favoino, E.; Sakakura, K.; Favole, A.; Tsuchikawa, T.; Silver, S.; Watkins, S.C.; et al. Functional characterization of an scFv-Fc antibody that immunotherapeutically targets the common cancer cell surface proteoglycan CSPG4. Cancer Res. 2011, 71, 7410–7422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, A.K.; Lavrovsky, Y.; Song, C.S.; Chen, S.; Jung, M.H.; Velu, N.K.; Bi, B.Y.; Chatterjee, B. Regulation of Androgen Action. Vitam. Horm. 1998, 55, 309–352. [Google Scholar] [CrossRef]
- Pietri, E.; Conteduca, V.; Andreis, D.; Massa, I.; Melegari, E.; Sarti, S.; Cecconetto, L.; Schirone, A.; Bravaccini, S.; Serra, P.; et al. Androgen receptor signaling pathways as a target for breast cancer treatment. Endocr. Relat. Cancer 2016, 23, R485–R498. [Google Scholar] [CrossRef] [Green Version]
- Basile, D.; Cinausero, M.; Iacono, D.; Pelizzari, G.; Bonotto, M.; Vitale, M.G.; Gerratana, L.; Puglisi, F. Androgen receptor in estrogen receptor positive breast cancer: Beyond expression. Cancer Treat. Rev. 2017, 61, 15–22. [Google Scholar] [CrossRef]
- Rampurwala, M.; Wisinski, K.B.; O’Regan, R. Role of the Androgen Receptor in Triple-Negative Breast Cancer. Clin. Adv. Hematol. Oncol. 2016, 14, 186–193. [Google Scholar]
- Zuo, T.; Wilson, P.; Cicek, A.F.; Harigopal, M. Androgen receptor expression is a favorable prognostic factor in triple-negative breast cancers. Hum. Pathol. 2018, 80, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Duan, J.J.; Bian, X.W.; Yu, S.C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef]
- Gucalp, A.; Traina, T.A. Triple-Negative Breast Cancer: Adjuvant Therapeutic Options. Chemother. Res. Pract. 2011, 2011, 696208. [Google Scholar] [CrossRef] [PubMed]
- Moran, M.S. Should Triple-Negative Breast Cancer (TNBC) Subtype Affect Local-Regional Therapy Decision Making? Am. Soc. Clin. Oncol. Educ. B 2014, 34, e32–e36. [Google Scholar] [CrossRef] [PubMed]
- Won, K.A.; Spruck, C. Triple-negative breast cancer therapy: Current and future perspectives. Int. J. Oncol. 2020, 57, 1245–1261. [Google Scholar] [CrossRef]
- Wahba, H.A.; El-Hadaad, H.A. Current approaches in treatment of triple-negative breast cancer. Cancer Biol. Med. 2015, 12, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.K.; Aqil, F.; Jeyabalan, J.; Spencer, W.A.; Beck, J.; Gachuki, B.W.; Alhakeem, S.S.; Oben, K.; Munagala, R.; Bondada, S.; et al. Milk-derived exosomes for oral delivery of paclitaxel. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 1627–1636. [Google Scholar] [CrossRef]
- Kandimalla, R.; Aqil, F.; Alhakeem, S.S.; Jeyabalan, J.; Tyagi, N.; Agrawal, A.; Yan, J.; Spencer, W.; Bondada, S.; Gupta, R.C. Targeted oral delivery of paclitaxel using colostrum-derived exosomes. Cancers 2021, 13, 3700. [Google Scholar] [CrossRef]
- Mustacchi, G.; De Laurentiis, M. The role of taxanes in triple-negative breast cancer: Literature review. Drug Des. Dev. Ther. 2015, 9, 4303–4318. [Google Scholar] [CrossRef] [Green Version]
- Venkatesh, P.; Kasi, A. Anthracyclines; StatPearls Publishing LLC: Treasure Island, FL, USA, 2021. [Google Scholar]
- Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: An overview of the randomised trials. Lancet 2005, 365, 1687–1717. [Google Scholar] [CrossRef]
- Nakatsukasa, K.; Koyama, H.; Oouchi, Y.; Imanishi, S.; Mizuta, N.; Sakaguchi, K.; Fujita, Y.; Fujiwara, I.; Kotani, T.; Matsuda, T.; et al. Docetaxel and cyclophosphamide as neoadjuvant chemotherapy in HER2-negative primary breast cancer. Breast Cancer 2017, 24, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.E.; Chen, S.C.; Lin, Y.C.; Lo, Y.F.; Hsueh, S.; Chang, H.K. Identification of patients with node-negative, triple-negative breast cancer who benefit from adjuvant cyclophosphamide, methotrexate, and 5-fluorouracil chemotherapy. Anticancer Res. 2014, 34, 1301–1306. [Google Scholar] [PubMed]
- Masuda, H.; Baggerly, K.A.; Wang, Y.; Zhang, Y.; Gonzalez-Angulo, A.M.; Meric-Bernstam, F.; Valero, V.; Lehmann, B.D.; Pietenpol, J.A.; Hortobagyi, G.N.; et al. Differential response to neoadjuvant chemotherapy among 7 triple-negative breast cancer molecular subtypes. Clin. Cancer Res. 2013, 19, 5533–5540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Tanaka, M.; Inagaki, A.; Wanibuchi, H.; Izumi, Y.; Miura, K.; Nagayama, K.; Shiota, M.; Iwao, H. Establishment of a 5-fluorouracil-resistant triple-negative breast cancer cell line. Int. J. Oncol. 2013, 43, 1985–1991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humeniuk, R.; Menon, L.G.; Mishra, P.J.; Gorlick, R.; Sowers, R.; Rode, W.; Pizzorno, G.; Cheng, Y.C.; Kemeny, N.; Bertino, J.R.; et al. Decreased levels of UMP kinase as a mechanism of fluoropyrimidine resistance. Mol. Cancer Ther. 2009, 8, 1037–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gmeiner, W. Novel Chemical Strategies for Thymidylate Synthase Inhibition. Curr. Med. Chem. 2012, 12, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Kunz, C.; Focke, F.; Saito, Y.; Schuermann, D.; Lettieri, T.; Selfridge, J.; Schär, P. Base Excision by Thymine DNA Glycosylase Mediates DNA-Directed Cytotoxicity of 5-Fluorouracil. PLoS Biol. 2009, 7, e1000091. [Google Scholar] [CrossRef]
- Li, Q.; Li, Q.; Zhang, P.; Yuan, P.; Wang, J.; Ma, F.; Luo, Y.; Fan, Y.; Cai, R.; Xu, B. A phase II study of capecitabine plus cisplatin in metastatic triple-negative breast cancer patients pretreated with anthracyclines and taxanes. Cancer Biol. Ther. 2015, 16, 1746–1753. [Google Scholar] [CrossRef] [Green Version]
- Soffietti, R.; Trevisan, E.; Rudà, R. Neurologic complications of chemotherapy and other newer and experimental approaches. Handb. Clin. Neurol. 2014, 121, 1199–1218. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Z.; Hu, X.; Wang, B.; Wang, L.; Yang, W.; Liu, Y.; Liu, G.; Di, G.; Hu, Z.; et al. Cisplatin and gemcitabine as the first line therapy in metastatic triple negative breast cancer. Int. J. Cancer 2015, 136, 204–211. [Google Scholar] [CrossRef]
- Jovanovic, B.; Mayer, I.A.; Mayer, E.L.; Abramson, V.G.; Bardia, A.; Sanders, M.E.; Kuba, M.G.; Estrada, M.V.; Beeler, J.S.; Shaver, T.M.; et al. A randomized phase II neoadjuvant study of cisplatin, paclitaxel with or without everolimus in patients with stage II/III triple-negative breast cancer (TNBC): Responses and long-term outcome correlated with increased frequency of DNA damage response gene. Clin. Cancer Res. 2017, 23, 4035–4045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Minckwitz, G.; Schneeweiss, A.; Loibl, S.; Salat, C.; Denkert, C.; Rezai, M.; Blohmer, J.U.; Jackisch, C.; Paepke, S.; Gerber, B.; et al. Neoadjuvant carboplatin in patients with triple-negative and HER2-positive early breast cancer (GeparSixto; GBG 66): A randomised phase 2 trial. Lancet Oncol. 2014, 15, 747–756. [Google Scholar] [CrossRef]
- Park, J.H.; Ahn, J.H.; Kim, S.B. How shall we treat early triple-negative breast cancer (TNBC): From the current standard to upcoming immuno-molecular strategies. ESMO Open 2018, 3, e000357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giuli, M.V.; Giuliani, E.; Screpanti, I.; Bellavia, D.; Checquolo, S. Notch Signaling Activation as a Hallmark for Triple-Negative Breast Cancer Subtype. J. Oncol. 2019, 2019, 8707053. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Wu, H.; Xu, H.; Xiong, H.; Chu, Q.; Yu, S.; Wu, G.S.; Wu, K. Notch signaling: An emerging therapeutic target for cancer treatment. Cancer Lett. 2015, 369, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Locatelli, M.A.; Aftimos, P.; Claire Dees, E.; LoRusso, P.M.; Pegram, M.D.; Awada, A.; Huang, B.; Cesari, R.; Jiang, Y.; Shaik, M.N.; et al. Phase I study of the gamma secretase inhibitor PF-03084014 in combination with docetaxel in patients with advanced triplenegative breast cancer. Oncotarget 2017, 8, 2320–2328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sardesai, S.; Badawi, M.; Mrozek, E.; Morgan, E.; Phelps, M.; Stephens, J.; Wei, L.; Kassem, M.; Ling, Y.; Lustberg, M.; et al. A phase I study of an oral selective gamma secretase (GS) inhibitor RO4929097 in combination with neoadjuvant paclitaxel and carboplatin in triple negative breast cancer. Investig. New Drugs 2020, 38, 1400–1410. [Google Scholar] [CrossRef]
- Chaudhuri, A.R.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Shi, J.; Liu, F.; Song, Y. Progress: Targeted therapy, immunotherapy, and new chemotherapy strategies in advanced triple-negative breast cancer. Cancer Manag. Res. 2020, 12, 9375–9387. [Google Scholar] [CrossRef]
- Gonzalez-Angulo, A.M.; Timms, K.M.; Liu, S.; Chen, H.; Litton, J.K.; Potter, J.; Lanchbury, J.S.; Stemke-Hale, K.; Hennessy, B.T.; Arun, B.K.; et al. Incidence and outcome of BRCA mutations in unselected patients with triple receptor-negative breast cancer. Clin. Cancer Res. 2011, 17, 1082–1089. [Google Scholar] [CrossRef] [Green Version]
- McCann, K.E.; Hurvitz, S.A.; McAndrew, N. Advances in Targeted Therapies for Triple-Negative Breast Cancer. Drugs 2019, 79, 1217–1230. [Google Scholar] [CrossRef] [PubMed]
- Medina, M.A.; Oza, G.; Sharma, A.; Arriaga, L.G.; Hernández, J.M.H.; Rotello, V.M.; Ramirez, J.T. Triple-negative breast cancer: A review of conventional and advanced therapeutic strategies. Int. J. Environ. Res. Public Health 2020, 17, 2078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marmé, F.; Schneeweiss, A. Targeted Therapies in Triple-Negative Breast Cancer. Breast Care 2015, 10, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Abramson, V.G.; Lehmann, B.D.; Ballinger, T.J.; Pietenpol, J.A. Subtyping of triple-negative breast cancer: Implications for therapy. Cancer 2015, 121, 8–16. [Google Scholar] [CrossRef] [Green Version]
- López-Knowles, E.; O’Toole, S.A.; McNeil, C.M.; Millar, E.K.A.; Qiu, M.R.; Crea, P.; Daly, R.J.; Musgrove, E.A.; Sutherland, R.L. PI3K pathway activation in breast cancer is associated with the basal-like phenotype and cancer-specific mortality. Int. J. Cancer 2010, 126, 1121–1131. [Google Scholar] [CrossRef]
- Chan, J.J.; Tan, T.J.Y.; Dent, R.A. Novel therapeutic avenues in triple-negative breast cancer: PI3K/AKT inhibition, androgen receptor blockade, and beyond. Ther. Adv. Med. Oncol. 2019, 11, 1758835919880429. [Google Scholar] [CrossRef]
- Zaytseva, Y.Y.; Valentino, J.D.; Gulhati, P.; Mark Evers, B. MTOR inhibitors in cancer therapy. Cancer Lett. 2012, 319, 1–7. [Google Scholar] [CrossRef]
- Garrido-Castro, A.C.; Saura, C.; Barroso-Sousa, R.; Guo, H.; Ciruelos, E.; Bermejo, B.; Gavilá, J.; Serra, V.; Prat, A.; Paré, L.; et al. Phase 2 study of buparlisib (BKM120), a pan-class I PI3K inhibitor, in patients with metastatic triple-negative breast cancer. Breast Cancer Res. 2020, 22, 120. [Google Scholar] [CrossRef]
- Xu, S.; Li, S.; Guo, Z.; Luo, J.; Ellis, M.J.; Ma, C.X. Combined targeting of mTOR and AKT Is an effective strategy for basal-like breast cancer in patient-derived xenograft models. Mol. Cancer Ther. 2013, 12, 1665–1675. [Google Scholar] [CrossRef] [Green Version]
- Peddi, P.F.; Ellis, M.J.; Ma, C. Molecular Basis of Triple Negative Breast Cancer and Implications for Therapy. Int. J. Breast Cancer 2012, 2012, 217185. [Google Scholar] [CrossRef]
- Von Minckwitz, G.; Jonat, W.; Fasching, P.; Du Bois, A.; Kleeberg, U.; Lück, H.J.; Kettner, E.; Hilfrich, J.; Eiermann, W.; Torode, J.; et al. A multicentre phase II study on gefitinib in taxane- and anthracycline-pretreated metastatic breast cancer. Breast Cancer Res. Treat. 2005, 89, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Albanell, J.; Ruiz, A.; Lluch, A.; Gascón, P.; Guillém, V.; González, S.; Sauleda, S.; Marimón, I.; Tabernero, J.M.; et al. Phase II and tumor pharmacodynamic study of gefitinib in patients with advanced breast cancer. J. Clin. Oncol. 2005, 23, 5323–5333. [Google Scholar] [CrossRef]
- Dickler, M.N.; Cobleigh, M.A.; Miller, K.D.; Klein, P.M.; Winer, E.P. Efficacy and safety of erlotinib in patients with locally advanced or metastatic breast cancer. Breast Cancer Res. Treat. 2009, 115, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Layman, R.M.; Ruppert, A.S.; Lynn, M.; Mrozek, E.; Ramaswamy, B.; Lustberg, M.B.; Wesolowski, R.; Ottman, S.; Carothers, S.; Bingman, A.; et al. Severe and prolonged lymphopenia observed in patients treated with bendamustine and erlotinib for metastatic triple negative breast cancer. Cancer Chemother. Pharmacol. 2013, 71, 1183–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapiro, G.I.; Bell-McGuinn, K.M.; Molina, J.R.; Bendell, J.; Spicer, J.; Kwak, E.L.; Pandya, S.S.; Millham, R.; Borzillo, G.; Pierce, K.J.; et al. First-in-human study of PF-05212384 (PKI-587), a small-molecule, intravenous, dual inhibitor of PI3K and mTOR in patients with advanced cancer. Clin. Cancer Res. 2015, 21, 1888–1895. [Google Scholar] [CrossRef] [Green Version]
- Gelman, I.H. Src-family tyrosine kinases as therapeutic targets in advanced cancer. Front. Biosci. 2011, E3, 801–807. [Google Scholar] [CrossRef] [Green Version]
- Yeatman, T.J. A renaissance for SRC. Nat. Rev. Cancer 2004, 4, 470–480. [Google Scholar] [CrossRef]
- Finn, R.S.; Dering, J.; Ginther, C.; Wilson, C.A.; Glaspy, P.; Tchekmedyian, N.; Slamon, D.J. Dasatinib, an orally active small molecule inhibitor of both the src and abl kinases, selectively inhibits growth of basal-type/”triple- negative” breast cancer cell lines growing in vitro. Breast Cancer Res. Treat. 2007, 105, 319–326. [Google Scholar] [CrossRef] [Green Version]
- Gucalp, A.; Sparano, J.A.; Caravelli, J.; Santamauro, J.; Patil, S.; Abbruzzi, A.; Pellegrino, C.; Bromberg, J.; Dang, C.; Theodoulou, M.; et al. Phase II trial of saracatinib (AZD0530), an oral SRC-inhibitor for the treatment of patients with hormone receptor-negative metastatic breast cancer. Clin. Breast Cancer 2011, 11, 306–311. [Google Scholar] [CrossRef] [Green Version]
- Thomas, R.; Al-Khadairi, G.; Decock, J. Immune Checkpoint Inhibitors in Triple Negative Breast Cancer Treatment: Promising Future Prospects. Front. Oncol. 2021, 10, 3464. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voorwerk, L.; Slagter, M.; Horlings, H.M.; Sikorska, K.; van de Vijver, K.K.; de Maaker, M.; Nederlof, I.; Kluin, R.J.C.; Warren, S.; Ong, S.F.; et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: The TONIC trial. Nat. Med. 2019, 25, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Rugo, H.S.; Adams, S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Henschel, V.; Molinero, L.; Chui, S.Y.; et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): Updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 44–59. [Google Scholar] [CrossRef]
- Bonnefoi, H.; Grellety, T.; Tredan, O.; Saghatchian, M.; Dalenc, F.; Mailliez, A.; L’Haridon, T.; Cottu, P.; Abadie-Lacourtoisie, S.; You, B.; et al. A phase II trial of abiraterone acetate plus prednisone in patients with triple-negative androgen receptor positive locally advanced or metastatic breast cancer (UCBG 12-1). Ann. Oncol. 2016, 27, 812–818. [Google Scholar] [CrossRef]
- my Huynh, M.; Pambid, M.R.; Jayanthan, A.; Dorr, A.; Los, G.; Dunn, S.E. The dawn of targeted therapies for triple negative breast cancer (TNBC): A snapshot of investigational drugs in phase I and II trials. Expert Opin. Investig. Drugs 2020, 29, 1199–1208. [Google Scholar] [CrossRef]
- Robles, A.J.; Cai, S.; Cichewicz, R.H.; Mooberry, S.L. Selective activity of deguelin identifies therapeutic targets for androgen receptor-positive breast cancer. Breast Cancer Res. Treat. 2016, 157, 475–488. [Google Scholar] [CrossRef]
- A Combination Study of Rucaparib and Atezolizumab in Participants with Advanced Gynecologic Cancers and Triple-Negative Breast Cancer. (n.d.). Available online: https://clinicaltrials.gov/ct2/show/study/NCT03101280 (accessed on 19 July 2021).
- Li, A.; Labrie, M.; Vuky, J.; Lim, J.Y.; Johnson, B.; Sivagnanam, S.; Betts, C.; Coussens, L.; Corless, C.L.; Bergan, R.C.; et al. Feasibility of real-time serial comprehensive tumor analytics: Pilot study of olaparib and durvalumab in metastatic triple negative breast cancer (mTNBC). J. Clin. Oncol. 2020, 38, e13092. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Waggoner, S.E.; Vidal, G.A.; Mita, M.M.; Fleming, G.F.; Holloway, R.W.; Van Le, L.; Sachdev, J.C.; Davis, E.C.; Colon-Otero, G.; et al. TOPACIO/Keynote-162 (NCT02657889): A phase 1/2 study of niraparib + pembrolizumab in patients (pts) with advanced triple-negative breast cancer or recurrent ovarian cancer (ROC)—Results from ROC cohort. J. Clin. Oncol. 2018, 36, 2016–2018. [Google Scholar] [CrossRef]
- Sammons, S.; Tan, T.J.Y.; Traina, T.A.; Kim, S.-B.; Im, Y.-H.; Bachelder, C.; Marcom, P.K.; Dent, R.A. Dora: A randomized phase II multicenter maintenance study of olaparib alone or olaparib in combination with durvalumab in platinum responsive advanced triple-negative breast cancer (aTNBC). J. Clin. Oncol. 2019, 37, TPS1113. [Google Scholar] [CrossRef]
- Abraham, J.; Vallier, A.-L.; Qian, W.; Machin, A.; Grybowicz, L.; Thomas, S.; Harvey, C.; McAdam, K.; Hughes-Davies, L.; Roylance, R.; et al. PARTNER: Randomised, phase II/III trial to evaluate the safety and efficacy of the addition of olaparib to platinum-based neoadjuvant chemotherapy in triple negative and/or germline BRCA mutated breast cancer patients. J. Clin. Oncol. 2018, 36, TPS605. [Google Scholar] [CrossRef]
- Assessing the Efficacy of Paclitaxel and Olaparib in Comparison to Paclitaxel/Carboplatin Followed by Epirubicin/Cyclophosphamide as Neoadjuvant Chemotherapy in Patients with HER2-Negative Early Breast Cancer and Homologous Recombination Deficiency (Gep, Clinicaltrials.Gov.). 2016. Available online: https://clinicaltrials.gov/ct2/show/NCT02789332 (accessed on 19 July 2021).
- Loibl, S.; Shaughnessy, J.O.; Untch, M.; Sikov, W.M.; Rugo, H.S.; Mckee, M.D.; Huober, J.; Golshan, M.; Von Minckwitz, G.; Maag, D.; et al. Addition of the PARP inhibitor veliparib plus carboplatin or carboplatin alone to standard neoadjuvant chemotherapy in triple-negative breast cancer (BrighTNess): A randomised, phase 3 trial. Lancet Oncol. 2018, 19, 497–509. [Google Scholar] [CrossRef]
- Study to Evaluate Sacituzumab Govitecan in Combination with Talazoparib in Patients with Metastatic Breast Cancer. Gov. 2019. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04039230 (accessed on 19 July 2021).
- TTAC-0001 and Pembrolizumab Phase Ib Combination Trial in Metastatic Triple-Negative Breast Cancer. Gov. 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT03720431 (accessed on 19 July 2021).
- Radovich, M.; Solzak, J.; Hancock, B.; Storniolo, A.; Schneider, B.; Miller, K. Abstract OT3-06-02: An initial safety study of gedatolisib plus PTK7-ADC for metastatic triple-negative breast cancer. Cancer Res. 2019, 79, OT3-06-02. [Google Scholar] [CrossRef]
- Liu, J.; Jiang, Z.; Li, Q.; Li, Y.; Liu, Q.; Song, E. Efficacy and safety of anti-PD-1 antibody SHR-1210 combined with apatinib in patients with advanced triple-negative breast cancer. J. Clin. Oncol. 2019, 37, 1066. [Google Scholar] [CrossRef]
- López-Miranda, E.; Gávila, J.; Pernas, S.; Saura, C.; Oliveira, M.; Serra, V.; Schmid, P.; Lord, S.; Paez, D.; Perez, J.; et al. Abstract OT1-01-06: PIQHASSO: Open label, non-randomized, multicenter phase 1/2b study investigating safety and efficacy of PQR309 and eribulin combination in patients (pts) with locally advanced (LA) or metastatic HER2 (-) and triple-negative breast canc. Cancer Res. 2017, 77, OT1-01-06. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Abramson, V.G.; Sanders, M.E.; Mayer, E.L.; Haddad, T.C.; Nanda, R.; Van Poznak, C.; Storniolo, A.M.; Nangia, J.R.; Gonzalez-Ericsson, P.I.; et al. TBCRC 032 IB/II Multicenter Study: Molecular Insights to AR Antagonist and PI3K Inhibitor Efficacy in Patients with AR+ Metastatic Triple-Negative Breast Cancer. Clin. Cancer Res. 2020, 26, 2111–2123. [Google Scholar] [CrossRef]
- Schmid, P.; Abraham, J.; Chan, S.; Wheatley, D.; Brunt, M.; Nemsadze, G.; Baird, R.; Park, Y.H.; Hall, P.; Perren, T.; et al. AZD5363 plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (PAKT): A randomised, double-blind, placebo-controlled, phase II trial. J. Clin. Oncol. 2018, 36, 1007. [Google Scholar] [CrossRef]
- A Study of AZD2014 in Combination with Selumetinib in Patients with Advanced Cancers (TORCMEK). Clinicaltrial.Gov. 2015. Available online: https://clinicaltrials.gov/ct2/show/NCT02583542 (accessed on 19 July 2021).
- Nab-Paclitaxel and Bevacizumab Followed by Bevacizumab and Erlotinib in Metastatic Breast Cancer. Clinicaltrials.Gov. 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT00733408 (accessed on 19 July 2021).
- Rodriguez, A.A.; Salazar, N.; Patel, T.A.; Mejia, J.A.; Froehlich, A.; Amin, N.S.; Weiss, H.; Rivera, E.; Chang, J.C.N. A randomized, parallel-arm, phase II trial to assess the efficacy of preoperative ixabepilone with or without cetuximab in patients with triple-negative breast cancer (TNBC). J. Clin. Oncol. 2014, 32, 1133. [Google Scholar] [CrossRef]
- Gucalp, A.; Boyle, L.A.; Alano, T.; Arumov, A.; Gounder, M.M.; Patil, S.; Feigin, K.; Edelweiss, M.; D’Andrea, G.; Bomberg, J.; et al. Phase II trial of bicalutamide in combination with palbociclib for the treatment of androgen receptor (+) metastatic breast cancer. J. Clin. Investig. 2020, 38, 1017. [Google Scholar] [CrossRef]
- Tolaney, S.M.; Kalinsky, K.; Kaklamani, V.G.; D’Adamo, D.R.; Aktan, G.; Tsai, M.L.; O’Regan, R.; Kaufman, P.A.; Wilks, S.; Andreopoulou, E.; et al. A phase Ib/II study of eribulin (ERI) plus pembrolizumab (PEMBRO) in metastatic triple-negative breast cancer (mTNBC) (ENHANCE 1). J. Clin. Oncol. 2020, 38, 1015. [Google Scholar] [CrossRef]
- Litton, J.; Moulder, S.; Helgason, T.; Clayborn, A.; Rauch, G.; Gilcrease, M.; Adrada, B.; Huo, L.; Hess, K.; Symmans, W.F.; et al. Abstract OT2-01-14: Triple-negative first-line study: Neoadjuvant trial of nab-paclitaxel and atezolizumab, a PD-L1 inhibitor, in patients with triple negative breast cancer (TNBC) (NCT02530489). Cancer Res. 2017, 77, OT2-01-14. [Google Scholar] [CrossRef]
- Kwa, M.J.; Iwano, A.; Esteva, F.J.; Novik, Y.; Speyer, J.L.; Oratz, R.; Meyers, M.I.; Axelrod, D.; Hogan, R.; Mendoza, S.; et al. Phase II trial of pembrolizumab in combination with nab-paclitaxel in patients with metastatic HER2-negative breast cancer. J. Clin. Oncol. 2017, 35, TPS1124. [Google Scholar] [CrossRef]
- Galunisertib and Paclitaxel in Treating Patients with Metastatic Androgen Receptor Negative (AR-) Triple Negative Breast Cancer, Clinicaltrials.Gov. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT02672475 (accessed on 20 July 2021).
- Damodaran, S.; Hess, K.; Rauch, M.; Astrada, B.; Santiago, L.; Litton, J.; Mittendorf, E.; Lim, B.; Ueno, N.; Tripathy, D.; et al. Abstract CT062: NCT02456857: A phase II trial of liposomal doxorubicin, bevacizumab and everolimus (DAE) in patients with localized triple-negative breast cancer (TNBC) with tumors predicted insensitive to standard neoadjuvant chemotherapy. Am. Assoc. Cancer Res. 2017, 77, CT062. [Google Scholar] [CrossRef]
- Pawar, A.; Prabhu, P. Nanosoldiers: A promising strategy to combat triple negative breast cancer. Biomed. Pharmacother. 2019, 110, 319–341. [Google Scholar] [CrossRef] [PubMed]
- Aqil, F.; Munagala, R.; Agrawal, A.K.; Gupta, R. Anticancer Phytocompounds: Experimental and Clinical Updates. In New Look to Phytomedicine: Advancements in Herbal Products as Novel Drug Leads; Elsevier Inc.: Philadelphia, PA, USA, 2018; pp. 237–272. ISBN 9780128146200. [Google Scholar]
- Prabhu, P.; Patravale, V. The upcoming field of theranostic nanomedicine: An overview. J. Biomed. Nanotechnol. 2012, 8, 859–882. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.K.; Agrawal, A.K.; Singh, S. Preformulation Challenges: The Concept Behind the Selection, Design and Preparation of Nanoformulations. In Nanoformulations in Human Health; Springer: Cham, Switzerland, 2020. [Google Scholar]
- Jain, S.; Raza, K.; Agrawal, A.K.; Vaidya, A. Nanotechnology Applications for Cancer Chemotherapy; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Kumar, D.N.; Chaudhuri, A.; Aqil, F.; Dehari, D.; Munagala, R.; Singh, S.; Gupta, R.C.; Agrawal, A.K. Exosomes as Emerging Drug Delivery and Diagnostic Modality for Breast Cancer: Recent Advances in Isolation and Application. Cancers 2022, 14, 1435. [Google Scholar] [CrossRef]
- Jain, S.; Kumar, S.; Agrawal, A.K.; Thanki, K.; Banerjee, U.C. Enhanced transfection efficiency and reduced cytotoxicity of novel lipid-polymer hybrid nanoplexes. Mol. Pharm. 2013, 10, 2416–2425. [Google Scholar] [CrossRef]
- Negi, S.; Chaudhuri, A.; Kumar, D.N.; Dehari, D.; Singh, S.; Agrawal, A.K. Nanotherapeutics in autophagy: A paradigm shift in cancer treatment. Drug Deliv. Transl. Res. 2022. [Google Scholar] [CrossRef]
- Pérez-Herrero, E.; Fernández-Medarde, A. Advanced targeted therapies in cancer: Drug nanocarriers, the future of chemotherapy. Eur. J. Pharm. Biopharm. 2015, 93, 52–79. [Google Scholar] [CrossRef] [Green Version]
- Jain, S.; Sharma, J.M.; Agrawal, A.K.; Mahajan, R.R. Surface stabilized efavirenz nanoparticles for oral bioavailability enhancement. J. Biomed. Nanotechnol. 2013, 9, 1862–1874. [Google Scholar] [CrossRef]
- Agrawal, A.K.; Harde, H.; Thanki, K.; Jain, S. Improved stability and antidiabetic potential of insulin containing folic acid functionalized polymer stabilized multilayered liposomes following oral administration. Biomacromolecules 2014, 15, 350–360. [Google Scholar] [CrossRef]
- van Elk, M.; Murphy, B.P.; Eufrásio-da-Silva, T.; O’Reilly, D.P.; Vermonden, T.; Hennink, P.W.E.; Duffy, G.P.; Ruiz-Hernández, E. Nanomedicines for advanced cancer treatments: Transitioning towards responsive systems. Int. J. Pharm. 2016, 515, 132–164. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Liu, T.; Xie, Y.; Sun, Z.; Liu, H.; Lin, J.; Liu, C.; Mao, Z.W.; Nie, S. Chitosan layered gold nanorods as synergistic therapeutics for photothermal ablation and gene silencing in triple-negative breast cancer. Acta Biomater. 2015, 25, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Shi, K.; Qu, Y.; Chu, B.; Qian, Z. Engineering Nanoparticles for Targeted Delivery of Nucleic Acid Therapeutics in Tumor. Mol. Ther.—Methods Clin. Dev. 2019, 12, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.S.; Lee, M.Y.; David, A.E.; Park, Y.S. Nanoparticles for gene delivery: Therapeutic and toxic effects. Mol. Cell. Toxicol. 2014, 10, 1–8. [Google Scholar] [CrossRef]
- Mohammad, A.S.; Griffith, J.I.; Adkins, C.E.; Shah, N.; Sechrest, E.; Dolan, E.L.; Terrell-Hall, T.B.; Hendriks, B.S.; Lee, H.; Lockman, P.R. Liposomal Irinotecan Accumulates in Metastatic Lesions, Crosses the Blood-Tumor Barrier (BTB), and Prolongs Survival in an Experimental Model of Brain Metastases of Triple Negative Breast Cancer. Pharm. Res. 2019, 35, 31. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Cai, P.; Li, J.; Zhang, T.; Lin, L.; Abbasi, A.Z.; Henderson, J.T.; Rauth, A.M.; Wu, X.Y. Blood-brain barrier-penetrating amphiphilic polymer nanoparticles deliver docetaxel for the treatment of brain metastases of triple negative breast cancer. J. Control. Release 2017, 246, 98–109. [Google Scholar] [CrossRef]
- Iorio, A.L.; Ros, M.; Fantappiè, O.; Lucchesi, M.; Facchini, L.; Stival, A.; Becciani, S.; Guidi, M.; Favre, C.; De Martino, M.; et al. Blood-Brain Barrier and Breast Cancer Resistance Protein: A Limit to the Therapy of CNS Tumors and Neurodegenerative Diseases. Anti-Cancer Agent Med. Chem. 2016, 16, 810–815. [Google Scholar]
- Li, J.; Cai, P.; Shalviri, A.; Henderson, J.T.; He, C.; Foltz, W.D.; Prasad, P.; Brodersen, P.M.; Chen, Y.; Dacosta, R.; et al. A multifunctional polymeric nanotheranostic system delivers doxorubicin and imaging agents across the blood-brain barrier targeting brain metastases of breast cancer. ACS Nano 2014, 8, 9925–9940. [Google Scholar] [CrossRef]
- Barenholz, Y. Doxil®—The first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef]
- Chen, N.; Brachmann, C.; Liu, X.; Pierce, D.W.; Dey, J.; Kerwin, W.S.; Li, Y.; Zhou, S.; Hou, S.; Carleton, M.; et al. Albumin-bound nanoparticle (nab) paclitaxel exhibits enhanced paclitaxel tissue distribution and tumor penetration. Cancer Chemother. Pharmacol. 2015, 76, 699–712. [Google Scholar] [CrossRef] [Green Version]
- Bosetti, R.; Jones, S.L. Cost-effectiveness of nanomedicine: Estimating the real size of nano-costs. Nanomedicine 2019, 14, 1367–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent Rajkumar, S. The high cost of prescription drugs: Causes and solutions. Blood Cancer J. 2020, 10, 71. [Google Scholar] [CrossRef] [PubMed]
- Kushwah, V.; Jain, D.K.; Agrawal, A.K.; Jain, S. Improved antitumor efficacy and reduced toxicity of docetaxel using anacardic acid functionalized stealth liposomes. Colloids Surf. B Biointerfaces 2018, 172, 213–223. [Google Scholar] [CrossRef]
- Jain, V.; Kumar, H.; Anod, H.V.; Chand, P.; Gupta, N.V.; Dey, S.; Kesharwani, S.S. A review of nanotechnology-based approaches for breast cancer and triple-negative breast cancer. J. Control. Release 2020, 326, 628–647. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.; Ju, R.J.; Liu, L.; Xie, H.J.; Mu, L.M.; Zhao, Y.; Yan, Y.; Hu, Y.J.; Wu, J.S.; Lu, W.L. Application of functional vincristine plus dasatinib liposomes to deletion of vasculogenic mimicry channels in triple-negative breast cancer. Oncotarget 2015, 6, 36625–36642. [Google Scholar] [CrossRef] [Green Version]
- Parvani, J.G.; Gujrati, M.D.; Mack, M.A.; Schiemann, W.P.; Lu, Z.R. Silencing β3 integrin by targeted ECO/siRNA nanoparticles inhibits EMT and metastasis of triple-negative breast cancer. Cancer Res. 2015, 75, 2316–2325. [Google Scholar] [CrossRef] [Green Version]
- Doddapaneni, R.; Patel, K.; Owaid, I.H.; Singh, M. Tumor neovasculature-targeted cationic PEGylated liposomes of gambogic acid for the treatment of triple-negative breast cancer. Drug Deliv. 2016, 23, 1232–1241. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Li, X.Q.; Duan, J.L.; Bao, C.J.; Cui, Y.N.; Su, Z.B.; Xu, J.R.; Luo, Q.; Chen, M.; Xie, Y.; et al. Nanosized functional miRNA liposomes and application in the treatment of TNBC by silencing slug gene. Int. J. Nanomed. 2019, 14, 3645–3667. [Google Scholar] [CrossRef] [Green Version]
- Burande, A.S.; Viswanadh, M.K.; Jha, A.; Mehata, A.K.; Shaik, A.; Agrawal, N.; Poddar, S.; Mahto, S.K.; Muthu, M.S. EGFR Targeted Paclitaxel and Piperine Co-loaded Liposomes for the Treatment of Triple Negative Breast Cancer. AAPS PharmSciTech 2020, 21, 151. [Google Scholar] [CrossRef]
- Thakur, V.; Kutty, R.V. Recent advances in nanotheranostics for triple negative breast cancer treatment. J. Exp. Clin. Cancer Res. 2019, 38, 430. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Mahira, S.; Majoral, J.P.; Bryszewska, M.; Khan, W.; Ionov, M. Dendrimer mediated targeting of siRNA against polo-like kinase for the treatment of triple negative breast cancer. J. Biomed. Mater. Res.—Part A 2019, 107, 1933–1944. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Gao, H.; Zhao, Z.; Rostami, I.; Wang, C.; Zhu, L.; Yang, Y. Improved tumor targeting and penetration by a dual-functional poly(amidoamine) dendrimer for the therapy of triple-negative breast cancer. J. Mater. Chem. B 2019, 7, 3724–3736. [Google Scholar] [CrossRef]
- Torres-Pérez, S.A.; del Ramos-Godínez, M.P.; Ramón-Gallegos, E. Glycosylated one-step PAMAM dendrimers loaded with methotrexate for target therapy in breast cancer cells MDA-MB-231. J. Drug Deliv. Sci. Technol. 2020, 58, 101769. [Google Scholar] [CrossRef]
- Mendes, L.P.; Pan, J.; Torchilin, V.P. Dendrimers as nanocarriers for nucleic acid and drug delivery in cancer therapy. Molecules 2017, 22, 1401. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, A.K.; Urimi, D.; Jain, S. Targeted Drug Delivery: Concepts and Design; Springer International Publishing: Cham, Switzerland, 2015; ISBN 9783319113548. [Google Scholar] [CrossRef]
- Varela-Moreira, A.; Shi, Y.; Fens, M.H.A.M.; Lammers, T.; Hennink, W.E.; Schiffelers, R.M. Clinical application of polymeric micelles for the treatment of cancer. Mater. Chem. Front. 2017, 1, 1485–1501. [Google Scholar] [CrossRef]
- Majumder, N.; Das, N.G.; Das, S.K. Polymeric micelles for anticancer drug delivery. Ther. Deliv. 2020, 11, 613–635. [Google Scholar] [CrossRef]
- Aqil, F.; Munagala, R.; Jeyabalan, J.; Agrawal, A.K.; Gupta, R. Exosomes for the Enhanced Tissue Bioavailability and Efficacy of Curcumin. AAPS J. 2017, 19, 1691–1702. [Google Scholar] [CrossRef]
- Kutty, R.V.; Tay, C.Y.; Lim, C.S.; Feng, S.S.; Leong, D.T. Anti-migratory and increased cytotoxic effects of novel dual drug-loaded complex hybrid micelles in triple negative breast cancer cells. Nano Res. 2015, 8, 2533–2547. [Google Scholar] [CrossRef] [Green Version]
- Su, S.; Ding, Y.; Li, Y.; Wu, Y.; Nie, G. Integration of photothermal therapy and synergistic chemotherapy by a porphyrin self- assembled micelle confers chemosensitivity in triple-negative breast cancer. Biomaterials 2016, 80, 169–178. [Google Scholar] [CrossRef]
- Paulmurugan, R.; Bhethanabotla, R.; Mishra, K.; Devulapally, R.; Foygel, K.; Sekar, T.V.; Ananta, J.S.; Massoud, T.F.; Joy, A. Folate Receptor Targeted Polymeric Micellar Nanocarriers for Delivery of Orlistat as a Repurposed Drug against Triple Negative Breast Cancer. Mol. Cancer Ther. 2016, 15, 221–231. [Google Scholar] [CrossRef] [Green Version]
- Brinkman, A.M.; Chen, G.; Wang, Y.; Hedman, C.J.; Sherer, N.M.; Havighurst, T.C.; Gong, S.; Xu, W. Aminoflavone-loaded EGFR-targeted unimolecular micelle nanoparticles exhibit anti-cancer effects in triple negative breast cancer. Biomaterials 2016, 101, 20–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martey, O.; Nimick, M.; Taurin, S.; Sundararajan, V.; Greish, K.; Rrosengren, R.J. Styrene maleic acid-encapsulated RL71 micelles suppress tumor growth in a murine xenograft model of triple negative breast cancer. Int. J. Nanomed. 2017, 12, 7225–7237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godugu, C.; Doddapaneni, R.; Singh, M. Honokiol nanomicellar formulation produced increased oral bioavailability and anticancer effects in triple negative breast cancer (TNBC). Colloids Surf. B Biointerfaces 2017, 153, 208–219. [Google Scholar] [CrossRef] [Green Version]
- Chida, T.; Miura, Y.; Cabral, H.; Nomoto, T.; Kataoka, K.; Nishiyama, N. Epirubicin-loaded polymeric micelles effectively treat axillary lymph nodes metastasis of breast cancer through selective accumulation and pH-triggered drug release. J. Control. Release 2018, 292, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Zuo, R.; Zhang, J.; Song, X.; Hu, S.; Gao, X.; Wang, J.; Ji, H.; Ji, C.; Peng, L.; Si, H.; et al. Encapsulating halofuginone hydrobromide in tpgs polymeric micelles enhances efficacy against triple-negative breast cancer cells. Int. J. Nanomed. 2021, 16, 1587–1600. [Google Scholar] [CrossRef]
- Prabhu, R.H.; Patravale, V.B.; Joshi, M.D. Polymeric nanoparticles for targeted treatment in oncology: Current insights. Int. J. Nanomed. 2015, 10, 1001–1018. [Google Scholar] [CrossRef] [Green Version]
- Kushwah, V.; Katiyar, S.S.; Dora, C.P.; Kumar Agrawal, A.; Lamprou, D.A.; Gupta, R.C.; Jain, S. Co-delivery of docetaxel and gemcitabine by anacardic acid modified self-assembled albumin nanoparticles for effective breast cancer management. Acta Biomater. 2018, 73, 424–436. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Joshi, A.; Toor, A.P.; Verma, G. Drug Delivery: Advancements and Challenges; Elsevier Inc.: Philadelphia, PA, USA, 2017. [Google Scholar]
- Patel, K.K.; Agrawal, A.K.; Anjum, M.M.; Tripathi, M.; Pandey, N.; Bhattacharya, S.; Tilak, R.; Singh, S. DNase-I functionalization of ciprofloxacin-loaded chitosan nanoparticles overcomes the biofilm-mediated resistance of Pseudomonas aeruginosa. Appl. Nanosci. 2020, 10, 563–575. [Google Scholar] [CrossRef]
- Urimi, D.; Agrawal, A.K.; Kushwah, V.; Jain, S. Polyglutamic Acid Functionalization of Chitosan Nanoparticles Enhances the Therapeutic Efficacy of Insulin Following Oral Administration. AAPS PharmSciTech 2019, 20, 131. [Google Scholar] [CrossRef]
- Patel, K.K.; Tripathi, M.; Pandey, N.; Agrawal, A.K.; Gade, S.; Anjum, M.M.; Tilak, R.; Singh, S. Alginate lyase immobilized chitosan nanoparticles of ciprofloxacin for the improved antimicrobial activity against the biofilm associated mucoid P. aeruginosa infection in cystic fibrosis. Int. J. Pharm. 2019, 563, 30–42. [Google Scholar] [CrossRef]
- Shilpi, D.; Kushwah, V.; Agrawal, A.K.; Jain, S. Improved Stability and Enhanced Oral Bioavailability of Atorvastatin Loaded Stearic Acid Modified Gelatin Nanoparticles. Pharm. Res. 2017, 34, 1505–1516. [Google Scholar] [CrossRef] [PubMed]
- Kushwah, V.; Katiyar, S.S.; Agrawal, A.K.; Gupta, R.C.; Jain, S. Co-delivery of docetaxel and gemcitabine using PEGylated self-assembled stealth nanoparticles for improved breast cancer therapy. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 1629–1641. [Google Scholar] [CrossRef] [PubMed]
- Grewal, I.K.; Singh, S.; Arora, S.; Sharma, N. Polymeric nanoparticles for breast cancer therapy: A comprehensive review. Biointerface Res. Appl. Chem. 2021, 11, 11151–11171. [Google Scholar] [CrossRef]
- Gupta, U.; Sharma, S.; Khan, I.; Gothwal, A.; Sharma, A.K.; Singh, Y.; Chourasia, M.K.; Kumar, V. Enhanced apoptotic and anticancer potential of paclitaxel loaded biodegradable nanoparticles based on chitosan. Int. J. Biol. Macromol. 2017, 98, 810–819. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Kennell, C.; Jafari, M.; Lee, J.Y.; Ruiz-Torres, S.J.; Waltz, S.E.; Lee, J.H. Sequential delivery of erlotinib and doxorubicin for enhanced triple negative Breast cancer treatment using polymeric nanoparticle. Int. J. Pharm. 2017, 530, 300–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Prasad, P.; Cai, P.; He, C.; Shan, D.; Rauth, A.M.; Wu, X.Y. Dual-Targeted hybrid nanoparticles of synergistic drugs for treating lung metastases of triple negative breast cancer in mice. Acta Pharmacol. Sin. 2017, 38, 835–847. [Google Scholar] [CrossRef] [Green Version]
- Rad, J.G.; Hoskin, D.W. Delivery of apoptosis-inducing piperine to triple-negative breast cancer cells via co-polymeric nanoparticles. Anticancer Res. 2020, 40, 689–694. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, D.; Xue, G.; Yu, S.; Yuan, C.; Huang, M.; Jiang, L. Improved therapeutic efficacy of quercetin-loaded polymeric nanoparticles on triple-negative breast cancer by inhibiting uPA. RSC Adv. 2020, 10, 34517–34526. [Google Scholar] [CrossRef]
- He, H.; Pham-Huy, L.A.; Dramou, P.; Xiao, D.; Zuo, P.; Pham-Huy, C. Carbon nanotubes: Applications in pharmacy and medicine. Biomed Res. Int. 2013, 2013, 578290. [Google Scholar] [CrossRef] [Green Version]
- Badea, M.A.; Prodana, M.; Dinischiotu, A.; Crihana, C.; Ionita, D.; Balas, M. Cisplatin loaded multiwalled carbon nanotubes induce resistance in triple negative breast cancer cells. Pharmaceutics 2018, 10, 228. [Google Scholar] [CrossRef] [Green Version]
- Eatemadi, A.; Daraee, H.; Karimkhanloo, H.; Kouhi, M.; Zarghami, N.; Akbarzadeh, A.; Abasi, M.; Hanifehpour, Y.; Joo, S.W. Carbon nanotubes: Properties, synthesis, purification, and medical applications. Nanoscale Res. Lett. 2014, 9, 393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahrenholtz, C.D.; Ding, S.; Bernish, B.W.; Wright, M.; Bierbach, U.; Singh, R.N. Self-assembling platinum-acridine loaded carbon nanotubes for triple-negative breast cancer chemotherapy. Mol. Cancer Res. 2016, 14, B05. [Google Scholar] [CrossRef]
- Singhai, N.J.; Maheshwari, R.; Ramteke, S. CD44 receptor targeted ‘smart’ multi-walled carbon nanotubes for synergistic therapy of triple-negative breast cancer. Colloids Interface Sci. Commun. 2020, 35, 100235. [Google Scholar] [CrossRef]
- Luo, X.; Wang, H.; Ji, D. Carbon nanotubes (CNT)-loaded ginsenosides Rb3 suppresses the PD-1/PD-L1 pathway in triple-negative breast cancer. Aging 2021, 13, 17177–17189. [Google Scholar] [CrossRef] [PubMed]
- Kaler, A.; Mittal, A.K.; Katariya, M.; Harde, H.; Agrawal, A.K.; Jain, S.; Banerjee, U.C. An investigation of in vivo wound healing activity of biologically synthesized silver nanoparticles. J. Nanopart. Res. 2014, 16, 2605. [Google Scholar] [CrossRef]
- Patel, K.K.; Surekha, D.B.; Tripathi, M.; Anjum, M.M.; Muthu, M.S.; Tilak, R.; Agrawal, A.K.; Singh, S. Antibiofilm Potential of Silver Sulfadiazine-Loaded Nanoparticle Formulations: A Study on the Effect of DNase-I on Microbial Biofilm and Wound Healing Activity. Mol. Pharm. 2019, 16, 3916–3925. [Google Scholar] [CrossRef]
- Sarkar, S.; Konar, S.; Prasad, P.N.; Rajput, S.; Kumar, B.N.P.; Rao, R.R.; Pathak, A.; Fisher, P.B.; Mandal, M. Micellear Gold Nanoparticles as Delivery Vehicles for Dual Tyrosine Kinase Inhibitor ZD6474 for Metastatic Breast Cancer Treatment. Langmuir 2017, 33, 7649–7659. [Google Scholar] [CrossRef]
- Laha, D.; Pal, K.; Chowdhuri, A.R.; Parida, P.K.; Sahu, S.K.; Jana, K.; Karmakar, P. Fabrication of curcumin-loaded folic acid-tagged metal organic framework for triple negative breast cancer therapy in in vitro and in vivo systems. New J. Chem. 2019, 43, 217–229. [Google Scholar] [CrossRef]
- Swanner, J.; Fahrenholtz, C.D.; Tenvooren, I.; Bernish, B.W.; Sears, J.J.; Hooker, A.; Furdui, C.M.; Alli, E.; Li, W.; Donati, G.L.; et al. Silver nanoparticles selectively treat triple-negative breast cancer cells without affecting non-malignant breast epithelial cells in vitro and in vivo. FASEB BioAdv. 2019, 1, 639–660. [Google Scholar] [CrossRef]
- Kumar, K.H.; Venkatesh, N.; Bhowmik, H.; Kuila, A. Metallic Nanoparticle: A Review. Biomed. J. Sci. Tech. Res. 2018, 4, 3765–3775. [Google Scholar] [CrossRef] [Green Version]
- Nirmala, M.J.; Nagarajan, R. Nanoemulsions in Cancer Therapeutics. J. Nanomed. Nanotechnol. 2016, 7, 7439. [Google Scholar] [CrossRef]
- Jain, S.; Garg, T.; Kushwah, V.; Thanki, K.; Agrawal, A.K.; Dora, C.P. α-Tocopherol as functional excipient for resveratrol and coenzyme Q10-loaded SNEDDS for improved bioavailability and prophylaxis of breast cancer. J. Drug Target. 2017, 25, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Rai, S.; Bhattacharya, S. A Conceptual Analysis of solid Self-emulsifying drug Delivery System and its Associate Patents for the Treatment of Cancer. Recent Pat. Nanotechnol. 2020, 14, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Jain, R.; Das, M.; Agrawal, A.K.; Thanki, K.; Kushwah, V. Combinatorial bio-conjugation of gemcitabine and curcumin enables dual drug delivery with synergistic anticancer efficacy and reduced toxicity. RSC Adv. 2014, 4, 29193–29201. [Google Scholar] [CrossRef]
- Sánchez-López, E.; Guerra, M.; Dias-Ferreira, J.; Lopez-Machado, A.; Ettcheto, M.; Cano, A.; Espina, M.; Camins, A.; Garcia, M.L.; Souto, E.B. Current applications of nanoemulsions in cancer therapeutics. Nanomaterials 2019, 9, 821. [Google Scholar] [CrossRef] [Green Version]
- Lonappan, D.; Krishnakumar, K.; Dineshkumar, B. Nanoemulsion In Pharmaceuticals. Am. J. PharmTech Res. 2018, 8, 1–14. [Google Scholar] [CrossRef]
- Zhao, T. Self-Nanoemulsifying Drug Delivery Systems (SNEDDS) for the Oral Delivery of Lipophilic Drugs. Ph.D. Thesis, University of Trento, Trento, Italy, 2015. [Google Scholar]
- Pereira, G.G.; Rawling, T.; Pozzoli, M.; Pazderka, C.; Chen, Y.; Dunstan, C.R.; Murray, M.; Sonvico, F. Nanoemulsion-enabled oral delivery of novel anticancer ω-3 fatty acid derivatives. Nanomaterials 2018, 8, 825. [Google Scholar] [CrossRef] [Green Version]
- Timur, S.S.; Yöyen-Ermiş, D.; Esendağlı, G.; Yonat, S.; Horzum, U.; Esendağlı, G.; Gürsoy, R.N. Efficacy of a novel LyP-1-containing self-microemulsifying drug delivery system (SMEDDS) for active targeting to breast cancer. Eur. J. Pharm. Biopharm. 2019, 136, 138–146. [Google Scholar] [CrossRef]
- Kim, B.; Pena, C.D.; Auguste, D.T. Targeted Lipid Nanoemulsions Encapsulating Epigenetic Drugs Exhibit Selective Cytotoxicity on CDH1-/FOXM1+ Triple Negative Breast Cancer Cells. Mol. Pharm. 2019, 16, 1813–1826. [Google Scholar] [CrossRef]
- Saraiva, S.M.; Gutiérrez-Lovera, C.; Martínez-Val, J.; Lores, S.; Bouzo, B.L.; Díez-Villares, S.; Alijas, S.; Pensado-López, A.; Vázquez-Ríos, A.J.; Sánchez, L.; et al. Edelfosine nanoemulsions inhibit tumor growth of triple negative breast cancer in zebrafish xenograft model. Sci. Rep. 2021, 11, 9873. [Google Scholar] [CrossRef]
- Mendes Miranda, S.E.; de Alcântara Lemos, J.; Fernandes, R.S.; de Silva, J.O.; Ottoni, F.M.; Townsend, D.M.; Rubello, D.; Alves, R.J.; Cassali, G.D.; Ferreira, L.A.M.; et al. Enhanced antitumor efficacy of lapachol-loaded nanoemulsion in breast cancer tumor model. Biomed. Pharmacother. 2021, 133, 110936. [Google Scholar] [CrossRef] [PubMed]
- Karunanidhi, P.; Verma, N.; Kumar, D.N.; Agrawal, A.K.; Singh, S. Triphenylphosphonium functionalized Ficus religiosa L. extract loaded nanoparticles improve the mitochondrial function in oxidative stress induced diabetes. AAPS PharmSciTech 2021, 22, 158. [Google Scholar] [CrossRef] [PubMed]
- Anjum, M.M.; Patel, K.K.; Dehari, D.; Pandey, N.; Tilak, R.; Agrawal, A.K.; Singh, S. Anacardic acid encapsulated solid lipid nanoparticles for Staphylococcus aureus biofilm therapy: Chitosan and DNase coating improves antimicrobial activity. Drug Deliv. Transl. Res. 2021, 11, 305–317. [Google Scholar] [CrossRef]
- Patel, K.K.; Gade, S.; Anjum, M.M.; Singh, S.K.; Maiti, P.; Agrawal, A.K.; Singh, S. Effect of penetration enhancers and amorphization on transdermal permeation flux of raloxifene-encapsulated solid lipid nanoparticlean ex vivo study on human skin. Appl. Nanosci. 2019, 9, 1383–1394. [Google Scholar] [CrossRef]
- Bayón-Cordero, L.; Alkorta, I.; Arana, L. Application of solid lipid nanoparticles to improve the efficiency of anticancer drugs. Nanomaterials 2019, 9, 474. [Google Scholar] [CrossRef] [Green Version]
- Harde, H.; Agrawal, A.K.; Katariya, M.; Kale, D.; Jain, S. Development of a topical adapalene-solid lipid nanoparticle loaded gel with enhanced efficacy and improved skin tolerability. RSC Adv. 2015, 5, 43917–43929. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, L.; Chen, T.; Guo, W.; Bao, X.; Wang, D.; Ren, B.; Wang, H.; Li, Y.; Wang, Y.; et al. Anticancer effects of resveratrol-loaded solid lipid nanoparticles on human breast cancer cells. Molecules 2017, 22, 1814. [Google Scholar] [CrossRef]
- Siddhartha, V.T.; Pindiprolu, S.K.S.S.; Chintamaneni, P.K.; Tummala, S.; Nandha Kumar, S. RAGE receptor targeted bioconjuguate lipid nanoparticles of diallyl disulfide for improved apoptotic activity in triple negative breast cancer: In vitro studies. Artif. Cells Nanomed. Biotechnol. 2018, 46, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Pindiprolu, S.K.S.S.; Chintamaneni, P.K.; Krishnamurthy, P.T.; Ratna Sree Ganapathineedi, K. Formulation-optimization of solid lipid nanocarrier system of STAT3 inhibitor to improve its activity in triple negative breast cancer cells. Drug Dev. Ind. Pharm. 2019, 45, 304–313. [Google Scholar] [CrossRef]
- Eskiler, G.G.; Cecener, G.; Dikmen, G.; Egeli, U.; Tunca, B. Talazoparib Loaded Solid Lipid Nanoparticles: Preparation, Characterization and Evaluation of the Therapeutic Efficacy In vitro. Curr. Drug Deliv. 2019, 16, 511–529. [Google Scholar] [CrossRef]
- Da Rocha, M.C.O.; Da Silva, P.B.; Radicchi, M.A.; Andrade, B.Y.G.; De Oliveira, J.V.; Venus, T.; Merker, C.; Estrela-Lopis, I.; Longo, J.P.F.; Báo, S.N. Docetaxel-loaded solid lipid nanoparticles prevent tumor growth and lung metastasis of 4T1 murine mammary carcinoma cells. J. Nanobiotechnol. 2020, 18, 43. [Google Scholar] [CrossRef] [PubMed]
- Ghasemiyeh, P.; Mohammadi-Samani, S. Solid lipid nanoparticles and nanostructured lipid carriers as novel drug delivery systems: Applications, advantages and disadvantages. Res. Pharm. Sci. 2018, 13, 288–303. [Google Scholar] [CrossRef] [PubMed]
- Gade, S.; Patel, K.K.; Gupta, C.; Anjum, M.M.; Deepika, D.; Agrawal, A.K.; Singh, S. An Ex Vivo Evaluation of Moxifloxacin Nanostructured Lipid Carrier Enriched In Situ Gel for Transcorneal Permeation on Goat Cornea. J. Pharm. Sci. 2019, 108, 2905–2916. [Google Scholar] [CrossRef] [PubMed]
- Nordin, N.; Yeap, S.K.; Rahman, H.S.; Zamberi, N.R.; Mohamad, N.E.; Abu, N.; Masarudin, M.J.; Abdullah, R.; Alitheen, N.B. Antitumor and Anti-Metastatic Effects of Citral-Loaded Nanostructured Lipid Carrier in 4T1-Induced Breast Cancer Mouse Model. Molecules 2020, 25, 2670. [Google Scholar] [CrossRef] [PubMed]
- Godugu, C.; Doddapaneni, R.; Safe, S.H.; Singh, M. Novel diindolylmethane derivatives based NLC formulations to improve the oral bioavailability and anticancer effects in triple negative breast cancer. Eur. J. Pharm. Biopharm. 2016, 108, 168–179. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Neupane, Y.R.; Panda, B.P.; Kohli, K. Lipid Based nanoformulation of lycopene improves oral delivery: Formulation optimization, ex vivo assessment and its efficacy against breast cancer. J. Microencapsul. 2017, 34, 416–429. [Google Scholar] [CrossRef]
- Ong, Y.S.; Yazan, L.S.; Ng, W.K.; Abdullah, R.; Mustapha, N.M.; Sapuan, S.; Foo, J.B.; Tor, Y.S.; How, C.W.; Rahman, N.A.; et al. Thymoquinone loaded in nanostructured lipid carrier showed enhanced anticancer activity in 4T1 tumor-bearing mice. Nanomedicine 2018, 13, 1567–1582. [Google Scholar] [CrossRef] [Green Version]
- Kebebe, D.; Wu, Y.; Zhang, B.; Yang, J.; Liu, Y.; Li, X.; Ma, Z.; Lu, P.; Liu, Z.; Li, J. Dimeric c(RGD) peptide conjugated nanostructured lipid carriers for efficient delivery of Gambogic acid to breast cancer. Int. J. Nanomed. 2019, 14, 6179–6195. [Google Scholar] [CrossRef] [Green Version]
- van der Meel, R.; Sulheim, E.; Shi, Y.; Kiessling, F.; Mulder, W.J.M.; Lammers, T. Smart cancer nanomedicine. Nat. Nanotechnol. 2019, 14, 1007–1017. [Google Scholar] [CrossRef]
- Cicha, I.; Chauvierre, C.; Texier, I.; Cabella, C.; Metselaar, J.M.; Szebeni, J.; Dézsi, L.; Alexiou, C.; Rouzet, F.; Storm, G.; et al. From design to the clinic: Practical guidelines for translating cardiovascular nanomedicine. Cardiovasc. Res. 2018, 114, 1714–1727. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Castranova, V. Toxicology of nanomaterials used in nanomedicine. J. Toxicol. Environ. Health—Part B Crit. Rev. 2011, 14, 593–632. [Google Scholar] [CrossRef] [PubMed]
- Pandey, G.; Jain, P. Assessing the nanotechnology on the grounds of costs, benefits, and risks. Beni-Suef Univ. J. Basic Appl. Sci. 2020, 9, 63. [Google Scholar] [CrossRef]
- Sharma, A.; Madhunapantula, S.V.; Robertson, G.P. Toxicological considerations when creating nanoparticle based drugs and drug delivery systems? Expert Opin. Drug Metab. Toxicol. 2012, 8, 47–69. [Google Scholar] [CrossRef] [PubMed]
- Mocan, T.; Matea, C.T.; Iancu, C.; Agoston-Coldea, L.; Mocan, L.; Orasan, R. Hypersensitivity and nanoparticles: Update and research trends. Clujul Med. 2016, 89, 216–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yingchoncharoen, P.; Kalinowski, D.S.; Richardson, D.R. Lipid-based drug delivery systems in cancer therapy: What is available and what is yet to come. Pharmacol. Rev. 2016, 68, 701–787. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
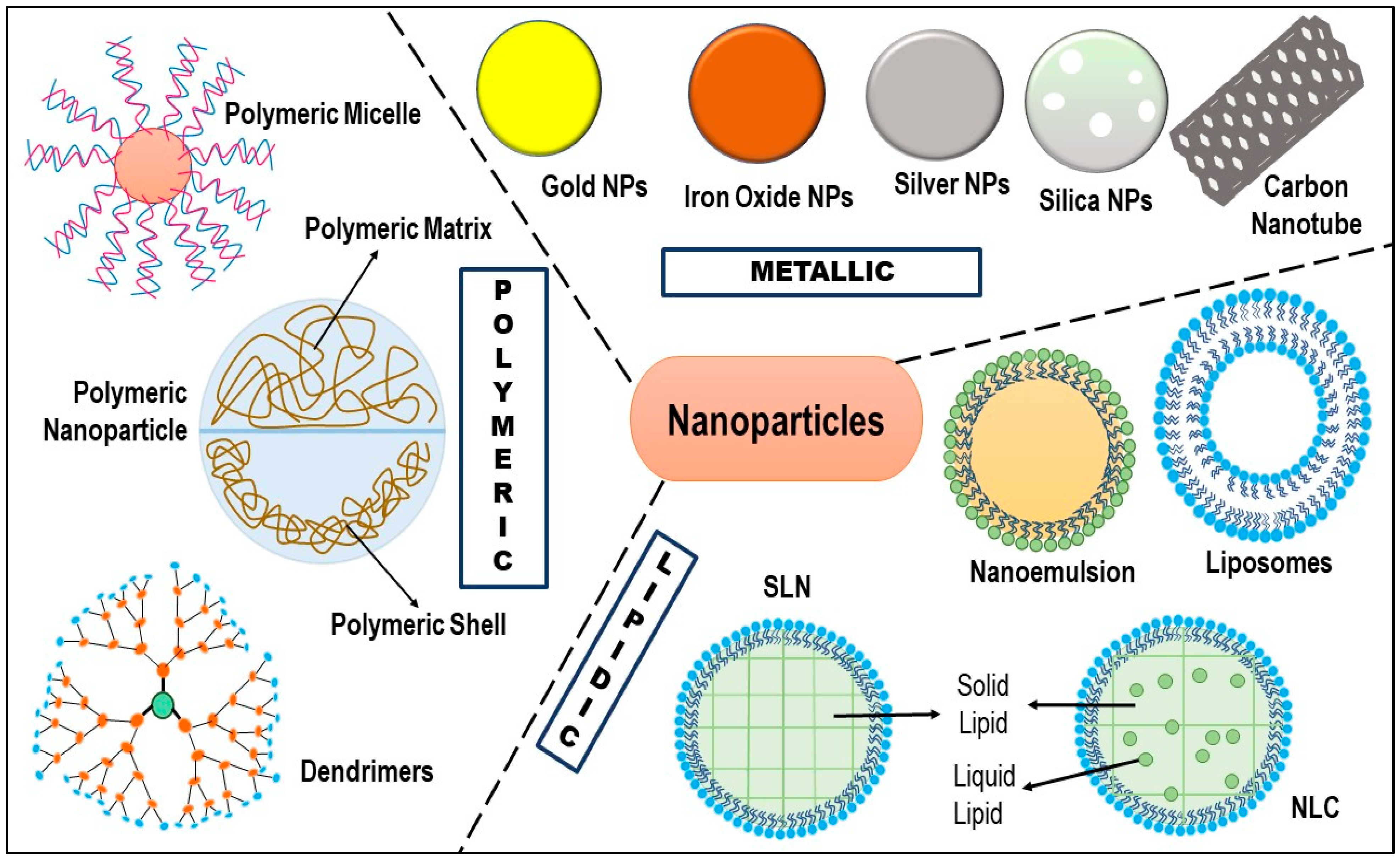{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TNBC Subtypes | Therapeutic Approaches | Therapeutic Classes | Examples |
|---|---|---|---|
| Basal-like 1 (BL-1) | Inhibits cell division, and interfere with DNA responses | Taxanes | Paclitaxel, Docetaxel |
| Platinum agents | Cisplatin, Carboplatin, Oxaliplatin | ||
| Anthracyclines | Doxorubicin, Daunorubicin, Etoposide | ||
| PARP inhibitors | Olaparib, Rucaparib, Talazoparib, Niraparib | ||
| Basal-like 2 (BL-2) | Inhibits signaling of EGFR, and MET | Platinum agents | Cisplatin, Carboplatin, Eptaplatin, and Oxaliplatin |
| PARP inhibitors | Olaparib, Rucaparib, Talazoparib and Niraparib, | ||
| Growth factor inhibitors | Erlotinib, Gefitinib, Afatinib, Cetuximab, Panitumumab, Bevacizumab, and Pertuzumab | ||
| mTOR inhibitors | Rapamycin, Everolimus, and RapaLink-1 | ||
| Immunomodulatory (IM) | Interferes or inhibits immune responses or signaling | Platinum agents | Cisplatin, Carboplatin, Eptaplatin Nedaplatin, and Oxaliplatin |
| PARP inhibitors | Olaparib, Rucaparib, Talazoparib and Niraparib | ||
| Immune checkpoint inhibitors | Ipilimumab, Nivolumab, Pembrolizumab, Cemiplimab, Atezolizumab, Avelumab and Durvalumab | ||
| Mesenchymal (M) | Inhibit signaling pathways including Notch, Wnt, IGFR1, PI3K/AKT/mTOR, TGFβ, EGFR, Src, and EMT | Growth factor inhibitors | Erlotinib, Gefitinib, Afatinib, Avitinib, lapatinib, Cetuximab, Panitumumab, Vandetanib, Bevacizumab, and Pertuzumab |
| mTOR inhibitors | Rapamycin, Everolimus, and RapaLink-1 | ||
| Src inhibitors | Dasatinib, Bosutinib, | ||
| PI3K inhibitors | Idelalisib, Alpelisib | ||
| AKT inhibitor | Ipatasertib, and Capivasertib | ||
| Mesenchymal stem-like (MSL) | Inhibit Wnt, PI3K/mTOR, EGFR, MAPK, TGFβ, Src, and EMT | Growth Factor inhibitors | Erlotinib, Gefitinib, Afatinib, Osimertinib, lapatinib, Cetuximab, Panitumumab, Vandetanib, Bevacizumab, and Pertuzumab |
| mTOR inhibitors | Rapamycin, Everolimus | ||
| PI3K inhibitors | Idelalisib, Alpelisib | ||
| MAPK inhibitors | Trametinib, Dabrafenib | ||
| Scr inhibitors | Bosutinib, Dasatinib | ||
| Luminal Androgen Receptor (LAR) | Inhibit AR signaling, PI3K/AKT/mTOR, and MAPK signaling, and FOXA1 signaling | Nonsteroidal antiandrogens | Enzalutamide, bicalutamide, orteronel |
| mTOR inhibitors | Rapamycin, Everolimus | ||
| PI3K inhibitors | Idelalisib, Taselisib |
| Clinical Trial Identifier | Treatment Regimen | Start Year/End Year | Stage of TNBC | Phase | Trial Status/Interim Results | References |
|---|---|---|---|---|---|---|
| NCT03101280 | Rucaparib (PARP inhibitor) + Atezolizumab (PD-1 inhibitor) | 2017/2020 | Advanced TNBC | I | Completed | [189] |
| NCT03544125 | Olaparib (PARP inhibitor) + Durvalumab (PD-1 inhibitor) | 2018/2020 | Metastatic TNBC | I | Completed | [190] |
| NCT02657889 | Niraparib (PARP inhibitor) + Pembrolizumab (PD-1 inhibitor) | 2016/2021 | Advanced and metastatic TNBC | I/II | Active; ORR 29% | [191] |
| NCT03167619 | Olaparib (PARP inhibitor) + Durvalumab (PD-1 inhibitor) | 2017/2021 | Advanced, platinum treated TNBC | II | Active | [192] |
| NCT03150576 | Olaparib (PARP inhibitor) + platinum-based Chemotherapy (Carboplatin) | 2017/2032 | TNBC | II/III | Recruiting | [193] |
| NCT02789332 | Olaparib (PARP inhibitor) + Paclitaxel (Chemotherapy) | 2016/2020 | Early TNBC | II | Completed –The pCR of the combination was found to be 55.1%, as compared to 48.6% with paclitaxel | [194] |
| NCT02032277 | Veliparib (PARP inhibitor) + standard neoadjuvant therapy (Carboplatin + paclitaxel + Cyclophospha- mide/doxorubicin) | 2014/2020 | Early TNBC | II-III | Completed –The pCR of the combina-tion was found to be 53% | [195] |
| NCT04039230 | Sacituzumab govitecan (antibody-drug conjugate) + talazoparib (PARP inhibitor) | 2019/2024 | Metastatic TNBC | I-II | Recruiting | [196] |
| NCT03720431 | TTAC0001 (mAb targeting VEGFR 2) + Pembrolizumab (PD-1 inhibitor) | 2018/2022 | Metastatic TNBC | I | Active, not recruiting | [197] |
| NCT03243331 | Gedatolisib (dual PI3K/mTOR inhibitor) + PTK7-ADC (antibody-drug conjugate) | 2017/2020 | Metastatic TNBC | I | Completed | [198] |
| NCT03394287 | SHR1210 (Anti-PD1-inhibitor) + Apatinib (tyrosine kinase inhibitor) | 2018/2020 | Advanced TNBC | II | Completed | [199] |
| NCT02723877 | PQR309 (dual PI3K/mTOR inhibitor) + Eribulin | 2016/2018 | TNBC | I/II | Completed | [200] |
| NCT02457910 | Enzalutamide (Non-steroidal antiandrogen) + Taselisib (PI3K inhibitor) | 2015/2020 | AR+ metastatic TNBC | I/II | Active, not recruiting | [201] |
| NCT02423603 | Paclitaxel + AZD5363 (AKT inhibitor) | 2015/2020 | Advanced/metastatic TNBC | II | Active, not recruiting | [202] |
| NCT02583542 | AZD2014 (mTORC1/2 inhibitor) + selumetinib (kinase inhibitor) | 2015/2020 | Advanced TNBC | I/II | Active, not recruiting | [203] |
| NCT00733408 | Nab-paclitaxel (Chemotherapy) + Erlotinib (EGFR TKI) + Bevacizumab (VGEF mAb) | 2008/2018 | Metastatic TNBC | II | Completed | [204] |
| NCT01097642 | Ixabepilone (chemotherapy) + Cetuximab (EGFR mAb) | 2010/2019 | TNBC | II | Completed | [205] |
| NCT02605486 | Bicalutamide (AR inhibitor) + Palbocilib (CDK4/6 Inhibitor) | 2015/2022 | AR+ metastatic TNBC | I/II | Active, not recruiting | [206] |
| NCT02513472 | Eribulin Mesylate (microtubule inhibitor) + Pembrolizumab (PD-1 inhibitor) | 2015/2020 | Metastatic TNBC | I/II | Active, not recruiting | [207] |
| NCT02530489 | Nab-Paclitaxel (Microtubule inhibitor) + Atezolizumab (PD-L1 inhibitor) | 2015/2023 | TNBC | II | Active, not recruiting | [208] |
| NCT02752685 | Pembrolizumab (PD-L1 inhibitor) + Nab-paclitaxel (Microtubule inhibitor) | 2016/2021 | Metastatic TNBC | II | Recruiting | [209] |
| NCT02672475 | Galunisertib (TGF-b inhibitor) + Paclitaxel (Microtubule inhibitor) | 2016/2023 | AR- metastatic TNBC | I | Active, not recruiting | [210] |
| NCT02456857 | Liposomal Doxorubicin (intercalating agent) + Bevacizumab (VGEF mAb) + Everolimus (mTOR inhibitor) | 2015/2022 | Locally advanced TNBC | II | Active, not recruiting | [211] |
| Nanoparticles | System | Observation | References |
|---|---|---|---|
| Liposomes | Dasatinib and Vincristine loaded liposomes | Dasatinib and vincristine-loaded liposomes exhibited targeted annihilation of VM channels by inhibiting VM indicators, resulting in the prevention of TNBC relapse. Further, the liposomes showed a delayed-release profile that enabled maximum drug delivery at the tumor site, facilitating maximum apoptosis with minimum leakage in circulation. | [237] |
| F3 peptide targeted liposomes encapsulating β3 integrin siRNA | The β3 integrin siRNA-loaded liposomes silenced the expression of overexpressed β3 integrin in TNBC cells. Moreover, targeted liposomes showed no sign of metastasis and relapse even after 4 weeks of post-treatment, as compared to untreated cells. | [238] | |
| Cationic PEGylated liposomes loaded with gambogic acid (GA) | The liposomes showed >50% reduction of tumor volume and a 1.7-fold decrease in tumor weight when compared with GA alone. | [239] | |
| Irinotecan loaded liposomes | The irinotecan-loaded liposomes exhibited prolonged plasma drug exposure with 17.7 ± 3.8 h MRT, as compared to free irinotecan (3.67 ± 1.2 h). Further, irinotecan-loaded liposomes showed increased accumulation in the metastatic lesion, as compared to free irinotecan. | [227] | |
| DSPE-PEG2000-tLyp-1 peptide-functionalized liposomes encapsulated with miRNA | The spherical nanosized liposomes (120 nm) were effectively captured by the TNBC cells and were targeted to mitochondria where the miRNA silenced the expression of the slug gene and resulted in the inhibition of the TGF-β1/Smad pathway and invasiveness. | [240] | |
| Slug gene Paclitaxel and piperine co-loaded liposomes | The percent encapsulation of paclitaxel and piperine in the liposomes was found to be 31% and 73%, respectively. The targeted liposome showed increased cellular uptake and improved cytotoxicity profile, as compared to the non-targeted counterparts. | [241] | |
| Dendrimers | Phosphorus and polyamidoamine dendrimer loaded with PLK1 siRNA | The dendrimers showed enhanced internalization in tumor cells due to their cationic nature, which favors their interaction with the tumor cell, as compared to the solution. | [243] |
| Poly(amidoamine) dendrimer encapsulated with doxorubicin | The dendrimers showed increased internalization in the tumor cells, along with effective tumor growth inhibition and prolonged survival. | [244] | |
| PAMAM dendrimers loaded with methotrexate and D-glucose | The spherical nano-ranged dendrimers (∼30 nm) were taken up by the tumor cells through the EPR effect and showed 2-fold increased tumor cell internalization due to the presence of positive charge on their surface (13 to 19 mV), as well as glucose moiety. In addition, the dendrimers showed 20% less cell viability as compared to free methotrexate. | [245] | |
| Polymeric micelles | Suberoylanilide hydroxamic acid (SAHA) and paclitaxel co-loaded hybrid micelle | The polymeric micelles showed a rapid release profile, indicating a rapid onset of action. Moreover, a synergistic effect was observed from hybrid micelle (IC50 = 0.52 μg/mL), as compared to non-micellar combination (IC50 = 3.071 μg/mL). | [251] |
| Doxorubicin and docetaxel co-loaded poly (D, L-lactide-co-glycolide) based polymeric micelle | The drug-loaded micelles showed increased internalization and accumulation in the tumor site and offered an effective drug release profile in acidic organelles along with the synergistic anti-tumor activity. | [252] | |
| 2-hydroxy-ethylacrylate (HEA) and 2-ethylhexylacrylate (EHA) copolymerized orlistat loaded micelle. | The polymeric micelle exhibited increased apoptosis, and decreased tumor volume, as compared to free orlistat. | [253] | |
| Aminoflavone (AF)-loaded EGFR targeted polymeric micelle | The small-sized polymeric unimolecular micelle showed effective cellular uptake by endocytosis in the presence of endosomal pH as compared to blood pH, concomitant with increased stability. The polymeric micelles also exhibited increased targetability and apoptosis as compared to free AF. | [254] | |
| Curcumin derivative RL71 loaded styrene-maleic acid (SMA)-based micelles | SMA-RL71-micelle showed an improved biodistribution and 16-fold increased drug accumulation in the tumor site as compared to free RL71. In addition, the SMA-RL71-micelle exhibited increased apoptosis with no cytotoxicity. | [255] | |
| Honokiol-loaded nanomicellar system | Honokiol-loaded nanomicelles exhibited increased absorption that resulted in increased oral bioavailability (Cmax = 4.06 fold; AUC= 6.26), as compared to 40 mg/kg free drug. In addition, the nanomicelles also showed a significant reduction in tumor volume and weight as compared to free drugs. | [256] | |
| Epirubicin (EPI)-loaded polymeric micelles functionalized with pH triggered moiety | The polymeric micelles underwent selective accumulation and penetration in primary tumors and vascularized axillary lymph node metastasis. The pH-triggered moiety further facilitated drug release in the acidic tumor microenvironment, sparing the healthy tissues. | [257] | |
| Halofuginone hydrobromide (HF) loaded TGPS polymeric micelles | Polymeric micelles showed a sustained drug release profile with excellent stability and biocompatibility in vivo. They also exhibited enhanced tumor growth inhibition (68.17%) in comparison to free drug. | [258] | |
| Polymeric Nanoparticles | Paclitaxel loaded chitosan nanoparticles (PTX-CS-NP) | The hemolytic toxicity profile of PTX-CS-NP was found to be 4-fold less than the free PTX. PTX-CS-NP showed a sustained drug release profile where approximately 60% of the drug was released within 24 h. Furthermore, the IC50 of PTX-CS-NP and free PTX were found to be 9.36 ± 1.13 μM, and 14.755 ± 1.68, respectively. | [268] |
| PLA-b-PEG nanoparticle loaded with erlotinib (Ei), and DOPA- doxorubicin (DOPA-Dox) | The drug release profile showed that approximately 80% of Ei was released within 4 h while only 20% of Dox was released up to 24 h of administration. Also, NPs initiated accumulation inside the tumor within 1 h of administration, which continued up to 24 h due to the EPR. | [269] | |
| RGD-conjugated polymer-lipid hybrid nanoparticles co-loaded with doxorubicin (DOX) and mitomycin C (MMC) | NPs exhibited a 31-fold decrement in the burden of lung metastases, concomitant with a 57% longer median survival time. | [270] | |
| mPEG-PLGA co-polymers based piperine-loaded nanoparticle | The polymeric nanoparticles undergo passive diffusion into the tumor site without affecting the kidney, liver, and spleen. The piperine-loaded polymeric NPs inhibited the growth of TNBC and induced apoptosis while sparing normal fibroblast. | [271] | |
| PLGA-TPGS NPs loaded with quercetin | Polymeric NPs exhibited a sustained drug release profile, as compared to free quercetin. In addition, NPs inhibited the growth of TNBC and induced apoptosis while sparing normal fibroblast, as supported by an increased tumor inhibition ratio of 67.88%, along with fewer lung metastasis colonies. Furthermore, NPs provided an inhibitory effect upon the migration of uPA (Urokinase-type plasminogen activator) knockdown on TNBC cells. | [272] | |
| Metallic Nanoparticles | Gold nanoparticles (AuNM) functionalized with a tyrosine kinase inhibitor, ZD6474 | AuNM exhibited a slow and sustained release of ZD6474 (82% following 45 h) at pH 5.5. AuMN showed targeting due to their low cytotoxicity and immunogenicity. Moreover, AuNM inhibited tumor growth and prevented metastasis without causing any haemotoxicity. | [281] |
| Curcumin loaded metal-organic framework (NMOF-3) tagged by folic acid (IRMOF-3@CCM@FA) | IRMOF-3@CCM@FA showed 55% drug release in pH 5.5, as compared to physiological pH (31%). Further, IRMOF-3@CCM@FA showed enhance apoptosis and targeted delivery of curcumin, as compared to free curcumin. | [282] | |
| Silver nanoparticles (AgNPs) | AgNPs exhibited accumulation to both TNBC and non-malignant breast cells, but facilitate rapid degradation only in TNBC cells. Moreover, the internalization of AgNPs within the TNBC showed depletion of cellular antioxidants, causing endoplasmic reticulum stress and apoptosis. | [283] | |
| Nanoemulsions | Omega 3-fatty acid derivative loaded nanoemulsion (NE) | The NE showed 99.9 ± 2.3% entrapment efficiency, enhanced tumor cell accumulation and a 50% reduced tumor weight as compared to the free derivative of omega 3 fatty acids. | [292] |
| Doxorubicin hydrochloride and LyP-1 co-loaded self-micro-emulsifying drug delivery system (SMEDDS) | SMEDDS exhibited lymphatic uptake, thereby increasing bioavailability. Moreover, the SMEDDS showed enhanced in vivo cytotoxicity in p32 expressing TNBC cells, along with reduced tumor growth and metastasis. | [293] | |
| Decitabine (DAC) and panobinostat (PAN) co-loaded lipid nanoemulsion (LNEs) | LNEs showed increased stability, enhanced internalization in tumor cells, without affective liver, and spleen, and a controlled-release delivery system. Furthermore, LNEs showed increased inhibition of tumor growth of M subtype TNBC. | [294] | |
| Edelfosine nanoemulsions (ET-NEs) | ET-NE exhibited an enhanced passive targeting into the tumor site via the EPR effect, with fewer chances of opsonization, and prolonged circulation time in the body. ET-NE also showed enhanced tumor growth inhibition, as compared to free ET. | [295] | |
| Lapachol—loaded nanoemulsion (LAP-NE) | LAP-NE showed increased internalization in tumor cells via EPR, with enhanced stability. NEs exhibited a sustained release profile, as compared to free drugs. Moreover, in comparison to free lapachol (IC50 = 6.60 ± 3.1 μM, relative tumor volume = 5.51), LAP-NE showed increased cytotoxicity (IC50 = 7.29 ± 1.79 μM) and reduced relative tumor volume (3.22). | [296] | |
| SLNs | Resveratrol-loaded SLNs (Res-SLN) | SLN showed enhanced physical stability, increased tumor site internalization, and targeted lymphatic uptake. Also, Res-SLNs exhibited a superior inhibitory activity over the growth, invasion, and migration of MDA-MB-231 cells | [302] |
| Di-allyl-disulfide loaded SLN, surface-functionalized with RAGE antibody (DADS-RAGE-SLN) | DADS-RAGE-SLN experienced flexible encapsulation, which resulted in sustained drug release. SLN showed increased accumulation in the acidic tumor microenvironment, and in comparison, to free DAD (15%), DAD-RAGE-SLN (61.8%) showed enhanced cytotoxic effect over TNBC cells (MDA-MB231). | [303] | |
| Niclosamide loaded SLN (Niclo-SLN) | SLN showed enhanced entrapment efficiency, with an enhanced internalization within tumor cells. The SLN showed initial burst release followed by sustained drug release. Further, Niclo-SLN (70%) showed an increased apoptotic rate as compared to free Niclo (50%). | [304] | |
| Talazoparib loaded SLNs | SLNs showed improved entrapment efficiency (85%). The SLN exhibited a sustained drug release profile (51%), as compared to free talazoparib (89%). Moreover, talazoparib-SLN (85.56%) showed enhanced apoptosis to BRCA1 deficient TNBC cells (HCC1937-R cells), as compared to free talazoparib (25.86%). | [305] | |
| Docetaxel loaded SLN (SLN-DTX) | SLN showed enhanced accumulation in tumor cells. The SLN-DTX showed an initial burst effect release, followed by controlled release. SLN showed enhanced cytotoxicity (IC50 = 0.08 µg/mL) as compared to free DTX (IC50 = 10 µg/mL). Further, SLN showed reduced lung metastasis as compared to free DTX. | [306] | |
| NLCs | Diindolylmethane (DIM) derivatives loaded NLC | The NLC showed enhanced internalization in tumor cells, a significant increment in oral bioavailability (4.73-fold increase in Cmax; 11.19-fold increase in AUC), and a decrease of tumor volume and tumor weight in an MDA-MB-231 cell line, as compared to free DIM derivative. | [310] |
| Lycopene loaded NLC | The NLC showed enhanced tumor site accumulation and increased encapsulation. Moreover, the NLC exhibited an initial burst release followed by sustained release with cumulative % drug release of 82.33 ± 3.67%. The lycopene-loaded NLC also exhibited enhanced cytotoxicity, as compared to free lycopene. | [311] | |
| Thymoquinone (TQ) loaded NLC | NLC showed improved entrapment efficiency and drug loading, along with enhanced site-specific internalization via EPR. In addition, the TQ-NLC showed enhanced apoptosis and anti-metastatic effect as compared to free TQ. | [312] | |
| Gambogic acid (GA) loaded NLC, functionalized with dimeric c (RGD) | GA-cRGD-NLC showed enhanced tumor site accumulation, enhanced encapsulation along with improved stability. In addition, GA-cRGD-NLC (0.25 µg/mL) showed increased cytotoxicity, as compared to untargeted GA-NLC (0.5 µg/mL) | [313] | |
| Citral loaded NLC | Citral-NLC showed increased internalization in the tumor cells and improved entrapment efficiency. Moreover, citral-NLC showed superiority in apoptosis, migration, invasion, and wound healing assay, as compared to free citral. | [309] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaudhuri, A.; Kumar, D.N.; Dehari, D.; Singh, S.; Kumar, P.; Bolla, P.K.; Kumar, D.; Agrawal, A.K. Emergence of Nanotechnology as a Powerful Cavalry against Triple-Negative Breast Cancer (TNBC). Pharmaceuticals 2022, 15, 542. https://doi.org/10.3390/ph15050542
Chaudhuri A, Kumar DN, Dehari D, Singh S, Kumar P, Bolla PK, Kumar D, Agrawal AK. Emergence of Nanotechnology as a Powerful Cavalry against Triple-Negative Breast Cancer (TNBC). Pharmaceuticals. 2022; 15(5):542. https://doi.org/10.3390/ph15050542
Chicago/Turabian StyleChaudhuri, Aiswarya, Dulla Naveen Kumar, Deepa Dehari, Sanjay Singh, Pradeep Kumar, Pradeep Kumar Bolla, Dinesh Kumar, and Ashish Kumar Agrawal. 2022. "Emergence of Nanotechnology as a Powerful Cavalry against Triple-Negative Breast Cancer (TNBC)" Pharmaceuticals 15, no. 5: 542. https://doi.org/10.3390/ph15050542