
Design, Synthesis, and In Vitro, In Silico and In Cellulo Evaluation of New Pyrimidine and Pyridine Amide and Carbamate Derivatives as Multi-Functional Cholinesterase Inhibitors

 ,
,  ,
,  , ,
, ,  ,
,  , and
, and
Abstract
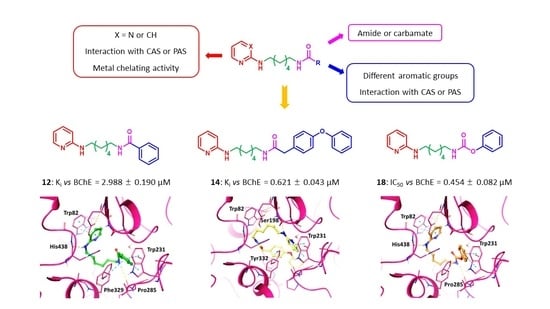
:
1. Introduction
2. Results and Discussion
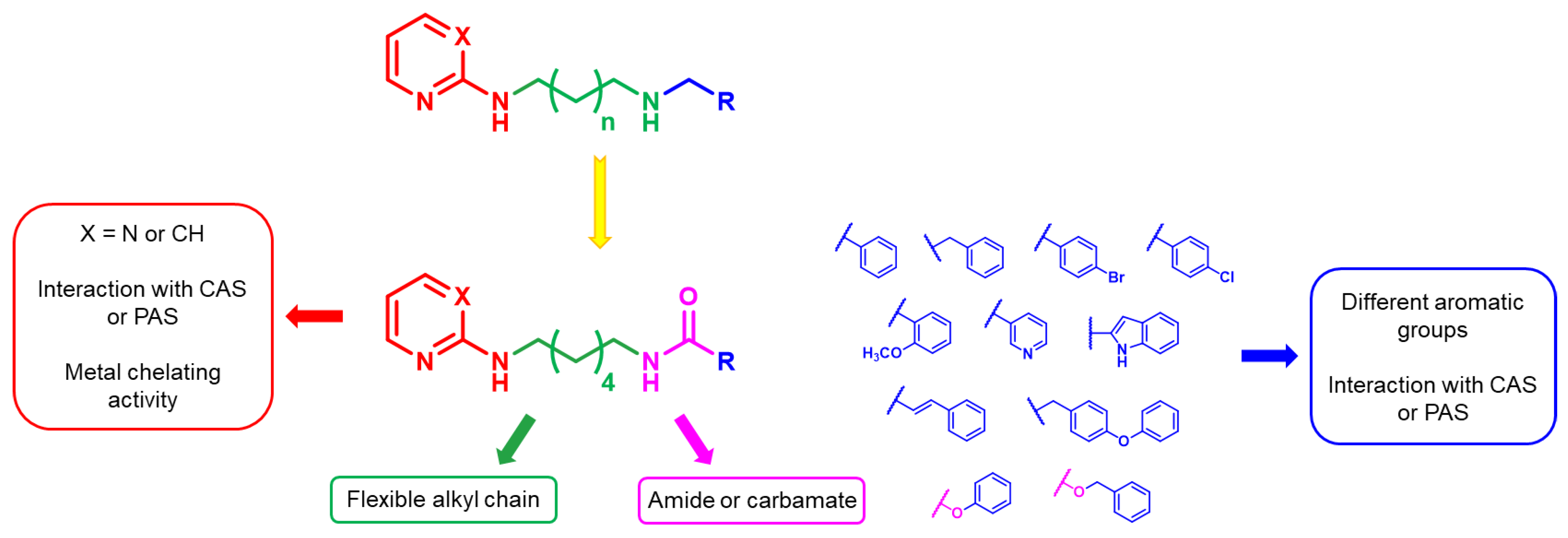
2.1. Chemistry
2.2. Enzymatic Assays
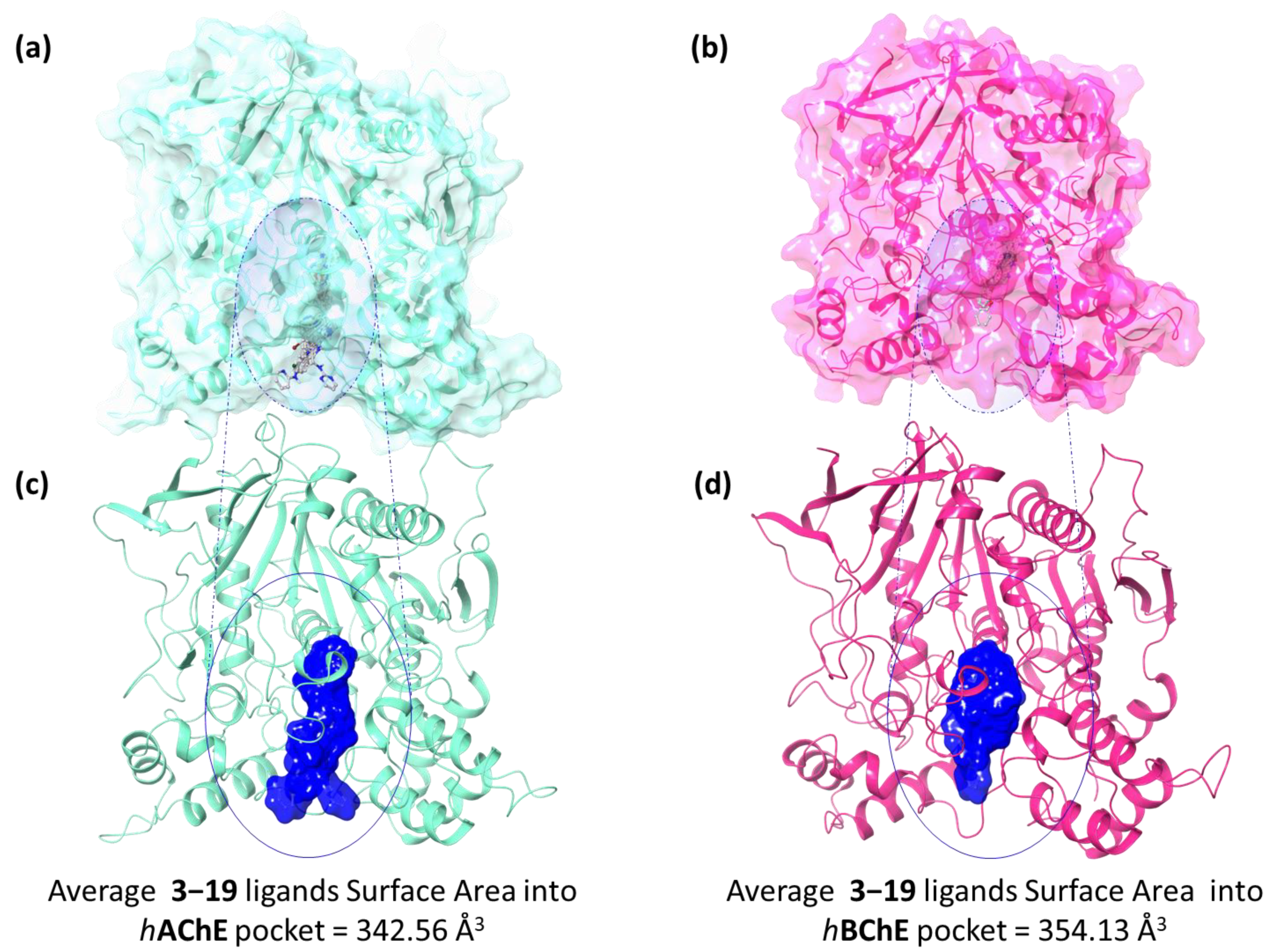
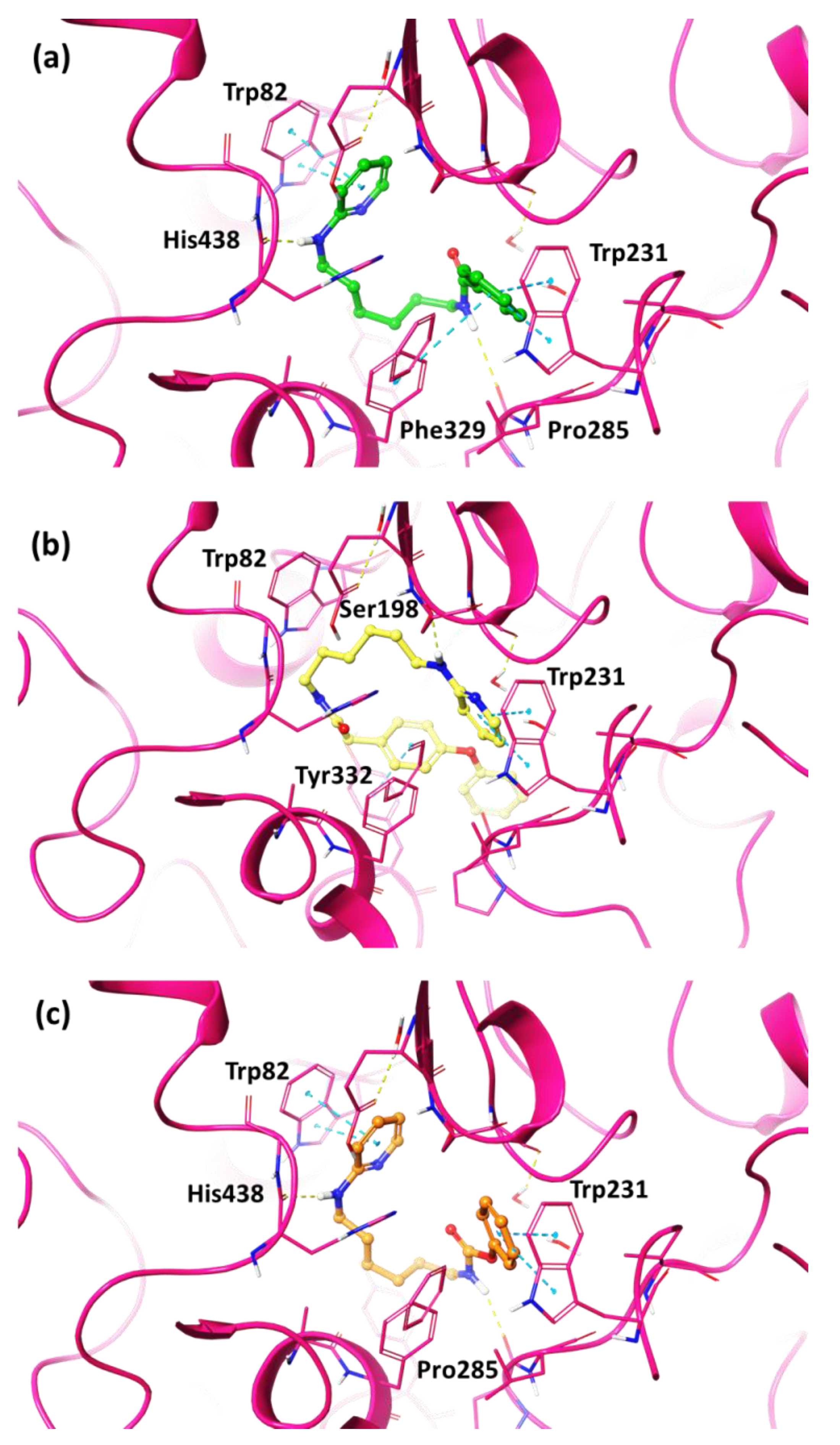
2.3. In Silico Studies
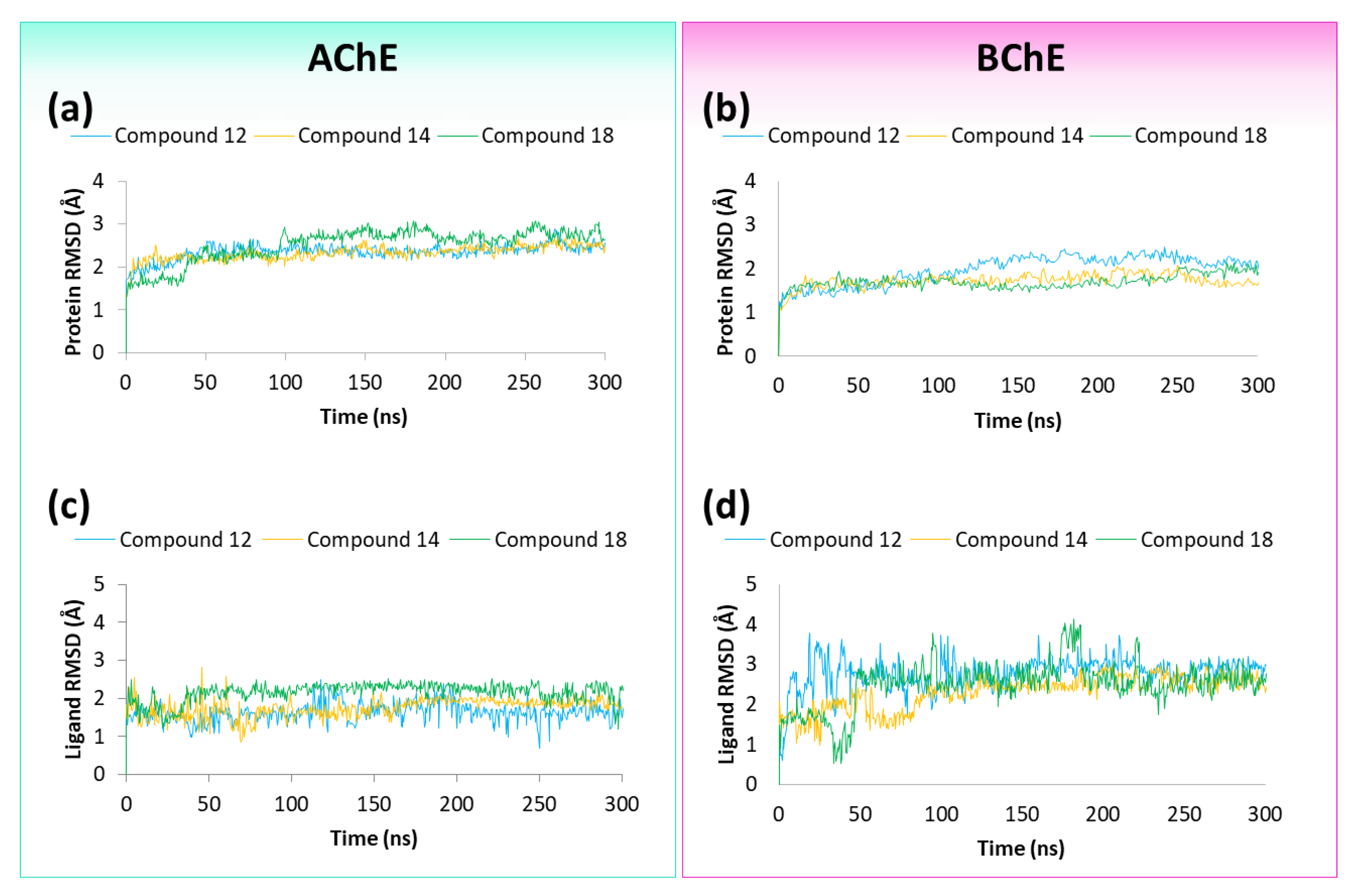
2.4. Molecular Dynamics Studies
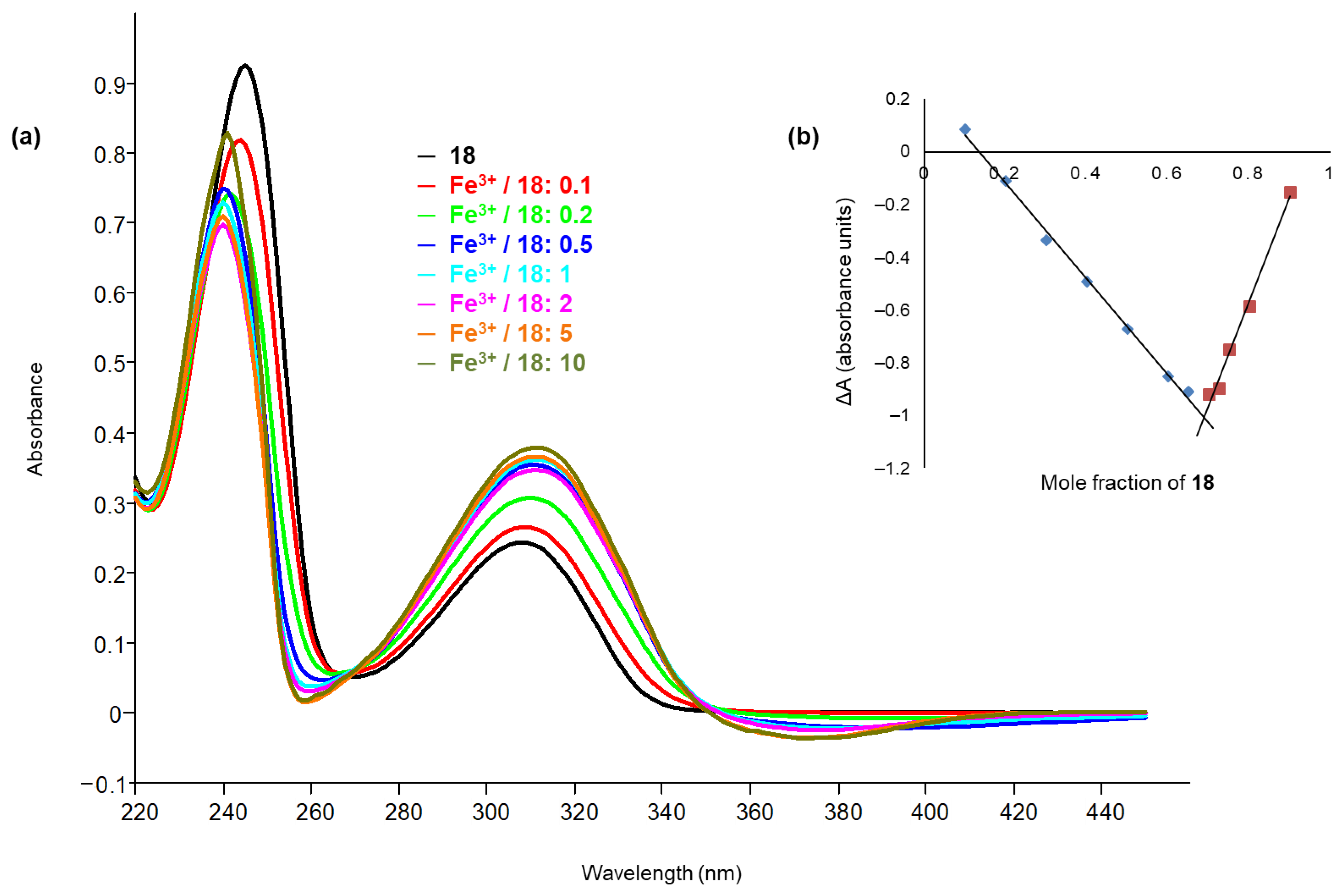
2.5. Chelation Studies
2.6. Inhibition of Amyloid and Tau Aggregation
2.7. Computation of Physicochemical Descriptors and ADME Parameters
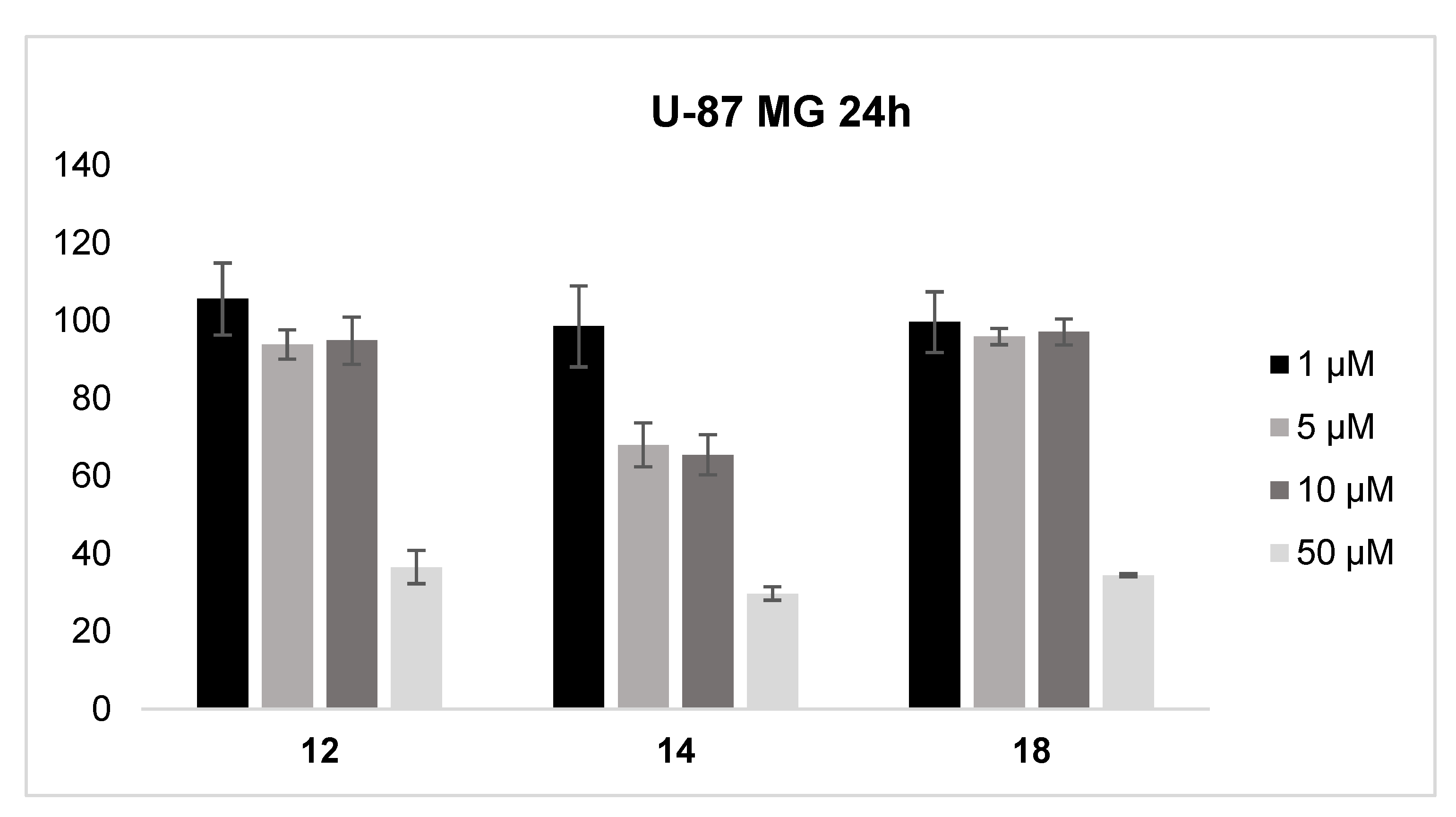
2.8. Cell Viability Studies
3. Materials and Methods
3.1. Chemistry
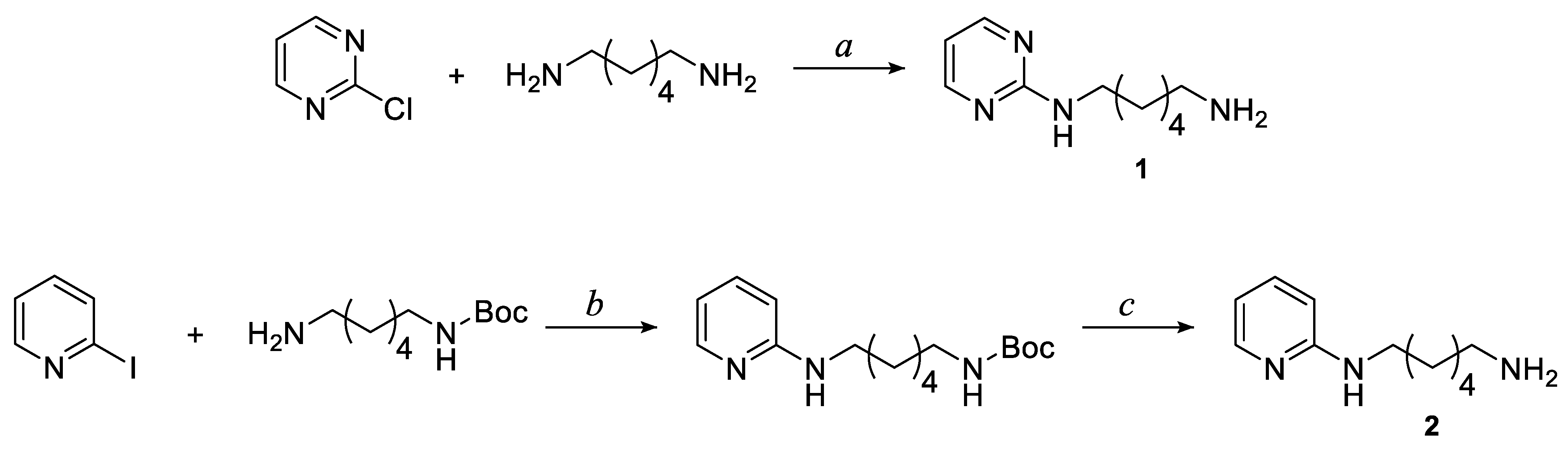
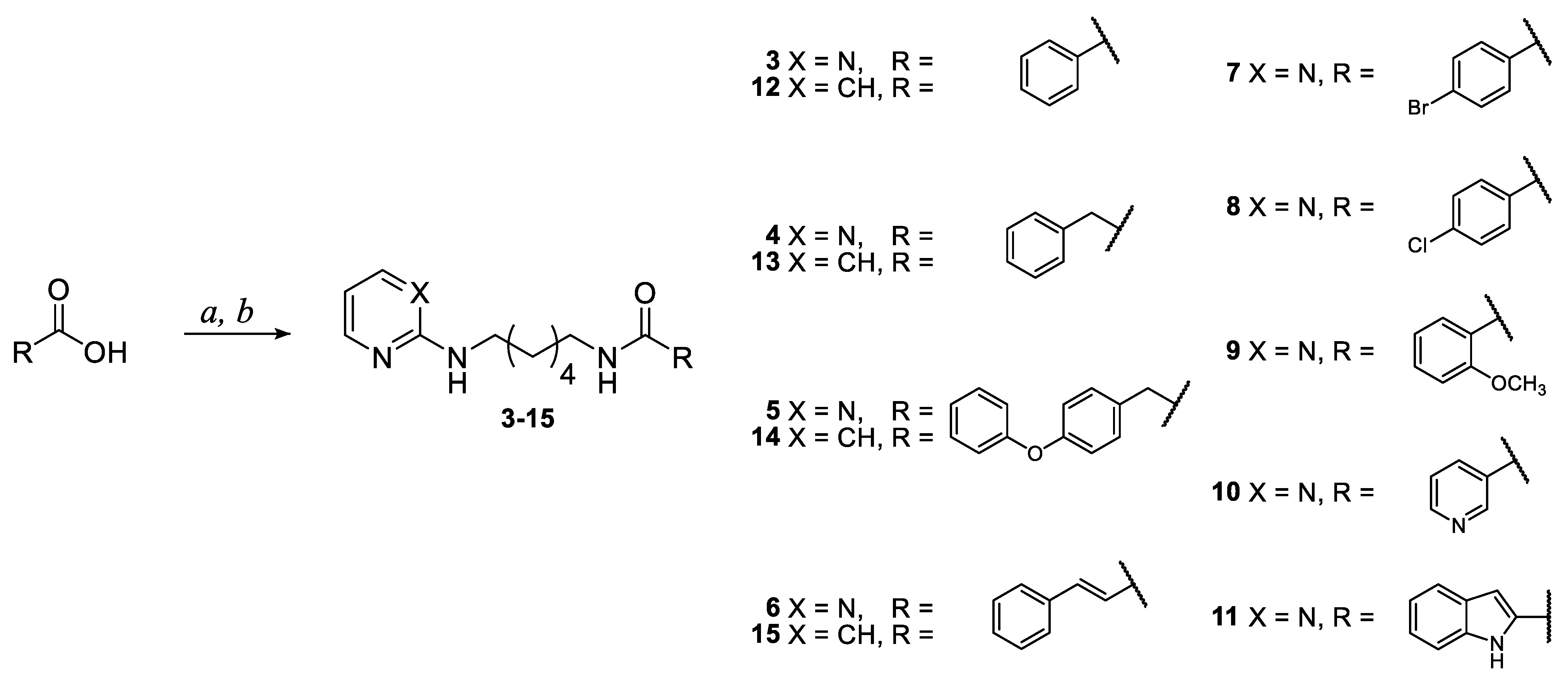
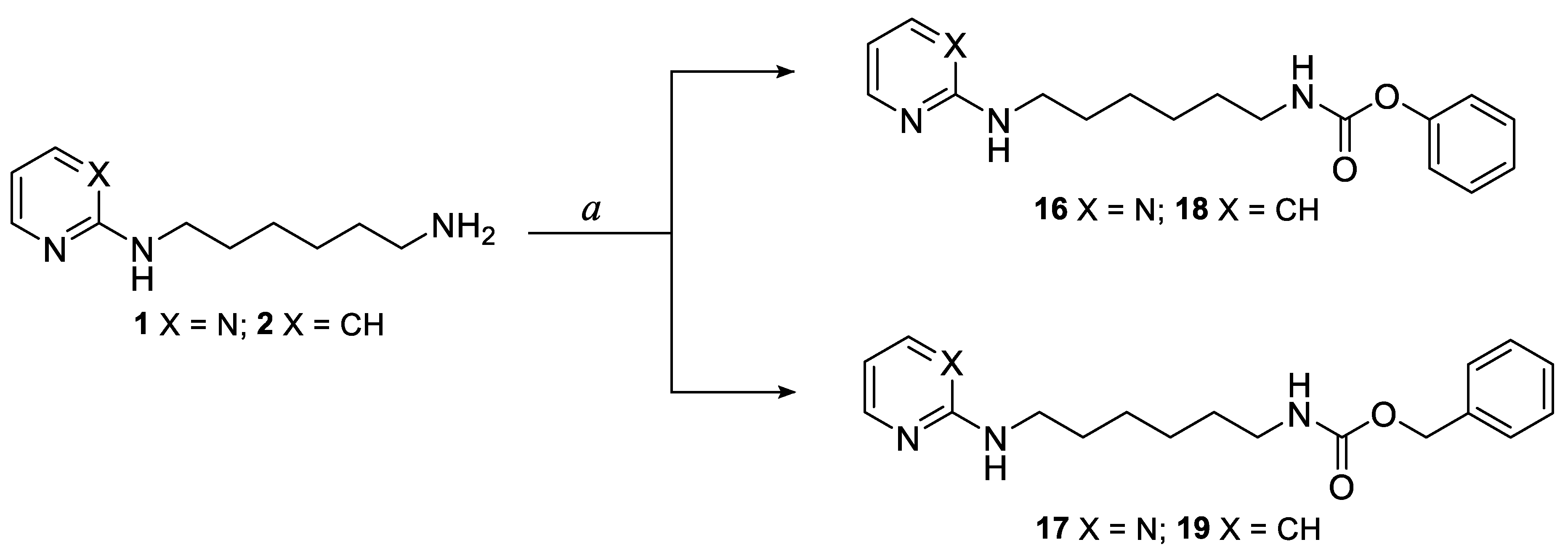
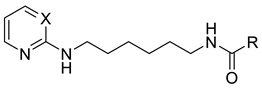
3.1.1. General Procedure for the Synthesis of Pyrimidine and Pyridine Amide Derivatives 3–15
3.1.2. General Procedure for the Synthesis of Pyrimidine and Pyridine Carbamate Derivatives 16–19
3.2. Enzymatic Assays
3.2.1. Percent Inhibition of EeAChE and eqBChE
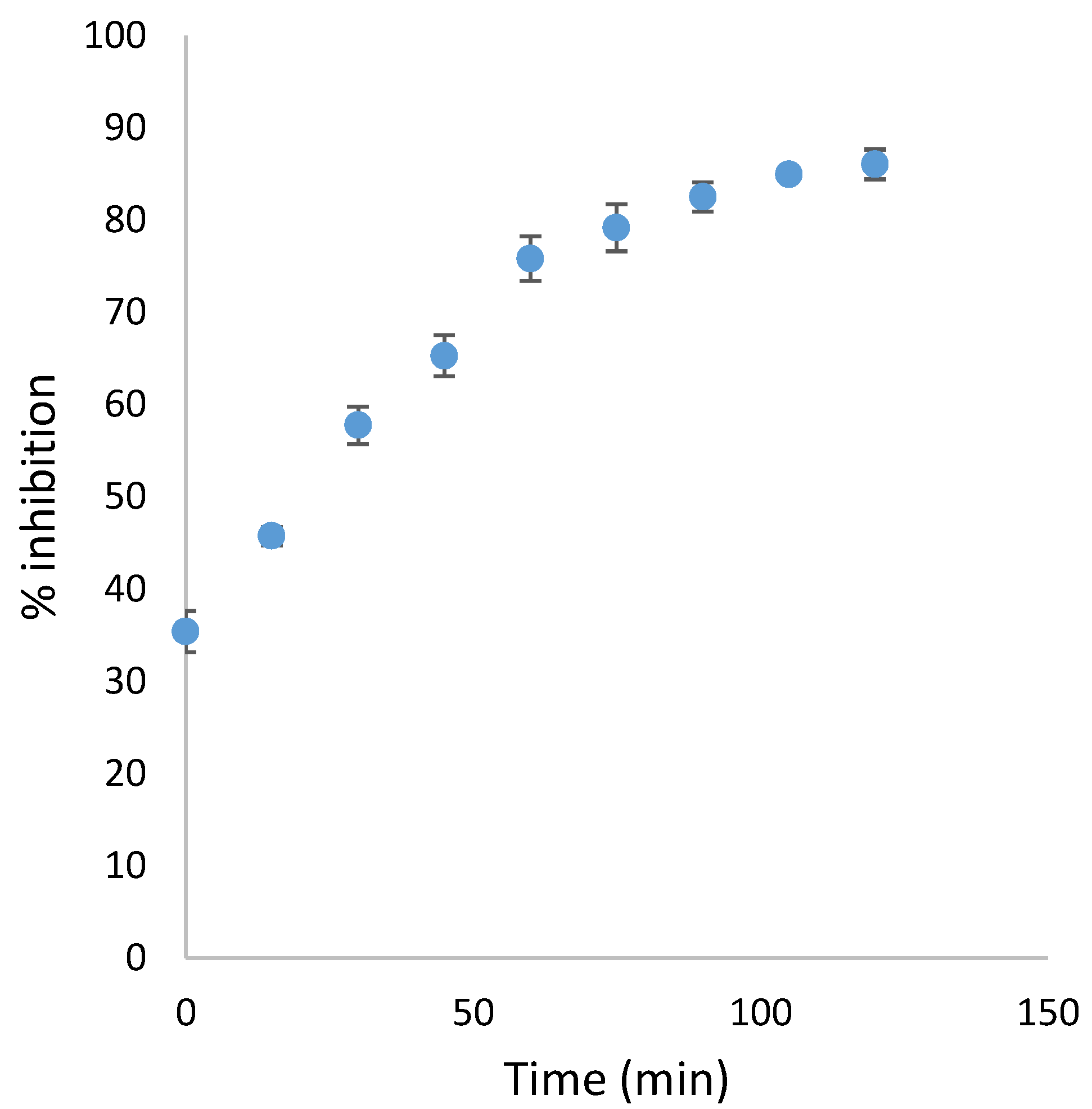
3.2.2. Time Dependent Inhibition Assay for Carbamate Derivatives 16–19
3.2.3. Determination of Constant and Mechanism of Inhibition vs. eqBChE
3.2.4. Determination of IC50 vs. eqBChE for Compound 18
3.2.5. Study of Reversibility of Inhibition of Compound 18 vs. eqBChE
- three were prepared with 2.5 mL of eqBChE solution (0.50 UmL−1) and 5 µL of inhibitor solution;
- three were prepared with 2.5 mL of eqBChE solution (0.50 UmL−1) and 5 µL of DMSO.
3.3. Molecular Docking Studies and ADME Prediction
3.4. Chelation Studies
3.4.1. UV-Vis Titration
3.4.2. Job’s Plot
3.5. Inhibition of Amyloid and Tau Aggregation
3.5.1. Cloning and Overexpression of the Aβ42 Peptide
3.5.2. Cloning and Overexpression of the Tau Protein
3.5.3. Thioflavin S (Th-S) Steady-State Fluorescence
3.6. Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- 2022 Alzheimer’s disease facts and figures. J. Alzheimer’s Assoc. 2022, 18, 700–789. [CrossRef] [PubMed]
- World Health Organization. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 15 March 2022).
- Ju, Y.; Tam, K.Y. Pathological mechanisms and therapeutic strategies for Alzheimer’s disease. Neural Regen. Res. 2022, 17, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Savelieff, M.G.; Nam, G.; Kang, J.; Lee, H.J.; Lee, M.; Lim, M.H. Development of Multifunctional Molecules as Potential Therapeutic Candidates for Alzheimer’s Disease, Parkinson’s Disease, and Amyotrophic Lateral Sclerosis in the Last Decade. Chem. Rev. 2019, 119, 1221–1322. [Google Scholar] [CrossRef] [PubMed]
- Albertini, C.; Salerno, A.; de Sena Murteira Pinheiro, P.; Bolognesi, M.L. From combinations to multitarget-directed ligands: A continuum in Alzheimer’s disease polypharmacology. Med. Res. Rev. 2021, 41, 2606–2633. [Google Scholar] [CrossRef]
- Kumar, N.; Kumar, V.; Anand, P.; Kumar, V.; Ranjan Dwivedi, A.; Kumar, V. Advancements in the development of multi-target directed ligands for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. 2022, 61, 116742. [Google Scholar] [CrossRef]
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chételat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer disease. Nat. Rev. Dis. Primers 2021, 7, 33. [Google Scholar] [CrossRef]
- Roda, A.R.; Serra-Mir, G.; Montoliu-Gaya, L.; Tiessler, L.; Villegas, S. Amyloid-beta peptide and tau protein crosstalk in Alzheimer’s disease. Neural. Regen. Res. 2022, 17, 1666–1674. [Google Scholar] [CrossRef]
- Chen, Y.; Fu, A.K.Y.; Ip, N.Y. Synaptic dysfunction in Alzheimer’s disease: Mechanisms and therapeutic strategies. Pharmacol. Ther. 2019, 195, 186–198. [Google Scholar] [CrossRef]
- Roberts, B.R.; Ryan, T.M.; Bush, A.I.; Masters, C.L.; Duce, J.A. The role of metallobiology and amyloid-β peptides in Alzheimer’s disease. J. Neurochem. 2012, 120, 149–166. [Google Scholar] [CrossRef]
- Cheung, J.; Rudolph, M.J.; Burshteyn, F.; Cassidy, M.S.; Gary, E.N.; Love, J.; Franklin, M.C.; Height, J.J. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J. Med. Chem. 2012, 55, 10282–10286. [Google Scholar] [CrossRef]
- Brus, B.; Košak, U.; Turk, S.; Pišlar, A.; Coquelle, N.; Kos, J.; Stojan, J.; Colletier, J.-P.; Gobec, S. Discovery, biological evaluation, and crystal structure of a novel nanomolar selective butyrylcholinesterase inhibitor. J. Med. Chem. 2014, 57, 8167–8179. [Google Scholar] [CrossRef] [PubMed]
- Rosenberry, T.L.; Brazzolotto, X.; Macdonald, I.R.; Wandhammer, M.; Trovaslet-Leroy, M.; Darvesh, S.; Nachon, F. Comparison of the binding of reversible inhibitors to human butyrylcholinesterase and acetylcholinesterase: A crystallographic, kinetic and calorimetric study. Molecules 2017, 22, 2098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silman, I.; Sussman, J.L. Acetylcholinesterase: ‘classical’ and ‘nonclassical’ functions and pharmacology. Curr. Opin. Pharmacol. 2005, 5, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Pérez, D.I.; Martínez, A.; Gil, C.; Campillo, N.E. From Bitopic Inhibitors to Multitarget Drugs for the Future Treatment of Alzheimer’s Disease. Curr. Med. Chem. 2015, 22, 3789–3806. [Google Scholar] [CrossRef] [PubMed]
- Nazam, N.; Farhana, A.; Shaikh, S. Recent Advances in Alzheimer’s Disease in Relation to Cholinesterase Inhibitors and NMDA Receptor Antagonists. In Autism Spectrum Disorder and Alzheimer’s Disease; Ghulam, M.A., Athanasios, A., Eds.; Springer Nature: Singapore, 2021; pp. 135–151. [Google Scholar] [CrossRef]
- Greig, N.H.; Utsuki, T.; Yu, Q.-s.; Zhu, X.; Holloway, H.W.; Perry, T.; Lee, B.; Ingram, D.K.; Lahiri, D.K. A new therapeutic target in Alzheimer’s disease treatment: Attention to butyrylcholinesterase. Curr. Med. Res. Opin. 2001, 17, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Xing, S.; Li, Q.; Xiong, B.; Chen, Y.; Feng, F.; Liu, W.; Sun, H. Structure and therapeutic uses of butyrylcholinesterase: Application in detoxification, Alzheimer’s disease, and fat metabolism. Med. Res. Rev. 2021, 41, 858–901. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Yang, H.; Chen, Y.; Sun, H. Recent progress in the identification of selective butyrylcholinesterase inhibitors for Alzheimer’s disease. Eur. J. Med. Chem. 2017, 132, 294–309. [Google Scholar] [CrossRef]
- Zhou, S.; Huang, G. The biological activities of butyrylcholinesterase inhibitors. Biomed. Pharmacother. 2022, 146, 112556. [Google Scholar] [CrossRef]
- Das, N.; Raymick, J.; Sarkar, S. Role of metals in Alzheimer’s disease. Metab. Brain Dis. 2021, 36, 1627–1639. [Google Scholar] [CrossRef]
- Fasae, K.D.; Abolaji, A.O.; Faloye, T.R.; Odunsi, A.Y.; Oyetayo, B.O.; Enya, J.I.; Rotimi, J.A.; Akinyemi, R.O.; Whitworth, A.J.; Aschner, M. Metallobiology and therapeutic chelation of biometals (copper, zinc and iron) in Alzheimer’s disease: Limitations, and current and future perspectives. J. Trace Elem. Med. Biol. 2021, 67, 126779. [Google Scholar] [CrossRef]
- Ma, C.; Hong, F.; Yang, S. Amyloidosis in Alzheimer’s disease: Pathogeny, etiology, and related therapeutic directions. Molecules 2022, 27, 1210. [Google Scholar] [CrossRef] [PubMed]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments in Alzheimer disease: An update. J. Cent. Nerv. Syst. Dis. 2020, 12, 1179573520907397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bortolami, M.; Rocco, D.; Messore, A.; Di Santo, R.; Costi, R.; Madia, V.N.; Scipione, L.; Pandolfi, F. Acetylcholinesterase inhibitors for the treatment of Alzheimer’s disease—A patent review (2016-present). Expert Opin. Ther. Pat. 2021, 31, 399–420. [Google Scholar] [CrossRef] [PubMed]
- Przybyłowska, M.; Dzierzbicka, K.; Kowalski, S.; Chmielewska, K.; Inkielewicz-Stepniak, I. Therapeutic Potential of Multi-functional Derivatives of Cholinesterase Inhibitors. Curr. Neuropharmacol. 2021, 19, 1323–1344. [Google Scholar] [CrossRef]
- Mishra, P.; Kumar, A.; Panda, G. Anti-cholinesterase hybrids as multi-target-directed ligands against Alzheimer’s disease (1998–2018). Bioorg. Med. Chem. 2019, 27, 895–930. [Google Scholar] [CrossRef]
- Chaves, S.; Várnagy, K.; Santos, M.A. Recent Multi-target Approaches on the Development of Anti-Alzheimer’s Agents Integrating Metal Chelation Activity. Curr. Med. Chem. 2021, 28, 7247–7277. [Google Scholar] [CrossRef]
- Sharma, A.; Pachauri, V.; Flora, S. Advances in multi-functional ligands and the need for metal-related pharmacology for the management of Alzheimer disease. Front. Pharmacol. 2018, 9, 1247. [Google Scholar] [CrossRef] [Green Version]
- Pandolfi, F.; de Vita, D.; Bortolami, M.; Coluccia, A.; Di Santo, R.; Costi, R.; Andrisano, V.; Alabiso, F.; Bergamini, C.; Fato, R.; et al. New pyridine derivatives as inhibitors of acetylcholinesterase and amyloid aggregation. Eur. J. Med. Chem. 2017, 141, 197–210. [Google Scholar] [CrossRef]
- Bortolami, M.; Pandolfi, F.; de Vita, D.; Carafa, C.; Messore, A.; Di Santo, R.; Feroci, M.; Costi, R.; Chiarotto, I.; Bagetta, D.; et al. New deferiprone derivatives as multi-functional cholinesterase inhibitors: Design, synthesis and in vitro evaluation. Eur. J. Med. Chem. 2020, 198, 112350. [Google Scholar] [CrossRef]
- Bortolami, M.; Pandolfi, F.; Tudino, V.; Messore, A.; Madia, V.N.; de Vita, D.; Di Santo, R.; Costi, R.; Romeo, I.; Alcaro, S.; et al. New Pyrimidine and Pyridine Derivatives as Multitarget Cholinesterase Inhibitors: Design, Synthesis, and In Vitro and In Cellulo Evaluation. ACS Chem. Neurosci. 2021, 12, 4090–4112. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V., Jr.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Dixon, M. The determination of enzyme inhibitor constants. Biochem. J. 1953, 55, 170–171. [Google Scholar] [CrossRef] [PubMed]
- Greening, D.W.; Simpson, R.J. A centrifugal ultrafiltration strategy for isolating the low-molecular weight (≤25 K) component of human plasma proteome. J. Proteom. 2010, 73, 637–648. [Google Scholar] [CrossRef] [PubMed]
- De Vita, D.; Pandolfi, F.; Ornano, L.; Feroci, M.; Chiarotto, I.; Sileno, I.; Pepi, F.; Costi, R.; Di Santo, R.; Scipione, L. New N,N-dimethylcarbamate inhibitors of acetylcholinesterase: Design synthesis and biological evaluation. J. Enzyme Inhib. Med. Chem. 2016, 31 (Suppl. S4), 106–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavarria, D.; Da Silva, O.; Benfeito, S.; Barreiro, S.; Garrido, J.; Cagide, F.; Soares, P.; Remião, F.; Brazzolotto, X.; Nachon, F.; et al. Fine-tuning the biological profile of multitarget mitochondriotropic antioxidants for neurodegenerative diseases. Antioxidants 2021, 10, 329. [Google Scholar] [CrossRef]
- Warren, G.L.; Do, T.D.; Kelley, B.P.; Nicholls, A.; Warren, S.D. Essential considerations for using protein–ligand structures in drug discovery. Drug Discov. Today 2012, 17, 1270–1281. [Google Scholar] [CrossRef]
- Warren, G.L.; Andrews, C.W.; Capelli, A.M.; Clarke, B.; LaLonde, J.; Lambert, M.H.; Lindvall, M.; Nevins, N.; Semus, S.F.; Senger, S.; et al. A critical assessment of docking programs and scoring functions. J. Med.Chem. 2006, 49, 5912–5931. [Google Scholar] [CrossRef]
- Catapano, M.C.; Tvrdý, V.; Karlíčková, J.; Migkos, T.; Valentová, K.; Křen, V.; Mladěnka, P. The stoichiometry of isoquercitrin complex with iron or copper is highly dependent on experimental conditions. Nutrients 2017, 9, 1193. [Google Scholar] [CrossRef] [Green Version]
- Job, P. Formation and Stability of Inorganic Complexes in Solution. Ann. Chim. 1928, 9, 113–203. [Google Scholar]
- Espargaró, A.; Medina, A.; Di Pietro, O.; Muñoz-Torrero, D.; Sabate, R. Ultra rapid in vivo screening for anti-Alzheimer anti-amyloid drugs. Sci. Rep. 2016, 6, 23349. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–26. [Google Scholar] [CrossRef]
- Schrödinger Release 2018-1: Maestro; Schrödinger LLC: New York, NY, USA, 2018.
- Schrödinger Release 2018-1: Protein Preparation Wizard; Schrödinger LLC: New York, NY, USA, 2018.
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2018-1: Prime; Schrödinger LLC: New York, NY, USA, 2018.
- Hasel, W.; Hendrickson, T.F.; Still, W.C. A rapid approximation to the solvent accessible surface areas of atoms. Tetrahedron Comput. Methodol. 1988, 1, 103–116. [Google Scholar] [CrossRef]
- Schrödinger Release 2018-1: Glide; Schrödinger LLC: New York, NY, USA, 2018.
- Maruca, A.; Ambrosio, F.A.; Lupia, A.; Romeo, I.; Rocca, R.; Moraca, F.; Talarico, C.; Bagetta, D.; Catalano, R.; Costa, G.; et al. Computer-based techniques for lead identification and optimization I: Basics. In Volume 1 Fundamental Concepts; Ntie-Kang, F., Ed.; De Gruyter: Berlin, Germany; Boston, MA, USA, 2020; pp. 311–332. [Google Scholar] [CrossRef]
- Schrödinger Release 2018-1: Desmond Molecular Dynamics System; D. E. Shaw Research: New York, NY, USA, 2018.
- Lupia, A.; Moraca, F.; Bagetta, D.; Maruca, A.; Ambrosio, F.A.; Rocca, R.; Catalano, R.; Romeo, I.; Talarico, C.; Ortuso, F.; et al. Computer-based techniques for lead identification and optimization II: Advanced search methods. Phys. Sci. Rev. 2020, 5, 20180114. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | % Inhibition vs. EeAChE ± SD a | % Inhibition vs. eqBChE ± SD a | ||||
|---|---|---|---|---|---|---|
| Cmp | R | X | [I] 9 µM | [I] 900 nM | [I] 9 µM | [I] 900 nM |
| 3 | 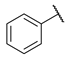 | N | 2.5 ± 4.1 | nd b | 8.5 ± 1.7 | nd b |
| 4 | 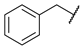 | N | 5.0 ± 4.4 | nd b | 5.1 ± 4.7 | nd b |
| 5 | 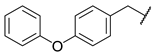 | N | 10.8 ± 2.7 | nd b | 16.6 ± 5.5 | nd b |
| 6 | 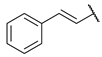 | N | ns c | 7.0 ± 1.6 | ns c | na d |
| 7 | 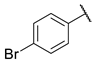 | N | 11.2 ± 5.0 | nd b | 7.4 ± 2.0 | nd b |
| 8 | 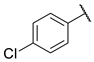 | N | 9.9 ± 0.8 | nd b | 2.1 ± 3.5 | nd b |
| 9 | 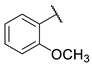 | N | 7.2 ± 0.8 | Nd b | 9.3 ± 2.4 | nd b |
| 10 | 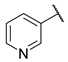 | N | na d | nd b | na d | nd b |
| 11 | 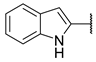 | N | ns c | 5.5 ± 1.4 | ns c | 14.1 ± 4.7 |
| 12 | 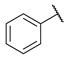 | CH | 34.0 ± 2.1 | nd b | 80.5 ± 1.1 | 22.3 ± 1.6 |
| 13 | 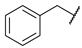 | CH | 46.7 ± 1.1 | nd b | 49.1 ± 3.2 | 4.3 ± 5.6 |
| 14 |  | CH | 42.6 ± 1.4 | nd b | 87.6 ± 1.5 | 45.2 ± 2.3 |
| 15 | 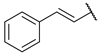 | CH | 45.4 ± 2.5 | nd b | 64.7 ± 2.9 | 31.5 ± 3.1 |
| Tacrine | 100 | 97.6 ± 0.1 | 100 | 99.7 ± 0.3 | ||
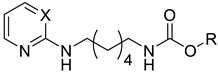 | % Inhibition vs. EeAChE ± SD a | % Inhibition vs. eqBChE ± SD a | |||||
|---|---|---|---|---|---|---|---|
| Cmp | R | X | [I] = 9 µM | [I] = 9 µM | [I] = 900 nM | ||
| t = 0 | t = 1 h | t = 0 | t = 0 | t = 1 h | |||
| 16 | 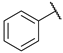 | N | na b | 7.2 ± 0.8 | 38.9 ± 2.5 | 6.4 ± 6.8 | 66.4 ± 5.9 |
| 17 | 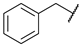 | N | na b | 10.9 ± 1.4 | 16.0 ± 3.7 | na b | 13.3 ± 3.9 |
| 18 | 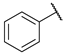 | CH | 29.4 ± 0.7 | 41.6 ± 0.7 | 85.8 ± 1.0 | 35.2 ± 0.3 | 79.6 ± 5.3 |
| 19 |  | CH | 8.4 ± 3.4 | 45.4 ± 2.7 | 80.7 ± 2.3 | 25.9 ± 2.9 | 32.4 ± 7.5 |
| eqBChE | |||
|---|---|---|---|
| Cmp | Mechanism | Ki ± SD (µM) | R2 |
| 12 | Competitive | 2.988 ± 0.190 | 0.987 |
| 14 | Competitive | 0.621 ± 0.043 | 0.980 |
| eqBChE | |
|---|---|
| Cmp | IC50 ± SE (nM) |
| 18 | 454.3 ± 82.4 |
| Tacrine | 3.7 ± 0.5 |
| Donepezil | 1727 ± 200 |
| Fe3+ | Cu2+ | |||||
|---|---|---|---|---|---|---|
| Cmp | λ (nm) | X | n | λ (nm) | X | n |
| 12 | 251 | 0.69 | 2 | - | - | - |
| 14 | 250 | 0.70 | 2 | - | - | - |
| 18 | 245 | 0.69 | 2 | 330 | 0.49 | 1 |
| Aβ42 Aggregation | Tau Aggregation | |||
|---|---|---|---|---|
| Cmp | % Inhibition [I] = 100 µM | SEM a | % Inhibition [I] = 100 µM | SEM a |
| 12 | 11.9 | 4.5 | 11.0 | 6.9 |
| 14 | 13.3 | 6.2 | 10.9 | 6.4 |
| 18 | 23.1 | 3.4 | 15.7 | 2.0 |
| Ref b | 98.8 | 1.0 | 94.7 | 3.1 |
| Cmp | MW | HBA | HBD | Heavy Atoms | RB | TPSA | MLogP | LogS ESOL | Sol Class | GI | BBB | Lipinski Viol |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 12 | 297.39 | 2 | 2 | 22 | 10 | 54.02 | 2.54 | −3.68 | Soluble | High | Yes | 0 |
| 14 | 403.52 | 3 | 2 | 30 | 12 | 63.25 | 3.29 | −5.10 | Moderately soluble | High | Yes | 0 |
| 18 | 313.39 | 3 | 2 | 23 | 11 | 63.25 | 2.53 | −3.88 | Soluble | High | Yes | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bortolami, M.; Pandolfi, F.; Tudino, V.; Messore, A.; Madia, V.N.; De Vita, D.; Di Santo, R.; Costi, R.; Romeo, I.; Alcaro, S.; et al. Design, Synthesis, and In Vitro, In Silico and In Cellulo Evaluation of New Pyrimidine and Pyridine Amide and Carbamate Derivatives as Multi-Functional Cholinesterase Inhibitors. Pharmaceuticals 2022, 15, 673. https://doi.org/10.3390/ph15060673
Bortolami M, Pandolfi F, Tudino V, Messore A, Madia VN, De Vita D, Di Santo R, Costi R, Romeo I, Alcaro S, et al. Design, Synthesis, and In Vitro, In Silico and In Cellulo Evaluation of New Pyrimidine and Pyridine Amide and Carbamate Derivatives as Multi-Functional Cholinesterase Inhibitors. Pharmaceuticals. 2022; 15(6):673. https://doi.org/10.3390/ph15060673
Chicago/Turabian StyleBortolami, Martina, Fabiana Pandolfi, Valeria Tudino, Antonella Messore, Valentina Noemi Madia, Daniela De Vita, Roberto Di Santo, Roberta Costi, Isabella Romeo, Stefano Alcaro, and et al. 2022. "Design, Synthesis, and In Vitro, In Silico and In Cellulo Evaluation of New Pyrimidine and Pyridine Amide and Carbamate Derivatives as Multi-Functional Cholinesterase Inhibitors" Pharmaceuticals 15, no. 6: 673. https://doi.org/10.3390/ph15060673