
Identification of Novel Small Molecule Ligands for JAK2 Pseudokinase Domain

 , , ,
, , , 
Abstract
:1. Introduction
2. Results
2.1. Small-Scale Pilot Screen Compared TSA and FP as Screening Methods
2.2. Summary of the Screens
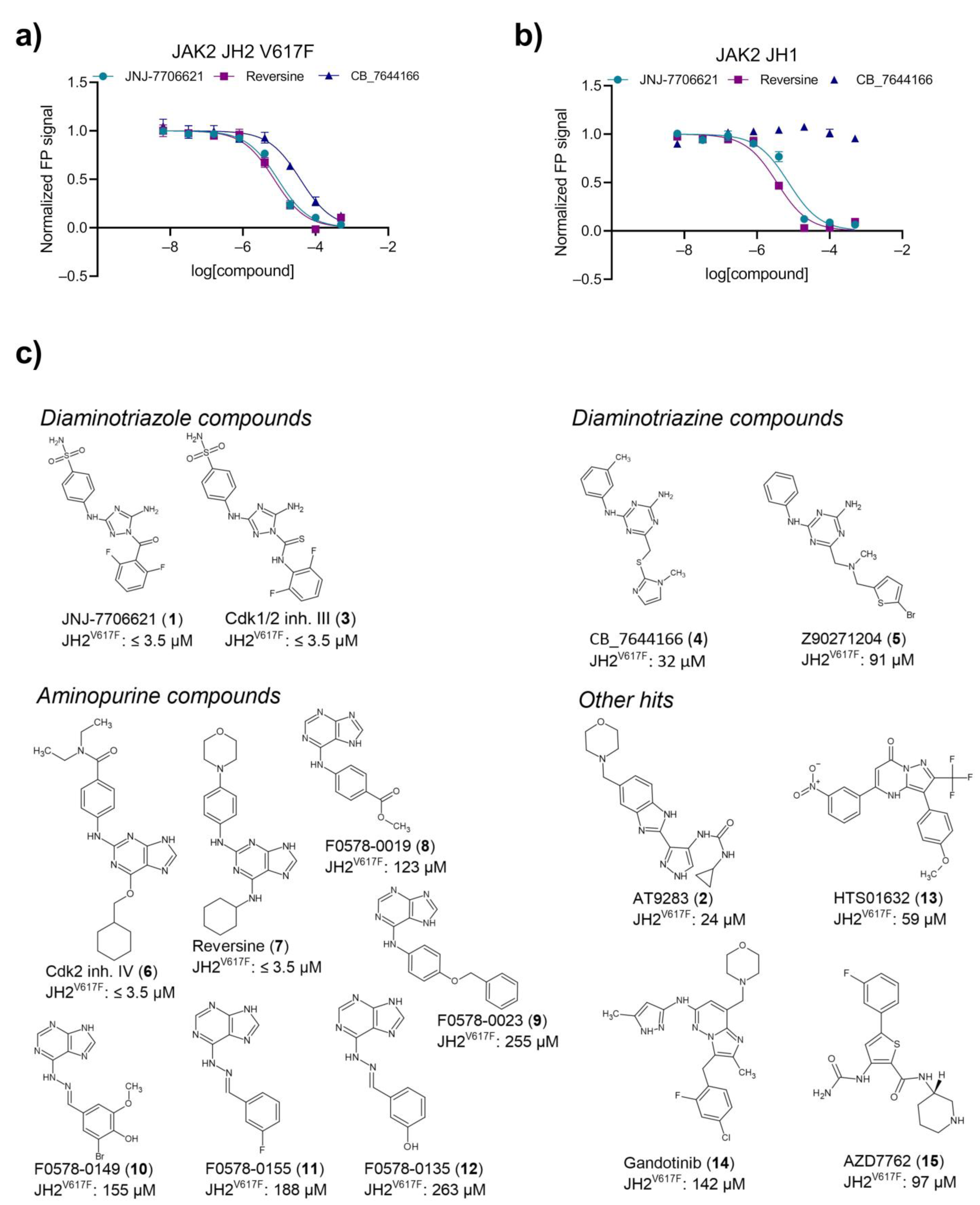
2.3. Primary Hit Follow-Up
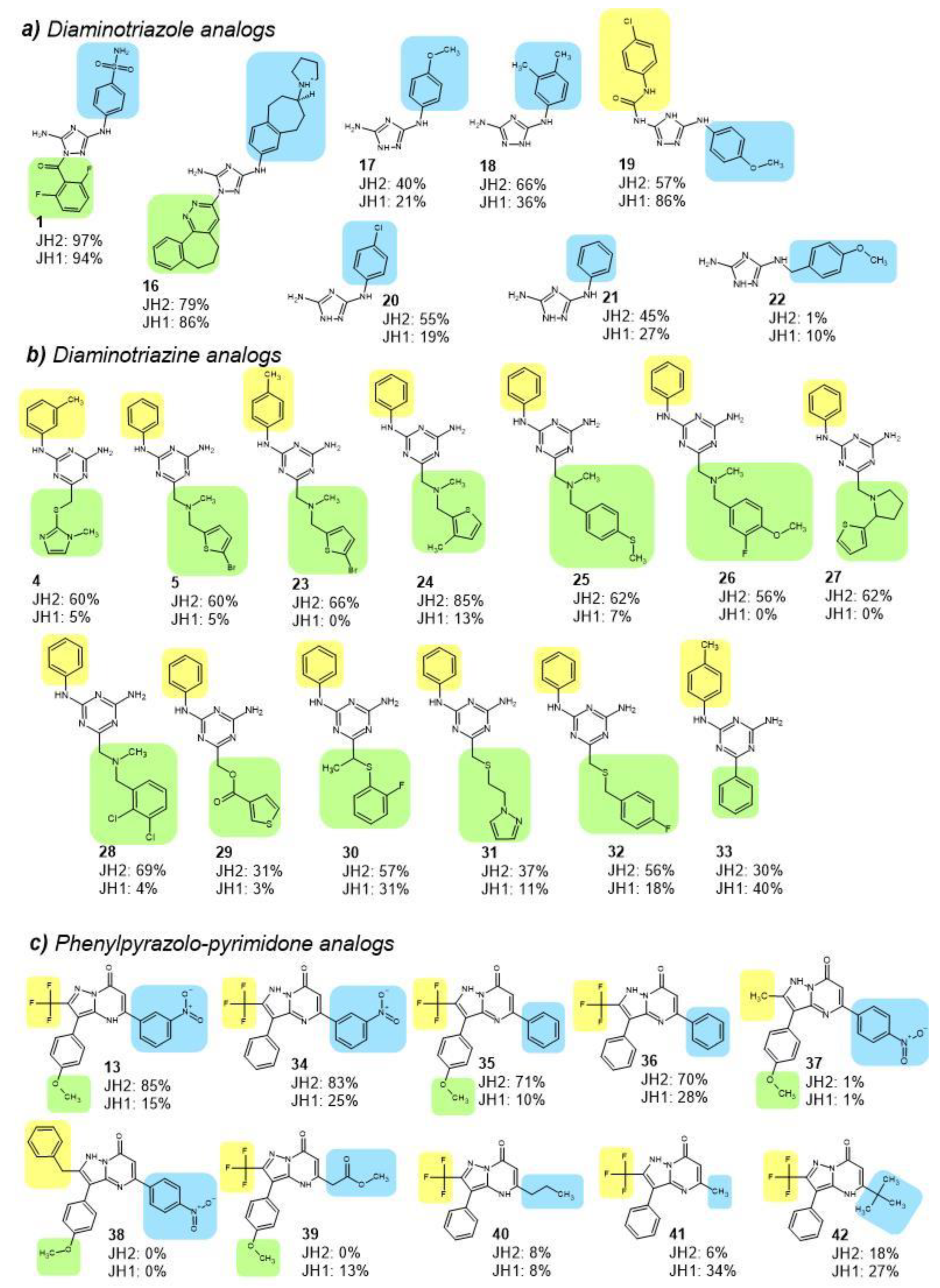
2.4. JH2 and JH1 Domain Targeting of Selected Hit Analogs
2.5. JAK Selectivity and Inhibition of MPN Cell Lines of Selected Binders
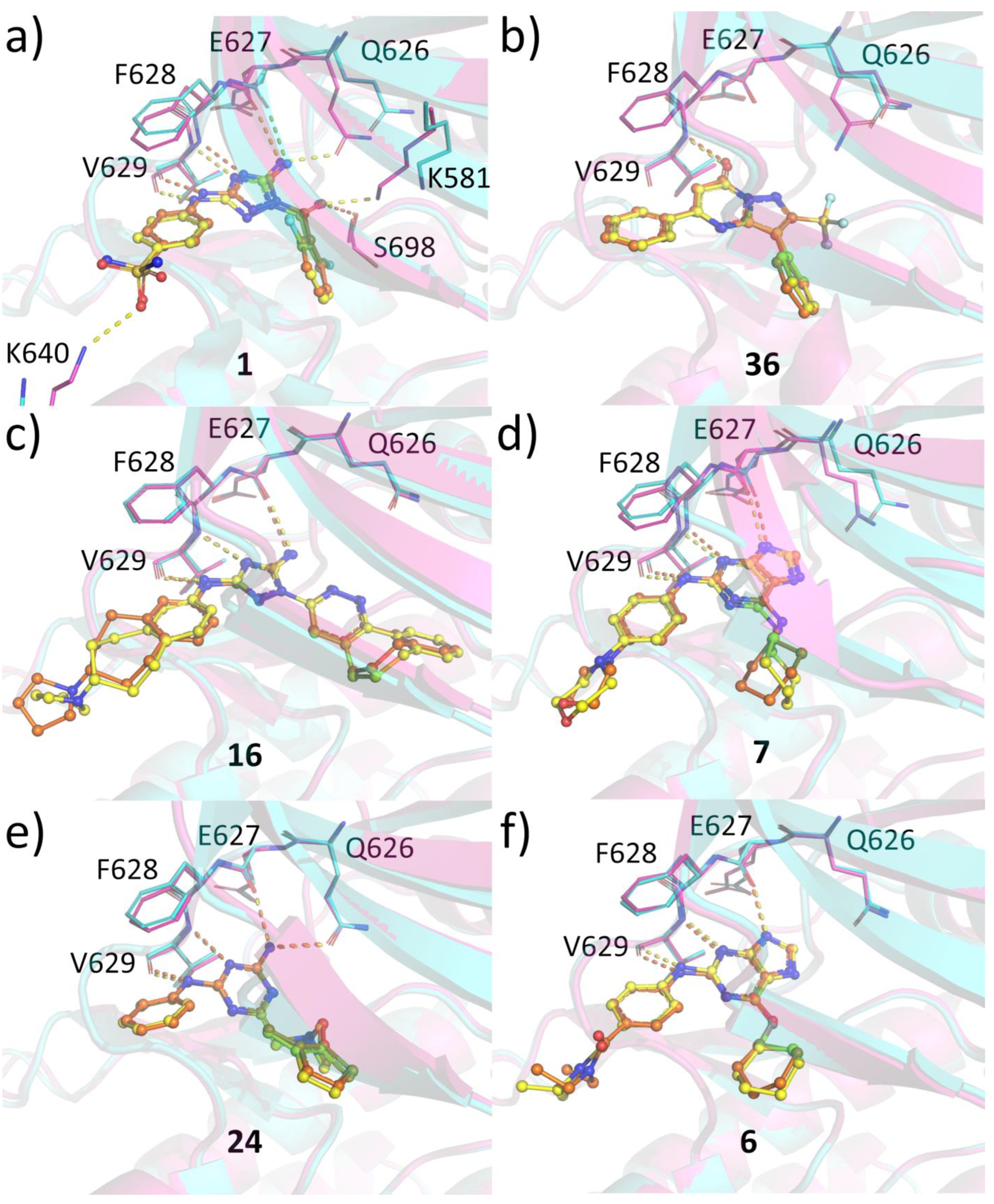
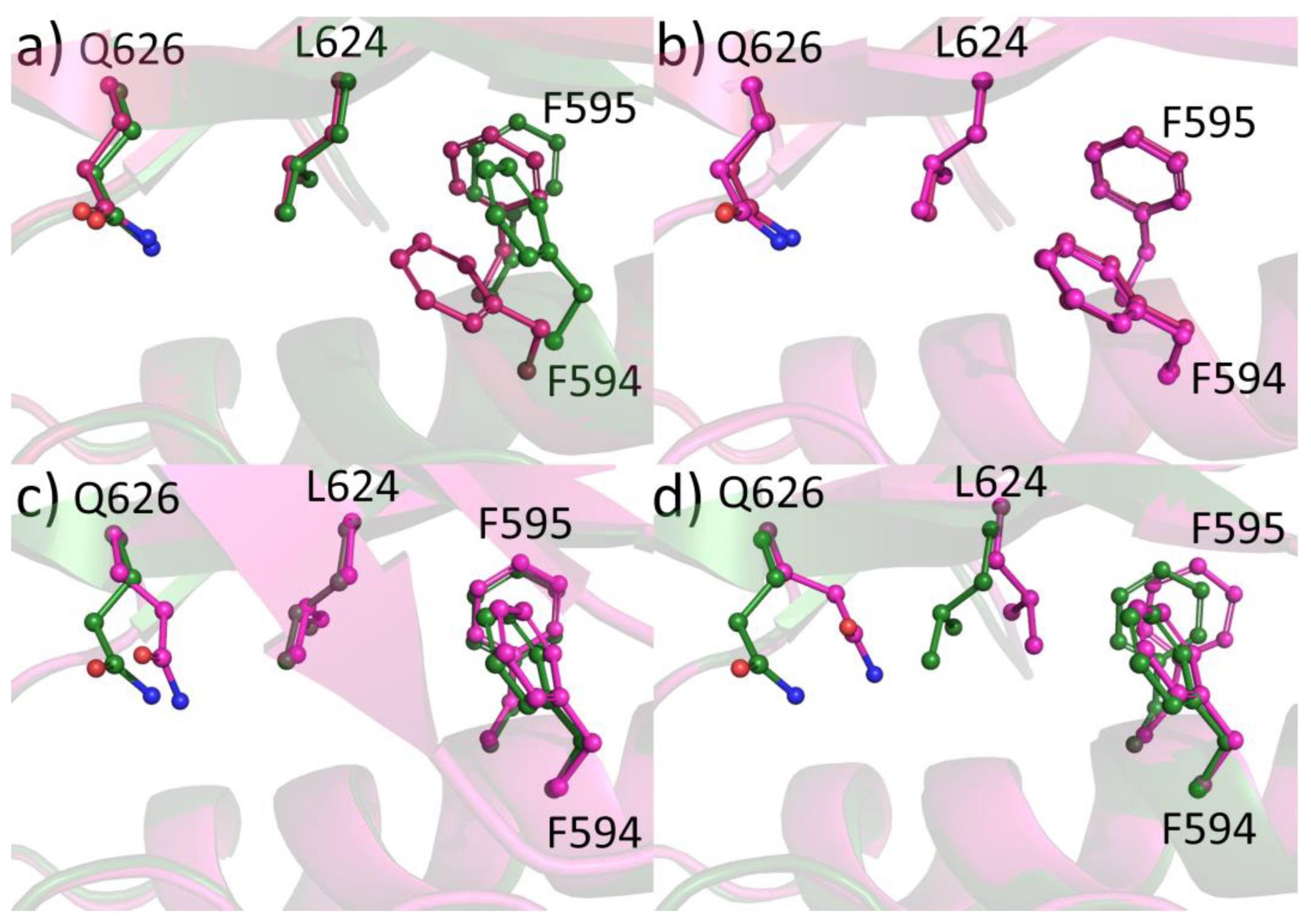
2.6. Protein-Ligand Crystallization
3. Discussion
4. Materials and Methods
4.1. Cloning, Production and Purification of Proteins Used in This Study
4.2. Screening
4.3. Compounds
4.4. FP Binding Assay
4.5. Cell Lines and Cell Viability Assay
4.6. Protein Crystallography
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- George Abraham, B.; Raivola, J.; Virtanen, A.; Silvennoinen, O. Janus Kinase. In Encyclopedia of Molecular Pharmacology; Offermanns, S., Rosenthal, W., Eds.; Springer International Publishing: Cham, Switzerland, 2021; pp. 893–902. ISBN 978-3-030-57401-7. [Google Scholar]
- Min, X.; Ungureanu, D.; Maxwell, S.; Hammarén, H.; Thibault, S.; Hillert, E.K.; Ayres, M.; Greenfield, B.; Eksterowicz, J.; Gabel, C.; et al. Structural and Functional Characterization of the JH2 Pseudokinase Domain of JAK Family Tyrosine Kinase 2 (TYK2). J. Biol. Chem. 2015, 290, 27261–27270. [Google Scholar] [CrossRef] [Green Version]
- Ungureanu, D.; Wu, J.; Pekkala, T.; Niranjan, Y.; Young, C.; Jensen, O.N.; Xu, C.F.; Neubert, T.A.; Skoda, R.C.; Hubbard, S.R.; et al. The Pseudokinase Domain of JAK2 Is a Dual-Specificity Protein Kinase That Negatively Regulates Cytokine Signaling. Nat. Struct. Mol. Biol. 2011, 18, 971–976. [Google Scholar] [CrossRef] [Green Version]
- Raivola, J.; Haikarainen, T.; Silvennoinen, O. Characterization of JAK1 Pseudokinase Domain in Cytokine Signaling. Cancers 2020, 12, 78. [Google Scholar] [CrossRef] [Green Version]
- Raivola, J.; Hammarén, H.M.; Virtanen, A.T.; Bulleeraz, V.; Ward, A.C.; Silvennoinen, O. Hyperactivation of Oncogenic JAK3 Mutants Depend on ATP Binding to the Pseudokinase Domain. Front. Oncol. 2018, 8, 560. [Google Scholar] [CrossRef] [Green Version]
- Saharinen, P.; Vihinen, M.; Silvennoinen, O. Autoinhibition of Jak2 Tyrosine Kinase Is Dependent on Specific Regions in its Pseudokinase Domain. Mol. Biol. Cell 2003, 14, 1448–1459. [Google Scholar] [CrossRef]
- Saharinen, P.; Silvennoinen, O. The Pseudokinase Domain Is Required for Suppression of Basal Activity of Jak2 and Jak3 Tyrosine Kinases and for Cytokine-Inducible Activation of Signal Transduction. J. Biol. Chem. 2002, 277, 47954–47963. [Google Scholar] [CrossRef] [Green Version]
- Saharinen, P.; Takaluoma, K.; Silvennoinen, O. Regulation of the Jak2 Tyrosine Kinase by Its Pseudokinase Domain. Mol. Cell. Biol. 2000, 20, 3387–3395. [Google Scholar] [CrossRef]
- Haan, C.; Behrmann, I.; Haan, S. Perspectives for the Use of Structural Information and Chemical Genetics to Develop Inhibitors of Janus Kinases. J. Cell. Mol. Med. 2010, 14, 504–527. [Google Scholar] [CrossRef]
- Skoda, R.C.; Duek, A.; Grisouard, J. Pathogenesis of Myeloproliferative Neoplasms. Exp. Hematol. 2015, 43, 599–608. [Google Scholar] [CrossRef] [Green Version]
- Hammarén, H.M.; Ungureanu, D.; Grisouard, J.; Skoda, R.C.; Hubbard, S.R.; Silvennoinen, O. ATP Binding to the Pseudokinase Domain of JAK2 Is Critical for Pathogenic Activation. Proc. Natl. Acad. Sci. USA 2015, 112, 4642–4647. [Google Scholar] [CrossRef]
- Wrobleski, S.T.; Moslin, R.; Lin, S.; Zhang, Y.; Spergel, S.; Kempson, J.; Tokarski, J.S.; Strnad, J.; Zupa-Fernandez, A.; Cheng, L.; et al. Highly Selective Inhibition of Tyrosine Kinase 2 (TYK2) for the Treatment of Autoimmune Diseases: Discovery of the Allosteric Inhibitor BMS-986165. J. Med. Chem. 2019, 62, 8973–8995. [Google Scholar] [CrossRef]
- Lo, M.C.; Aulabaugh, A.; Jin, G.; Cowling, R.; Bard, J.; Malamas, M.; Ellestad, G. Evaluation of Fluorescence-Based Thermal Shift Assays for Hit Identification in Drug Discovery. Anal. Biochem. 2004, 332, 153–159. [Google Scholar] [CrossRef]
- Murphy, J.M.; Zhang, Q.; Young, S.N.; Reese, M.L.; Bailey, F.P.; Eyers, P.A.; Ungureanu, D.; Hammaren, H.; Silvennoinen, O.; Varghese, L.N.; et al. A Robust Methodology to Subclassify Pseudokinases Based on Their Nucleotide-Binding Properties. Biochem. J. 2014, 457, 323–334. [Google Scholar] [CrossRef]
- Puleo, D.E.; Kucera, K.; Hammarén, H.M.; Ungureanu, D.; Newton, A.S.; Silvennoinen, O.; Jorgensen, W.L.; Schlessinger, J. Identification and Characterization of JAK2 Pseudokinase Domain Small Molecule Binders. ACS Med. Chem. Lett. 2017, 8, 618–621. [Google Scholar] [CrossRef]
- Howard, S.; Berdini, V.; Boulstridge, J.A.; Carr, M.G.; Cross, D.M.; Curry, J.; Devine, L.A.; Early, T.R.; Fazal, L.; Gill, A.L.; et al. Fragment-Based Discovery of the Pyrazol-4-Yl Urea (AT9283), a Multitargeted Kinase Inhibitor with Potent Aurora Kinase Activity. J. Med. Chem. 2009, 52, 379–388. [Google Scholar] [CrossRef]
- Lin, R.; Connolly, P.J.; Huang, S.; Wetter, S.K.; Lu, Y.; Murray, W.V.; Emanuel, S.L.; Gruninger, R.H.; Fuentes-Pesquera, A.R.; Rugg, C.A.; et al. 1-Acyl-1H-[1,2,4]Triazole-3,5-Diamine Analogues as Novel and Potent Anticancer Cyclin-Dependent Kinase Inhibitors: Synthesis and Evaluation of Biological Activities. J. Med. Chem. 2005, 48, 4208–4211. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New Data Content and Improved Web Interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Irwin, J.J.; Shoichet, B.K. ZINC--a Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Newton, A.S.; Deiana, L.; Puleo, D.E.; Cisneros, J.A.; Cutrona, K.J.; Schlessinger, J.; Jorgensen, W.L. JAK2 JH2 Fluorescence Polarization Assay and Crystal Structures for Complexes with Three Small Molecules. ACS Med. Chem. Lett. 2017, 8, 614–617. [Google Scholar] [CrossRef]
- Bandaranayake, R.M.; Ungureanu, D.; Shan, Y.; Shaw, D.E.; Silvennoinen, O.; Hubbard, S.R. Crystal Structures of the JAK2 Pseudokinase Domain and the Pathogenic Mutant V617F. Nat. Struct. Mol. Biol. 2012, 19, 754–759. [Google Scholar] [CrossRef]
- Wilmes, S.; Hafer, M.; Vuorio, J.; Tucker, J.A.; Winkelmann, H.; Löchte, S.; Stanly, T.A.; Pulgar Prieto, K.D.; Poojari, C.; Sharma, V.; et al. Mechanism of Homodimeric Cytokine Receptor Activation and Dysregulation by Oncogenic Mutations. Science 2020, 367, 643–652. [Google Scholar] [CrossRef] [Green Version]
- Tokarski, J.; Zupa-Fernandez, A.; Tredup, J.; Pike, K.; Chang, C.; Xie, D.; Cheng, L.; Pedicord, D.; Muckelbauer, J.; Johnson, S.; et al. Tyrosine Kinase 2-Mediated Signal Transduction in T Lymphocytes Is Blocked by Pharmacological Stabilization of Its Pseudokinase Domain. J. Biol. Chem. 2015, 290, 11061–11074. [Google Scholar] [CrossRef] [Green Version]
- Kavanagh, M.E.; Horning, B.D.; Khattri, R.; Roy, N.; Lu, J.P.; Whitby, L.R.; Ye, E.; Brannon, J.C.; Parker, A.; Chick, J.M.; et al. Selective Inhibitors of JAK1 Targeting an Isoform-Restricted Allosteric Cysteine. Nat. Chem. Biol. 2022, 18, 1388–1398. [Google Scholar] [CrossRef]
- McNally, R.; Li, Q.; Li, K.; Dekker, C.; Vangrevelinghe, E.; Jones, M.; Chène, P.; MacHauer, R.; Radimerski, T.; Eck, M.J. Discovery and Structural Characterization of ATP-Site Ligands for the Wild-Type and V617F Mutant JAK2 Pseudokinase Domain. ACS Chem. Biol. 2019, 14, 587–593. [Google Scholar] [CrossRef]
- Newton, A.S.; Liosi, M.E.; Henry, S.P.; Deiana, L.; Faver, J.C.; Krimmer, S.G.; Puleo, D.E.; Schlessinger, J.; Jorgensen, W.L. Indoloxytriazines as Binding Molecules for the JAK2 JH2 Pseudokinase Domain and Its V617F Variant. Tetrahedron Lett. 2021, 77, 153248. [Google Scholar] [CrossRef]
- Qi, J.; Zhang, F.; Mi, Y.; Fu, Y.; Xu, W.; Zhang, D.; Wu, Y.; Du, X.; Jia, Q.; Wang, K.; et al. Design, Synthesis and Biological Activity of Pyrazolo [1,5-a]Pyrimidin-7(4H)-Ones as Novel Kv7/KCNQ Potassium Channel Activators. Eur. J. Med. Chem. 2011, 46, 934–943. [Google Scholar] [CrossRef]
- Shan, Y.; Gnanasambandan, K.; Ungureanu, D.; Kim, E.T.; Hammarén, H.; Yamashita, K.; Silvennoinen, O.; Shaw, D.E.; Hubbard, S.R. Molecular Basis for Pseudokinase-Dependent Autoinhibition of JAK2 Tyrosine Kinase. Nat. Struct. Mol. Biol. 2014, 21, 579–584. [Google Scholar] [CrossRef] [Green Version]
- Liosi, M.-E.; Ippolito, J.A.; Henry, S.P.; Krimmer, S.G.; Newton, A.S.; Cutrona, K.J.; Olivarez, R.A.; Mohanty, J.; Schlessinger, J.; Jorgensen, W.L. Insights on JAK2 Modulation by Potent, Selective, and Cell-Permeable Pseudokinase-Domain Ligands. J. Med. Chem. 2022, 65, 8380–8400. [Google Scholar] [CrossRef]
- Hammarén, H.M.; Virtanen, A.T.; Abraham, B.G.; Peussa, H.; Hubbard, S.R.; Silvennoinen, O. Janus Kinase 2 Activation Mechanisms Revealed by Analysis of Suppressing Mutations. J. Allergy Clin. Immunol. 2019, 143, 1549–1559.e6. [Google Scholar] [CrossRef] [Green Version]
- Martin, P.; Papayannopoulou, T. HEL Cells: A New Human Erythroleukemia Cell Line with Spontaneous and Induced Globin Expression. Science 1982, 216, 1233–1235. [Google Scholar] [CrossRef]
- Uozumi, K.; Otsuka, M.; Ohno, N.; Moriyama, T.; Suzuki, S.; Shimotakahara, S.; Matsumura, I.; Hanada, S.; Arima, T. Establishment and Characterization of a New Human Megakaryoblastic Cell Line (SET-2) That Spontaneously Matures to Megakaryocytes and Produces Platelet-like Particles. Leukemia 2000, 14, 142–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otwinowski, Z.; Minor, W. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [Google Scholar] [CrossRef] [PubMed]
- Winter, G.; Lobley, C.M.C.; Prince, S.M. Decision Making in Xia2. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 1260–1273. [Google Scholar] [CrossRef] [Green Version]
- Evans, P.R. An Introduction to Data Reduction: Space-Group Determination, Scaling and Intensity Statistics. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 282–292. [Google Scholar] [CrossRef] [Green Version]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the Refinement of Macromolecular Crystal Structures. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards Automated Crystallographic Structure Refinement with Phenix.Refine. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 352–367. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and Development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Library Type | Provider | No. of Compounds | Screening Method | Screening Format | No. of Hits |
|---|---|---|---|---|---|
| Kinase-targeted | Uni Copenhagen Biomol SelleckChem Vitas-M | 4200 160 190 1000 | TSA TSA, FP TSA, FP FP | 384- 384- 384- 1536- | 4 5 1 0 |
| Nucleotide/ nucleoside analogs | Otava | 960 | TSA, FP | 384- | 22 |
| Clinical bioactives | NIH | 740 | TSA, FP | 384- | 2 |
| Known drugs | Prestwick | 1200 | FP | 1536- | 3 |
| Fragments | Org Pharm Chem | 500 | FP | 1536- | 1 |
| Natural derivatives | Timtec Analyticon | 3000 1000 | FP FP | 1536- 1536- | 0 0 |
| Diverse | Enamine MayBridge Elite Synergy Specs ChemBridge | 28,200 14,400 2300 1900 30,300 17,500 | FP FP FP FP FP FP | 1536- 1536- 1536- 1536- 1536- 1536- | 3 9 0 0 3 2 |
| SUM: | 107,600 | 55 |
| Compound | Binding IC50 [µM] | Cell Viability IC50 [µM] | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| JAK2 | JAK1 | JAK3 | TYK2 | HEL | SET-2 | |||||||
| JH2 V617F | JH2 | JH1 | JH2 | JH1 | JH2 | JH1 | JH2 | JH1 | ||||
| Diaminotriazole | 1 | 0.32 | 0.31 | 0.04 | 0.004 | 0.08 | 7.2 | 0.15 | 0.14 | 0.04 | 5.6 | 5.8 |
| 18 | 69 | 67 | ND | 41 | ND | 15 | ND | ND | ND | ND | ND | |
| Diaminotriazine | 4 | 26 | 68 | ND | 4.5 | ND | ND | ND | 27 | ND | ND | ND |
| 24 | 24 | 70 | ND | ND | ND | ND | ND | ND | ND | ND | ND | |
| Aminopurine | 7 | 1.9 | 1.3 | 0.68 | 0.15 | 0.35 | 4.7 | 0.30 | 0.06 | 0.12 | 9.3 | 4.0 |
| Phenylpyrazolo-pyrimidone | 34 | 39 | 14 | ND | ND | ND | 28 | 70 | 9.7 | 50 | ND | ND |
| 36 | 47 | 25 | ND | ND | ND | ND | ND | 32 | ND | ND | ND | |
| Pyrazolyl-formamide | 2 | 7.7 | 5.8 | 0.05 | 0.11 | 0.06 | 11 | 0.07 | 0.04 | 0.02 | 1.3 | 0.54 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Virtanen, A.T.; Haikarainen, T.; Sampathkumar, P.; Palmroth, M.; Liukkonen, S.; Liu, J.; Nekhotiaeva, N.; Hubbard, S.R.; Silvennoinen, O. Identification of Novel Small Molecule Ligands for JAK2 Pseudokinase Domain. Pharmaceuticals 2023, 16, 75. https://doi.org/10.3390/ph16010075
Virtanen AT, Haikarainen T, Sampathkumar P, Palmroth M, Liukkonen S, Liu J, Nekhotiaeva N, Hubbard SR, Silvennoinen O. Identification of Novel Small Molecule Ligands for JAK2 Pseudokinase Domain. Pharmaceuticals. 2023; 16(1):75. https://doi.org/10.3390/ph16010075
Chicago/Turabian StyleVirtanen, Anniina T., Teemu Haikarainen, Parthasarathy Sampathkumar, Maaria Palmroth, Sanna Liukkonen, Jianping Liu, Natalia Nekhotiaeva, Stevan R. Hubbard, and Olli Silvennoinen. 2023. "Identification of Novel Small Molecule Ligands for JAK2 Pseudokinase Domain" Pharmaceuticals 16, no. 1: 75. https://doi.org/10.3390/ph16010075
APA StyleVirtanen, A. T., Haikarainen, T., Sampathkumar, P., Palmroth, M., Liukkonen, S., Liu, J., Nekhotiaeva, N., Hubbard, S. R., & Silvennoinen, O. (2023). Identification of Novel Small Molecule Ligands for JAK2 Pseudokinase Domain. Pharmaceuticals, 16(1), 75. https://doi.org/10.3390/ph16010075