Antiproliferative Properties of Type I and Type II Interferon

Abstract
:1. Historical Perspective
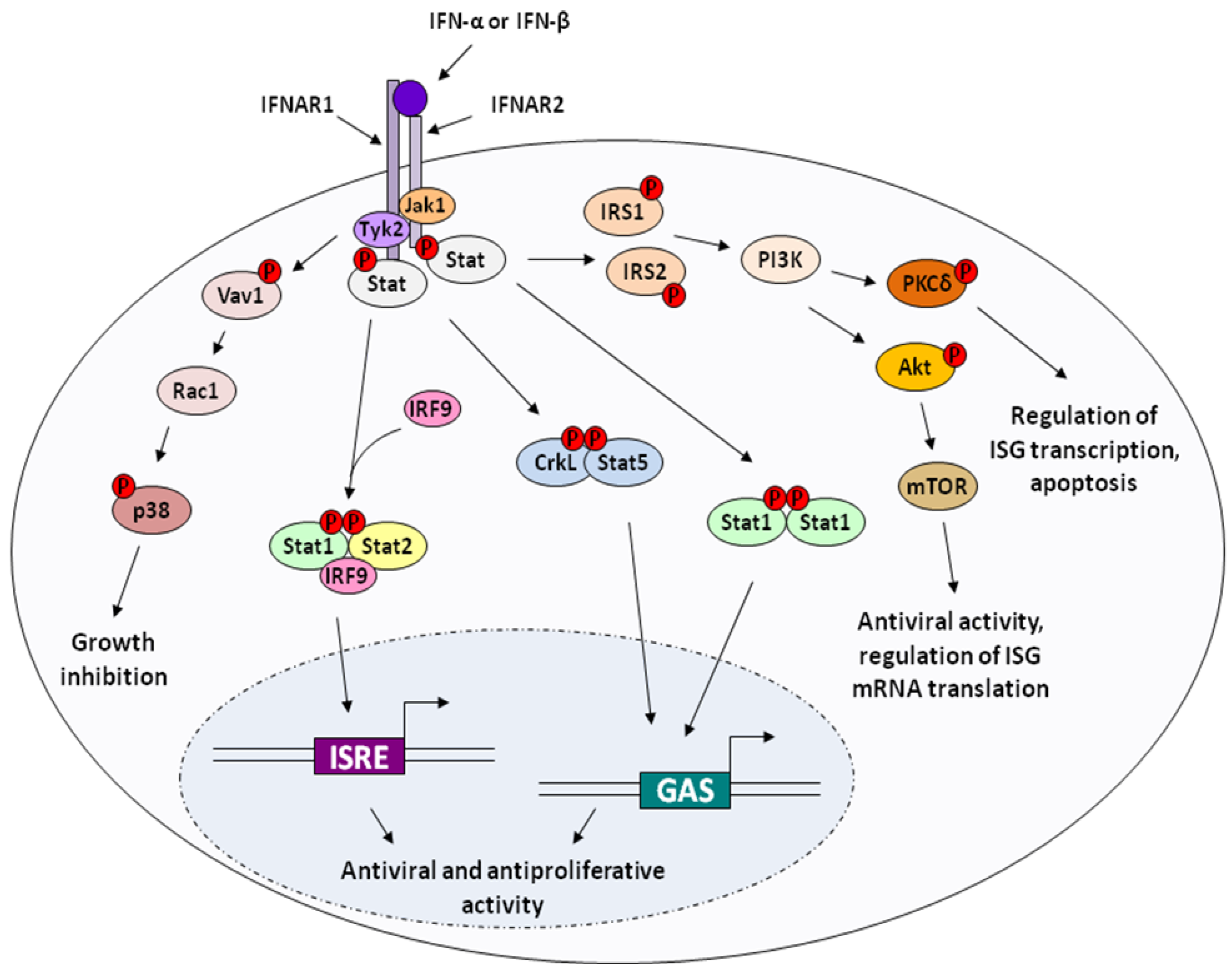
2. Molecular Mechanisms of IFN Action
2.1. Cell cycle inhibition and antiproliferation

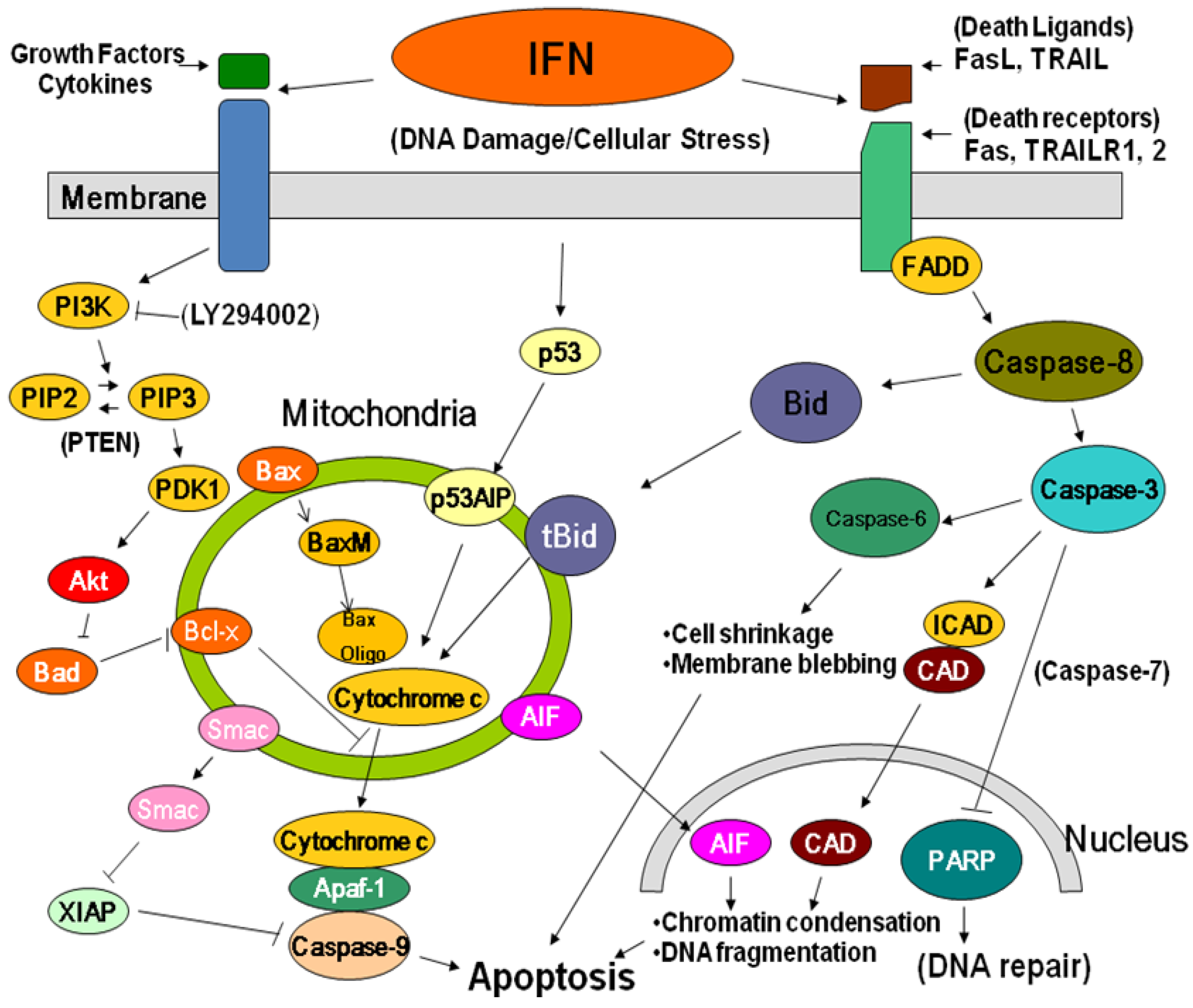
2.2. Apoptosis and Cytotoxicity

2.3. Anti-tumor
2.4. Indirect / Immunomodulatory effects of Interferon on cell growth and survival
3. Antitumor Activity of Interferon-Activated Monocytes in Vitro.
Effect of IFN-activated human monocytes on some tumor cell lines
4. Clinical Applications
4.1. Chronic Myelogenous Leukemia
4.2. Follicular Lymphoma
4.3. Malignant Melanoma
4.4. Hairy Cell Leukemia
4.5. AIDS-Related Kaposi’s Sarcoma


{kind=link}
{kind=link}
5. Pharmacokinetics of IFN-αs Licensed by the U.S. FDA
5.1. IFN-α2a
5.2. IFN-α2b
6. Antibodies to Interferon-α
7. Conclusions
References
- Isaacs, A.; Lindenmann, J. Virus interference. I. The interferon. Proc. R. Soc. Lond B.Biol. Sci. 1957, 147, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Paucker, K.; Cantell, K.; Henle, W. Quantitative studies on viral interference in suspended L cells : III. Effect of interfering viruses and interferon on the growth rate of cells. Virology 1962, 17, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Oxman, M.N.; Baron, S.; Black, P.H.; Takemoto, K.K.; Habel, K.; Rowe, W.P. The effect of interferon on SV-40 T antigen production in SV-40-transformed cells. Virology 1967, 32, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Gresser, I. Interferon and cancer: therapeutic prospects. Rev. Eur. Etud. Clin. Biol. 1970, 15, 23–27. [Google Scholar]
- Sarma, P.S.; Shiu, G.; Baron, S.; Huebner, R.J. Inhibitory effect of interferon on murine sarcoma and leukaemia virus infection in vitro. Nature 1969, 223, 845–846. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, G.R. The effect of interferon on focus formation and yield of murine sarcoma virus in vitro. Proc. Soc. Exp. Biol. Med. 1969, 130, 960–965. [Google Scholar] [PubMed]
- Chany, C.; Vignal, M. Effect of prolonged interferon treatment on mouse embryonic fibroblasts transformed by murine sarcoma virus. J. Gen. Virol. 1970, 7, 203–210. [Google Scholar]
- Gresser, I.; Bourali, C.; Levy, J.P.; Fontaine-Brouty-Boye, D.; Thomas, M.T. Increased survival in mice inoculated with tumor cells and treated with interferon preparations. Proc. Natl. Acad. Sci. U S A. 1969, 63, 51–57. [Google Scholar]
- Brouty-Boye, D.; Wybier-Franqui, J.; Suarez, H.G.; Gresser, I. Long-term effects of IFN on the phenotype of transformed and tumor cells. In The Interferon System; Dianzani, F., Rossi, G.B., Eds.; Raven Press: Rome, Italy, 1985; Volume 24, pp. 303–308. [Google Scholar]
- Came, P.E.; Moore, D.H. Effect of exogenous interferon treatment on mouse mammary tumors. J. Natl. Cancer Inst. 1972, 48, 1151–1154. [Google Scholar]
- Balkwill, F.; Taylor-Papadimitriou, J.; Fantes, K.H.; Sebesteny, A. Human lymphoblastoid interferon can inhibit the growth of human breast cancer xenografts in athymic (nude) mice. Eur. J. Cancer 1980, 16, 569–573. [Google Scholar] [PubMed]
- Roos, G.; Leanderson, T.; Lundgren, E. Interferon-induced cell cycle changes in human hematopoietic cell lines and fresh leukemic cells. Cancer Res. 1984, 44, 2358–2362. [Google Scholar]
- Grander, D.; Sangfelt, O.; Erickson, S. How does interferon exert its cell growth inhibitory effect? Eur. J. Haematol. 1997, 59, 129–135. [Google Scholar] [PubMed]
- Matsuoka, M.; Tani, K.; Asano, S. Interferon-alpha-induced G1 phase arrest through up-regulated expression of CDK inhibitors, p19Ink4D and p21Cip1 in mouse macrophages. Oncogene 1998, 16, 2075–2086. [Google Scholar]
- Sangfelt, O.; Erickson, S.; Einhorn, S.; Grander, D. Induction of Cip/Kip and Ink4 cyclin dependent kinase inhibitors by interferon-alpha in hematopoietic cell lines. Oncogene 1997, 14, 415–423. [Google Scholar]
- Sangfelt, O.; Erickson, S.; Castro, J.; Heiden, T.; Gustafsson, A.; Einhorn, S.; Grander, D. Molecular mechanisms underlying interferon-alpha-induced G0/G1 arrest: CKI-mediated regulation of G1 Cdk-complexes and activation of pocket proteins. Oncogene 1999, 18, 2798–2810. [Google Scholar]
- Sangfelt, O.; Erickson, S.; Grander, D. Mechanisms of interferon-induced cell cycle arrest. Front. Biosci. 2000, 5, D479–D487. [Google Scholar] [CrossRef] [PubMed]
- Sangfelt, O.; Erickson, S.; Castro, J.; Heiden, T.; Einhorn, S.; Grander, D. Induction of apoptosis and inhibition of cell growth are independent responses to interferon-alpha in hematopoietic cell lines. Cell Growth Differ. 1997, 8, 343–352. [Google Scholar]
- Tiefenbrun, N.; Melamed, D.; Levy, N.; Resnitzky, D.; Hoffman, I.; Reed, S.I.; Kimchi, A. Alpha interferon suppresses the cyclin D3 and cdc25A genes, leading to a reversible G0-like arrest. Mol. Cell. Biol. 1996, 16, 3934–3944. [Google Scholar]
- Einat, M.; Resnitzky, D.; Kimchi, A. Close link between reduction of c-myc expression by interferon and, G0/G1 arrest. Nature 1985, 313, 597–600. [Google Scholar] [CrossRef] [PubMed]
- Jonak, G.J.; Knight, E., Jr. Selective reduction of c-myc mRNA in Daudi cells by human beta interferon. Proc. Natl. Acad. Sci. U S A. 1984, 81, 1747–1750. [Google Scholar]
- Knight, E., Jr.; Anton, E.D.; Fahey, D.; Friedland, B.K.; Jonak, G.J. Interferon regulates c-myc gene expression in Daudi cells at the post-transcriptional level. Proc. Natl. Acad. Sci. U S A 1985, 82, 1151–1154. [Google Scholar]
- Sarkar, D.; Park, E.S.; Fisher, P.B. Defining the mechanism by which IFN-beta dowregulates c-myc expression in human melanoma cells: pivotal role for human polynucleotide phosphorylase (hPNPaseold-35). Cell Death Differ. 2006, 13, 1541–1553. [Google Scholar]
- Rosenwald, I.B.; Rhoads, D.B.; Callanan, L.D.; Isselbacher, K.J.; Schmidt, E.V. Increased expression of eukaryotic translation initiation factors eIF-4E and eIF-2 alpha in response to growth induction by c-myc. Proc. Natl. Acad. Sci. U S A. 1993, 90, 6175–6178. [Google Scholar]
- Jeffrey, I.W.; Elia, A.; Bornes, S.; Tilleray, V.J.; Gengatharan, K.; Clemens, M.J. Interferon-alpha induces sensitization of cells to inhibition of protein synthesis by tumour necrosis factor-related apoptosis-inducing ligand. FEBS. J. 2006, 273, 3698–3708. [Google Scholar]
- Xiao, S.; Li, D.; Zhu, H.Q.; Song, M.G.; Pan, X.R.; Jia, P.M.; Peng, L.L.; Dou, A.X.; Chen, G.Q.; Chen, S.J.; Chen, Z.; Tong, J.H. RIG-G as a key mediator of the antiproliferative activity of interferon-related pathways through enhancing p21 and p27 proteins. Proc. Natl. Acad. Sci. U S A. 2006, 103, 16448–16453. [Google Scholar]
- Kumar, R.; Atlas, I. Interferon alpha induces the expression of retinoblastoma gene product in human Burkitt lymphoma Daudi cells: role in growth regulation. Proc. Natl. Acad. Sci. U S A. 1992, 89, 6599–6603. [Google Scholar]
- Thomas, N.S.; Pizzey, A.R.; Tiwari, S.; Williams, C.D.; Yang, J. p130, p107, and pRb are differentially regulated in proliferating cells and during cell cycle arrest by alpha-interferon. J. Biol. Chem. 1998, 273, 23659–23667. [Google Scholar] [PubMed]
- Burke, L.C.; Bybee, A.; Thomas, N.S. The retinoblastoma protein is partially phosphorylated during early G1 in cycling cells but not in G1 cells arrested with alpha-interferon. Oncogene 1992, 7, 783–788. [Google Scholar]
- Uddin, S.; Sweet, M.; Colamonici, O.R.; Krolewski, J.J.; Platanias, L.C. The vav proto-oncogene product (p95vav) interacts with the Tyk-2 protein tyrosine kinase. FEBS. Lett. 1997, 403, 31–34. [Google Scholar]
- Uddin, S.; Lekmine, F.; Sharma, N.; Majchrzak, B.; Mayer, I.; Young, P.R.; Bokoch, G.M.; Fish, E.N.; Platanias, L.C. The Rac1/p38 mitogen-activated protein kinase pathway is required for interferon alpha-dependent transcriptional activation but not serine phosphorylation of Stat proteins. J. Biol. Chem. 2000, 275, 27634–27640. [Google Scholar]
- Katsoulidis, E.; Li, Y.; Mears, H.; Platanias, L.C. The p38 mitogen-activated protein kinase pathway in interferon signal transduction. J. Interferon Cytokine Res. 2005, 25, 749–756. [Google Scholar]
- Platanias, L.C. The p38 mitogen-activated protein kinase pathway and its role in interferon signaling. Pharmacol. Ther. 2003, 98, 129–142. [Google Scholar]
- Kovarik, P.; Stoiber, D.; Eyers, P.A.; Menghini, R.; Neininger, A.; Gaestel, M.; Cohen, P.; Decker, T. Stress-induced phosphorylation of STAT1 at Ser727 requires p38 mitogen-activated protein kinase whereas IFN-gamma uses a different signaling pathway. Proc. Natl. Acad. Sci. U S A. 1999, 96, 13956–13961. [Google Scholar]
- Ono, K.; Han, J. The p38 signal transduction pathway: activation and function. Cell Signal. 2000, 12, 1–13. [Google Scholar]
- Brancho, D.; Tanaka, N.; Jaeschke, A.; Ventura, J.J.; Kelkar, N.; Tanaka, Y.; Kyuuma, M.; Takeshita, T.; Flavell, R.A.; Davis, R.J. Mechanism of p38 MAP kinase activation in vivo. Genes Dev. 2003, 17, 1969–1978. [Google Scholar] [CrossRef] [PubMed]
- Bulavin, D.V.; Amundson, S.A.; Fornace, A.J. p38 and Chk1 kinases: different conductors for the G(2)/M checkpoint symphony. Curr. Opin. Genet. Dev. 2002, 12, 92–97. [Google Scholar]
- Ahmad, S.; Alsayed, Y.M.; Druker, B.J.; Platanias, L.C. The type I interferon receptor mediates tyrosine phosphorylation of the CrkL adaptor protein. J. Biol. Chem. 1997, 272, 29991–29994. [Google Scholar]
- Platanias, L.C.; Uddin, S.; Bruno, E.; Korkmaz, M.; Ahmad, S.; Alsayed, Y.; Van Den Berg, D.; Druker, B.J.; Wickrema, A.; Hoffman, R. CrkL and CrkII participate in the generation of the growth inhibitory effects of interferons on primary hematopoietic progenitors. Exp. Hematol. 1999, 27, 1315–1321. [Google Scholar]
- Fish, E.N.; Uddin, S.; Korkmaz, M.; Majchrzak, B.; Druker, B.J.; Platanias, L.C. Activation of a CrkL-stat5 signaling complex by type I interferons. J. Biol. Chem. 1999, 274, 571–573. [Google Scholar]
- Rodriguez-Villanueva, J.; McDonnell, T.J. Induction of apoptotic cell death in non-melanoma skin cancer by interferon-alpha. Int. J. Cancer 1995, 61, 110–114. [Google Scholar]
- Sangfelt; Strander, H. Apoptosis and cell growth inhibition as antitumor effector functions of interferons. Med. Oncol. 2001, 18, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Milner, A.E.; Grand, R.J.; Gregory, C.D. Effects of interferon-alpha on human B cells: repression of apoptosis and prevention of cell growth are independent responses of Burkitt lymphoma lines. Int. J. Cancer 1995, 61, 348–354. [Google Scholar]
- Jewell, A.P.; Worman, C.P.; Lydyard, P.M.; Yong, K.L.; Giles, F.J.; Goldstone, A.H. Interferon-alpha up-regulates bcl-2 expression and protects B-CLL cells from apoptosis in vitro and in vivo. Br. J. Haematol. 1994, 88, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Spets, H.; Georgii-Hemming, P.; Siljason, J.; Nilsson, K.; Jernberg-Wiklund, H. Fas/APO-1 (CD95)-mediated apoptosis is activated by interferon-gamma and interferon- in interleukin-6 (IL-6)-dependent and IL-6-independent multiple myeloma cell lines. Blood 1998, 92, 2914–2923. [Google Scholar]
- Selleri, C.; Sato, T.; Del Vecchio, L.; Luciano, L.; Barrett, A.J.; Rotoli, B.; Young, N.S.; Maciejewski, J.P. Involvement of Fas-mediated apoptosis in the inhibitory effects of interferon-alpha in chronic myelogenous leukemia. Blood 1997, 89, 957–964. [Google Scholar]
- Buechner, S.A.; Wernli, M.; Harr, T.; Hahn, S.; Itin, P.; Erb, P. Regression of basal cell carcinoma by intralesional interferon-alpha treatment is mediated by CD95 (Apo-1/Fas)-CD95 ligand-induced suicide. J. Clin. Invest. 1997, 100, 2691–2696. [Google Scholar]
- Chawla-Sarkar, M.; Leaman, D.W.; Borden, E.C. Preferential induction of apoptosis by interferon (IFN)-beta compared with IFN-alpha2: correlation with TRAIL/Apo2L induction in melanoma cell lines. Clin. Cancer Res. 2001, 7, 1821–1831. [Google Scholar]
- Chen, Q.; Gong, B.; Mahmoud-Ahmed, A.S.; Zhou, A.; Hsi, E.D.; Hussein, M.; Almasan, A. Apo2L/TRAIL and Bcl-2-related proteins regulate type I interferon-induced apoptosis in multiple myeloma. Blood 2001, 98, 2183–2192. [Google Scholar]
- Kayagaki, N.; Yamaguchi, N.; Nakayama, M.; Eto, H.; Okumura, K.; Yagita, H. Type I interferons (IFNs) regulate tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) expression on human T cells: A novel mechanism for the antitumor effects of type I IFNs. J. Exp. Med. 1999, 189, 1451–1460. [Google Scholar]
- Balachandran, S.; Roberts, P.C.; Kipperman, T.; Bhalla, K.N.; Compans, R.W.; Archer, D.R.; Barber, G.N. Alpha/beta interferons potentiate virus-induced apoptosis through activation of the FADD/Caspase-8 death signaling pathway. J. Virol. 2000, 74, 1513–1523. [Google Scholar]
- Tsuno, T.; Mejido, J.; Zhao, T.; Schmeisser, H.; Morrow, A.; Zoon, K.C. IRF9 is a key factor for eliciting the antiproliferative activity of IFN-alpha. J. Immunother. 2009, 32, 803–816. [Google Scholar]
- Thyrell, L.; Erickson, S.; Zhivotovsky, B.; Pokrovskaja, K.; Sangfelt, O.; Castro, J.; Einhorn, S.; Grander, D. Mechanisms of Interferon-alpha induced apoptosis in malignant cells. Oncogene 2002, 21, 1251–1262. [Google Scholar]
- Minami, R.; Muta, K.; Ilseung, C.; Abe, Y.; Nishimura, J.; Nawata, H. Interleukin-6 sensitizes multiple myeloma cell lines for apoptosis induced by interferon-alpha. Exp. Hematol. 2000, 28, 244–255. [Google Scholar]
- Chawla-Sarkar, M.; Lindner, D.J.; Liu, Y.F.; Williams, B.R.; Sen, G.C.; Silverman, R.H.; Borden, E.C. Apoptosis and interferons: role of interferon-stimulated genes as mediators of apoptosis. Apoptosis 2003, 8, 237–249. [Google Scholar]
- Ossina, N.K.; Cannas, A.; Powers, V.C.; Fitzpatrick, P.A.; Knight, J.D.; Gilbert, J.R.; Shekhtman, E.M.; Tomei, L.D.; Umansky, S.R.; Kiefer, M.C. Interferon-gamma modulates a p53-independent apoptotic pathway and apoptosis-related gene expression. J. Biol. Chem. 1997, 272, 16351–16357. [Google Scholar]
- Herzer, K.; Hofmann, T.G.; Teufel, A.; Schimanski, C.C.; Moehler, M.; Kanzler, S.; Schulze-Bergkamen, H.; Galle, P.R. IFN-alpha-induced apoptosis in hepatocellular carcinoma involves promyelocytic leukemia protein and TRAIL independently of p53. Cancer Res. 2009, 69, 855–862. [Google Scholar]
- Stark, G.R.; Kerr, I.M.; Williams, B.R.; Silverman, R.H.; Schreiber, R.D. How cells respond to interferons. Annu. Rev. Biochem. 1998, 67, 227–264. [Google Scholar]
- Silverman, R.H.; Cayley, P.J.; Knight, M.; Gilbert, C.S.; Kerr, I.M. Control of the ppp(a2'p)nA system in HeLa cells. Effects of interferon and virus infection. Eur. J. Biochem. 1982, 124, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Garcia, R.; Franklin, R.A.; McCubrey, J.A. Cell death of MCF-7 human breast cancer cells induced by EGFR activation in the absence of other growth factors. Cell Cycle 2006, 5, 1840–1846. [Google Scholar]
- Uddin, S.; Yenush, L.; Sun, X.J.; Sweet, M.E.; White, M.F.; Platanias, L.C. Interferon-alpha engages the insulin receptor substrate-1 to associate with the phosphatidylinositol 3'-kinase. J. Biol. Chem. 1995, 270, 15938–15941. [Google Scholar]
- Uddin, S.; Fish, E.N.; Sher, D.A.; Gardziola, C.; White, M.F.; Platanias, L.C. Activation of the phosphatidylinositol 3-kinase serine kinase by IFN-alpha. J. Immunol. 1997, 158, 2390–2397. [Google Scholar]
- Uddin, S.; Sassano, A.; Deb, D.K.; Verma, A.; Majchrzak, B.; Rahman, A.; Malik, A.B.; Fish, E.N.; Platanias, L.C. Protein kinase C-delta (PKC-delta ) is activated by type I interferons and mediates phosphorylation of Stat1 on serine 727. J. Biol. Chem. 2002, 277, 14408–14416. [Google Scholar]
- DeVries, T.A.; Kalkofen, R.L.; Matassa, A.A.; Reyland, M.E. Protein kinase Cdelta regulates apoptosis via activation of STAT1. J. Biol. Chem. 2004, 279, 45603–45612. [Google Scholar] [PubMed]
- Kaur, S.; Uddin, S.; Platanias, L.C. The PI3' kinase pathway in interferon signaling. J. Interferon Cytokine Res. 2005, 25, 780–787. [Google Scholar]
- Thyrell, L.; Hjortsberg, L.; Arulampalam, V.; Panaretakis, T.; Uhles, S.; Dagnell, M.; Zhivotovsky, B.; Leibiger, I.; Grander, D.; Pokrovskaja, K. Interferon alpha-induced apoptosis in tumor cells is mediated through the phosphoinositide 3-kinase/mammalian target of rapamycin signaling pathway. J. Biol. Chem. 2004, 279, 24152–24162. [Google Scholar]
- Rosner, D.; Stoneman, V.; Littlewood, T.; McCarthy, N.; Figg, N.; Wang, Y.; Tellides, G.; Bennett, M. Interferon-gamma induces Fas trafficking and sensitization to apoptosis in vascular smooth muscle cells via a PI3K- and Akt-dependent mechanism. Am. J. Pathol. 2006, 168, 2054–2063. [Google Scholar] [CrossRef] [PubMed]
- Kusaba, H.; Ghosh, P.; Derin, R.; Buchholz, M.; Sasaki, C.; Madara, K.; Longo, D.L. Interleukin-12-induced interferon-gamma production by human peripheral blood T cells is regulated by mammalian target of rapamycin (mTOR). J. Biol. Chem. 2005, 280, 1037–1043. [Google Scholar]
- Le, J.; Yip, Y.K.; Vilcek, J. Cytolytic activity of interferon-gamma and its synergism with 5-fluorouracil. Int. J. Cancer 1984, 34, 495–500. [Google Scholar]
- Grander, D.; Xu, B.; Einhorn, S. Cytotoxic effect of interferon on primary malignant tumour cells. Studies in various malignancies. Eur. J. Cancer 1993, 29A, 1940–1943. [Google Scholar] [PubMed]
- Singh, R.K.; Gutman, M.; Bucana, C.D.; Sanchez, R.; Llansa, N.; Fidler, I.J. Interferons alpha and beta down-regulate the expression of basic fibroblast growth factor in human carcinomas. Proc. Natl. Acad. Sci. USA 1995, 92, 4562–4566. [Google Scholar]
- von Marschall, Z.; Scholz, A.; Cramer, T.; Schafer, G.; Schirner, M.; Oberg, K.; Wiedenmann, B.; Hocker, M.; Rosewicz, S. Effects of interferon alpha on vascular endothelial growth factor gene transcription and tumor angiogenesis. J. Natl. Cancer Inst. 2003, 95, 437–448. [Google Scholar]
- Tang, N.; Wang, L.; Esko, J.; Giordano, F.J.; Huang, Y.; Gerber, H.P.; Ferrara, N.; Johnson, R.S. Loss of HIF-1alpha in endothelial cells disrupts a hypoxia-driven VEGF autocrine loop necessary for tumorigenesis. Cancer Cell 2004, 6, 485–495. [Google Scholar]
- Raig, E.T.; Jones, N.B.; Varker, K.A.; Benniger, K.; Go, M.R.; Biber, J.L.; Lesinski, G.B.; Carson, W.E., 3rd. VEGF secretion is inhibited by interferon-alpha in several melanoma cell lines. J. Interferon Cytokine Res. 2008, 28, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Sidky, Y.A.; Borden, E.C. Inhibition of angiogenesis by interferons: effects on tumor- and lymphocyte-induced vascular responses. Cancer Res. 1987, 47, 5155–5161. [Google Scholar]
- Wang, Q.; Miyakawa, Y.; Fox, N.; Kaushansky, K. Interferon-alpha directly represses megakaryopoiesis by inhibiting thrombopoietin-induced signaling through induction of SOCS-1. Blood 2000, 96, 2093–2099. [Google Scholar]
- Yamane, A.; Nakamura, T.; Suzuki, H.; Ito, M.; Ohnishi, Y.; Ikeda, Y.; Miyakawa, Y. Interferon-alpha 2b-induced thrombocytopenia is caused by inhibition of platelet production but not proliferation and endomitosis in human megakaryocytes. Blood 2008, 112, 542–550. [Google Scholar]
- Caraglia, M.; Abbruzzese, A.; Leardi, A.; Pepe, S.; Budillon, A.; Baldassare, G.; Selleri, C.; Lorenzo, S.D.; Fabbrocini, A.; Giuberti, G.; Vitale, G.; Lupoli, G.; Bianco, A.R.; Tagliaferri, P. Interferon-alpha induces apoptosis in human KB cells through a stress-dependent mitogen activated protein kinase pathway that is antagonized by epidermal growth factor. Cell Death Differ. 1999, 6, 773–780. [Google Scholar]
- Caraglia, M.; Leardi, A.; Corradino, S.; Ciardiello, F.; Budillon, A.; Guarrasi, R.; Bianco, A.R.; Tagliaferri, P. alpha-Interferon potentiates epidermal growth factor receptor-mediated effects on human epidermoid carcinoma KB cells. Int. J. Cancer 1995, 61, 342–347. [Google Scholar]
- Caraglia, M.; Vitale, G.; Marra, M.; Budillon, A.; Tagliaferri, P.; Abbruzzese, A. Alpha-interferon and its effects on signalling pathways within cells. Curr. Protein Pept. Sci. 2004, 5, 475–485. [Google Scholar]
- Hakansson, A.; Gustafsson, B.; Krysander, L.; Hakansson, L. Tumour-infiltrating lymphocytes in metastatic malignant melanoma and response to interferon alpha treatment. Br. J. Cancer 1996, 74, 670–676. [Google Scholar]
- Bracarda, S.; Eggermont, A.M.; Samuelsson, J. Redefining the role of interferon in the treatment of malignant diseases. Eur. J. Cancer 2009. [Google Scholar]
- Prasanna, S.J.; Saha, B.; Nandi, D. Involvement of oxidative and nitrosative stress in modulation of gene expression and functional responses by IFNgamma. Int. Immunol. 2007, 19, 867–879. [Google Scholar]
- Maher, S.G.; Romero-Weaver, A.L.; Scarzello, A.J.; Gamero, A.M. Interferon: cellular executioner or white knight? Curr. Med. Chem. 2007, 14, 1279–1289. [Google Scholar] [CrossRef] [PubMed]
- Dianzani, F.; Antonelli, G.; Capobianchi, M.R. Biological activity of gamma interferon. Ann. 1st Super Sanita. 1990, 26, 255–261. [Google Scholar]
- Le Page, C.; Genin, P.; Baines, M.G.; Hiscott, J. Interferon activation and innate immunity. Rev. Immunogenet. 2000, 2, 374–386. [Google Scholar]
- Dudley, M.E.; Rosenberg, S.A. Adoptive-cell-transfer therapy for the treatment of patients with cancer. Nat. Rev. Cancer 2003, 3, 666–675. [Google Scholar]
- Gerrard, T.L.; Jurgensen, C.H.; Fauci, A.S. Differential effect of monoclonal anti-DR antibody on monocytes in antigen- and mitogen-stimulated responses: mechanism of inhibition and relationship to interleukin 1 secretion. Cell. Immunol. 1983, 82, 394–402. [Google Scholar]
- Hibbs, J.B., Jr.; Chapman, H.A., Jr.; Weinberg, J.B. The macrophage as an antineoplastic surveillance cell: biological perspectives. J. Reticuloendothel. Soc. 1978, 24, 549–570. [Google Scholar] [PubMed]
- Russell, S.W.; Doe, W.F.; McIntosh, A.T. Functional characterization of a stable, noncytolytic stage of macrophage activation in tumors. J. Exp. Med. 1977, 146, 1511–1520. [Google Scholar]
- Suzuki, T.; Yoshie, H.; Jeannel, D.; Tortevoye, P.; Fournier, S.; Dupont, B.; de The, G.; Hara, K. Detection of intracellular p24-positive macrophages in gingival crevicular fluid from periodontal lesions of stage IV AIDS patients. AIDS 1996, 10, 804–805. [Google Scholar]
- Ballinger, M.N.; Paine, R., 3rd; Serezani, C.H.; Aronoff, D.M.; Choi, E.S.; Standiford, T.J.; Toews, G.B.; Moore, B.B. Role of granulocyte macrophage colony-stimulating factor during gram-negative lung infection with Pseudomonas aeruginosa. Am. J. Respir. Cell. Mol .Biol. 2006, 34, 766–774. [Google Scholar] [CrossRef] [PubMed]
- Boyle, M.D.; Ormerod, M.G. Destruction of allogeneic tumour cells by peritoneal macrophages. Transplantation 1976, 21, 242–246. [Google Scholar]
- Burke, D.S.; Fowler, A.K.; Redfield, R.R.; Dilworth, S.; Oster, C.N. Isolation of HIV-1 from the blood of seropositive adults: patient stage of illness and sample inoculum size are major determinants of a positive culture. The Walter Reed Retroviral Research Group. J. Acquir. Immune. Defic. Syndr. 1990, 3, 1159–1167. [Google Scholar] [PubMed]
- Isaacs, A.; Westwood, M.A. Inhibition by interferon of the growth of vaccinia virus in the rabbit skin. Lancet 1959, 2, 324–325. [Google Scholar]
- Chan, C.W.; Crafton, E.; Fan, H.N.; Flook, J.; Yoshimura, K.; Skarica, M.; Brockstedt, D.; Dubensky, T.W.; Stins, M.F.; Lanier, L.L.; Pardoll, D.M.; Housseau, F. Interferon-producing killer dendritic cells provide a link between innate and adaptive immunity. Nat. Med. 2006, 12, 207–213. [Google Scholar]
- Marshall, J.D.; Heeke, D.S.; Abbate, C.; Yee, P.; Van Nest, G. Induction of interferon-gamma from natural killer cells by immunostimulatory CpG DNA is mediated through plasmacytoid-dendritic-cell-produced interferon-alpha and tumour necrosis factor-alpha. Immunology 2006, 117, 38–46. [Google Scholar]
- Buhtoiarov, I.N.; Lum, H.D.; Berke, G.; Sondel, P.M.; Rakhmilevich, A.L. Synergistic activation of macrophages via CD40 and TLR9 results in T cell independent antitumor effects. J. Immunol. 2006, 176, 309–318. [Google Scholar]
- Alexander, P.; Evans, R. Endotoxin and double stranded RNA render macrophages cytotoxic. Nat. New Biol. 1971, 232, 76–78. [Google Scholar]
- Fan, D.; Liaw, A.; Denkins, Y.M.; Collins, J.H.; Van Arsdall, M.; Chang, J.L.; Chakrabarty, S.; Nguyen, D.; Kruzel, E.; Fidler, I.J. Type-1 transforming growth factor-beta differentially modulates tumoricidal activity of murine peritoneal macrophages against metastatic variants of the B16 murine melanoma. J. Exp. Ther. Oncol. 2002, 2, 286–297. [Google Scholar]
- Tsung, K.; Dolan, J.P.; Tsung, Y.L.; Norton, J.A. Macrophages as effector cells in interleukin 12-induced T cell-dependent tumor rejection. Cancer Res. 2002, 62, 5069–5075. [Google Scholar]
- Berdel, W.E.; Bausert, W.R.; Weltzien, H.U.; Modolell, M.L.; Widmann, K.H.; Munder, P.G. The influence of alkyl-lysophospholipids and lysophospholipid-activated macrophages on the development of metastasis of 3-Lewis lung carcinoma. Eur. J. Cancer 1980, 16, 1199–1204. [Google Scholar]
- Du, C.; Feng, N.; Jin, H.; Lee, V.; Wang, M.; Wright, J.A.; Young, A.H. Macrophages play a critical role in the anti-tumor activity of Virulizin. Int. J. Oncol. 2003, 23, 1341–1346. [Google Scholar]
- Kaplan, A.M.; Morahan, P.S. Macrophage mediated tumor cell cytotoxicity. Ann. N Y. Acad. Sci. 1976, 276, 134–145. [Google Scholar]
- Andreesen, R.; Hennemann, B. Adoptive immunotherapy with autologous macrophages: current status and future perspectives. Pathobiology 1991, 59, 259–263. [Google Scholar]
- Andreesen, R.; Scheibenbogen, C.; Brugger, W.; Krause, S.; Meerpohl, H.G.; Leser, H.G.; Engler, H.; Lohr, G.W. Adoptive transfer of tumor cytotoxic macrophages generated in vitro from circulating blood monocytes: a new approach to cancer immunotherapy. Cancer Res. 1990, 50, 7450–7456. [Google Scholar] [PubMed]
- Baron, S.; Hernandez, J.; Bekisz, J.; Poast, J.; Goldman, N.; Clouse, K.; Fields, K.; Bacot, S.; Wang, J.; Zoon, K. Clinical model: interferons activate human monocytes to an eradicative tumor cell level in vitro. J. Interferon Cytokine Res. 2007, 27, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Webb, D.S.; Gerrard, T.L. IFN-alpha and IFN-gamma can affect both monocytes and tumor cells to modulate monocyte-mediated cytotoxicity. J. Immunol. 1990, 144, 3643–3648. [Google Scholar]
- Klimp, A.H.; de Vries, E.G.; Scherphof, G.L.; Daemen, T. A potential role of macrophage activation in the treatment of cancer. Crit. Rev. Oncol. Hematol. 2002, 44, 143–161. [Google Scholar]
- Larabi, M.; Legrand, P.; Appel, M.; Gil, S.; Lepoivre, M.; Devissaguet, J.; Puisieux, F.; Barratt, G. Reduction of no synthase expression and tumor necrosis factor alpha production in macrophages by amphotericin B lipid carriers. Antimicrob. Agents Chemother. 2001, 45, 553–562. [Google Scholar]
- Bartholeyns, J.; Lombard, Y.; Poindron, P. Immunotherapy of murine sarcoma by adoptive transfer of resident peritoneal macrophages proliferating in culture. Anticancer Res. 1988, 8, 145–151. [Google Scholar]
- Bartholeyns, J.; Lopez, M. Immune control of neoplasia by adoptive transfer of macrophages: potentiality for antigen presentation and gene transfer. Anticancer Res. 1994, 14, 2673–2676. [Google Scholar]
- Ebina, T.; Fujimiya, Y.; Yamaguchi, T.; Ogama, N.; Sasaki, H.; Isono, N.; Suzuki, Y.; Katakura, R.; Tanaka, K.; Nagata, K.; Takano, S.; Tamura, K.; Uno, K.; Kishida, T. The use of BRM-activated killer cells in adoptive immunotherapy: a pilot study with nine advanced cancer patients. Biotherapy 1998, 11, 241–253. [Google Scholar]
- Eymard, J.C.; Lopez, M.; Cattan, A.; Bouche, O.; Adjizian, J.C.; Bernard, J. Phase I/II trial of autologous activated macrophages in advanced colorectal cancer. Eur. J. Cancer 1996, 32A, 1905–1911. [Google Scholar]
- Fidler, I.J. Inhibition of pulmonary metastasis by intravenous injection of specifically activated macrophages. Cancer Res. 1974, 34, 1074–1078. [Google Scholar]
- Hibbs, J.B., Jr.; Lambert, L.H., Jr.; Remington, J.S. Possible role of macrophage mediated nonspecific cytotoxicity in tumour resistance. Nat. New Biol. 1972, 235, 48–50. [Google Scholar] [PubMed]
- Lasek, W.; Basak, G.; Switaj, T.; Jakubowska, A.B.; Wysocki, P.J.; Mackiewicz, A.; Drela, N.; Jalili, A.; Kaminski, R.; Kozar, K.; Jakobisiak, M. Complete tumour regressions induced by vaccination with IL-12 gene-transduced tumour cells in combination with IL-15 in a melanoma model in mice. Cancer Immunol. Immunother. 2004, 53, 363–372. [Google Scholar]
- Talpaz, M.; Kantarjian, H.M.; McCredie, K.B.; Keating, M.J.; Trujillo, J.; Gutterman, J. Clinical investigation of human alpha interferon in chronic myelogenous leukemia. Blood 1987, 69, 1280–1288. [Google Scholar]
- Talpaz, M.; Shah, N.P.; Kantarjian, H.; Donato, N.; Nicoll, J.; Paquette, R.; Cortes, J.; O'Brien, S.; Nicaise, C.; Bleickardt, E.; Blackwood-Chirchir, M.A.; Iyer, V.; Chen, T.T.; Huang, F.; Decillis, A.P.; Sawyers, C.L. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N. Engl. J. Med. 2006, 354, 2531–2541. [Google Scholar]
- Kikawa, Y.; Fuji, Y.; Takano, T.; Shigematsu, Y.; Sudo, M.; Okamoto, M.; Mizutani, S. A long-term suppression by alpha-interferon of Philadelphia chromosome in children with chronic myelogenous leukemia. Acta. Paediatr. Jpn. 1993, 35, 361–364. [Google Scholar]
- Guilhot, F.; Roy, L.; Guilhot, J.; Millot, F. Interferon therapy in chronic myelogenous leukemia. Hematol. Oncol. Clin. North Am. 2004, 18, 585–603, viii. [Google Scholar] [CrossRef] [PubMed]
- Essers, M.A.; Offner, S.; Blanco-Bose, W.E.; Waibler, Z.; Kalinke, U.; Duchosal, M.A.; Trumpp, A. IFN-alpha activates dormant haematopoietic stem cells in vivo. Nature 2009, 458, 904–908. [Google Scholar] [PubMed]
- Archuleta, T.D.; Armitage, J.O. Advances in follicular lymphoma. Semin. Oncol. 2004, 31, 66–71. [Google Scholar]
- Aurora, V.; Winter, J.N. Follicular lymphoma: today's treatments and tomorrow's targets. Expert Opin. Pharmacother. 2006, 7, 1273–1290. [Google Scholar]
- Sabel, M.S.; Sondak, V.K. Pros and cons of adjuvant interferon in the treatment of melanoma. Oncologist 2003, 8, 451–458. [Google Scholar]
- Fanta, P.T.; Saven, A. Hairy cell leukemia. Cancer Treat Res 2008, 142, 193–209. [Google Scholar]
- Cannon, T.; Mobarek, D.; Wegge, J.; Tabbara, I.A. Hairy cell leukemia: current concepts. Cancer Invest. 2008, 26, 860–865. [Google Scholar]
- Krown, S.E.; Li, P.; Von Roenn, J.H.; Paredes, J.; Huang, J.; Testa, M.A. Efficacy of low-dose interferon with antiretroviral therapy in Kaposi's sarcoma: a randomized phase II AIDS clinical trials group study. J. Interferon Cytokine Res. 2002, 22, 295–303. [Google Scholar]
- Chawla-Sarkar, M.; Masci, P.; Borden, EC. Interferons and Cytokines for Anti-Infective and Cancer Therapy. In Biotechnology and Biopharmaceuticals; John Wiley and Sons, Inc.: Hoboken N.J, USA, 2003; pp. 161–208. [Google Scholar]
- Oberg, K.; Alm, G. The incidence and clinical significance of antibodies to interferon-a in patients with solid tumors. Biotherapy 1997, 10, 1–5. [Google Scholar]
- McKenna, R.M.; Oberg, K.E. Antibodies to interferon-alpha in treated cancer patients: incidence and significance. J. Interferon Cytokine Res. 1997, 17, 141–143. [Google Scholar]
- Steis, R.G.; Smith, J.W., 2nd; Urba, W.J.; Venzon, D.J.; Longo, D.L.; Barney, R.; Evans, L.M.; Itri, L.M.; Ewel, C.H. Loss of interferon antibodies during prolonged continuous interferon-alpha 2a therapy in hairy cell leukemia. Blood 1991, 77, 792–798. [Google Scholar] [PubMed]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bekisz, J.; Baron, S.; Balinsky, C.; Morrow, A.; Zoon, K.C. Antiproliferative Properties of Type I and Type II Interferon. Pharmaceuticals 2010, 3, 994-1015. https://doi.org/10.3390/ph3040994
Bekisz J, Baron S, Balinsky C, Morrow A, Zoon KC. Antiproliferative Properties of Type I and Type II Interferon. Pharmaceuticals. 2010; 3(4):994-1015. https://doi.org/10.3390/ph3040994
Chicago/Turabian StyleBekisz, Joseph, Samuel Baron, Corey Balinsky, Angel Morrow, and Kathryn C. Zoon. 2010. "Antiproliferative Properties of Type I and Type II Interferon" Pharmaceuticals 3, no. 4: 994-1015. https://doi.org/10.3390/ph3040994