
Alvespimycin Exhibits Potential Anti-TGF-β Signaling in the Setting of a Proteasome Activator in Rats with Bleomycin-Induced Pulmonary Fibrosis: A Promising Novel Approach
 , , , ,
, , , ,  and
and 
Abstract
:1. Introduction
2. Results
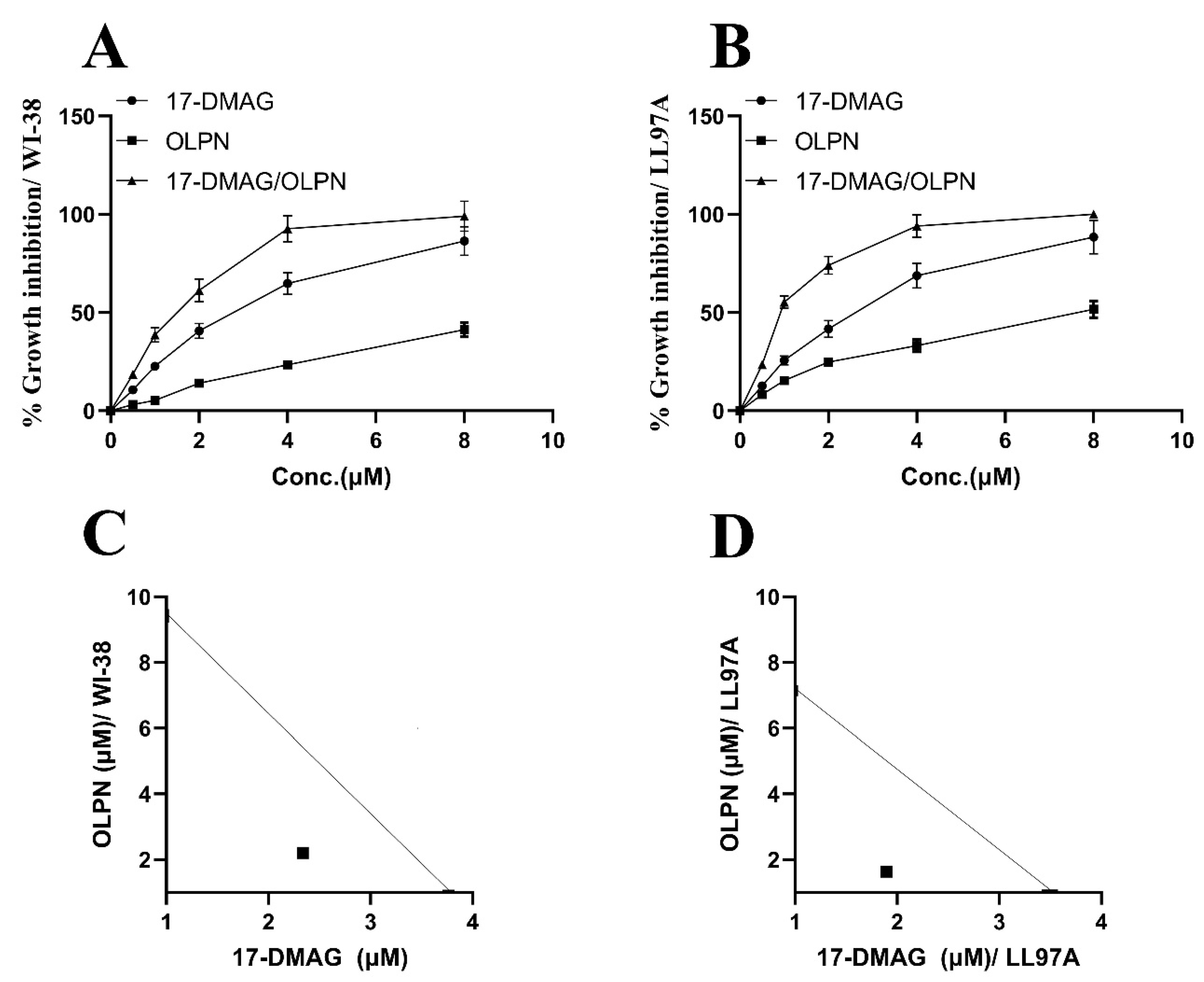
2.1. Effect of Alvespimycin and OLPN on Cell Viability
2.2. Effect on the Combination Index between Alvespimycin and OLPN
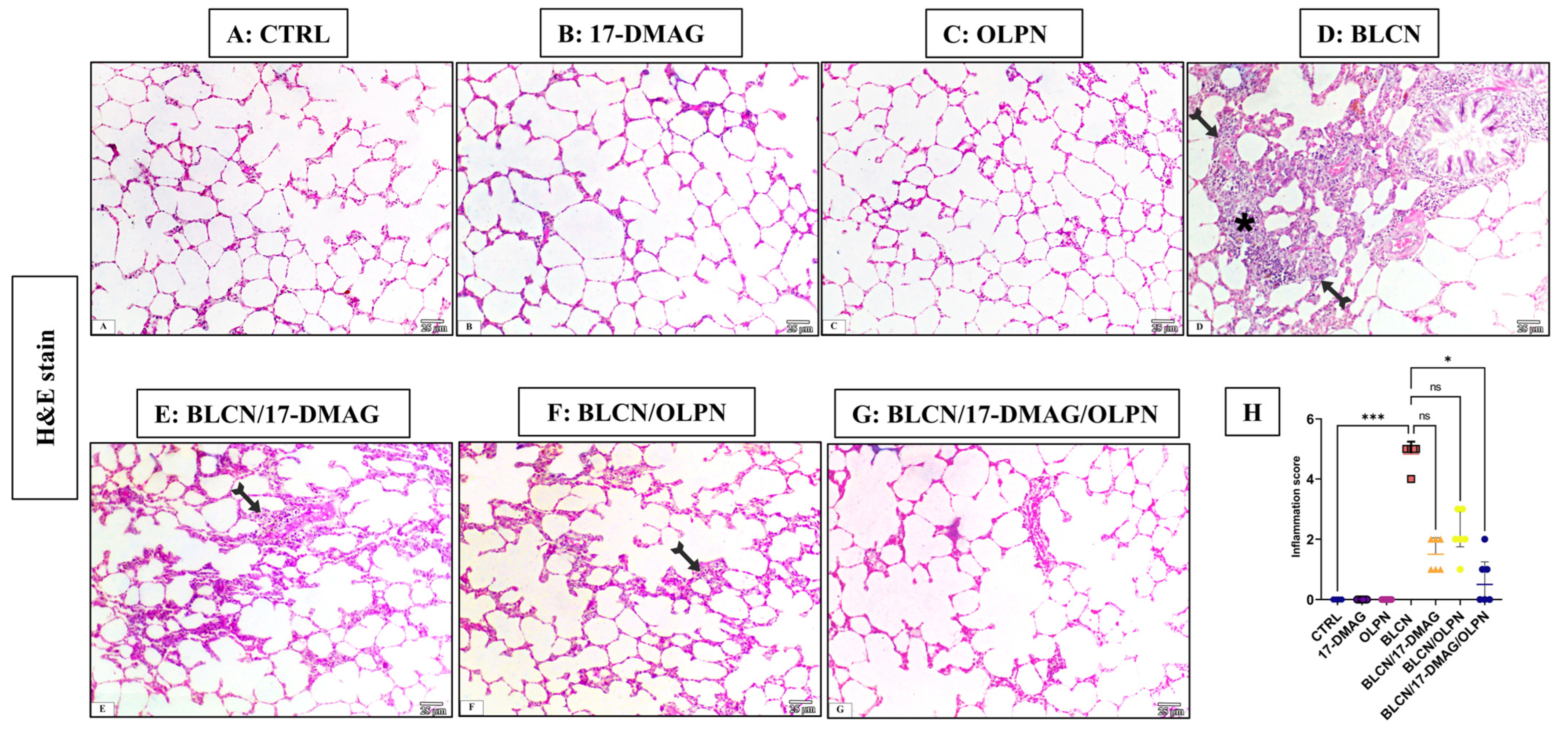
2.3. Histopathological Examination of Lung Tissue Sections
2.3.1. H&E Stain
2.3.2. Masson’s Trichrome Stain
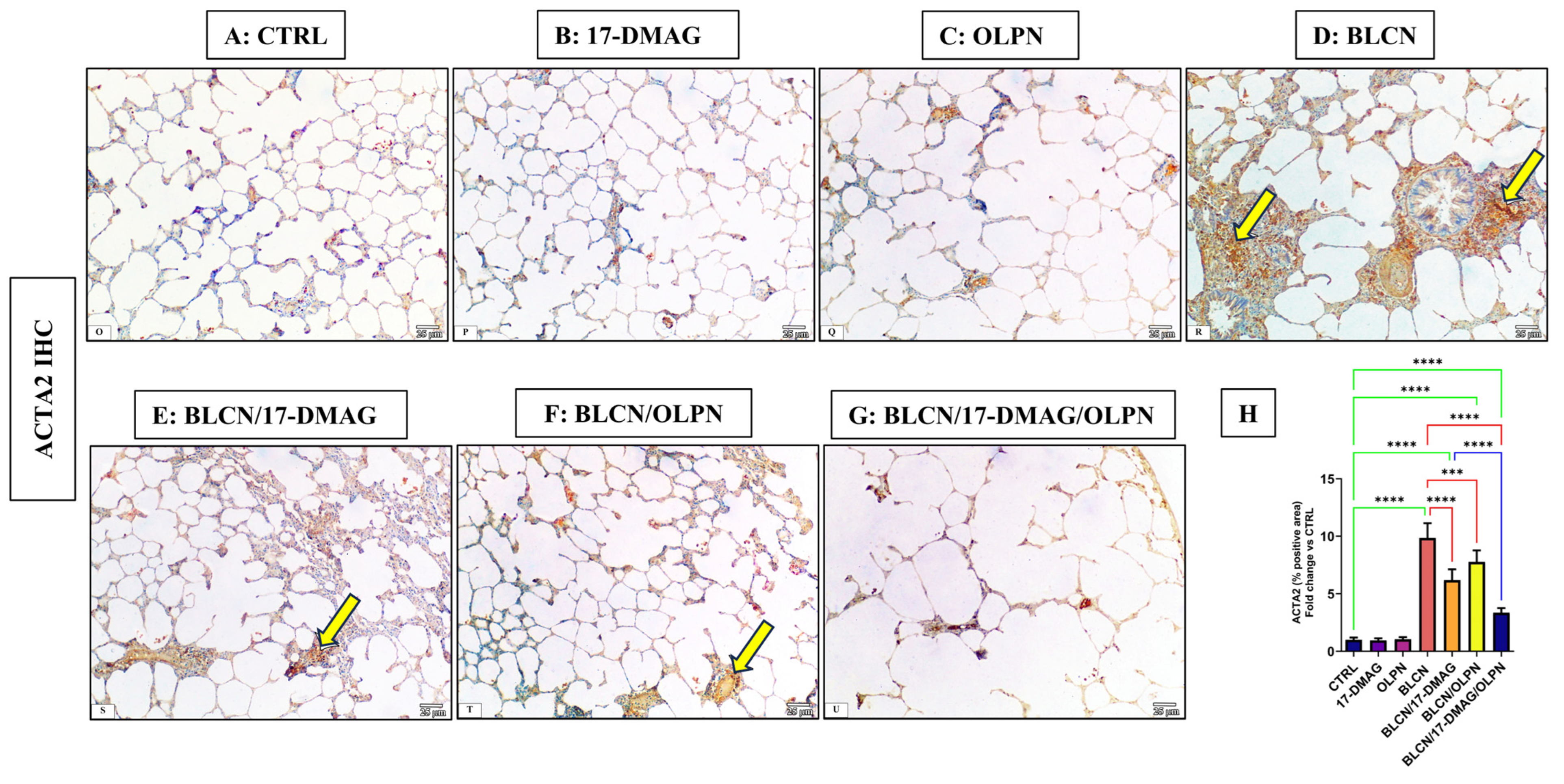
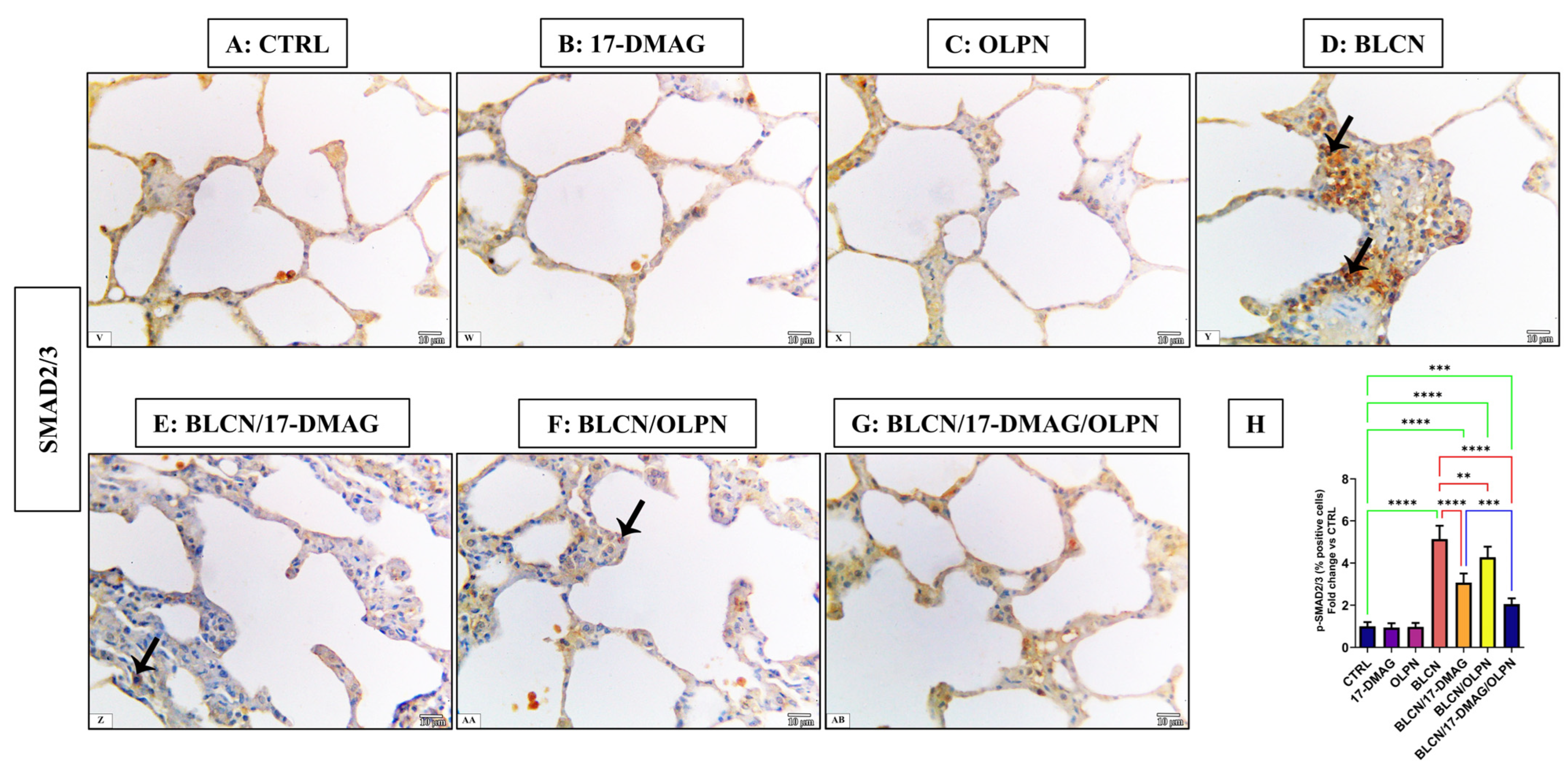
2.4. Effect on the Immune Expression of ACTA2 and p-SMAD2/3
2.4.1. Immuno-Expression of ACTA2
2.4.2. Immuno-Expression of p-SMAD2/3
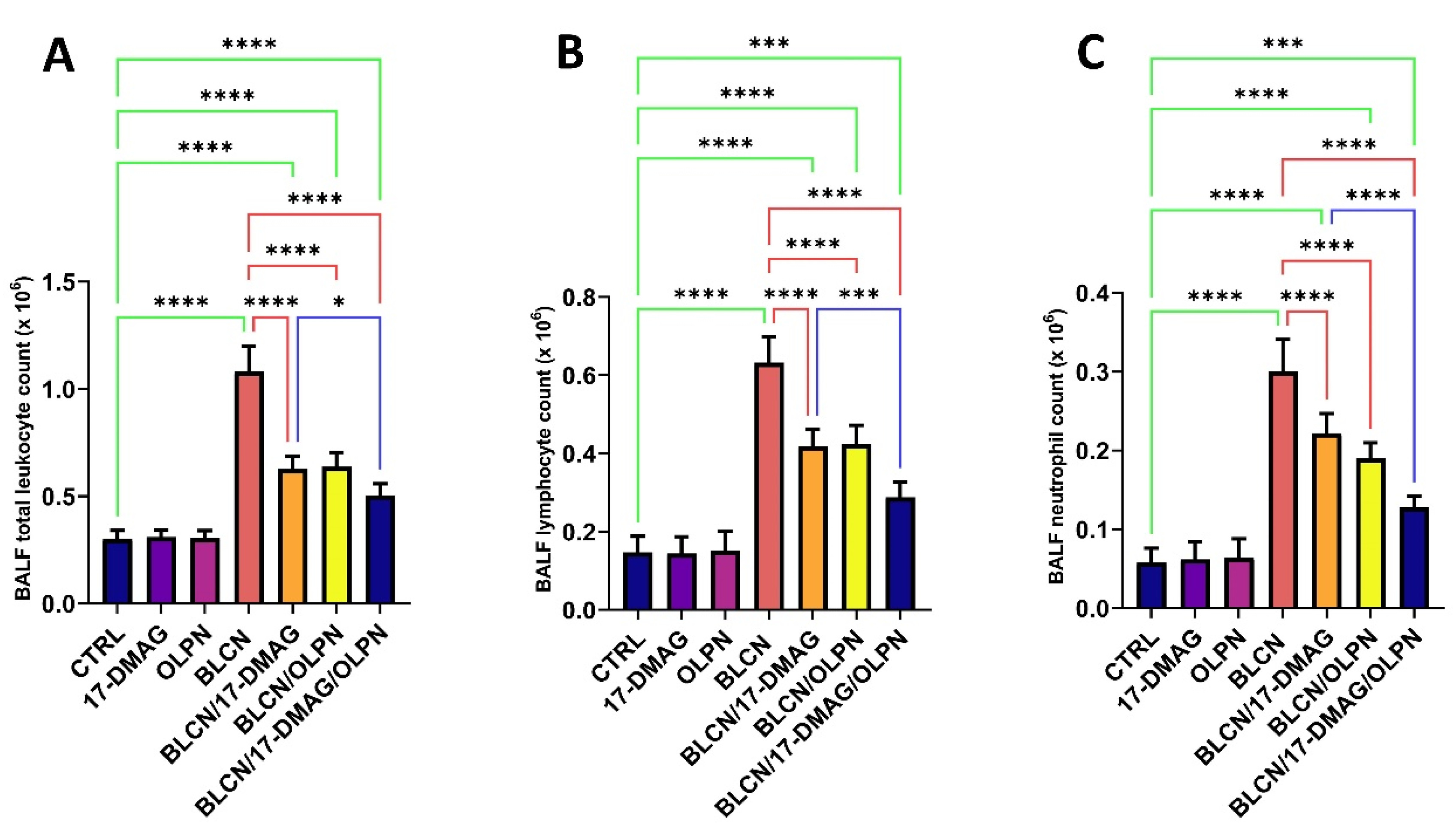
2.5. Effect on Total Leukocyte Count, Lymphocyte Count, and Neutrophil Count in the BALF
2.6. Effect on BALF Total Protein, LDH, and NOx
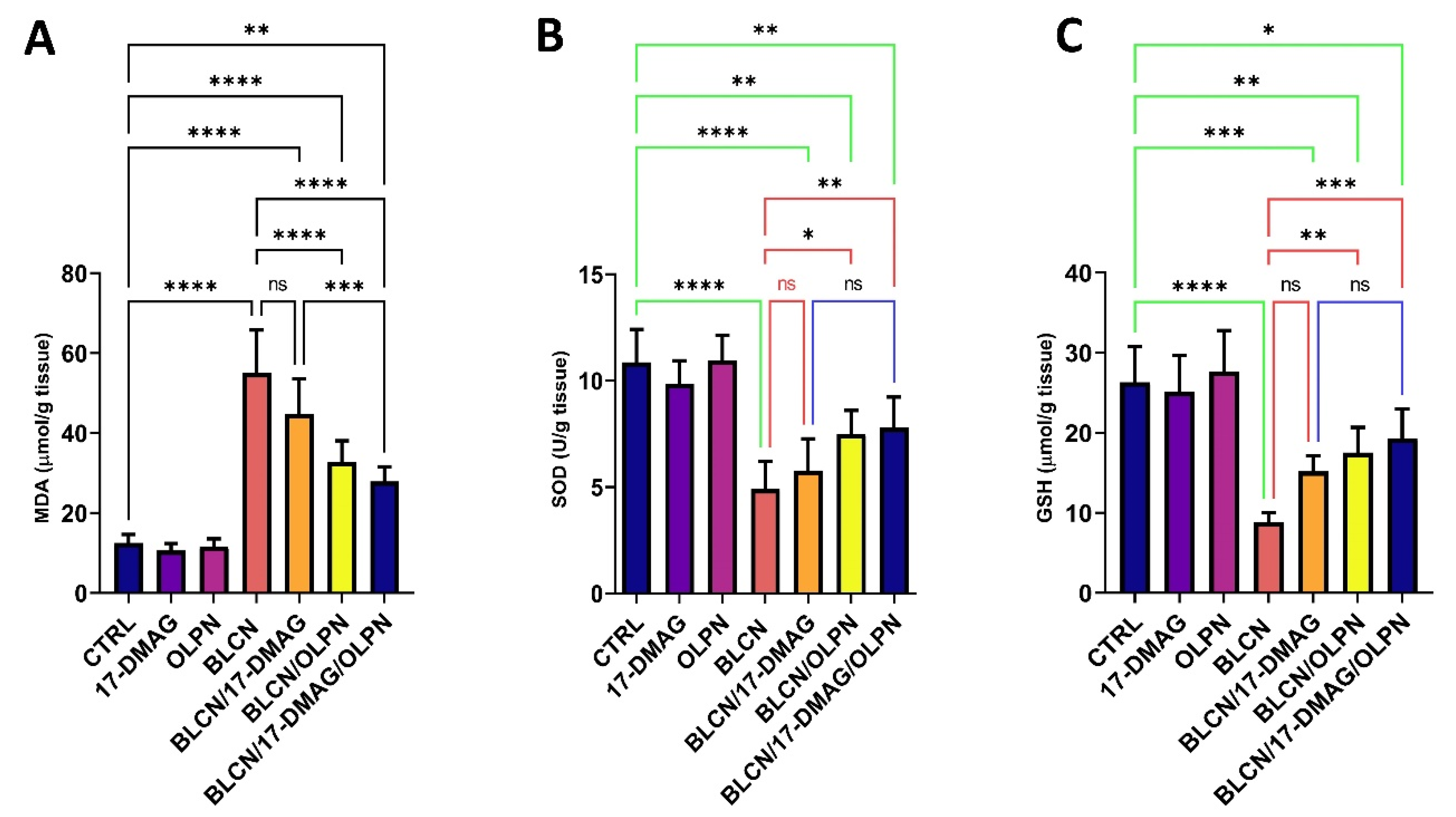
2.7. Effect on MDA, SOD, and GSH
2.8. Effect on PDGF-BB, TIMP-1, COL1A1 mRNA, and Hydroxyproline
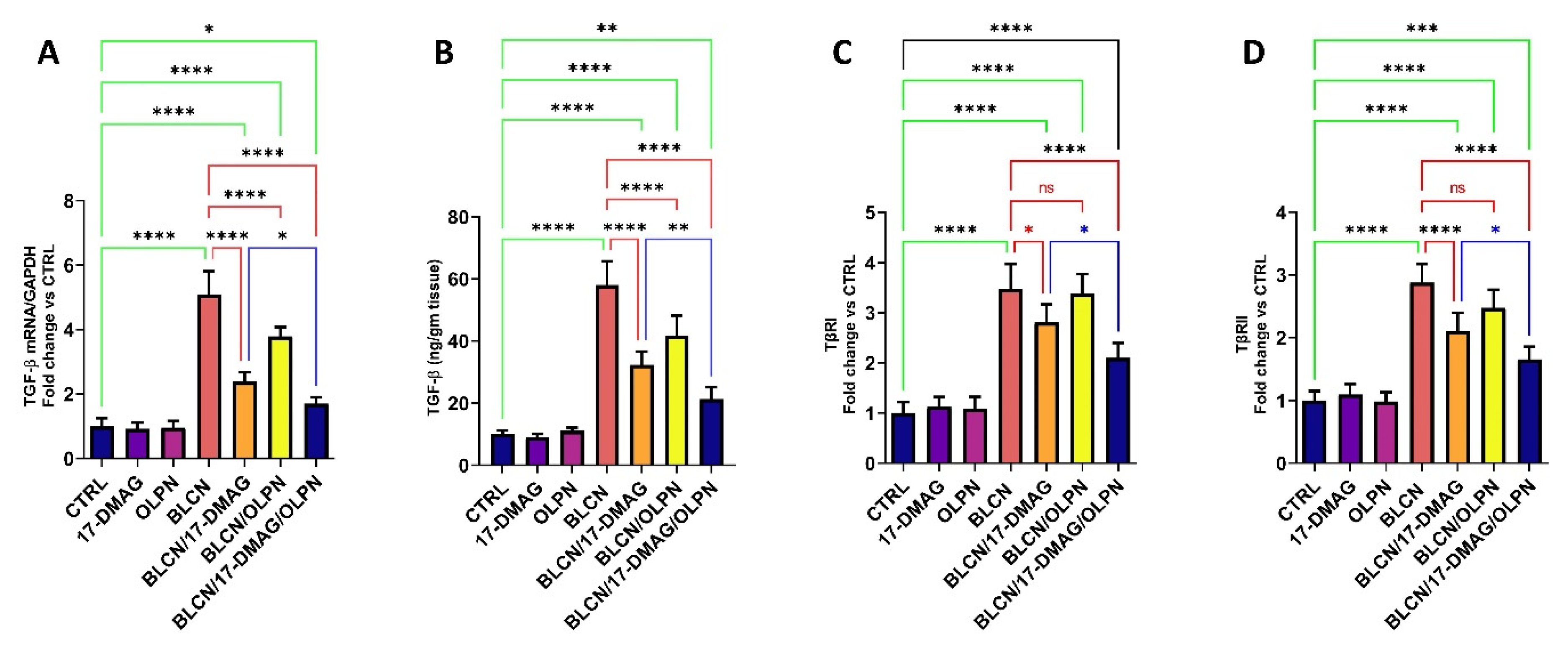
2.9. Effect on TGF-β mRNA, TGF-β, TβRI, and TβRII
2.10. Effect of 17-DMAG and OLPN on 20S Proteasomal Activity in Lung Tissue
2.11. Effect on HSP70 and HSP90
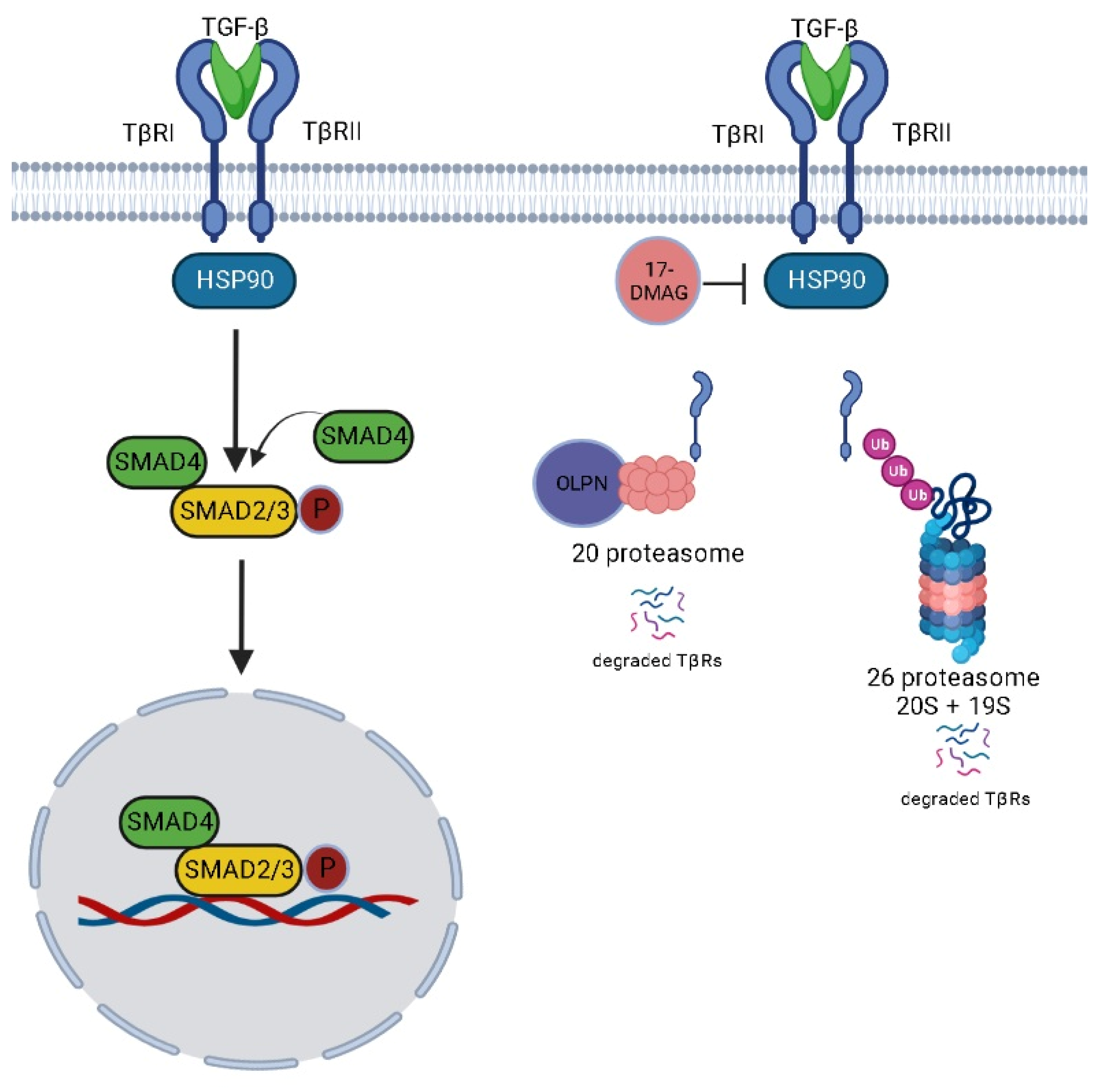
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Cell Viability
4.2. Determination of the Index of Combination
4.3. Induction of Pulmonary Fibrosis in Rats
4.4. Study Design
4.5. Rationale of Drug Dosing
4.6. Histological Examination of Lung Tissue Sections
4.7. Immunohistochemical Examination of Lung Tissue Sections
4.8. Determination of Bronchoalveolar Lavage Fluid (BALF) Total Leukocyte, Neutrophil, and Lymphocyte Count
4.9. Determination of Lactate Dehydrogenase (LDH) Activity, BALF Total Protein, and Total Nitrite and Nitrate (NOx)
4.10. Determination of Malondialdehyde (MDA), Superoxide Dismutase (SOD), and Reduced Glutathione (GSH)
4.11. Determination of Platelet-Derived Growth Factor BB (PDGF-BB), Tissue Inhibitor of Metalloproteinase (TIMP-1), and Hydroxyproline
4.12. Determination of TGF-β mRNA and Collagen Type I, Alpha 1 (COL1A1) mRNA
4.13. Determination of TGF-β, TβRI, and TβRII
4.14. Determination of HSP70 and HSP90
4.15. Determination of the Proteasomal Activity in Lung Tissue
4.16. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- El-Kashef, D.H.; Youssef, M.E.; Nasr, M.; Alrouji, M.; Alhajlah, S.; AlOmeir, O.; El Adle Khalaf, N.; Ghaffar, D.M.A.; Jamil, L.; Abdel-Nasser, Z.M.; et al. Pimitespib, an HSP90 inhibitor, augments nifuroxazide-induced disruption in the IL-6/STAT3/HIF-1α autocrine loop in rats with bleomycin-challenged lungs: Evolutionary perspective in managing pulmonary fibrosis. Biomed. Pharmacother. 2022, 153, 113487. [Google Scholar] [CrossRef] [PubMed]
- Wolters, P.J.; Blackwell, T.S.; Eickelberg, O.; Loyd, J.E.; Kaminski, N.; Jenkins, G.; Maher, T.M.; Molina-Molina, M.; Noble, P.W.; Raghu, G.; et al. Time for a change: Is idiopathic pulmonary fibrosis still idiopathic and only fibrotic? Lancet Respir. Med. 2018, 6, 154–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selman, M.; Pardo, A. Revealing the pathogenic and aging-related mechanisms of the enigmatic idiopathic pulmonary fibrosis. an integral model. Am. J. Respir. Crit. Care Med. 2014, 189, 1161–1172. [Google Scholar] [CrossRef]
- Miao, H.; Wu, X.-Q.; Zhang, D.-D.; Wang, Y.-N.; Guo, Y.; Li, P.; Xiong, Q.; Zhao, Y.-Y. Deciphering the cellular mechanisms underlying fibrosis-associated diseases and therapeutic avenues. Pharmacol. Res. 2021, 163, 105316. [Google Scholar] [CrossRef]
- Herrera, J.; Henke, C.A.; Bitterman, P.B. Extracellular matrix as a driver of progressive fibrosis. J. Clin. Investig. 2018, 128, 45–53. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.; Dalurzo, M.; Panse, P.; Parish, J.; Leslie, K. Usual interstitial pneumonia-pattern fibrosis in surgical lung biopsies. Clinical, radiological and histopathological clues to aetiology. J. Clin. Pathol. 2013, 66, 896–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Confalonieri, P.; Volpe, M.C.; Jacob, J.; Maiocchi, S.; Salton, F.; Ruaro, B.; Confalonieri, M.; Braga, L. Regeneration or Repair? The Role of Alveolar Epithelial Cells in the Pathogenesis of Idiopathic Pulmonary Fibrosis (IPF). Cells 2022, 11, 2095. [Google Scholar] [CrossRef]
- Biernacka, A.; Dobaczewski, M.; Frangogiannis, N.G. TGF-β signaling in fibrosis. Growth Factors 2011, 29, 196–202. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.-F.; Lin, H.; Liu, D.-C.; Zhu, X.-Y.; Huang, N.; Wei, Y.-X.; Li, L. Heat shock protein 90 inhibitor ameliorates pancreatic fibrosis by degradation of transforming growth factor-β receptor. Cell. Signal. 2021, 84, 110001. [Google Scholar] [CrossRef]
- Kasuya, Y.; Kim, J.-D.; Hatano, M.; Tatsumi, K.; Matsuda, S. Pathophysiological Roles of Stress-Activated Protein Kinases in Pulmonary Fibrosis. Int. J. Mol. Sci. 2021, 22, 6041. [Google Scholar] [CrossRef]
- Saber, S.; Abd El-Kader, E.M.; Sharaf, H.; El-Shamy, R.; El-Saeed, B.; Mostafa, A.; Ezzat, D.; Shata, A. Celastrol augments sensitivity of NLRP3 to CP-456773 by modulating HSP-90 and inducing autophagy in dextran sodium sulphate-induced colitis in rats. Toxicol. Appl. Pharmacol. 2020, 400, 115075. [Google Scholar] [CrossRef] [PubMed]
- Shaaban, A.A.; Abdelhamid, A.M.; Shaker, M.E.; Cavalu, S.; Maghiar, A.M.; Alsayegh, A.A.; Babalghith, A.O.; El-Ahwany, E.; Amin, N.A.; Mohammed, O.A.; et al. Combining the HSP90 inhibitor TAS-116 with metformin effectively degrades the NLRP3 and attenuates inflammasome activation in rats: A new management paradigm for ulcerative colitis. Biomed. Pharmacother. 2022, 153, 113247. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, Q.; You, Q. Targeting the HSP90–CDC37–kinase chaperone cycle: A promising therapeutic strategy for cancer. Med. Res. Rev. 2022, 42, 156–182. [Google Scholar] [CrossRef] [PubMed]
- Bellaye, P.-S.; Burgy, O.; Bonniaud, P.; Kolb, M. HSP47: A potential target for fibrotic diseases and implications for therapy. Expert Opin. Ther. Targets 2021, 25, 49–62. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, X.; Huang, W.; Ge, X. The role of heat shock proteins in the regulation of fibrotic diseases. Biomed. Pharmacother. 2021, 135, 111067. [Google Scholar] [CrossRef]
- Liu, J.; Jin, J.; Liang, T.; Feng, X.-H. To Ub or not to Ub: A regulatory question in TGF-β signaling. Trends Biochem. Sci. 2022, 47, 1059–1072. [Google Scholar] [CrossRef]
- Bickel, D.; Gohlke, H. C-terminal modulators of heat shock protein of 90 kDa (HSP90): State of development and modes of action. Bioorg. Med. Chem. 2019, 27, 115080. [Google Scholar] [CrossRef]
- Radovanac, K.; Morgner, J.; Schulz, J.-N.; Blumbach, K.; Patterson, C.; Geiger, T.; Mann, M.; Krieg, T.; Eckes, B.; Fässler, R.; et al. Stabilization of integrin-linked kinase by the Hsp90-CHIP axis impacts cellular force generation, migration and the fibrotic response. EMBO J. 2013, 32, 1409–1424. [Google Scholar] [CrossRef] [Green Version]
- Datta, R.; Bansal, T.; Rana, S.; Datta, K.; Chattopadhyay, S.; Chawla-Sarkar, M.; Sarkar, S. Hsp90/Cdc37 assembly modulates TGFβ receptor-II to act as a profibrotic regulator of TGFβ signaling during cardiac hypertrophy. Cell. Signal. 2015, 27, 2410–2424. [Google Scholar] [CrossRef]
- Yun, C.W.; Kim, H.J.; Lim, J.H.; Lee, S.H. Heat Shock Proteins: Agents of Cancer Development and Therapeutic Targets in Anti-Cancer Therapy. Cells 2020, 9, 60. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Zhang, X.-D.; Cheng, G.; Hu, Y.-H.; Wang, H.-Y. Inhibition of hepatic stellate cell proliferation by heat shock protein 90 inhibitors in vitro. Mol. Cell. Biochem. 2009, 330, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.-I.; Tanaka, Y.; Namba, T.; Azuma, A.; Mizushima, T. Heat shock protein 70 protects against bleomycin-induced pulmonary fibrosis in mice. Biochem. Pharmacol. 2010, 80, 920–931. [Google Scholar] [CrossRef] [PubMed]
- Sellares, J.; Veraldi, K.L.; Thiel, K.J.; Cárdenes, N.; Alvarez, D.; Schneider, F.; Pilewski, J.M.; Rojas, M.; Feghali-Bostwick, C.A. Intracellular Heat Shock Protein 70 Deficiency in Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2018, 60, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Namba, T.; Tanaka, K.-I.; Hoshino, T.; Azuma, A.; Mizushima, T. Suppression of Expression of Heat Shock Protein 70 by Gefitinib and Its Contribution to Pulmonary Fibrosis. PLoS ONE 2011, 6, e27296. [Google Scholar] [CrossRef] [Green Version]
- Wrighton, K.H.; Lin, X.; Feng, X.-H. Critical regulation of TGFβ signaling by Hsp90. Proc. Natl. Acad. Sci. USA 2008, 105, 9244–9249. [Google Scholar] [CrossRef]
- Guswanto, A.; Nugraha, A.B.; Tuvshintulga, B.; Tayebwa, D.S.; Rizk, M.A.; Batiha, G.E.; Gantuya, S.; Sivakumar, T.; Yokoyama, N.; Igarashi, I. 17-DMAG inhibits the multiplication of several Babesia species and Theileria equi on in vitro cultures, and Babesia microti in mice. Int. J. Parasitol. Drugs Drug Resist. 2018, 8, 104–111. [Google Scholar] [CrossRef]
- Chondrogianni, N.; Gonos, E.S. Proteasome dysfunction in mammalian aging: Steps and factors involved. Exp. Gerontol. 2005, 40, 931–938. [Google Scholar] [CrossRef]
- Wang, Y.L.; Shen, H.H.; Cheng, P.Y.; Chu, Y.J.; Hwang, H.R.; Lam, K.K.; Lee, Y.M. 17-DMAG, an HSP90 Inhibitor, Ameliorates Multiple Organ Dysfunction Syndrome via Induction of HSP70 in Endotoxemic Rats. PLoS ONE 2016, 11, e0155583. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.; Chen, Y.-G. Regulation of TGF-β receptor activity. Cell Biosci. 2012, 2, 9. [Google Scholar] [CrossRef] [Green Version]
- Katsiki, M.; Chondrogianni, N.; Chinou, I.; Rivett, A.J.; Gonos, E.S. The olive constituent oleuropein exhibits proteasome stimulatory properties in vitro and confers life span extension of human embryonic fibroblasts. Rejuvenation Res. 2007, 10, 157–172. [Google Scholar] [CrossRef]
- El Demerdash, N.; Chen, M.W.; O’Brien, C.E.; Adams, S.; Kulikowicz, E.; Martin, L.J.; Lee, J.K. Oleuropein Activates Neonatal Neocortical Proteasomes, but Proteasome Gene Targeting by AAV9 Is Variable in a Clinically Relevant Piglet Model of Brain Hypoxia-Ischemia and Hypothermia. Cells 2021, 10, 2120. [Google Scholar] [CrossRef] [PubMed]
- Visioli, F.; Bellosta, S.; Galli, C. Oleuropein, the bitter principle of olives, enhances nitric oxide production by mouse macrophages. Life Sci. 1998, 62, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Visioli, F.; Poli, A.; Gall, C. Antioxidant and other biological activities of phenols from olives and olive oil. Med. Res. Rev. 2002, 22, 65–75. [Google Scholar] [CrossRef]
- Carluccio, M.A.; Siculella, L.; Ancora, M.A.; Massaro, M.; Scoditti, E.; Storelli, C.; Visioli, F.; Distante, A.; De Caterina, R. Olive oil and red wine antioxidant polyphenols inhibit endothelial activation: Antiatherogenic properties of Mediterranean diet phytochemicals. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 622–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripoli, E.; Giammanco, M.; Tabacchi, G.; Di Majo, D.; Giammanco, S.; La Guardia, M. The phenolic compounds of olive oil: Structure, biological activity and beneficial effects on human health. Nutr. Res. Rev. 2005, 18, 98–112. [Google Scholar] [CrossRef]
- Owen, R.W.; Giacosa, A.; Hull, W.E.; Haubner, R.; Würtele, G.; Spiegelhalder, B.; Bartsch, H. Olive-oil consumption and health: The possible role of antioxidants. Lancet Oncol. 2000, 1, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Andreadou, I.; Sigala, F.; Iliodromitis, E.K.; Papaefthimiou, M.; Sigalas, C.; Aligiannis, N.; Savvari, P.; Gorgoulis, V.; Papalabros, E.; Kremastinos, D.T. Acute doxorubicin cardiotoxicity is successfully treated with the phytochemical oleuropein through suppression of oxidative and nitrosative stress. J. Mol. Cell. Cardiol. 2007, 42, 549–558. [Google Scholar] [CrossRef]
- Bazoti, F.N.; Bergquist, J.; Markides, K.E.; Tsarbopoulos, A. Noncovalent interaction between amyloid-beta-peptide (1-40) and oleuropein studied by electrospray ionization mass spectrometry. J. Am. Soc. Mass Spectrom. 2006, 17, 568–575. [Google Scholar] [CrossRef] [Green Version]
- Andreadou, I.; Iliodromitis, E.K.; Mikros, E.; Constantinou, M.; Agalias, A.; Magiatis, P.; Skaltsounis, A.L.; Kamber, E.; Tsantili-Kakoulidou, A.; Kremastinos, D.T. The olive constituent oleuropein exhibits anti-ischemic, antioxidative, and hypolipidemic effects in anesthetized rabbits. J. Nutr. 2006, 136, 2213–2219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, R.J.; Guillen-Guio, B.; Oldham, J.M.; Ma, S.F.; Dressen, A.; Paynton, M.L.; Kraven, L.M.; Obeidat, M.; Li, X.; Ng, M.; et al. Genome-Wide Association Study of Susceptibility to Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2020, 201, 564–574. [Google Scholar] [CrossRef]
- Spagnolo, P.; Kropski, J.A.; Jones, M.G.; Lee, J.S.; Rossi, G.; Karampitsakos, T.; Maher, T.M.; Tzouvelekis, A.; Ryerson, C.J. Idiopathic pulmonary fibrosis: Disease mechanisms and drug development. Pharmacol. Ther. 2021, 222, 107798. [Google Scholar] [CrossRef] [PubMed]
- Sibinska, Z.; Tian, X.; Korfei, M.; Kojonazarov, B.; Kolb, J.S.; Klepetko, W.; Kosanovic, D.; Wygrecka, M.; Ghofrani, H.A.; Weissmann, N.J. Amplified canonical transforming growth factor-β signalling via heat shock protein 90 in pulmonary fibrosis. Eur. Respir. J. 2017, 49, 1501941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zohny, M.H.; Cavalu, S.; Youssef, M.E.; Kaddah, M.M.Y.; Mourad, A.A.E.; Gaafar, A.G.A.; El-Ahwany, E.; Amin, N.A.; Arakeep, H.M.; Shata, A.; et al. Coomassie brilliant blue G-250 dye attenuates bleomycin-induced lung fibrosis by regulating the NF-κB and NLRP3 crosstalk: A novel approach for filling an unmet medical need. Biomed. Pharmacother. 2022, 148, 112723. [Google Scholar] [CrossRef]
- Whitesell, L.; Lindquist, S.L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Jv, Y.; Cai, L.; Tian, X.; Huo, X.; Wang, C.; Zhang, B.; Sun, C.; Ning, J.; Feng, L.; et al. Gambogic acid attenuates liver fibrosis by inhibiting the PI3K/AKT and MAPK signaling pathways via inhibiting HSP90. Toxicol. Appl. Pharmacol. 2019, 371, 63–73. [Google Scholar] [CrossRef]
- Noh, H.; J Kim, H.; R Yu, M.; Kim, W.-Y.; Kim, J.; H Ryu, J.; H Kwon, S.; S Jeon, J.; C Han, D.; Ziyadeh, F. Heat shock protein 90 inhibitor attenuates renal fibrosis through degradation of transforming growth factor-β type II receptor. Lab. Investig. 2012, 92, 1583–1596. [Google Scholar] [CrossRef] [Green Version]
- Massagué, J.; Chen, Y.G. Controlling TGF-beta signaling. Genes Dev. 2000, 14, 627–644. [Google Scholar] [CrossRef]
- Wrighton, K.H.; Lin, X.; Feng, X.-H. Phospho-control of TGF-β superfamily signaling. Cell Res. 2009, 19, 8–20. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Chen, C.H. Proteasome regulators: Activators and inhibitors. Curr. Med. Chem. 2009, 16, 931–939. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.; Li, X.; Lin, J.; Zheng, W.; Hu, Z.; Xuan, J.; Ni, W.; Pan, X. Oleuropein inhibits the IL-1β-induced expression of inflammatory mediators by suppressing the activation of NF-κB and MAPKs in human osteoarthritis chondrocytes. Food Funct. 2017, 8, 3737–3744. [Google Scholar] [CrossRef]
- Xing, C.; Xu, L.; Yao, Y. Beneficial role of oleuropein in sepsis-induced myocardial injury. Possible Involvement of GSK-3β/NF-kB pathway. Acta Cir. Bras. 2021, 36, e360107. [Google Scholar] [CrossRef]
- Youssef, M.E.; Abd El-Fattah, E.E.; Abdelhamid, A.M.; Eissa, H.; El-Ahwany, E.; Amin, N.A.; Hetta, H.F.; Mahmoud, M.H.; Batiha, G.E.-S.; Gobba, N.; et al. Interference With the AMPKα/mTOR/NLRP3 Signaling and the IL-23/IL-17 Axis Effectively Protects Against the Dextran Sulfate Sodium Intoxication in Rats: A New Paradigm in Empagliflozin and Metformin Reprofiling for the Management of Ulcerative Colitis. Front. Pharmacol. 2021, 12, 719984. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Liu, C.; Zhou, D.; Zhang, L. TGF-β/SMAD Pathway and Its Regulation in Hepatic Fibrosis. J. Histochem. Cytochem. 2016, 64, 157–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-β-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Abdelhamid, A.M.; Youssef, M.E.; Abd El-Fattah, E.E.; Gobba, N.A.; Gaafar, A.G.A.; Girgis, S.; Shata, A.; Hafez, A.-M.; El-Ahwany, E.; Amin, N.A.; et al. Blunting p38 MAPKα and ERK1/2 activities by empagliflozin enhances the antifibrotic effect of metformin and augments its AMPK-induced NF-κB inactivation in mice intoxicated with carbon tetrachloride. Life Sci. 2021, 286, 120070. [Google Scholar] [CrossRef]
- Shin, Y.; Klucken, J.; Patterson, C.; Hyman, B.T.; McLean, P.J. The Co-chaperone Carboxyl Terminus of Hsp70-interacting Protein (CHIP) Mediates α-Synuclein Degradation Decisions between Proteasomal and Lysosomal Pathways*. J. Biol. Chem. 2005, 280, 23727–23734. [Google Scholar] [CrossRef] [Green Version]
- Mao, H.; Li, Z.; Zhou, Y.; Li, Z.; Zhuang, S.; An, X.; Zhang, B.; Chen, W.; Nie, J.; Wang, Z.; et al. HSP72 attenuates renal tubular cell apoptosis and interstitial fibrosis in obstructive nephropathy. Am. J. Physiol.-Ren. Physiol. 2008, 295, F202–F214. [Google Scholar] [CrossRef] [Green Version]
- Evans, C.G.; Chang, L.; Gestwicki, J.E. Heat Shock Protein 70 (Hsp70) as an Emerging Drug Target. J. Med. Chem. 2010, 53, 4585–4602. [Google Scholar] [CrossRef] [Green Version]
- Saber, S.; El-Fattah, E.E.A.; Abdelhamid, A.M.; Mourad, A.A.E.; Hamouda, M.A.M.; Elrabat, A.; Zakaria, S.; Haleem, A.A.; Mohamed, S.Z.; Elgharabawy, R.M.; et al. Innovative challenge for the inhibition of hepatocellular carcinoma progression by combined targeting of HSP90 and STAT3/HIF-1α signaling. Biomed. Pharmacother. 2023, 158, 114196. [Google Scholar] [CrossRef]
- Saber, S.; Hasan, A.M.; Mohammed, O.A.; Saleh, L.A.; Hashish, A.A.; Alamri, M.M.S.; Al-Ameer, A.Y.; Alfaifi, J.; Senbel, A.; Aboregela, A.M.; et al. Ganetespib (STA-9090) augments sorafenib efficacy via necroptosis induction in hepatocellular carcinoma: Implications from preclinical data for a novel therapeutic approach. Biomed. Pharmacother. 2023, 164, 114918. [Google Scholar] [CrossRef]
- Abdelhamid, A.M.; Youssef, M.E.; Cavalu, S.; Mostafa-Hedeab, G.; Youssef, A.; Elazab, S.T.; Ibrahim, S.; Allam, S.; Elgharabawy, R.M.; El-Ahwany, E.; et al. Carbocisteine as a Modulator of Nrf2/HO-1 and NFκB Interplay in Rats: New Inspiration for the Revival of an Old Drug for Treating Ulcerative Colitis. Front. Pharmacol. 2022, 13, 887233. [Google Scholar] [CrossRef] [PubMed]
- Egorin, M.J.; Lagattuta, T.F.; Hamburger, D.R.; Covey, J.M.; White, K.D.; Musser, S.M.; Eiseman, J.L. Pharmacokinetics, tissue distribution, and metabolism of 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin (NSC 707545) in CD2F1 mice and Fischer 344 rats. Cancer Chemother. Pharmacol. 2002, 49, 7–19. [Google Scholar] [CrossRef]
- Hadrich, F.; Mahmoudi, A.; Bouallagui, Z.; Feki, I.; Isoda, H.; Feve, B.; Sayadi, S. Evaluation of hypocholesterolemic effect of oleuropein in cholesterol-fed rats. Chem. Biol. Interact. 2016, 252, 54–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youssef, M.E.; El-Azab, M.F.; Abdel-Dayem, M.A.; Yahya, G.; Alanazi, I.S.; Saber, S. Electrocardiographic and histopathological characterizations of diabetic cardiomyopathy in rats. Environ. Sci. Pollut. Res. 2022, 29, 25723–25732. [Google Scholar] [CrossRef]
- Abd El-Fattah, E.E.; Saber, S.; Mourad, A.A.E.; El-Ahwany, E.; Amin, N.A.; Cavalu, S.; Yahya, G.; Saad, A.S.; Alsharidah, M.; Shata, A.; et al. The dynamic interplay between AMPK/NFκB signaling and NLRP3 is a new therapeutic target in inflammation: Emerging role of dapagliflozin in overcoming lipopolysaccharide-mediated lung injury. Biomed. Pharmacother. 2022, 147, 112628. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Exp. Groups | Day 1 | Day 2–21 |
|---|---|---|
| CTRL (n = 6) | Surgical operation Endotracheal saline | Vehicle |
| 17-DMAG (n = 6) | Surgical operation Endotracheal saline | 17-DMAG (10 mg/kg/thrice a week, p.o.) |
| OLPN (n = 6) | Surgical operation Endotracheal saline | OLPN (50 mg/kg/day, p.o.) |
| BLCN (n = 12) | Surgical procedure Endotracheal infusion of bleomycin (5 mg/kg) | - |
| BLCN/17-DMAG (n = 12) | Surgical operation Endotracheal infusion of bleomycin (5 mg/kg) | 17-DMAG (10 mg/kg/thrice a week, p.o.) |
| BLCN/OLPN (n = 12) | Surgical operation Endotracheal infusion of bleomycin (5 mg/kg) | OLPN (50 mg/kg/day, p.o.) |
| BLCN/17-DMAG/OLPN (n = 12) | Surgical operation Endotracheal infusion of bleomycin (5 mg/kg) | 17-DMAG (10 mg/kg/thrice a week, p.o.) + OLPN (50 mg/kg/day, p.o.) |
| Gene | GenBank Accession | Forward Primer | Reverse Primer | Amplicon Size (bp) |
|---|---|---|---|---|
| Col1a1 | NM_053304.1 | 5′-GACATGTTCAGCTTTGTGGACCC-3′ | 5′-AGGGACCCTTAGGCCATTGTGTA-3′ | 120 |
| TGF-β1 | NM_021578.2 | 5′-CTTCTCCACCAACTACTGCTTC- 3′ | 5′-GGGTCCCAGGCAGAAGTT-3′ | 139 |
| GAPDH | NM_001289726.1 | 5′-TCAAGAAGGTGGTGAAGCAG-3′ | 5′-AGGTGGAAGAATGGGAGTTG-3′ | 111 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohammed, O.A.; Abdel-Reheim, M.A.; Saleh, L.A.; Alamri, M.M.S.; Alfaifi, J.; Adam, M.I.E.; Farrag, A.A.; AlQahtani, A.A.J.; BinAfif, W.F.; Hashish, A.A.; et al. Alvespimycin Exhibits Potential Anti-TGF-β Signaling in the Setting of a Proteasome Activator in Rats with Bleomycin-Induced Pulmonary Fibrosis: A Promising Novel Approach. Pharmaceuticals 2023, 16, 1123. https://doi.org/10.3390/ph16081123
Mohammed OA, Abdel-Reheim MA, Saleh LA, Alamri MMS, Alfaifi J, Adam MIE, Farrag AA, AlQahtani AAJ, BinAfif WF, Hashish AA, et al. Alvespimycin Exhibits Potential Anti-TGF-β Signaling in the Setting of a Proteasome Activator in Rats with Bleomycin-Induced Pulmonary Fibrosis: A Promising Novel Approach. Pharmaceuticals. 2023; 16(8):1123. https://doi.org/10.3390/ph16081123
Chicago/Turabian StyleMohammed, Osama A., Mustafa Ahmed Abdel-Reheim, Lobna A. Saleh, Mohannad Mohammad S. Alamri, Jaber Alfaifi, Masoud I. E. Adam, Alshaimaa A. Farrag, AbdulElah Al Jarallah AlQahtani, Waad Fuad BinAfif, Abdullah A. Hashish, and et al. 2023. "Alvespimycin Exhibits Potential Anti-TGF-β Signaling in the Setting of a Proteasome Activator in Rats with Bleomycin-Induced Pulmonary Fibrosis: A Promising Novel Approach" Pharmaceuticals 16, no. 8: 1123. https://doi.org/10.3390/ph16081123