
Computational Chemistry for the Identification of Lead Compounds for Radiotracer Development



Abstract
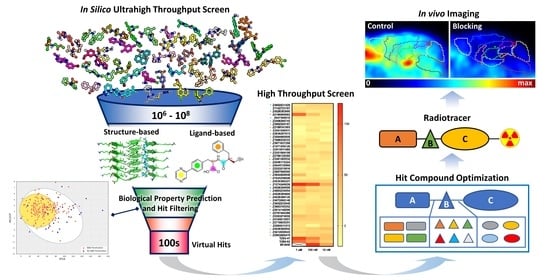
:
1. Introduction
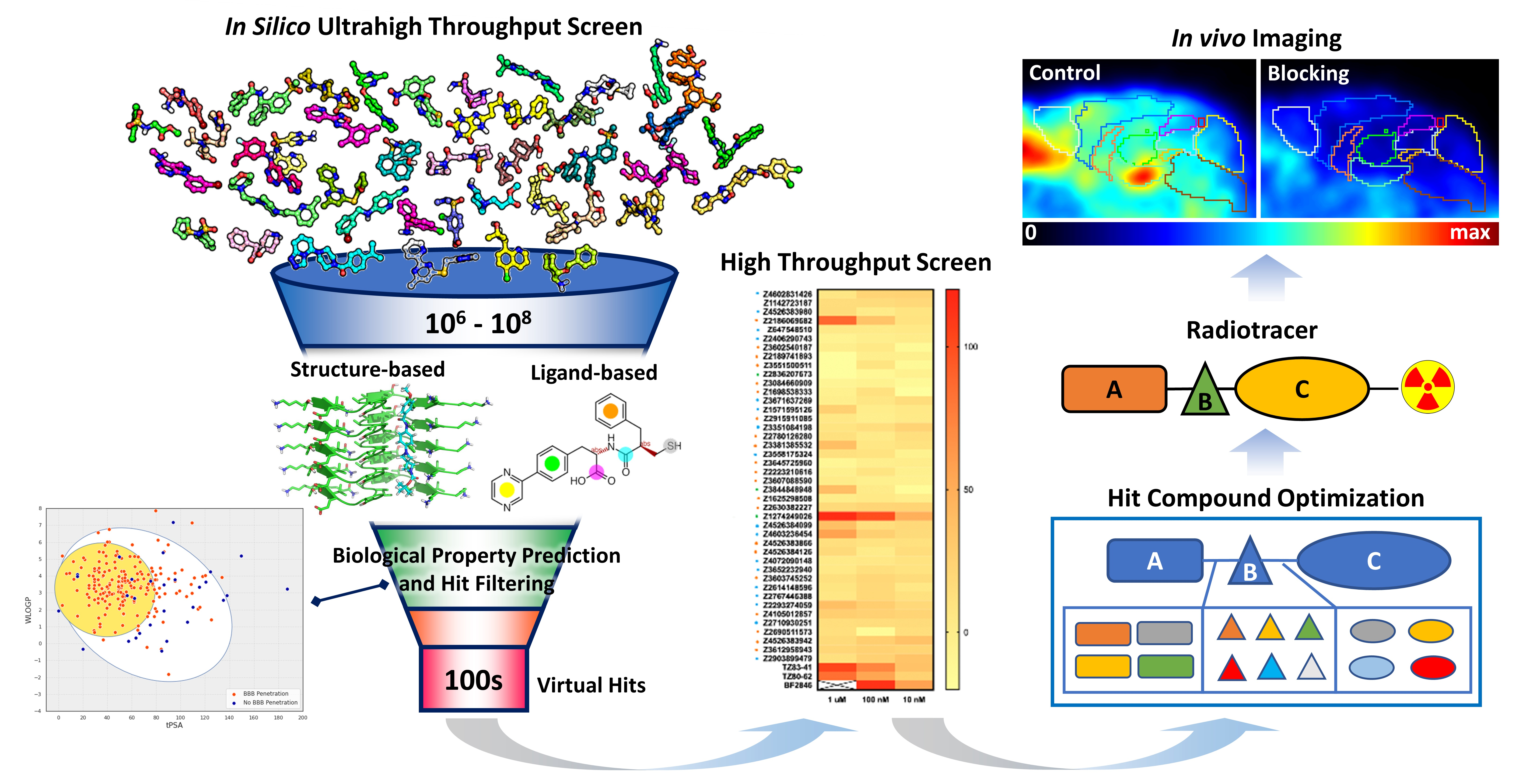
2. Virtual Screening
2.1. Virtual Screening Overview
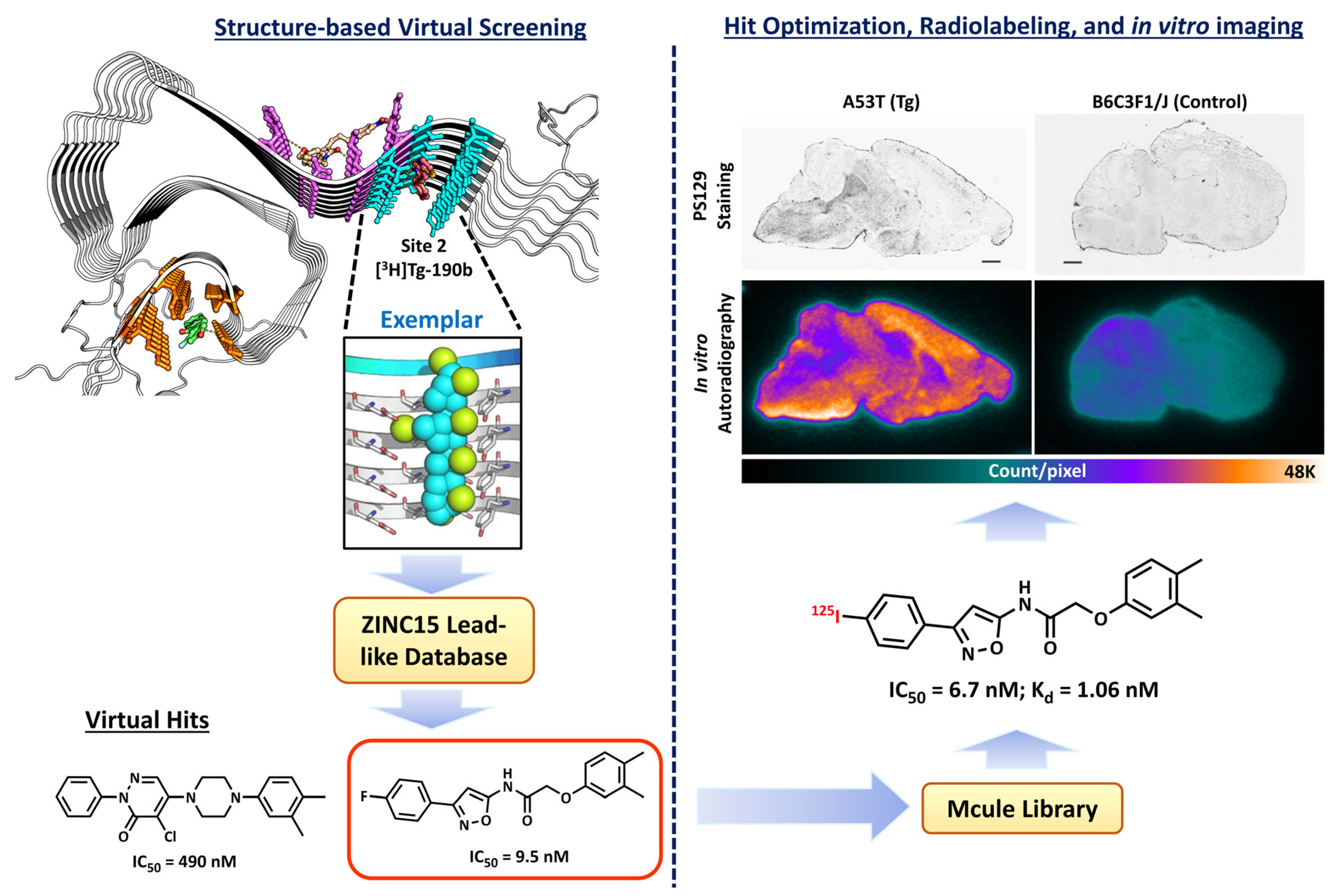
2.2. Structure-Based Virtual Screening
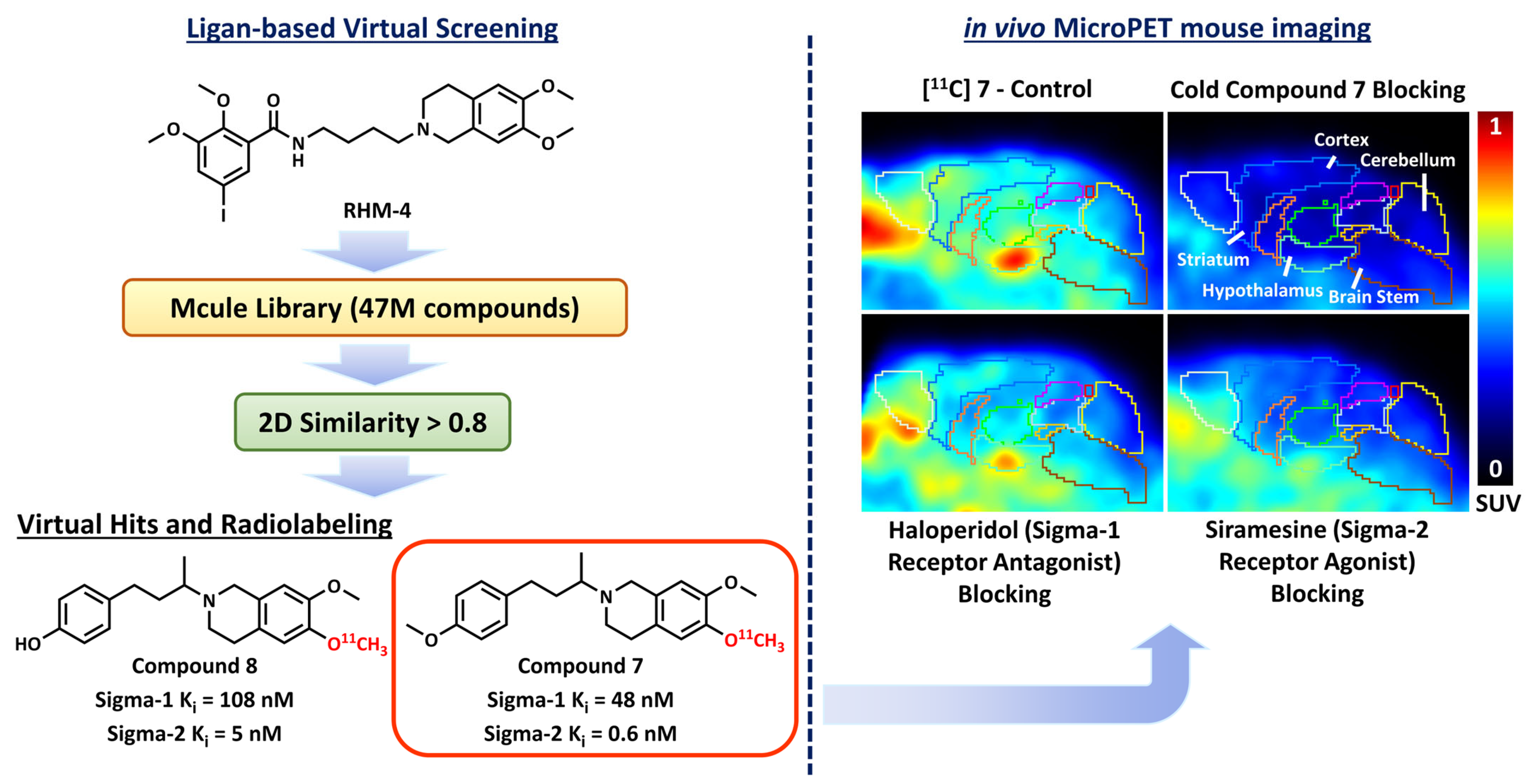
2.3. Ligand-Based Virtual Screening
3. Biological Property Prediction and Hit Filtering
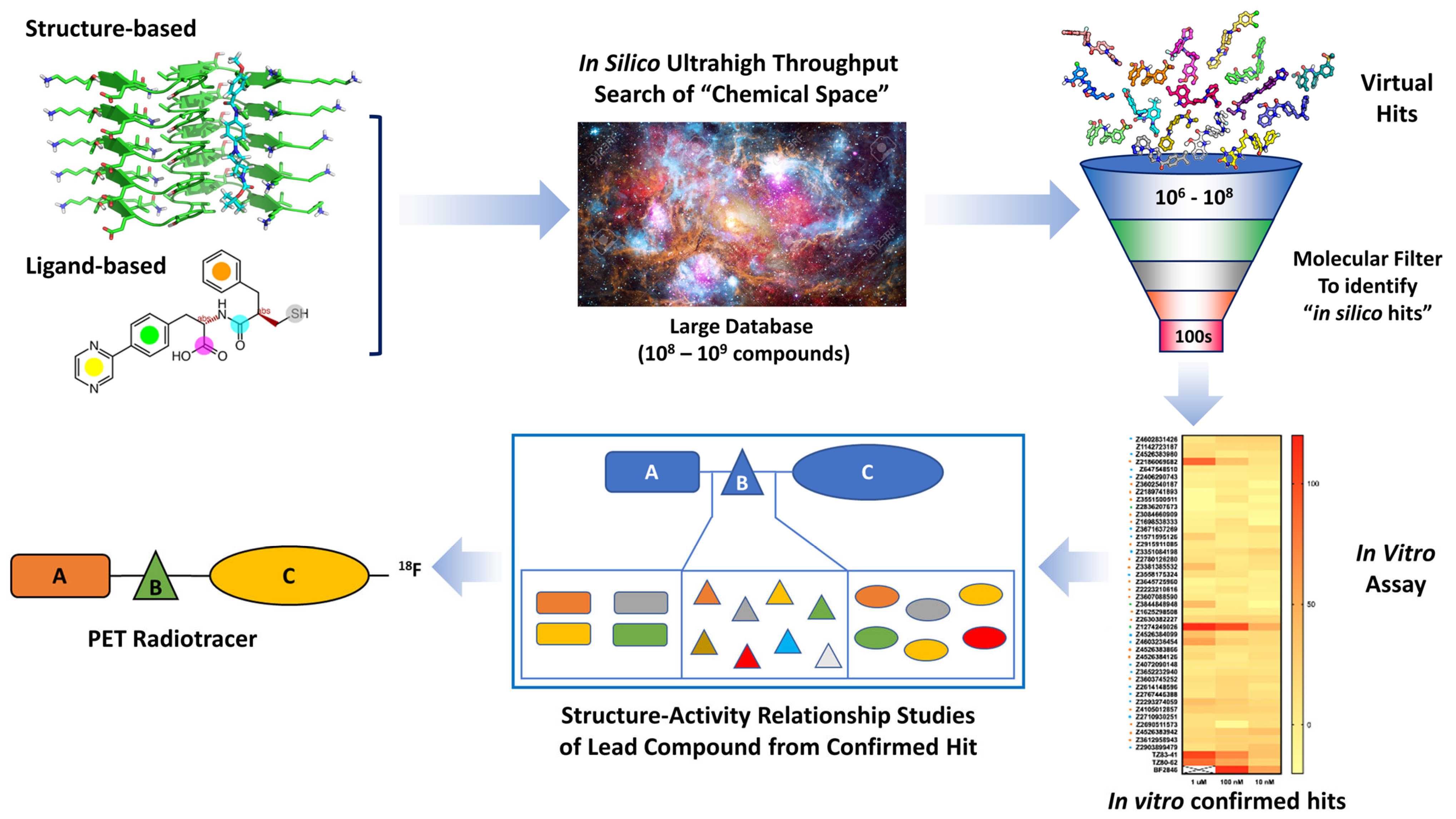
4. Hit Compound Optimization
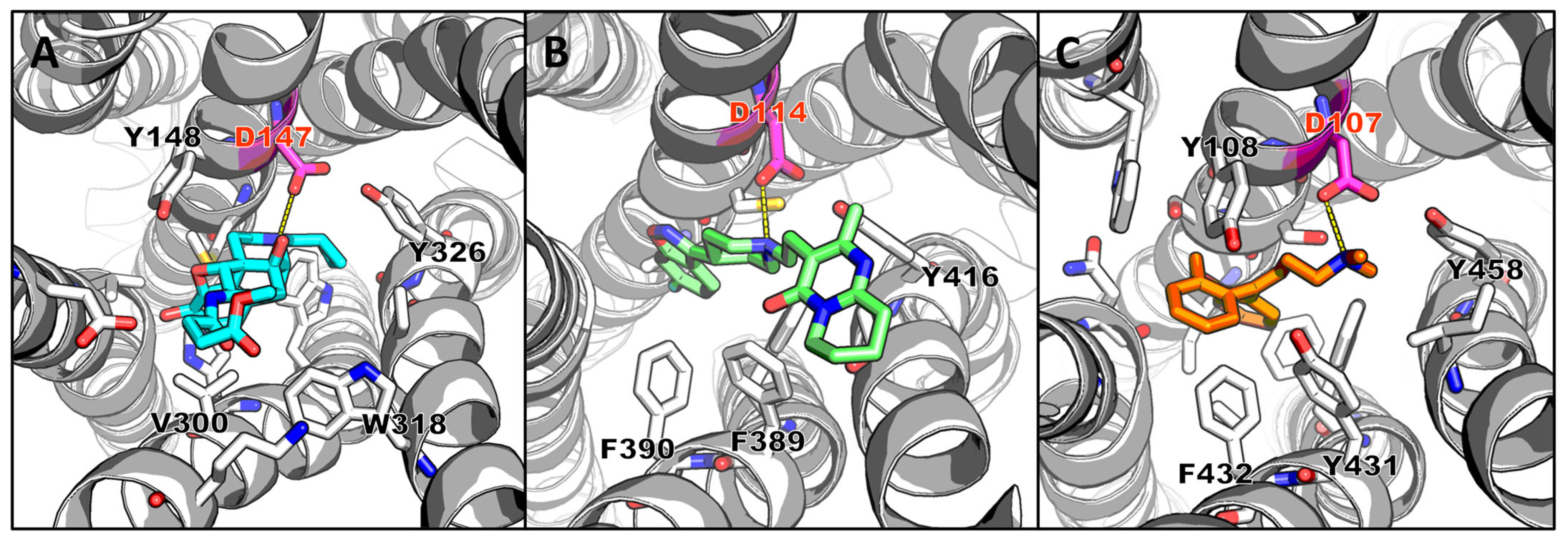
4.1. Structure-Based Hit Compound Optimization
4.2. Ligand-Based Hit Compound Optimization
5. Limitations and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hsieh, C.-J.; Ferrie, J.J.; Xu, K.; Lee, I.; Graham, T.J.; Tu, Z.; Yu, J.; Dhavale, D.; Kotzbauer, P.; Petersson, E.J.; et al. Alpha synuclein fibrils contain multiple binding sites for small molecules. ACS Chem. Neurosci. 2018, 9, 2521–2527. [Google Scholar] [CrossRef]
- Ferrie, J.J.; Lengyel-Zhand, Z.; Janssen, B.; Lougee, M.G.; Giannakoulias, S.; Hsieh, C.-J.; Pagar, V.V.; Weng, C.-C.; Xu, H.; Graham, T.J.; et al. Identification of a nanomolar affinity α-synuclein fibril imaging probe by ultra-high throughput in silico screening. Chem. Sci. 2020, 11, 12746–12754. [Google Scholar] [CrossRef]
- Kim, H.Y.; Lee, J.Y.; Hsieh, C.-J.; Riad, A.; Izzo, N.J.; Catalano, S.M.; Graham, T.J.; Mach, R.H. Screening of σ2 receptor ligands and in vivo evaluation of 11C-labeled 6, 7-Dimethoxy-2-[4-(4-methoxyphenyl) butan-2-yl]-1, 2, 3, 4-tetrahydroisoquinoline for potential use as a σ2 receptor brain PET tracer. J. Med. Chem. 2022, 65, 6261–6272. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, X.; Cui, M.; Zhang, J.; Guo, Z.; Li, Y.; Zhang, X.; Dai, J.; Liu, B. Preliminary characterization and in vivo studies of structurally identical 18F-and 125I-labeled benzyloxybenzenes for PET/SPECT imaging of β-amyloid plaques. Sci. Rep. 2015, 5, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational Methods in Drug Discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef] [Green Version]
- Vazquez, J.; Lopez, M.; Gibert, E.; Herrero, E.; Luque, F.J. Merging Ligand-Based and Structure-Based Methods in Drug Discovery: An Overview of Combined Virtual Screening Approaches. Molecules 2020, 25, 27. [Google Scholar] [CrossRef]
- Ricci-Lopez, J.; Aguila, S.A.; Gilson, M.K.; Brizuela, C.A. Improving structure-based virtual screening with ensemble docking and machine learning. J. Chem. Inf. Model. 2021, 61, 5362–5376. [Google Scholar] [CrossRef]
- Wojcikowski, M.; Ballester, P.J.; Siedlecki, P. Performance of machine-learning scoring functions in structure-based virtual screening. Sci. Rep. 2017, 7, 10. [Google Scholar] [CrossRef] [Green Version]
- Bahi, M.; Batouche, M. Deep learning for ligand-based virtual screening in drug discovery. In Proceedings of the 2018 3rd International Conference on Pattern Analysis and Intelligent Systems (PAIS), Tebessa, Algeria, 24–25 October 2018; pp. 1–5. [Google Scholar]
- Bonanno, E.; Ebejer, J.-P. Applying machine learning to ultrafast shape recognition in ligand-based virtual screening. Front. Pharmacol. 2020, 10, 1675. [Google Scholar] [CrossRef]
- Bustamam, A.; Hamzah, H.; Husna, N.A.; Syarofina, S.; Dwimantara, N.; Yanuar, A.; Sarwinda, D. Artificial intelligence paradigm for ligand-based virtual screening on the drug discovery of type 2 diabetes mellitus. J. Big Data 2021, 8, 74. [Google Scholar] [CrossRef]
- Lin, X.Q.; Li, X.; Lin, X.B. A Review on Applications of Computational Methods in Drug Screening and Design. Molecules 2020, 25, 17. [Google Scholar] [CrossRef] [Green Version]
- Maia, E.H.B.; Assis, L.C.; de Oliveira, T.A.; da Silva, A.M.; Taranto, A.G. Structure-Based Virtual Screening: From Classical to Artificial Intelligence. Front. Chem. 2020, 8, 18. [Google Scholar] [CrossRef]
- Eckert, H.; Bojorath, J. Molecular similarity analysis in virtual screening: Foundations, limitations and novel approaches. Drug Discov. Today 2007, 12, 225–233. [Google Scholar] [CrossRef]
- Kontoyianni, M. Library size in virtual screening: Is it truly a number’s game? Expert Opin. Drug Discov. 2022, 17, 1177–1179. [Google Scholar] [CrossRef]
- Manglik, A.; Lin, H.; Aryal, D.K.; McCorvy, J.D.; Dengler, D.; Corder, G.; Levit, A.; Kling, R.C.; Bernat, V.; Hübner, H.; et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature 2016, 537, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Lansu, K.; Karpiak, J.; Liu, J.; Huang, X.-P.; McCorvy, J.D.; Kroeze, W.K.; Che, T.; Nagase, H.; Carroll, F.I.; Jin, J.; et al. In silico design of novel probes for the atypical opioid receptor MRGPRX2. Nat. Chem. Biol. 2017, 13, 529–536. [Google Scholar] [CrossRef] [Green Version]
- De Graaf, C.; Kooistra, A.J.; Vischer, H.F.; Katritch, V.; Kuijer, M.; Shiroishi, M.; Iwata, S.; Shimamura, T.; Stevens, R.C.; De Esch, I.J. Crystal structure-based virtual screening for fragment-like ligands of the human histamine H1 receptor. J. Med. Chem. 2011, 54, 8195–8206. [Google Scholar] [CrossRef] [Green Version]
- Kiss, R.; Kiss, B.; Könczöl, Á.; Szalai, F.; Jelinek, I.; László, V.; Noszál, B.; Falus, A.; Keseru, G.M. Discovery of novel human histamine H4 receptor ligands by large-scale structure-based virtual screening. J. Med. Chem. 2008, 51, 3145–3153. [Google Scholar] [CrossRef]
- Levoin, N.; Labeeuw, O.; Billot, X.; Calmels, T.; Danvy, D.; Krief, S.; Berrebi-Bertrand, I.; Lecomte, J.-M.; Schwartz, J.-C.; Capet, M. Discovery of nanomolar ligands with novel scaffolds for the histamine H4 receptor by virtual screening. Eur. J. Med. Chem. 2017, 125, 565–572. [Google Scholar] [CrossRef]
- Clark, D.E.; Higgs, C.; Wren, S.P.; Dyke, H.J.; Wong, M.; Norman, D.; Lockey, P.M.; Roach, A.G. A virtual screening approach to finding novel and potent antagonists at the melanin-concentrating hormone 1 receptor. J. Med. Chem. 2004, 47, 3962–3971. [Google Scholar] [CrossRef]
- Cavasotto, C.N.; Orry, A.J.; Murgolo, N.J.; Czarniecki, M.F.; Kocsi, S.A.; Hawes, B.E.; O’Neill, K.A.; Hine, H.; Burton, M.S.; Voigt, J.H. Discovery of novel chemotypes to a G-protein-coupled receptor through ligand-steered homology modeling and structure-based virtual screening. J. Med. Chem. 2008, 51, 581–588. [Google Scholar] [CrossRef] [Green Version]
- Kellenberger, E.; Springael, J.-Y.; Parmentier, M.; Hachet-Haas, M.; Galzi, J.-L.; Rognan, D. Identification of nonpeptide CCR5 receptor agonists by structure-based virtual screening. J. Med. Chem. 2007, 50, 1294–1303. [Google Scholar] [CrossRef]
- Carlsson, J.; Yoo, L.; Gao, Z.-G.; Irwin, J.J.; Shoichet, B.K.; Jacobson, K.A. Structure-based discovery of A2A adenosine receptor ligands. J. Med. Chem. 2010, 53, 3748–3755. [Google Scholar] [CrossRef]
- Katritch, V.; Jaakola, V.-P.; Lane, J.R.; Lin, J.; IJzerman, A.P.; Yeager, M.; Kufareva, I.; Stevens, R.C.; Abagyan, R. Structure-based discovery of novel chemotypes for adenosine A2A receptor antagonists. J. Med. Chem. 2010, 53, 1799–1809. [Google Scholar] [CrossRef] [Green Version]
- Kolb, P.; Rosenbaum, D.M.; Irwin, J.J.; Fung, J.J.; Kobilka, B.K.; Shoichet, B.K. Structure-based discovery of β2-adrenergic receptor ligands. Proc. Natl. Acad. Sci. USA 2009, 106, 6843–6848. [Google Scholar] [CrossRef] [Green Version]
- Kaczor, A.A.; Silva, A.G.; Loza, M.I.; Kolb, P.; Castro, M.; Poso, A. Structure-Based Virtual Screening for Dopamine D2 Receptor Ligands as Potential Antipsychotics. ChemMedChem 2016, 11, 718–729. [Google Scholar] [CrossRef]
- Kumar, R.; Kumar, A.; Långström, B.; Darreh-Shori, T. Discovery of novel choline acetyltransferase inhibitors using structure-based virtual screening. Sci. Rep. 2017, 7, 16287. [Google Scholar] [CrossRef] [Green Version]
- Seidler, P.M.; Murray, K.A.; Boyer, D.R.; Ge, P.; Sawaya, M.R.; Hu, C.J.; Cheng, X.; Abskharon, R.; Pan, H.; DeTure, M.A.; et al. Structure-based discovery of small molecules that disaggregate Alzheimer’s disease tissue derived tau fibrils in vitro. Nat. Commun. 2022, 13, 5451. [Google Scholar] [CrossRef]
- Jin, H.; Wu, C.; Su, R.; Sun, T.; Li, X.; Guo, C. Identifying Dopamine D3 Receptor Ligands through Virtual Screening and Exploring the Binding Modes of Hit Compounds. Molecules 2023, 28, 527. [Google Scholar] [CrossRef]
- Olah, M.M.; Bologa, C.G.; Oprea, T.I. Strategies for compound selection. Curr. Drug Discov. Technol. 2004, 1, 211–220. [Google Scholar] [CrossRef] [Green Version]
- Edwards, B.S.; Bologa, C.; Young, S.M.; Balakin, K.V.; Prossnitz, E.R.; Savchuck, N.P.; Sklar, L.A.; Oprea, T.I. Integration of virtual screening with high-throughput flow cytometry to identify novel small molecule formylpeptide receptor antagonists. Mol. Pharmacol. 2005, 68, 1301–1310. [Google Scholar] [CrossRef]
- Klabunde, T.; Giegerich, C.; Evers, A. Sequence-derived three-dimensional pharmacophore models for G-protein-coupled receptors and their application in virtual screening. J. Med. Chem. 2009, 52, 2923–2932. [Google Scholar] [CrossRef]
- Ko, K.; Kim, H.-J.; Ho, P.-S.; Lee, S.O.; Lee, J.-E.; Min, C.-R.; Kim, Y.C.; Yoon, J.-H.; Park, E.-J.; Kwon, Y.-J.; et al. Discovery of a novel highly selective histamine H4 receptor antagonist for the treatment of atopic dermatitis. J. Med. Chem. 2018, 61, 2949–2961. [Google Scholar] [CrossRef]
- Vettorazzi, M.; Angelina, E.; Lima, S.; Gonec, T.; Otevrel, J.; Marvanova, P.; Padrtova, T.; Mokry, P.; Bobal, P.; Acosta, L.M.; et al. An integrative study to identify novel scaffolds for sphingosine kinase 1 inhibitors. Eur. J. Med. Chem. 2017, 139, 461–481. [Google Scholar] [CrossRef]
- Manepalli, S.; Geffert, L.M.; Surratt, C.K.; Madura, J.D. Discovery of novel selective serotonin reuptake inhibitors through development of a protein-based pharmacophore. J. Chem. Inf. Model. 2011, 51, 2417–2426. [Google Scholar] [CrossRef] [Green Version]
- Engel, S.; Skoumbourdis, A.P.; Childress, J.; Neumann, S.; Deschamps, J.R.; Thomas, C.J.; Colson, A.-O.; Costanzi, S.; Gershengorn, M.C. A virtual screen for diverse ligands: Discovery of selective G protein-coupled receptor antagonists. J. Am. Chem. Soc. 2008, 130, 5115–5123. [Google Scholar] [CrossRef]
- Evers, A.; Klabunde, T. Structure-based drug discovery using GPCR homology modeling: Successful virtual screening for antagonists of the alpha1A adrenergic receptor. J. Med. Chem. 2005, 48, 1088–1097. [Google Scholar] [CrossRef]
- Evers, A.; Klebe, G. Successful virtual screening for a submicromolar antagonist of the neurokinin-1 receptor based on a ligand-supported homology model. J. Med. Chem. 2004, 47, 5381–5392. [Google Scholar] [CrossRef]
- Adeshina, Y.O.; Deeds, E.J.; Karanicolas, J. Machine learning classification can reduce false positives in structure-based virtual screening. Proc. Natl. Acad. Sci. USA 2020, 117, 18477–18488. [Google Scholar] [CrossRef]
- Renner, S.; Noeske, T.; Parsons, C.G.; Schneider, P.; Weil, T.; Schneider, G. New allosteric modulators of metabotropic glutamate receptor 5 (mGluR5) found by ligand-based virtual screening. ChemBioChem 2005, 6, 620–625. [Google Scholar] [CrossRef]
- Noeske, T.; Jirgensons, A.; Starchenkovs, I.; Renner, S.; Jaunzeme, I.; Trifanova, D.; Hechenberger, M.; Bauer, T.; Kauss, V.; Parsons, C.G. Virtual screening for selective allosteric mGluR1 antagonists and structure–activity relationship investigations for coumarine derivatives. ChemMedChem: Chem. Enabling Drug Discov. 2007, 2, 1763–1773. [Google Scholar] [CrossRef]
- Yu, Y.; Dong, H.; Peng, Y.; Welsh, W.J. QSAR-Based Computational Approaches to Accelerate the Discovery of Sigma-2 Receptor (S2R) Ligands as Therapeutic Drugs. Molecules 2021, 26, 5270. [Google Scholar] [CrossRef]
- Floresta, G.; Amata, E.; Barbaraci, C.; Gentile, D.; Turnaturi, R.; Marrazzo, A.; Rescifina, A. A structure-and ligand-based virtual screening of a database of “Small” marine natural products for the identification of “Blue” Sigma-2 receptor ligands. Mar. Drugs 2018, 16, 384. [Google Scholar] [CrossRef] [Green Version]
- Tikhonova, I.G.; Sum, C.S.; Neumann, S.; Engel, S.; Raaka, B.M.; Costanzi, S.; Gershengorn, M.C. Discovery of novel agonists and antagonists of the free fatty acid receptor 1 (FFAR1) using virtual screening. J. Med. Chem. 2008, 51, 625–633. [Google Scholar] [CrossRef] [Green Version]
- Staroń, J.; Kurczab, R.; Warszycki, D.; Satała, G.; Krawczyk, M.; Bugno, R.; Lenda, T.; Popik, P.; Hogendorf, A.S.; Hogendorf, A.; et al. Virtual screening-driven discovery of dual 5-HT6/5-HT2A receptor ligands with pro-cognitive properties. Eur. J. Med. Chem. 2020, 185, 111857. [Google Scholar] [CrossRef]
- Kurczab, R.; Nowak, M.; Chilmonczyk, Z.; Sylte, I.; Bojarski, A.J. The development and validation of a novel virtual screening cascade protocol to identify potential serotonin 5-HT7R antagonists. Bioorganic Med. Chem. Lett. 2010, 20, 2465–2468. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Vyas, V.K.; Ukawala, R.D.; Ghate, M.; Chintha, C. Homology Modeling a Fast Tool for Drug Discovery: Current Perspectives. Indian J. Pharm. Sci. 2012, 74, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, K.W.; Lemmon, G.H.; DeLuca, S.L.; Sheehan, J.H.; Meiler, J. Practically Useful: What the ROSETTA Protein Modeling Suite Can Do for You. Biochemistry 2010, 49, 2987–2998. [Google Scholar] [CrossRef]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 2006, 22, 195–201. [Google Scholar] [CrossRef] [Green Version]
- Skolnick, J.; Gao, M.; Zhou, H.Y.; Singh, S. AlphaFold 2: Why It Works and Its Implications for Understanding the Relationships of Protein Sequence, Structure, and Function. J. Chem. Inf. Model. 2021, 61, 4827–4831. [Google Scholar] [CrossRef]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.Z.; Lopez, R.; Magrane, M.; et al. UniProt: The Universal Protein knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef]
- Sehnal, D.; Svobodová Vařeková, R.; Berka, K.; Pravda, L.; Navrátilová, V.; Banáš, P.; Ionescu, C.-M.; Otyepka, M.; Koča, J. MOLE 2.0: Advanced approach for analysis of biomacromolecular channels. J. Cheminformatics 2013, 5, 39. [Google Scholar] [CrossRef] [Green Version]
- Volkamer, A.; Kuhn, D.; Rippmann, F.; Rarey, M. DoGSiteScorer: A web server for automatic binding site prediction, analysis and druggability assessment. Bioinformatics 2012, 28, 2074–2075. [Google Scholar] [CrossRef] [Green Version]
- Innis, C.A. siteFiNDER| 3D: A web-based tool for predicting the location of functional sites in proteins. Nucleic Acids Res. 2007, 35, W489–W494. [Google Scholar] [CrossRef] [Green Version]
- Krasowski, A.; Muthas, D.; Sarkar, A.; Schmitt, S.; Brenk, R. DrugPred: A structure-based approach to predict protein druggability developed using an extensive nonredundant data set. J. Chem. Inf. Model. 2011, 51, 2829–2842. [Google Scholar] [CrossRef]
- Irwin, J.J.; Shoichet, B.K. ZINC—A free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A free tool to discover chemistry for biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15–ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- Moroz, Y.; Chuprina, A.; Mykytenko, D. Enamine REAL DataBase—An instrumental and practical vehicle for charting new regions of the relevant drug discovery chemical space. Abstr. Pap. Am. Chem. Soc. 2016, 251, 2. [Google Scholar]
- Enaine REAL Space. Available online: https://enamine.net/compound-collections/real-compounds (accessed on 13 February 2023).
- WuXi AppTec. Available online: https://www.wuxiapptec.com/ (accessed on 13 February 2023).
- ChemDiv Compound Libraries. Available online: https://www.chemdiv.com/catalog/complete-list-of-compounds-libraries/ (accessed on 13 February 2023).
- Asinex Screening Libraries. Available online: https://www.asinex.com/screening-libraries-(all-libraries) (accessed on 13 February 2023).
- ChemBridge Lead-Like and Drug-Like Compound Database. Available online: https://chembridge.com/screening-compounds/lead-like-drug-like-compounds/ (accessed on 13 February 2023).
- Mcule Databse. Available online: https://mcule.com/database/ (accessed on 13 February 2023).
- Goodsell, D.S.; Morris, G.M.; Olson, A.J. Automated docking of flexible ligands: Applications of AutoDock. J. Mol. Recognit. 1996, 9, 1–5. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Quiroga, R.; Villarreal, M.A. Vinardo: A Scoring Function Based on Autodock Vina Improves Scoring, Docking, and Virtual Screening. PLoS ONE 2016, 11, 18. [Google Scholar] [CrossRef] [Green Version]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Allen, W.J.; Balius, T.E.; Mukherjee, S.; Brozell, S.R.; Moustakas, D.T.; Lang, P.T.; Case, D.A.; Kuntz, I.D.; Rizzo, R.C. DOCK 6: Impact of new features and current docking performance. J. Comput. Chem. 2015, 36, 1132–1156. [Google Scholar] [CrossRef] [Green Version]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [Green Version]
- Mcgann, M.R.; Almond, H.R.; Nicholls, A.; Grant, J.A.; Brown, F.K. Gaussian docking functions. Biopolym. Orig. Res. Biomol. 2003, 68, 76–90. [Google Scholar] [CrossRef] [Green Version]
- Meiler, J.; Baker, D. ROSETTALIGAND: Protein-small molecule docking with full side-chain flexibility. Proteins 2006, 65, 538–548. [Google Scholar] [CrossRef]
- Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Mathiesen, J.M.; Sunahara, R.K.; Pardo, L.; Weis, W.I.; Kobilka, B.K.; Granier, S. Crystal structure of the µ-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321–326. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Che, T.; Levit, A.; Shoichet, B.K.; Wacker, D.; Roth, B.L. Structure of the D2 dopamine receptor bound to the atypical antipsychotic drug risperidone. Nature 2018, 555, 269–273. [Google Scholar] [CrossRef]
- Shimamura, T.; Shiroishi, M.; Weyand, S.; Tsujimoto, H.; Winter, G.; Katritch, V.; Abagyan, R.; Cherezov, V.; Liu, W.; Han, G.W. Structure of the human histamine H1 receptor complex with doxepin. Nature 2011, 475, 65–70. [Google Scholar] [CrossRef] [Green Version]
- Weiss, D.R.; Karpiak, J.; Huang, X.-P.; Sassano, M.F.; Lyu, J.; Roth, B.L.; Shoichet, B.K. Selectivity challenges in docking screens for GPCR targets and antitargets. J. Med. Chem. 2018, 61, 6830–6845. [Google Scholar] [CrossRef] [Green Version]
- Ramírez, D.; Caballero, J. Is it reliable to use common molecular docking methods for comparing the binding affinities of enantiomer pairs for their protein target? Int. J. Mol. Sci. 2016, 17, 525. [Google Scholar] [CrossRef] [Green Version]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef]
- Kurogi, Y.; Guner, O.F. Pharmacophore modeling and three-dimensional database searching for drug design using catalyst. Curr. Med. Chem. 2001, 8, 1035–1055. [Google Scholar] [CrossRef]
- Grant, J.A.; Pickup, B.T. A GAUSSIAN DESCRIPTION OF MOLECULAR SHAPE. J. Phys. Chem. 1995, 99, 3503–3510. [Google Scholar] [CrossRef]
- Tresadern, G.; Bemporad, D.; Howe, T. A comparison of ligand based virtual screening methods and application to corticotropin releasing factor 1 receptor. J. Mol. Graph. Model. 2009, 27, 860–870. [Google Scholar] [CrossRef]
- Johnson, D.K.; Karanicolas, J. Ultra-high-throughput structure-based virtual screening for small-molecule inhibitors of protein–protein interactions. J. Chem. Inf. Model. 2016, 56, 399–411. [Google Scholar] [CrossRef] [Green Version]
- Weber, L. JChem Base—ChemAxon. Chem. World 2008, 5, 65–66. [Google Scholar]
- Durrant, J.D.; McCammon, J.A. BINANA: A novel algorithm for ligand-binding characterization. J. Mol. Graph. Model. 2011, 29, 888–893. [Google Scholar] [CrossRef] [Green Version]
- Gramatica, P. Principles of QSAR models validation: Internal and external. QSAR Comb. Sci. 2007, 26, 694–701. [Google Scholar] [CrossRef]
- Vogt, M.; Bajorath, J. Chemoinformatics: A view of the field and current trends in method development. Bioorganic Med. Chem. 2012, 20, 5317–5323. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Hutchison, G.R. Cinfony—Combining Open Source cheminformatics toolkits behind a common interface. Chem. Cent. J. 2008, 2, 10. [Google Scholar] [CrossRef] [Green Version]
- Willett, P. Similarity-based virtual screening using 2D fingerprints. Drug Discov. Today 2006, 11, 1046–1053. [Google Scholar] [CrossRef] [Green Version]
- Fisanick, W.; Lipkus, A.H.; Rusinko, A. Similarity searching on cas registry substances. 2. 2d structural similarity. J. Chem. Inf. Comput. Sci. 1994, 34, 130–140. [Google Scholar] [CrossRef]
- Jain, A.K.; Duin, R.P.W.; Mao, J.C. Statistical pattern recognition: A review. IEEE Trans. Pattern Anal. Mach. Intell. 2000, 22, 4–37. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [Green Version]
- van de Waterbeemd, H.; Gifford, E. ADMET in silico modelling: Towards prediction paradise? Nat. Rev. Drug Discov. 2003, 2, 192–204. [Google Scholar] [CrossRef]
- Stéen, E.J.L.; Vugts, D.J.; Windhorst, A.D. The application of in silico methods for prediction of blood-brain barrier permeability of small molecule PET tracers. Front. Nucl. Med. 2022, 2, 12. [Google Scholar] [CrossRef]
- Wager, T.T.; Hou, X.; Verhoest, P.R.; Villalobos, A. Moving beyond rules: The development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem. Neurosci. 2010, 1, 435–449. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Villalobos, A.; Beck, E.M.; Bocan, T.; Chappie, T.A.; Chen, L.; Grimwood, S.; Heck, S.D.; Helal, C.J.; Hou, X.; et al. Design and selection parameters to accelerate the discovery of novel central nervous system positron emission tomography (PET) ligands and their application in the development of a novel phosphodiesterase 2A PET ligand. J. Med. Chem. 2013, 56, 4568–4579. [Google Scholar] [CrossRef]
- Daina, A.; Zoete, V. A boiled-egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Sharma, A.; Alexiou, A.; Bilgrami, A.L.; Kamal, M.A.; Ashraf, G.M. DeePred-BBB: A Blood Brain Barrier Permeability Prediction Model With Improved Accuracy. Front. Neurosci. 2022, 16, 11. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Chen, L.; Beck, E.M.; Chappie, T.A.; Coelho, R.V.; Doran, S.D.; Fan, K.-H.; Helal, C.J.; Humphrey, J.M.; Hughes, Z.; et al. The discovery of a novel phosphodiesterase (PDE) 4B-preferring radioligand for positron emission tomography (PET) imaging. J. Med. Chem. 2017, 60, 8538–8551. [Google Scholar] [CrossRef]
- Van der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Brown, B.P.; Mendenhall, J.; Geanes, A.R.; Meiler, J. General Purpose Structure-Based Drug Discovery Neural Network Score Functions with Human-Interpretable Pharmacophore Maps. J. Chem. Inf. Model. 2021, 61, 603–620. [Google Scholar] [CrossRef]
- Qi, Y.; Li, Y.; Fang, Y.; Gao, H.; Qiang, B.; Wang, S.; Zhang, H. Design, synthesis, biological evaluation, and molecular docking of 2, 4-diaminopyrimidine derivatives targeting focal adhesion kinase as tumor radiotracers. Mol. Pharm. 2021, 18, 1634–1642. [Google Scholar] [CrossRef]
- Fang, Y.; Wang, D.; Xu, X.; Dava, G.; Liu, J.; Li, X.; Xue, Q.; Wang, H.; Zhang, J.; Zhang, H. Preparation, in vitro and in vivo evaluation, and molecular dynamics (MD) simulation studies of novel F-18 labeled tumor imaging agents targeting focal adhesion kinase (FAK). RSC Adv. 2018, 8, 10333–10345. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, C.-J.; Riad, A.; Lee, J.Y.; Sahlholm, K.; Xu, K.; Luedtke, R.R.; Mach, R.H. Interaction of ligands for pet with the dopamine D3 receptor: In silico and in vitro methods. Biomolecules 2021, 11, 529. [Google Scholar] [CrossRef]
- Xu, K.; Hsieh, C.-J.; Lee, J.Y.; Riad, A.; Izzo, N.J.; Look, G.; Catalano, S.; Mach, R.H. Exploration of Diazaspiro Cores as Piperazine Bioisosteres in the Development of σ2 Receptor Ligands. Int. J. Mol. Sci. 2022, 23, 8259. [Google Scholar] [CrossRef]
- Ågren, R.; Zeberg, H.; Stępniewski, T.M.; Free, R.B.; Reilly, S.W.; Luedtke, R.R.; Århem, P.; Ciruela, F.; Sibley, D.R.; Mach, R.H.; et al. Ligand with Two Modes of Interaction with the Dopamine D2 Receptor–An Induced-Fit Mechanism of Insurmountable Antagonism. ACS Chem. Neurosci. 2020, 11, 3130–3143. [Google Scholar] [CrossRef]
- Chen, P.-J.; Taylor, M.; Griffin, S.A.; Amani, A.; Hayatshahi, H.; Korzekwa, K.; Ye, M.; Mach, R.H.; Liu, J.; Luedtke, R.R.; et al. Design, synthesis, and evaluation of N-(4-(4-phenyl piperazin-1-yl) butyl)-4-(thiophen-3-yl) benzamides as selective dopamine D3 receptor ligands. Bioorganic Med. Chem. Lett. 2019, 29, 2690–2694. [Google Scholar] [CrossRef]
- Hayatshahi, H.S.; Xu, K.; Griffin, S.A.; Taylor, M.; Mach, R.H.; Liu, J.; Luedtke, R.R. Analogues of arylamide phenylpiperazine ligands to investigate the factors influencing D3 dopamine receptor bitropic binding and receptor subtype selectivity. ACS Chem. Neurosci. 2018, 9, 2972–2983. [Google Scholar] [CrossRef]
- Moritz, A.E.; Bonifazi, A.; Guerrero, A.M.; Kumar, V.; Free, R.B.; Lane, J.R.; Verma, R.K.; Shi, L.; Newman, A.H.; Sibley, D.R. Evidence for a stereoselective mechanism for bitopic activity by extended-length antagonists of the D3 dopamine receptor. ACS Chem. Neurosci. 2020, 11, 3309–3320. [Google Scholar] [CrossRef]
- Shaik, A.B.; Boateng, C.A.; Battiti, F.O.; Bonifazi, A.; Cao, J.; Chen, L.; Chitsazi, R.; Ravi, S.; Lee, K.H.; Shi, L.; et al. Structure Activity Relationships for a Series of Eticlopride-Based Dopamine D2/D3 Receptor Bitopic Ligands. J. Med. Chem. 2021, 64, 15313–15333. [Google Scholar] [CrossRef]
- Kim, H.Y.; Lee, J.Y.; Hsieh, C.-J.; Taylor, M.; Luedtke, R.R.; Mach, R.H. Design and Synthesis of Conformationally Flexible Scaffold as Bitopic Ligands for Potent D3-Selective Antagonists. Int. J. Mol. Sci. 2022, 24, 432. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.; Hou, T. End-point binding free energy calculation with MM/PBSA and MM/GBSA: Strategies and applications in drug design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Zheng, G.; Yang, F.; Fu, T.; Tu, G.; Chen, Y.; Yao, X.; Xue, W.; Zhu, F. Computational characterization of the selective inhibition of human norepinephrine and serotonin transporters by an escitalopram scaffold. Phys. Chem. Chem. Phys. 2018, 20, 29513–29527. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, G.; Fu, T.; Hong, J.; Li, F.; Yao, X.; Xue, W.; Zhu, F. The binding mode of vilazodone in the human serotonin transporter elucidated by ligand docking and molecular dynamics simulations. Phys. Chem. Chem. Phys. 2020, 22, 5132–5144. [Google Scholar] [CrossRef]
- Künze, G.; Kümpfel, R.; Rullmann, M.; Barthel, H.; Brendel, M.; Patt, M.; Sabri, O. Molecular Simulations Reveal Distinct Energetic and Kinetic Binding Properties of [18F] PI-2620 on Tau Filaments from 3R/4R and 4R Tauopathies. ACS Chem. Neurosci. 2022, 13, 2222–2234. [Google Scholar] [CrossRef]
- Murugan, N.A.; Nordberg, A.; Ågren, H. Cryptic sites in tau fibrils explain the preferential binding of the AV-1451 PET tracer toward Alzheimer’s tauopathy. ACS Chem. Neurosci. 2021, 12, 2437–2447. [Google Scholar] [CrossRef]
- Kuang, G.; Murugan, N.A.; Zhou, Y.; Nordberg, A.; Ågren, H. Computational insight into the binding profile of the second-generation PET tracer PI2620 with tau fibrils. ACS Chem. Neurosci. 2020, 11, 900–908. [Google Scholar] [CrossRef]
- Murugan, N.A.; Nordberg, A.; Ågren, H. Different positron emission tomography tau tracers bind to multiple binding sites on the tau fibril: Insight from computational modeling. ACS Chem. Neurosci. 2018, 9, 1757–1767. [Google Scholar] [CrossRef] [Green Version]
- Thai, N.Q.; Bednarikova, Z.; Gancar, M.; Linh, H.Q.; Hu, C.K.; Li, M.S.; Gazova, Z. Compound CID 9998128 Is a Potential Multitarget Drug for Alzheimer’s Disease. Acs Chem. Neurosci. 2018, 9, 2588–2598. [Google Scholar] [CrossRef]
- Lougee, M.G.; Pagar, V.V.; Kim, H.J.; Pancoe, S.X.; Chia, W.K.; Mach, R.H.; Garcia, B.A.; Petersson, E.J. Harnessing the intrinsic photochemistry of isoxazoles for the development of chemoproteomic crosslinking methods. Chem. Commun. 2022, 58, 9116–9119. [Google Scholar] [CrossRef]
- Janssen, B.; Tian, G.; Lengyel-Zhand, Z.; Hsieh, C.-J.; Lougee, M.G.; Riad, A.; Xu, K.; Hou, C.; Weng, C.-C.; Lopresti, B.J.; et al. A Novel radioligand for in vitro and in vivo α-synuclein imaging. Submitted.
- Pancoe, S.X.; Wang, Y.J.; Shimogawa, M.; Perez, R.M.; Giannakoulias, S.; Petersson, E.J. Effects of Mutations and Post-Translational Modifications on α-Synuclein In Vitro Aggregation. J. Mol. Biol. 2022, 434, 167859. [Google Scholar] [CrossRef]
- Zhao, K.; Lim, Y.-J.; Liu, Z.; Long, H.; Sun, Y.; Hu, J.-J.; Zhao, C.; Tao, Y.; Zhang, X.; Li, D.; et al. Parkinson’s disease-related phosphorylation at Tyr39 rearranges α-synuclein amyloid fibril structure revealed by cryo-EM. Proc. Natl. Acad. Sci. USA 2020, 117, 20305–20315. [Google Scholar] [CrossRef]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Falcon, B.; Zhang, W.J.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Vidal, R.; Crowther, R.A.; Ghetti, B.; Scheres, S.H.W.; Goedert, M. Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature 2018, 561, 137–140. [Google Scholar] [CrossRef]
- Falcon, B.; Zivanov, J.; Zhang, W.J.; Murzin, A.G.; Garringer, H.J.; Vidal, R.; Crowther, R.A.; Newell, K.L.; Ghetti, B.; Goedert, M.; et al. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature 2019, 568, 420–423. [Google Scholar] [CrossRef]
- Zhang, W.J.; Tarutani, A.; Newell, K.L.; Murzin, A.G.; Matsubara, T.; Falcon, B.; Vidal, R.; Garringer, H.J.; Shi, Y.; Ikeuchi, T.; et al. Novel tau filament fold in corticobasal degeneration. Nature 2020, 580, 283–287. [Google Scholar] [CrossRef]
- Arakhamia, T.; Lee, C.E.; Carlomagno, Y.; Duong, D.M.; Kundinger, S.R.; Wang, K.; Williams, D.; DeTure, M.; Dickson, D.W.; Cook, C.N.; et al. Posttranslational Modifications Mediate the Structural Diversity of Tauopathy Strains. Cell 2020, 180, 633–644. [Google Scholar] [CrossRef]
- Hocke, C.; Prante, O.; Salama, I.; Hübner, H.; Löber, S.; Kuwert, T.; Gmeiner, P. 18F-labeled FAUC 346 and BP 897 derivatives as subtype-selective potential PET radioligands for the dopamine D3 receptor. ChemMedChem: Chem. Enabling Drug Discov. 2008, 3, 788–793. [Google Scholar] [CrossRef]
- López, L.; Selent, J.; Ortega, R.; Masaguer, C.F.; Domínguez, E.; Areias, F.; Brea, J.; Loza, M.I.; Sanz, F.; Pastor, M. Synthesis, 3D-QSAR, and Structural Modeling of Benzolactam Derivatives with Binding Affinity for the D2 and D3 Receptors. ChemMedChem 2010, 5, 1300–1317. [Google Scholar] [CrossRef]
- Wang, Q.; Mach, R.H.; Luedtke, R.R.; Reichert, D.E. Subtype selectivity of dopamine receptor ligands: Insights from structure and ligand-based methods. J. Chem. Inf. Model. 2010, 50, 1970–1985. [Google Scholar] [CrossRef] [Green Version]
- De Simone, A.; Russo, D.; Ruda, G.F.; Micoli, A.; Ferraro, M.; Di Martino, R.M.C.; Ottonello, G.; Summa, M.; Armirotti, A.; Bandiera, T.; et al. Design, Synthesis, Structure–Activity Relationship Studies, and Three-Dimensional Quantitative Structure–Activity Relationship (3D-QSAR) Modeling of a Series of O-Biphenyl Carbamates as Dual Modulators of Dopamine D3 Receptor and Fatty Acid Amide Hydrolase. J. Med. Chem. 2017, 60, 2287–2304. [Google Scholar] [CrossRef]
- Li, A.; Mishra, Y.; Malik, M.; Wang, Q.; Li, S.; Taylor, M.; Reichert, D.E.; Luedtke, R.R.; Mach, R.H. Evaluation of N-phenyl homopiperazine analogs as potential dopamine D3 receptor selective ligands. Bioorganic Med. Chem. 2013, 21, 2988–2998. [Google Scholar] [CrossRef] [Green Version]
- De, P.; Roy, K. QSAR modeling of PET imaging agents for the diagnosis of Parkinson’s disease targeting dopamine receptor. Theor. Chem. Acc. 2020, 139, 176. [Google Scholar] [CrossRef]
- Radan, M.; Ruzic, D.; Antonijevic, M.; Djikic, T.; Nikolic, K. In silico identification of novel 5-HT2A antagonists supported with ligand-and target-based drug design methodologies. J. Biomol. Struct. Dyn. 2021, 39, 1819–1837. [Google Scholar] [CrossRef]
- Rescifina, A.; Floresta, G.; Marrazzo, A.; Parenti, C.; Prezzavento, O.; Nastasi, G.; Dichiara, M.; Amata, E. Development of a Sigma-2 Receptor affinity filter through a Monte Carlo based QSAR analysis. Eur. J. Pharm. Sci. 2017, 106, 94–101. [Google Scholar] [CrossRef]
- Floresta, G.; Rescifina, A.; Marrazzo, A.; Dichiara, M.; Pistarà, V.; Pittalà, V.; Prezzavento, O.; Amata, E. Hyphenated 3D-QSAR statistical model-scaffold hopping analysis for the identification of potentially potent and selective sigma-2 receptor ligands. Eur. J. Med. Chem. 2017, 139, 884–891. [Google Scholar] [CrossRef]
- Ambure, P.; Roy, K. Exploring structural requirements of imaging agents against Aβ plaques in Alzheimer’s disease: A QSAR approach. Comb. Chem. High Throughput Screen. 2015, 18, 411–419. [Google Scholar] [CrossRef]
- Cisek, K.; Kuret, J. QSAR studies for prediction of cross-β sheet aggregate binding affinity and selectivity. Bioorganic Med. Chem. 2012, 20, 1434–1441. [Google Scholar] [CrossRef] [Green Version]
- Kovac, M.; Mavel, S.; Deuther-Conrad, W.; Méheux, N.; Glöckner, J.; Wenzel, B.; Anderluh, M.; Brust, P.; Guilloteau, D.; Emond, P. 3D QSAR study, synthesis, and in vitro evaluation of (+)-5-FBVM as potential PET radioligand for the vesicular acetylcholine transporter (VAChT). Bioorganic Med. Chem. 2010, 18, 7659–7667. [Google Scholar] [CrossRef]
- Sarhan, M.O.; Abd El-Karim, S.S.; Anwar, M.M.; Gouda, R.H.; Zaghary, W.A.; Khedr, M.A. Discovery of New Coumarin-Based Lead with Potential Anticancer, CDK4 Inhibition and Selective Radiotheranostic Effect: Synthesis, 2D & 3D QSAR, Molecular Dynamics, In Vitro Cytotoxicity, Radioiodination, and Biodistribution Studies. Molecules 2021, 26, 2273. [Google Scholar] [CrossRef]
- Wang, Q.; Mach, R.H.; Reichert, D.E. Docking and 3D-QSAR studies on isatin sulfonamide analogues as caspase-3 inhibitors. J. Chem. Inf. Model. 2009, 49, 1963–1973. [Google Scholar] [CrossRef] [Green Version]
- Munoz, C.; Adasme, F.; Alzate-Morales, J.H.; Vergara-Jaque, A.; Kniess, T.; Caballero, J. Study of differences in the VEGFR2 inhibitory activities between semaxanib and SU5205 using 3D-QSAR, docking, and molecular dynamics simulations. J. Mol. Graph. Model. 2012, 32, 39–48. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Target | # of Compounds/ Compound Library | Hit Rate a | Binding Affinity of Hits | Literature |
|---|---|---|---|---|---|
| Structure-based virtual screening | |||||
| Docking | μ-opioid receptor | 3 M/ZINC | 23/23 | 2.3–14 μM | Manglik et al., 2016 [16] |
| Docking | Mas-related G protein- coupled receptor X2 (MRGPRX2) | 3.7 M/ZINC | 20/20 | <10 μM | Lansu et al., 2017 [17] |
| Docking | Histamine H1 receptor | 100 K/ZINC | 19/26 (73%) | 6 nM–10 μM | De Graaf et al., 2011 [18] |
| Docking | Histamine H4 receptor | 8.7 M/ZINC | 16/255 (6%) | 85–1480 nM | Kiss et al., 2008 [19] |
| Docking | Histamine H4 receptor | 7 K/Bioprojet chemical library | 28/120 (23%) | 4 nM–16 μM | Levoin et al., 2017 [20] |
| Docking | Melanin-concentrating hormone receptor 1 (MCH-R1) | 187 K/In-house collection [21] | 6/129 (5%) | 7–20 μM | Cavasotto et al., 2008 [22] |
| Docking | Chemokine receptor CCR5 | 1.6 M/8 vendors | 10/59 (17%) | 5–200 μM | Kellenberger et al., 2007 [23] |
| Docking | Adenosine receptor A2A | 1.4 M/ZINC | 7/20 (35%) | 200 nM–9 μM | Carlsson et al., 2010 [24] |
| Docking | Adenosine receptor A2A | 4.3 M/Molsoft ScreenPub | 23/56 (41%) | <10 μM | Katritch et al., 2010 [25] |
| Docking | β2-adrenergic receptor | 1 M/ZINC | 6/25 (24%) | <4 μM | Kolb et al., 2009 [26] |
| Docking | Dopamine D2 receptor | 6.5 M/Enamine | 10/21 (48%) | 58 nM–25 μM | Kaczor et al., 2016 [27] |
| Docking | Choline acetyltransferase (ChAT) | 300 K/Asinex Gold and Platinum collection library | 3/35 (9%) | 7–26 μM | Kumar et al., 2017 [28] |
| Docking | Tau fibrils | 62 K/FDA-approved small molecule drugs and ChemBridge CNS-set | 4/46 (9%) | <5 μM | Seidler et al., 2022 [29] |
| Docking | Dopamine D3 receptor | 1.5 M/ChemDiv | 27/37(73%) | <10 μM | Jin et al., 2023 [30] |
| Pharmacophore | Formylpeptide receptor (FPR) | 480 K/Chemical Diversity Laboratories [31] | 30/4324 (0.7%) | 1–32 μM | Edwards et al., 2005 [32] |
| Pharmacophore | complement component 3a receptor 1 (C3AR1) | -/In-house collection | 4/157 (3%) | <10 μM | Klabunde et al., 2009 [33] |
| Pharmacophore | Alpha-synuclein fibrils | 10 M/ZINC15 | 2/17 (12%) | 10–490 nM | Ferrie et al., 2020 [2] |
| Pharmacophore | Histamine H4 receptor | 22 M/ZINC12 | 3/291 (1%) | <10 μM | Ko et al., 2018 [34] |
| Pharmacophore Docking | Sphingosine kinase 1 (SphK1) | 147/Custom-selected Library | 3/16 (19%) | 12–60 μM | Vettorazzi et al., 2017 [35] |
| Pharmacophore Docking | Serotonin transporter (SERT) | 1 M/ZINC | 2/15 (13%) | 17–38 μM | Manepalli et al., 2011 [36] |
| Pharmacophore Docking | Thyrotropin-releasing hormone receptor1 (TRH-R1) | 1 M/ZINC | 100/100 | Sub μM–μM | Engel et al., 2008 [37] |
| Pharmacophore Docking | Alpha1A adrenergic receptor | 23 K/MDL Drug Data Report | 37/80 (46%) | <10 μM | Evers et al., 2005 [38] |
| Pharmacophore Docking | Neurokinin-1 (NK1) receptor | 827 K/7 databases | 1/7 (14%) | 0.25 μM | Evers et al., 2004 [39] |
| Machine learning | Acetylcholinesterase (AchE) | 15 M/Enamine REAL database | 10/23(43%) | <50 μM | Adeshina et al., 2020 [40] |
| Ligand-based virtual screening | |||||
| Pharmacophore | Metabotropic glutamate receptor 5 (mGluR5) | 194 K/Asinex Gold compound collection | 9/27 (33%) | <70 μM | Renner et al., 2005 [41] |
| Pharmacophore | Metabotropic glutamate receptor 1 (mGluR1) | 201 K/Asinex Gold Collection | 6/23 (26%) | 0.75–>40 μM | Noeske et al., 2007 [42] |
| 2D-QSAR | Sigma 2 receptor | 2 K/DrugBank | 10/34 (29%) | 140 nM–μM | Yu et. al., 2021 [43] |
| 2D Fingerprint | Sigma 2 receptor | 47 M/MCule Inc. | 12/46 (26%) | 0.6–700 nM | Kim et al., 2022 [3] |
| Ligand- and structure-based virtual screening | |||||
| 2D/3D-QSAR Docking | Sigma 2 receptor | 1517/Seaweed Metabolite and ChEBI | 15/15 | 0.6–5.3 nM | Floresta et al., 2018 [44] |
| 2D Fingerprint Pharmacophore | Melanin-concentrating hormone 1 receptor (MCH-1) | 615 K/24 Vendors | 15/795 (1.9%) | 1–30 μM | Clark et al., 2004 [21] |
| Similarity Pharmacophore Docking | Free fatty acid receptor 1 (FFAR1) | 2.6 M/ZINC | 6/52 (12%) | <10 μM | Tikhonova et al., 2008 [45] |
| Pharmacophore Docking | Subtype six serotonin receptor (5-HT6) | -/Princeton BM and ChemBridge | 14/92 (15%) | <1 μM | Staron et al., 2020 [46] |
| Pharmacophore Docking | 5-HT7 receptor (5-HT7R) | 730 K/Enamine Screening Collection | 2/26 (8%) | 197–265 nM | Kurczab et al., 2010 [47] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsieh, C.-J.; Giannakoulias, S.; Petersson, E.J.; Mach, R.H. Computational Chemistry for the Identification of Lead Compounds for Radiotracer Development. Pharmaceuticals 2023, 16, 317. https://doi.org/10.3390/ph16020317
Hsieh C-J, Giannakoulias S, Petersson EJ, Mach RH. Computational Chemistry for the Identification of Lead Compounds for Radiotracer Development. Pharmaceuticals. 2023; 16(2):317. https://doi.org/10.3390/ph16020317
Chicago/Turabian StyleHsieh, Chia-Ju, Sam Giannakoulias, E. James Petersson, and Robert H. Mach. 2023. "Computational Chemistry for the Identification of Lead Compounds for Radiotracer Development" Pharmaceuticals 16, no. 2: 317. https://doi.org/10.3390/ph16020317
APA StyleHsieh, C.-J., Giannakoulias, S., Petersson, E. J., & Mach, R. H. (2023). Computational Chemistry for the Identification of Lead Compounds for Radiotracer Development. Pharmaceuticals, 16(2), 317. https://doi.org/10.3390/ph16020317