Lp-PLA2 Inhibition—The Atherosclerosis Panacea?

Abstract
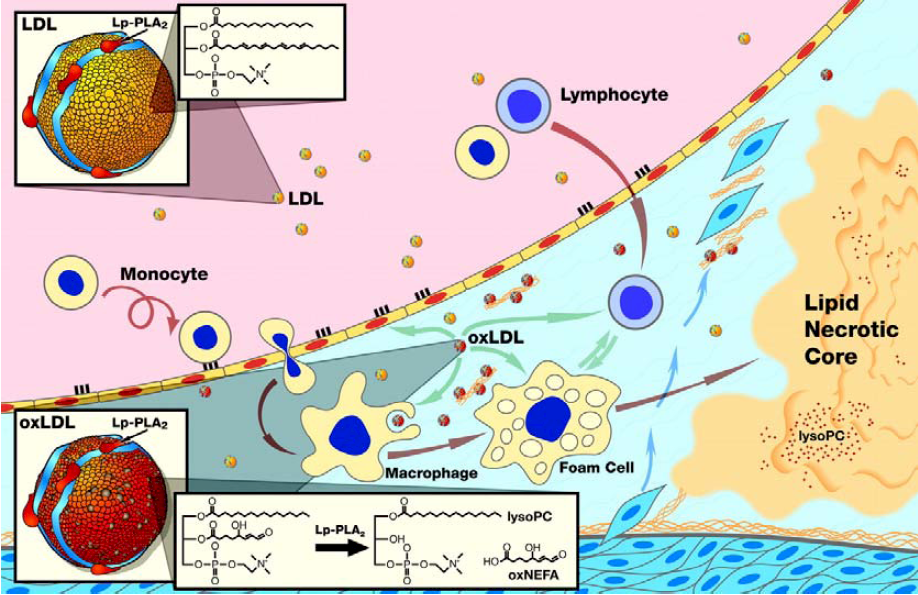
:1. Biochemistry and Biology

1.1. Pathoanatomical Evidence
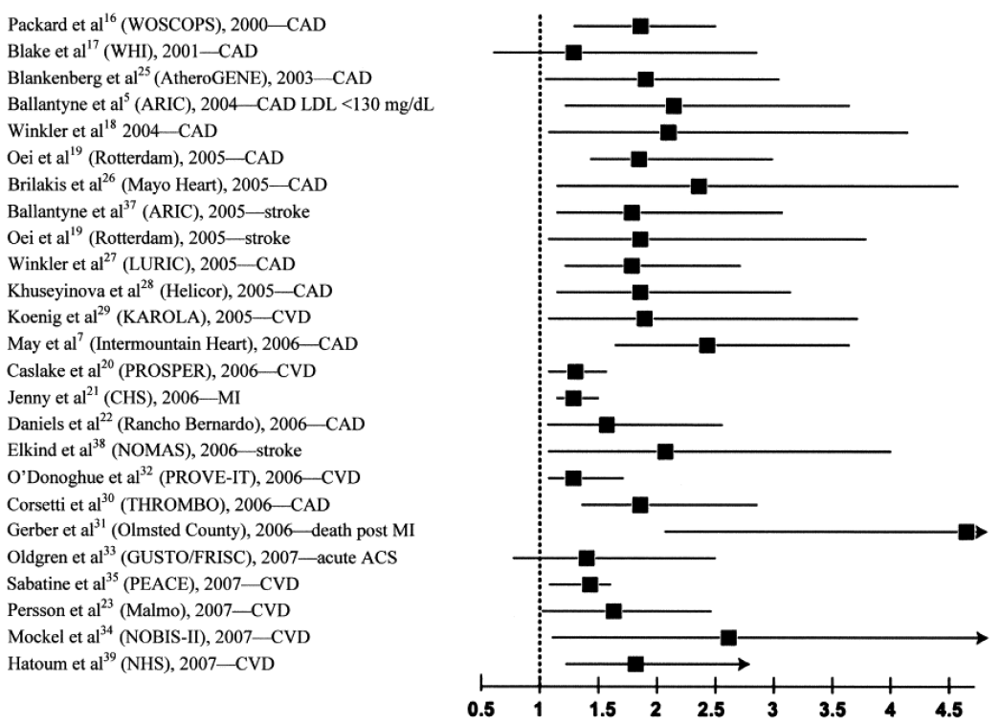
2. The Epidemiologic Evidence
2.1. Lp-PLA2 in Apparently Healthy, Middle-Aged Subjects

2.2. Lp-PLA2 in the Elderly Population
2.3. Lp-PLA2 in Patients with ACS
2.4. Lp-PLA2 in Patients with Stable CHD
3. Clinical Studies
3.1. Lp-PLA2 and Endothelial Dysfunction
3.2. Lp-PLA2 as A Target for Pharmacologic Intervention

{kind=link}
{kind=link}
| Author [Ref.] | Phase | Mass or activity | Cohort | N | Treatment | Primary Endpoint | Significant effects |
|---|---|---|---|---|---|---|---|
| Johnson et al. [56] | II | Lp-PLA2 activity | Patients before elective endarterectomy | 59 | 14 days | Lp-PLA2 activity | 80% reduction in enzyme activity |
| Mohler et al. [57] | II | Lp-PLA2 activity | CHD and CHD-risk equivalent patients receiving atorvastatin | 959 | 12 weeks | Lp-PLA2 activity and biomarkers | 43-66% reduction in enzyme activity, 13% reduction of IL-6, and CRP (p = 0.028 and p = 0.15) |
| Serruys et al. [58] | II | Lp-PLA2 activity | Patients with angiographically documented CHD | 330 | 12 months | Coronary atheroma plaque cap deformability; CRP | 59% reduction in enzyme activity, halt of necrotic core expansion (-5.2 mm3; p = 0.012) |
4. Disclosures
References and Notes
- Asano, K.; Okamoto, S.; Fukunaga, K.; Shiomi, T.; Mori, T.; Iwata, M.; Ikeda, Y.; Yamaguchi, K. Cellular sources of platelet-activating-factor acetylhydrolase activity in plasma. Biochem. Biophys. Res. Commun. 1999, 261, 511–514. [Google Scholar] [CrossRef] [PubMed]
- Stafforini, D.M.; Elstad, M.R.; McIntyre, T.M.; Zimmerman, G.A.; Prescott, S.M. Human macrophages secrete platelet-activating factor acetylhydrolase. J. Biol. Chem. 1990, 265, 9682–9687. [Google Scholar]
- Venable, M.E.; Zimmerman, G.A.; McIntyre, T.M.; Prescott, S.M. Platelet-activating factor: a phospholipid autacoid with diverse actions. J. Lipid Res. 1993, 34, 691–702. [Google Scholar]
- Min, J.H.; Jain, M.K.; Wilder, C.; Paul, L.; Apitz-Castro, R.; Aspleaf, D.C.; Gelb, M.H. Membrane-bound plasma platelet activating factor acetylhydrolase acts on substrate in the aqueous phase. Biochemistry 1999, 38, 12935–12942. [Google Scholar]
- Stafforini, D.M.; Tjoelker, L.W.; McCormick, S.P.; Vaitkus, D.; McIntyre, T.M.; Gray, P.W.; Young, S.G.; Prescott, S.M. Molecular basis of the interaction between plasma platelet-activating factor acetylhydrolase and low-density lipoprotein. J. Biol. Chem. 1999, 274, 7018–7024. [Google Scholar]
- Macphee, C.H.; Moores, K.E.; Boyd, H.F.; Dhanak, D.; Ife, R.J.; Leach, C.A.; Leake, D.S.; Milliner, K.J.; Patterson, R.A.; Suckling, K.E.; Tew, D.G.; Hickey, D.M. Lipoprotein-associated phospholipase A2, platelet-activating factor acetylhydrolase, generates two bioactive products during the oxidation of low-density lipoprotein: use of a novel inhibitor. Biochem. J. 1999, 338, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Hannson, G.K. Mechanisms of disease—Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef] [PubMed]
- Khan-Merchant, N.; Penumetcha, M.; Meilhac, O.; Parthasarathy, S. Oxidized fatty acids promote atherosclerosis only in the presence of dietary cholesterol in low-density lipoprotein receptor knockout mice. J. Nutr. 2002, 132, 3256–3262. [Google Scholar] [PubMed]
- Bäck, M. Leukotriene signaling in atherosclerosis and ischemia. Cardiovasc. Drugs Ther. 2009, 23, 41–48. [Google Scholar]
- Balsinde, J.; Winstead, M.V.; Dennis, E.A. Phospholipase A (2) regulation of arachidonic acid mobilization. FEBS Lett. 2002, 531, 2–6. [Google Scholar]
- Matsumo, T.; Kobayashi, T.; Kamata, K. Role of lysophoshatidylcholine (LPC) in atheroclerosis. Curr. Med. Chem. 2007, 14, 3209–3220. [Google Scholar]
- Schmitz, G.; Ruebsaamen, K. Metabolism and atherogenic disease association of lysophosphatidylcholine. Atherosclerosis 2009, 10–18. [Google Scholar]
- Hakkinen, T.; Luoma, J.S.; Hiltunen, M.O.; Macphee, C.H.; Milliner, K.J.; Patel, L.; Rice, S.Q.; Tew, D.G.; Karkola, K.; Ylä-Herttuala, S. Lipoprotein-associated phospholipase 2, a platelet-activating factor acetylhydrolase, is expressed by macrophages in human and rabbit atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2909–2917. [Google Scholar] [PubMed]
- Kume, N.; Cybulski, M.I.; Gimbrone, M.A., Jr. Lysophosphatidylcholine, a component of atherogenic lipoproteins, induces mononuclear leukocyte adhesion molecules in cultured human and rabbit arterial endothelial cells. J. Clin. Invest. 1992, 90, 1138–1144. [Google Scholar] [CrossRef] [PubMed]
- Rong, J.X.; Berman, J.W.; Taubman, M.B.; Fisher, E.A. Lysophosphatidylcholine stimulates monocyte chemoattractant protein-1 gene expression in rat aortic smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1617–1623. [Google Scholar]
- Takahara, N.; Kashiwaga, A.; Maegawa, H.; Shigeta, Y. Lysophosphatidylcholine stimulates the expression and production of MCP-1 by human vascular endothelial cells. Metabolism 1996, 45, 559–564. [Google Scholar]
- Liu-Wu, Y.; Hurt-Camejo, E.; Wiklund, O. Lysophosphatidylcholine induces the production of IL-1beta by human monocytes. Atherosclerosis 1998, 137, 351–357. [Google Scholar]
- Ousman, S.S.; David, S. Lysophosphatidylcholine induces rapid recruitment and activation of macrophages in the adult mouse spinal cord. Glia 2000, 30, 92–104. [Google Scholar]
- Macphee, C.H.; Nelsonj, J.J.; Zalewski, A. Lipoprotein-associated phospholipase A2 as a target of therapy. Curr. Opin. Lipidol. 2005, 16, 4442–4446. [Google Scholar]
- Tjoelker, L.W.; Wilder, C.; Eberhardt, C.; Stafforini, D.M.; Dietsch, G.; Schimpf, B.; Hooper, S.; Le Trong, H.; Cousens, L.S.; Zimmerman, G.A. Anti-inflammatory properties of a platelet-activating factor acetylhydrolase. Nature 1995, 374, 549–553. [Google Scholar]
- Lerman, A.; McConnell, J.P. Lipoprotein-associated phospholipase A2: a risk marker or a risk factor? Am. J. Cardiol. 2008, 101, 11F–22F. [Google Scholar] [PubMed]
- Theilmeier, G.; De Geest, B.; Van Veldhofen, P.P.; Stengel, D.; Michiels, C.; Lox, M.; Landeloos, M.; Chapman, M.J.; Ninio, E.; Collen, D.; Himpens, B.; Holvoet, P. HDL-associated PAF-AH reduces endothelial adhesiveness in apoE-/- mice. FASEB J. 2000, 14, 2032–2039. [Google Scholar]
- Chen, C.H.; Jiang, T.; Yang, J.H.; Jiang, W.; Lu, J.; Marathe, G.K.; Pownall, H.J.; Ballantyne, C.M.; McIntyre, T.M.; Henry, P.D.; Yang, C.Y. Low-density lipoprotein in hypercholesterolemic human plasma induces vascular endothelial cell apoptosis by inhibiting fibroblast growth factor 2 transcription. Circulation 2003, 107, 2102–2108. [Google Scholar]
- Zalewski, A.; Macphee, C. Role of lipoprotein-associated phospholipase A2 in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 923–931. [Google Scholar]
- Mohler, E.R., III; Sarov-Blat, L.; Shi, Y.; Hamamdzic, D.; Zalewski, A.; Macphee, C.; Llano, R.; Pelchovitz, D.; Mainigi, S.K.; Osman, H.; et al. Site-specific atherogenic gene expression correlates with subsequent variable lesion development in coronary and peripheral vasculature. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 850–855. [Google Scholar] [PubMed]
- Mannheim, D.; Herrmann, J.; Versari, D.; Gössl, M.; Meyer, F.B.; McConnell, J.P.; Lerman, L.O.; Lerman, A. Enhanced expression of Lp-PLA2 and lysophosphatidylcholine in symptomatic carotid atherosclerotic plaques. Stroke 2008, 39, 1448–1455. [Google Scholar]
- Herrmann, J.; Mannheim, D.; Wohlert, C.; Versari, D.; Meyer, F.B.; McConnell, J.P.; Gössl, M.; Lerman, L.O.; Lerman, A. Expression of lipoprotein-associated phospholipase A2 in carotid artery plaques predicts long-term cardiac outcome. Eur. Heart J. 2009, 30, 2930–2938. [Google Scholar]
- Kolodgie, F.D.; Burke, A.P.; Skorija, K.S.; Ladich, E.; Kutys, R.; Makuria, A.T.; Virmani, R. Lipoprotein-associated phospholipase A2 protein expression in the natural progression of human coronary atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2523–2529. [Google Scholar]
- Guerra, R.; Zhao, B.; Mooser, V.; Stafforini, D.; Johnston, J.M.; Cohen, J.C. Determinants of plasma platelet-activating factor acetylhydrolase: heritability and relationship to plasma lipoproteins. J. Lipid Res. 1997, 38, 2281–2288. [Google Scholar]
- Lenzini, L.; Antezza, K.; Caroccia, B.; Wolfert, R.L.; Szczech, R.; Cesari, M.; Narkiewicz, K.; Williams, C.J.; Rossi, G.P. A twin study of heritability of plasma lipoprotein-associated phospholipase A2 (Lp-PLA2) mass and activity. Atherosclerosis 2009, 205, 181–185. [Google Scholar]
- Garza, C.A.; Montori, V.M.; McConnell, J.P.; Somers, V.K.; Kullo, I.J.; Lopez-Jimenez, F. Association between lipoprotein-associated phospholipase A2 and cardiovascular disease: A systematic review. Mayo Clin. Proc. 2007, 82, 159–165. [Google Scholar]
- Packard, C.J.; O’Reilly, D.S.; Caslake, M.J.; McMahon, A.D.; Cooney, J.; Macphee, C.H.; Suckling, K.E.; Krishna, M.; Wilkinson, F.E.; Rumley, A.; Lowe, G.D. Lipoprotein-associated phospholipase A2 as an independent predictor of coronary heart disease. N. Engl. J. Med. 2000, 343, 1148–1155. [Google Scholar]
- Blake, G.J.; Dada, N.; Fox, J.C.; Manson, J.E.; Ridker, P.M. A prospective evaluation of lipoprotein-associated phospholipase A2 levels and the risk of future cardiovascular events in women. J. Am. Coll. Cardiol. 2001, 38, 1302–1306. [Google Scholar]
- Koenig, W.; Khuseyinova, N. Lipoprotein-associated and secretory phospholipase A2 in cardiovascular disease: the epidemiologic evidence. Cardiovasc. Drugs Ther. 2009, 23, 85–92. [Google Scholar]
- Ballantyne, C.M.; Hoogeveen, R.C.; Bang, H.; Coresh, J.; Folsom, A.R.; Heiss, G.; Sharrett, A.R. Lipoprotein-associated phospholipase A2, high-sensitivity C-reactive protein, and risk for incident coronary heart disease in middle-aged men and women in the Atherosclerosis Risk in Communities (ARIC) study. Circulation 2004, 109, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Koenig, W.; Khuseyinova, N.; Lowel, H.; Trischler, G.; Meisinger, C. Lipoprotein-associated phospholipase A2 adds to risk prediction of incident coronary events by C-reactive protein in apparently healthy middle-aged men from the general population: results from the 14-year follow-up of a large cohort from southern Germany. Circulation 2004, 110, 1903–1908. [Google Scholar]
- Oei, H.H.; van der Meer, I.M.; Hofman, A.; Koudstaal, P.J.; Stijnen, T.; Breteler, M.M.; Witteman, J.C. Lipoprotein-associated phospholipase A2 activity is associated with risk of coronary heart disease and ischemic stroke: the Rotterdam Study. Circulation 2005, 111, 570–575. [Google Scholar]
- Kiechl, S.; Willeit, J.; Mayr, M.; Viehweider, B.; Oberhollenzer, M.; Kronenberg, F.; Wiedemann, C.J.; Oberthaler, S.; Xu, Q.; Witztum, J.L.; Tsimikas, S. Oxidized phospholipids, lipoprotein (a), lipoprotein-associated phospholipase A2 activity, and 10-year cardiovascular outcomes: prospective results from the Bruneck study. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1788–1795. [Google Scholar] [PubMed]
- Daniels, L.B.; Laughlin, G.A.; Sarno, M.J.; Bettencourt, R.; Wolfert, R.L.; Barrett-Connor, E. Lipoprotein-associated phospholipase A2 is an independent predictor of incident coronary heart disease in an apparently healthy older population: the Rancho Bernardo Study. J. Am. Coll. Cardiol. 2008, 51, 913–919. [Google Scholar]
- Jenny, N.S.; Solomon, C.; Cushman, M.; Nelson, J.J.; Tracy, R.P.; Psaty, B.M.; Furberg, C.D. Lipoprotein-associated phospholipase A2 and cardiovascular disease: results from the Cardiovascular Health Study. Circulation 2006, 113, E332, Abstract. [Google Scholar]
- Caslake, M.J.; Cooney, J.; Murray, E.; Bedford, D.; Robertson, M.; Nelson, J.J; Packard, C.J. Lipoprotein-associated phospholipase A2 as a risk factor for coronary vascular disease in the elderly. Atherosclerosis. 2006, 7 (Suppl.), 484, Abstract. [Google Scholar]
- Möckel, M.; Müller, R.; Vollert, J.O.; Müller, C.; Danne, O.; Gareis, R.; Störk, T.; Dietz, R.; Koenig, W. Lipoprotein-associated phospholipase A2 for early risk stratification in patients with suspected acute coronary syndrome: a multi-marker approach: the North Wuerttemberg and Berlin Infarction Study-II (NOBIS-II). Clin. Res. Cardiol. 2007, 96, 604–612. [Google Scholar]
- Gerber, Y.; McConnell, J.P.; Jaffe, A.S.; Weston, S.A.; Killian, J.M.; Roger, V.L. Lipoprotein-associated phospholipase A2 and prognosis after myocardial infarction in the community. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2517–2522. [Google Scholar]
- O’Donoghue, M.; Morrow, D.A.; Sabatine, M.S.; Murphy, S.A.; McCabe, C.H.; Cannon, C.P.; Braunwald, E. Lipoprotein-associated phospholipase A2 and its association with cardiovascular outcomes in patients with acute coronary syndromes in the PROVE IT-TIMI 22 (PRavastatin Or atorVastatin Evaluation and Infection Therapy-Thrombolysis In Myocardial Infarction) trial. Circulation 2006, 113, 1745–1752. [Google Scholar]
- Oldgren, J.; James, S.K.; Siegbahn, A.; Wallentin, L. Lipoprotein-associated phospholipase A2 does not predict mortality or new ischaemic events in acute coronary syndrome patients. Eur. Heart. J. 2007, 28, 699–704. [Google Scholar]
- Brilakis, E.S.; McConnell, J.P.; Lennon, R.J.; Elesber, A.A.; Meyer, J.G.; Berger, P.B. Association of lipoprotein-associated phospholipase A2 levels with coronary artery disease risk factors, angiographic coronary artery disease, and major adverse events at follow-up. Eur. Heart J. 2005, 26, 137–144. [Google Scholar] [PubMed]
- Koenig, W.; Twardella, D.; Brenner, H.; Rothenbacher, D. Lipoprotein-associated phospholipase A2 predicts future cardiovascular events in patients with coronary heart disease independently of traditional risk factors, markers of inflammation, renal function and hemodynamic stress. Arterioscler Thromb. Vasc. Biol. 2006, 26, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Corsetti, J.P.; Rainwater, D.L.; Moss, A.J.; Zareba, W.; Sparks, C.E. High lipoprotein-associated phospholipase A2 is a risk factor for recurrent coronary events in postinfarction patients. Clin. Chem. 2006, 52, 1331–1338. [Google Scholar]
- Sabatine, M.S.; Morrow, D.A.; O’Donoghue, M.; Jablonski, K.; Rice, M.M.; Solomon, S.; Rosenberg, Y.; Domanski, M.J.; Hsia, J. PEACE Investigators. Prognostic utility of lipoprotein-associated phospholipase A2 for cardiovascular outcomes in patients with stable coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2463–2469. [Google Scholar] [CrossRef] [PubMed]
- Winkler, K.; Hoffmann, M.M.; Winkelmann, B.R.; Friedrich, I.; Schäfer, G.; Seelhorst, U.; Wellnitz, B.; Wieland, H.; Boehm, B.O.; März, W. Lipoprotein-associated phospholipase A2 predicts 5-year cardiac mortality independently of established risk factors and adds prognostic information in patients with low and medium high-sensitivity C-reactive protein (the Ludwigshafen risk and cardiovascular health study). Clin. Chem. 2007, 53, 1440–1447. [Google Scholar]
- Yang, E.H.; McConnell, J.P.; Lennon, R.J.; Barsness, G.W.; Pumper, G.; Hartman, S.J.; Rihal, C.S.; Lerman, L.O.; Lerman, A. Lipoprotein-associated phospholipase A2 is an independent marker for coronary endothelial dysfunction in humans. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 106–111. [Google Scholar]
- Lavi, S.; McConnell, J.; P Rihal, C.S.; Prasad, A.; Mathew, V.; Lerman, L.O.; Lerman, A. Local production of lipoprotein-associated phospholipase A2 and lysophosphatidlcholine in the coronary circulation: association with early coronary atherosclerosis and endothelial dysfunction in humans. Circulation 2007, 115, 2715–2721. [Google Scholar]
- Karakas, M.; Koenig, W. Phospholipase A2 as a therapeutic target for atherosclerosis. Clin. Lip. 2010, 5, 43–56. [Google Scholar]
- Rosenson, R.S. Future role for selective phospolipase A2 inhibitors in the prevention of atherosclerotic cardiovascular disease. Cardiovasc. Drugs Ther. 2009, 23, 93–101. [Google Scholar]
- Wilensky, R.L.; Shi, Y.; Mohler, E.R.; Hamamdzic, D.; Burgert, M.E.; Li, J.; Postle, A.; Fenning, R.S.; Bollinger, J.G.; Hoffmann, B.E.; et al. Inhibition of lipoprotein-associated phospholipase A2 reduces complex coronary atherosclerotic plaque development. Nat. Med. 2008, 14, 1015–1016. [Google Scholar] [PubMed]
- Johnson, A.; Zalewski, A.; Janmohamed, S.; Sawyer, J.; Rolfe, T.; Staszkiewicz, W.; Alvarez, S. Lipoprotein-associated phospholipase A2 (Lp-PLA2) activity, an emerging CV risk marker, can be inhibited in atherosclerotic lesions and plasma by novel pharmacologic intervention: the results of a multicenter clinical study. Circulation 2004, 110, III-590. [Google Scholar]
- Mohler, E.R.; Ballantyne, C.M.; Davidson, M.H.; Hanefeld, M.; Ruilope, L.M.; Johnson, J.L.; Zalewski, A. The effect of darapladib on plasma lipoprotein-associated phospholipase A2 activity and cardiovascular biomarkers in patients with stable coronary heart disease risk equivalent: the results of a multicenter, randomized, double-blind, placebo-controlled study. J. Am. Coll. Cardiol. 2008, 51, 1632–1641. [Google Scholar] [CrossRef] [PubMed]
- Serruys, P.W.; Garcia-Garcia, H.M.; Buszman, P.; Erne, P.; Verheye, S.; Aschermann, M.; Duckers, H.; Bleie, O.; Dudek, D.; Botker, H.E.; von Birgelen, C.; DÁmico, D.; Hutchinson, T.; Zambanini, A.; Mastik, F.; van Ees, G.A.; van der Steen, A.F.; Vince, D.G.; Ganz, P.; Hamm, C.W.; Wijns, W.; Zalewski, A. Effects of the direct lipoprotein-associated phospholipase A2 inhibitor darapladib on human coronary atherosclerotic plaque. Circulation 2008, 118, 1172–1182. [Google Scholar]
- McCullough, P.A. Darapladib and atherosclerotic plaque: should lipoprotein-associated phospholipase A2 be a therapeutic target? Curr. Atheroscler. Rep. 2009, 11, 334–337. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Karakas, M.; Koenig, W. Lp-PLA2 Inhibition—The Atherosclerosis Panacea? Pharmaceuticals 2010, 3, 1360-1373. https://doi.org/10.3390/ph3051360
Karakas M, Koenig W. Lp-PLA2 Inhibition—The Atherosclerosis Panacea? Pharmaceuticals. 2010; 3(5):1360-1373. https://doi.org/10.3390/ph3051360
Chicago/Turabian StyleKarakas, Mahir, and Wolfgang Koenig. 2010. "Lp-PLA2 Inhibition—The Atherosclerosis Panacea?" Pharmaceuticals 3, no. 5: 1360-1373. https://doi.org/10.3390/ph3051360
APA StyleKarakas, M., & Koenig, W. (2010). Lp-PLA2 Inhibition—The Atherosclerosis Panacea? Pharmaceuticals, 3(5), 1360-1373. https://doi.org/10.3390/ph3051360