NSAIDs: Old Drugs Reveal New Anticancer Targets

Abstract
:1. Introduction
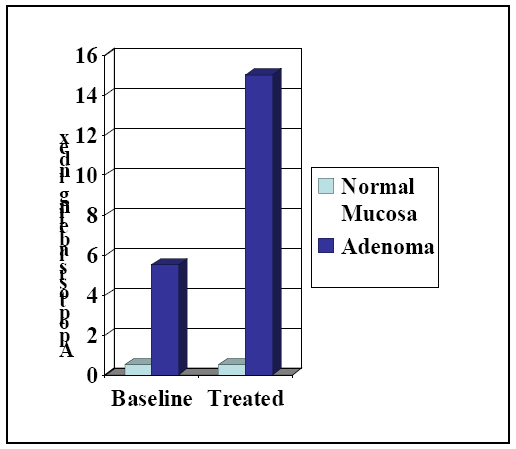
2. Colorectal Cancer
3. Cancer Chemopreventive Activity of NSAIDs
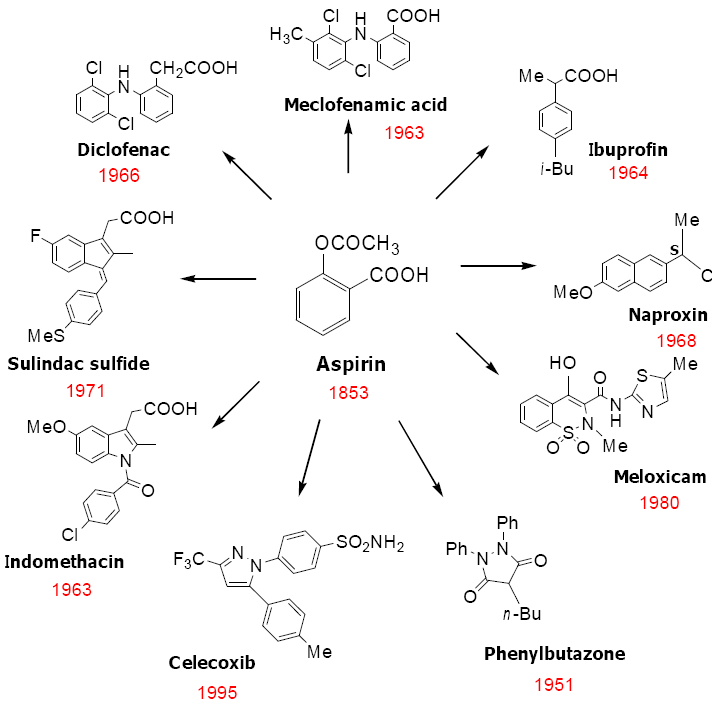
4. Classification of NSAIDs

5. Cyclooxygenase Independent Anticancer Activity of NSAIDs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 |
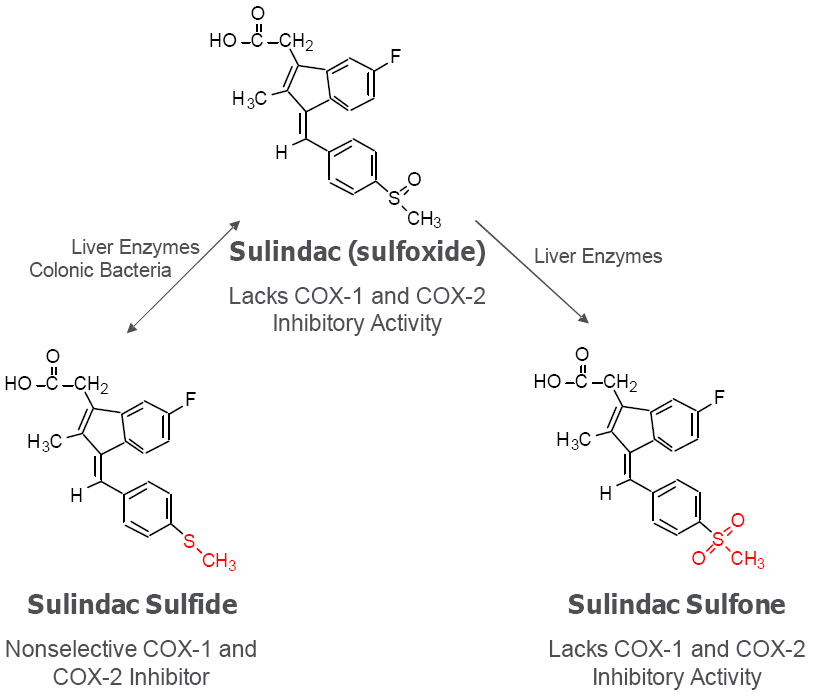
6. Cancer Chemopreventive Activity of Sulindac and Its Non-COX Inhibitory Sulfone Metabolite

 |

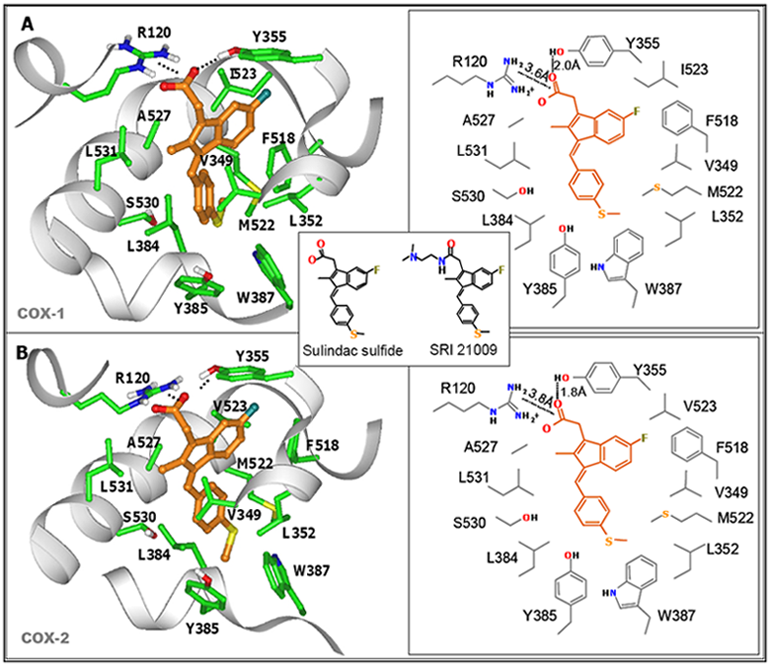
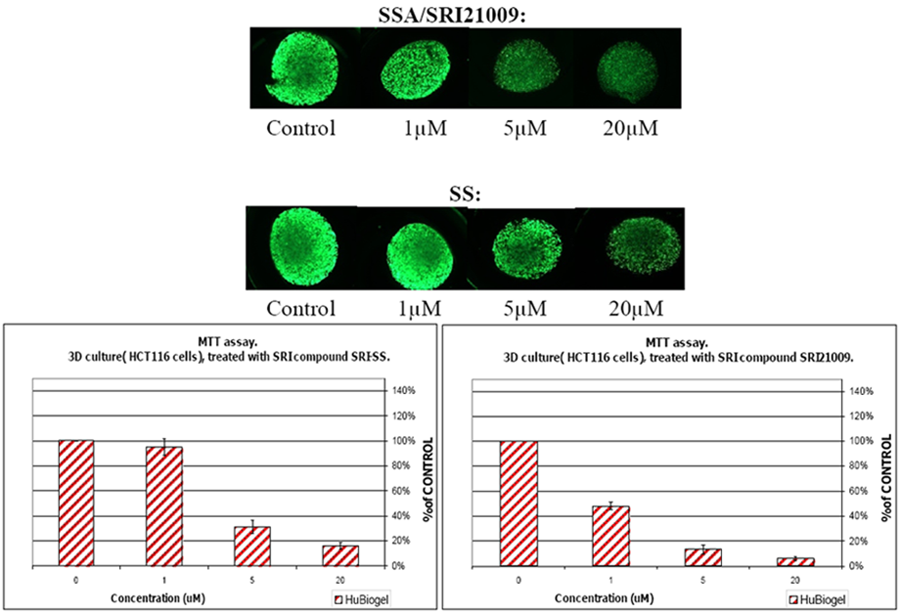
7. Design of a Novel Non-COX Inhibitory Sulindac Derivative with Potent Tumor Cell Growth Inhibitory Activity


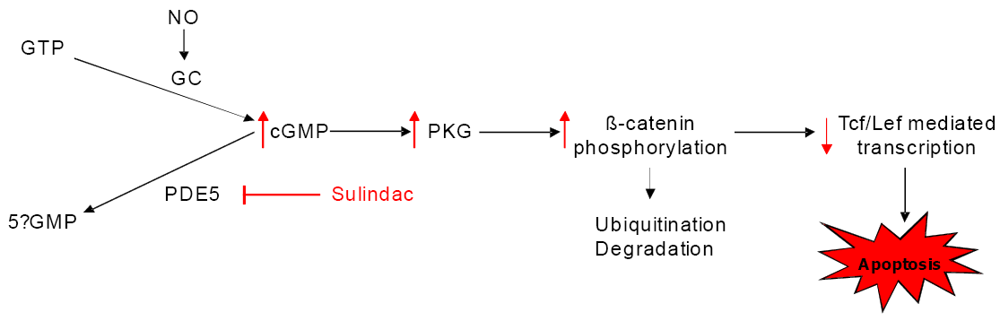
8. COX-Independent Targets of NSAIDs

9. Conclusions
References
- Thun, M.J.; Henley, S.J.; Patrono, C. Nonsteroidal anti-inflammatory drugs as anticancer agents: mechanistic, pharmacologic, and clinical issues. J. Natl. Cancer Inst. 2002, 94, 252–266. [Google Scholar]
- Chan, T.A. Nonsteroidal anti-inflammatory drugs, apoptosis, and colon-cancer chemoprevention. Lancet Oncol. 2002, 3, 166–174. [Google Scholar]
- Matsuhashi, N.; Nakajima, A.; Shinohara, K.; Oka, T.; Yazaki, Y. Rectal cancer after sulindac therapy for a sporadic adenomatous colonic polyp. Am. J. Gastroenterol. 1998, 93, 2261–2266. [Google Scholar]
- Waddell, W.R.; Loughry, R.W. Sulindac for polyposis of the colon. J Surg Oncol 1983, 24, 83–87. [Google Scholar]
- Rigau, J.; Pique, J.M.; Rubio, E.; Planas, R.; Tarrech, J.M.; Bordas, J.M. Effects of long-term sulindac therapy on colonic polyposis. Ann. Intern. Med. 1991, 115, 952–954. [Google Scholar]
- Giardiello, F.M.; Hamilton, S.R.; Krush, A.J.; Piantadosi, S.; Hylind, L.M.; Celano, P.; Booker, S.V.; Robinson, C.R.; Offerhaus, G.J. Treatment of colonic and rectal adenomas with sulindac in familial adenomatous polyposis. N. Engl. J. Med. 1993, 328, 1313–1316. [Google Scholar]
- Nugent, K.P.; Farmer, K.C.; Spigelman, A.D.; Williams, C.B.; Phillips, R.K. Randomized controlled trial of the effect of sulindac on duodenal and rectal polyposis and cell proliferation in patients with familial adenomatous polyposis. Br. J. Surg. 1993, 80, 1618–1619. [Google Scholar]
- Steinbach, G.; Lynch, P.M.; Phillips, R.K.; Wallace, M.H.; Hawk, E.; Gordon, G.B.; Wakabayashi, N.; Saunders, B.; Shen, Y.; Fujimura, T.; Su, L.K.; Levin, B. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N. Engl. J. Med. 2000, 342, 1946–1952. [Google Scholar] [PubMed]
- Vane, J.R.; Botting, R.M. Mechanism of action of antiinflammatory drugs. Int. J. Tissue React. 1998, 20, 3–15. [Google Scholar]
- Vane, J.R.; Bakhle, Y.S.; Botting, R.M. Cyclooxygenases 1 and 2. Annu. Rev. Pharmacol. Toxicol. 1998, 38, 97–120. [Google Scholar]
- Mukherjee, D. Selective cyclooxygenase-2 (COX-2) inhibitors and potential risk of cardiovascular events. Biochem. Pharmacol. 2002, 63, 817–821. [Google Scholar]
- Niv, Y.; Fraser, G.M. Adenocarcinoma in the rectal segment in familial polyposis coli is not prevented by sulindac therapy. Gastroenterology 1994, 107, 854–857. [Google Scholar]
- Brown, J.R.; DuBois, R.N. COX-2: a molecular target for colorectal cancer prevention. J. Clin. Oncol. 2005, 23, 2840–2855. [Google Scholar]
- Eberhart, C.E.; Coffey, R.J.; Radhika, A.; Giardiello, F.M.; Ferrenbach, S.; DuBois, R.N. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology 1994, 107, 1183–1188. [Google Scholar]
- Rigas, B.; Goldman, I.S.; Levine, L. Altered eicosanoid levels in human colon cancer. J. Lab. Clin. Med. 1993, 122, 518–523. [Google Scholar]
- Sano, H.; Kawahito, Y.; Wilder, R.L.; Hashiramoto, A.; Mukai, S.; Asai, K.; Kimura, S.; Kato, H.; Kondo, M.; Hla, T. Expression of cyclooxygenase-1 and -2 in human colorectal cancer. Cancer Res. 1995, 55, 3785–3789. [Google Scholar]
- Alberts, D.S.; Hixson, L.; Ahnen, D.; Bogert, C.; Einspahr, J.; Paranka, N.; Brendel, K.; Gross, P.H.; Pamukcu, R.; Burt, R.W. Do NSAIDs exert their colon cancer chemoprevention activities through the inhibition of mucosal prostaglandin synthetase? J. Cell Biochem. Suppl. 1995, 22, 18–23. [Google Scholar] [PubMed]
- Piazza, G.A.; Rahm, A.L.; Krutzsch, M.; Sperl, G.; Paranka, N.S.; Gross, P.H.; Brendel, K.; Burt, R.W.; Alberts, D.S.; Pamukcu, R.; Ahnen, D.J. Antineoplastic drugs sulindac sulfide and sulfone inhibit cell growth by inducing apoptosis. Cancer Res. 1995, 55, 3110–3116. [Google Scholar] [PubMed]
- Hanif, R.; Pittas, A.; Feng, Y.; Koutsos, M.I.; Qiao, L.; Staiano-Coico, L.; Shiff, S.I.; Rigas, B. Effects of nonsteroidal anti-inflammatory drugs on proliferation and on induction of apoptosis in colon cancer cells by a prostaglandin-independent pathway. Biochem. Pharmacol. 1996, 52, 237–245. [Google Scholar]
- Elder, D.J.; Halton, D.E.; Hague, A.; Paraskeva, C. Induction of apoptotic cell death in human colorectal carcinoma cell lines by a cyclooxygenase-2 (COX-2)-selective nonsteroidal anti-inflammatory drug: independence from COX-2 protein expression. Clin. Cancer Res. 1997, 3, 1679–1683. [Google Scholar]
- Jemal, A.; Murray, T.; Ward, E.; Samuels, A.; Tiwari, R.C.; Ghafoor, A.; Feuer, E.J.; Thun, M.J. Cancer statistics, 2005. CA Cancer J. Clin. 2005, 55, 10–30. [Google Scholar]
- Wingo, P.A.; Ries, L.A.; Parker, S.L.; Heath, C.W., Jr. Long-term cancer patient survival in the United States. Cancer Epidemiol. Biomarkers Prev. 1998, 7, 271–282. [Google Scholar]
- Greenlee, R.T.; Hill-Harmon, M.B.; Murray, T.; Thun, M. Cancer statistics, 2001. CA Cancer J. Clin. 2001, 51, 15–36. [Google Scholar]
- O'Shaughnessy, J.A.; Kelloff, G.J.; Gordon, G.B.; Dannenberg, A.J.; Hong, W.K.; Fabian, C.J.; Sigman, C.C.; Bertagnolli, M.M.; Stratton, S.P.; Lam, S.; Nelson, W.G.; Meyskens, F.L.; Alberts, D.S.; Follen, M.; Rustgi, A.K.; Papadimitrakopoulou, V.; Scardino, P.T.; Gazdar, A.F.; Wattenberg, L.W.; Sporn, M.B.; Sakr, W.A.; Lippman, S.M.; Von Hoff, D.D. Treatment and prevention of intraepithelial neoplasia: an important target for accelerated new agent development. Clin. Cancer Res. 2002, 8, 314–346. [Google Scholar]
- Oshima, M.; Oshima, H.; Kitagawa, K.; Kobayashi, M.; Itakura, C.; Taketo, M. Loss of Apc heterozygosity and abnormal tissue building in nascent intestinal polyps in mice carrying a truncated Apc gene. Proc. Natl. Acad. Sci. USA 1995, 92, 4482–4486. [Google Scholar]
- Levy, D.B.; Smith, K.J.; Beazer-Barclay, Y.; Hamilton, S.R.; Vogelstein, B.; Kinzler, K.W. Inactivation of both APC alleles in human and mouse tumors. Cancer Res. 1994, 54, 5953–5958. [Google Scholar]
- Peifer, M.; Polakis, P. Wnt signaling in oncogenesis and embryogenesis--a look outside the nucleus. Science 2000, 287, 1606–1609. [Google Scholar]
- Bafico, A.; Liu, G.; Goldin, L.; Harris, V.; Aaronson, S.A. An autocrine mechanism for constitutive Wnt pathway activation in human cancer cells. Cancer Cell 2004, 6, 497–506. [Google Scholar]
- Smalley, W.; Ray, W.A.; Daugherty, J.; Griffin, M.R. Use of nonsteroidal anti-inflammatory drugs and incidence of colorectal cancer: a population-based study. Arch. Intern. Med. 1999, 159, 161–166. [Google Scholar]
- Piazza, G.A.; Alberts, D.S.; Hixson, L.J.; Paranka, N.S.; Li, H.; Finn, T.; Bogert, C.; Guillen, J.M.; Brendel, K.; Gross, P.H.; Sperl, G.; Ritchie, J.; Burt, R.W.; Ellsworth, L.; Ahnen, D.J.; Pamukcu, R. Sulindac sulfone inhibits azoxymethane-induced colon carcinogenesis in rats without reducing prostaglandin levels. Cancer Res. 1997, 57, 2909–2915. [Google Scholar]
- Beazer-Barclay, Y.; Levy, D.B.; Moser, A.R.; Dove, W.F.; Hamilton, S.R.; Vogelstein, B.; Kinzler, K.W. Sulindac suppresses tumorigenesis in the Min mouse. Carcinogenesis 1996, 17, 1757–1760. [Google Scholar]
- Barker, N.; Clevers, H. Mining the Wnt pathway for cancer therapeutics. Nat. Rev. Drug Discov. 2006, 5, 997–1014. [Google Scholar]
- Mukherjee, D.; Nissen, S.E.; Topol, E.J. Risk of cardiovascular events associated with selective COX-2 inhibitors. JAMA 2001, 286, 954–959. [Google Scholar]
- Piazza, G.A.; Rahm, A.K.; Finn, T.S.; Fryer, B.H.; Li, H.; Stoumen, A.L.; Pamukcu, R.; Ahnen, D.J. Apoptosis primarily accounts for the growth-inhibitory properties of sulindac metabolites and involves a mechanism that is independent of cyclooxygenase inhibition, cell cycle arrest, and p53 induction. Cancer Res. 1997, 57, 2452–2459. [Google Scholar] [PubMed]
- Rigas, B.; Shiff, S.J. Is inhibition of cyclooxygenase required for the chemopreventive effect of NSAIDs in colon cancer? A model reconciling the current contradiction. Med. Hypotheses 2000, 54, 210–215. [Google Scholar]
- Kashfi, K.; Rigas, B. Non-COX-2 targets and cancer: expanding the molecular target repertoire of chemoprevention. Biochem. Pharmacol. 2005, 70, 969–986. [Google Scholar]
- Kusuhara, H.; Matsuyuki, H.; Matsuura, M.; Imayoshi, T.; Okumoto, T.; Matsui, H. Induction of apoptotic DNA fragmentation by nonsteroidal anti-inflammatory drugs in cultured rat gastric mucosal cells. Eur. J. Pharmacol. 1998, 360, 273–280. [Google Scholar]
- de Mello, M.C.; Bayer, B.M.; Beaven, M.A. Evidence that prostaglandins do not have a role in the cytostatic action of anti-inflammatory drugs. Biochem. Pharmacol. 1980, 29, 311–318. [Google Scholar]
- Williams, C.S.; Watson, A.J.; Sheng, H.; Helou, R.; Shao, J.; DuBois, R.N. Celecoxib prevents tumor growth in vivo without toxicity to normal gut: lack of correlation between in vitro and in vivo models. Cancer Res. 2000, 60, 6045–6051. [Google Scholar] [PubMed]
- Brideau, C.; Kargman, S.; Liu, S.; Dallob, A.L.; Ehrich, E.W.; Rodger, I.W.; Chan, C.C. A human whole blood assay for clinical evaluation of biochemical efficacy of cyclooxygenase inhibitors. Inflamm. Res. 1996, 45, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Cryer, B.; Feldman, M. Cyclooxygenase-1 and cyclooxygenase-2 selectivity of widely used nonsteroidal anti-inflammatory drugs. Am. J. Med. 1998, 104, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Song, X.; Lin, H.P.; Young, D.C.; Yan, S.; Marquez, V.E.; Chen, C.S. Using cyclooxygenase-2 inhibitors as molecular platforms to develop a new class of apoptosis-inducing agents. J. Natl. Cancer Inst. 2002, 94, 1745–1757. [Google Scholar]
- Myers, C.; Koki, A.; Pamukcu, R.; Wechter, W.; Padley, R.J. Proapoptotic anti-inflammatory drugs. Urology 2001, 57, 73–76. [Google Scholar]
- Elder, D.J.; Paraskeva, C. Induced apoptosis in the prevention of colorectal cancer by non-steroidal anti-inflammatory drugs. Apoptosis 1999, 4, 365–372. [Google Scholar]
- Rao, C.V.; Rivenson, A.; Simi, B.; Zang, E.; Kelloff, G.; Steele, V.; Reddy, B.S. Chemoprevention of colon carcinogenesis by sulindac, a nonsteroidal anti-inflammatory agent. Cancer Res. 1995, 55, 1464–1472. [Google Scholar]
- Moorghen, M.; Ince, P.; Finney, K.J.; Sunter, J.P.; Appleton, D.R.; Watson, A.J. A protective effect of sulindac against chemically-induced primary colonic tumours in mice. J. Pathol. 1988, 156, 341–347. [Google Scholar]
- Davies, N.M.; Watson, M.S. Clinical pharmacokinetics of sulindac. A dynamic old drug. Clin. Pharmacokinet. 1997, 32, 437–459. [Google Scholar] [CrossRef] [PubMed]
- Reddy, B.S.; Kawamori, T.; Lubet, R.A.; Steele, V.E.; Kelloff, G.J.; Rao, C.V. Chemopreventive efficacy of sulindac sulfone against colon cancer depends on time of administration during carcinogenic process. Cancer Res. 1999, 59, 3387–3391. [Google Scholar]
- de Jong, T.A.; Skinner, S.A.; Malcontenti-Wilson, C.; Vogiagis, D.; Bailey, M.; van Driel, I.R.; O'Brien, P.E. Inhibition of rat colon tumors by sulindac and sulindac sulfone is independent of K-ras (codon 12) mutation. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 278, G266–G272. [Google Scholar]
- Piazza, G.A.; Thompson, W.J.; Pamukcu, R.; Alila, H.W.; Whitehead, C.M.; Liu, L.; Fetter, J.R.; Gresh, W.E., Jr.; Klein-Szanto, A.J.; Farnell, D.R.; Eto, I.; Grubbs, C.J. Exisulind, a novel proapoptotic drug, inhibits rat urinary bladder tumorigenesis. Cancer Res. 2001, 61, 3961–3968. [Google Scholar] [PubMed]
- Malkinson, A.M.; Koski, K.M.; Dwyer-Nield, L.D.; Rice, P.L.; Rioux, N.; Castonguay, A.; Ahnen, D.J.; Thompson, H.; Pamukcu, R.; Piazza, G.A. Inhibition of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced mouse lung tumor formation by FGN-1 (sulindac sulfone). Carcinogenesis 1998, 19, 1353–1356. [Google Scholar]
- Thompson, H.J.; Briggs, S.; Paranka, N.S.; Piazza, G.A.; Brendel, K.; Gross, P.H.; Sperl, G.J.; Pamukcu, R.; Ahnen, D.J. Inhibition of mammary carcinogenesis in rats by sulfone metabolite of sulindac. J. Natl. Cancer Inst. 1995, 87, 1259–1260. [Google Scholar]
- Thompson, H.J.; Jiang, C.; Lu, J.; Mehta, R.G.; Piazza, G.A.; Paranka, N.S.; Pamukcu, R.; Ahnen, D.J. Sulfone metabolite of sulindac inhibits mammary carcinogenesis. Cancer Res. 1997, 57, 267–271. [Google Scholar]
- Stoner, G.D.; Budd, G.T.; Ganapathi, R.; DeYoung, B.; Kresty, L.A.; Nitert, M.; Fryer, B.; Church, J.M.; Provencher, K.; Pamukcu, R.; Piazza, G.; Hawk, E.; Kelloff, G.; Elson, P.; van Stolk, R.U. Sulindac sulfone induced regression of rectal polyps in patients with familial adenomatous polyposis. Adv. Exp. Med. Biol. 1999, 470, 45–53. [Google Scholar]
- Arber, N.; Kuwada, S.; Leshno, M.; Sjodahl, R.; Hultcrantz, R.; Rex, D. Sporadic adenomatous polyp regression with exisulind is effective but toxic: a randomised, double blind, placebo controlled, dose-response study. Gut 2006, 55, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Piazza, G.A.; Keeton, A.B.; Tinsley, H.N.; Gary, B.D.; Whitt, J.D.; Mathew, B.; Thaiparambil, J.; Coward, L.; Gorman, G.; Li, Y.; Sani, B.; Hobrath, J.V.; Maxuitenko, Y.Y.; Reynolds, R.C. A novel sulindac derivative that does not inhibit cyclooxygenases but potently inhibits colon tumor cell growth and induces apoptosis with antitumor activity. Cancer Prev. Res. (Phila. PA) 2009, 2, 572–580. [Google Scholar] [CrossRef]
- Yuan, K.; Kucik, D.; Singh, R.K.; Listinsky, C.M.; Listinsky, J.J.; Siegal, G.P. Alterations in human breast cancer adhesion-motility in response to changes in cell surface glycoproteins displaying alpha-L-fucose moieties. Int. J. Oncol. 2008, 32, 797–807. [Google Scholar]
- Shureiqi, I.; Chen, D.; Lotan, R.; Yang, P.; Newman, R.A.; Fischer, S.M.; Lippman, S.M. 15-Lipoxygenase-1 mediates nonsteroidal anti-inflammatory drug-induced apoptosis independently of cyclooxygenase-2 in colon cancer cells. Cancer Res. 2000, 60, 6846–6850. [Google Scholar]
- Herrmann, C.; Block, C.; Geisen, C.; Haas, K.; Weber, C.; Winde, G.; Moroy, T.; Muller, O. Sulindac sulfide inhibits Ras signaling. Oncogene 1998, 17, 1769–1776. [Google Scholar]
- He, T.C.; Chan, T.A.; Vogelstein, B.; Kinzler, K.W. PPARdelta is an APC-regulated target of nonsteroidal anti-inflammatory drugs. Cell 1999, 99, 335–345. [Google Scholar]
- Yamamoto, Y.; Yin, M.J.; Lin, K.M.; Gaynor, R.B. Sulindac inhibits activation of the NF-kappaB pathway. J. Biol. Chem. 1999, 274, 27307–27314. [Google Scholar]
- Zhu, J.; Huang, J.W.; Tseng, P.H.; Yang, Y.T.; Fowble, J.; Shiau, C.W.; Shaw, Y.J.; Kulp, S.K.; Chen, C.S. From the cyclooxygenase-2 inhibitor celecoxib to a novel class of 3-phosphoinositide-dependent protein kinase-1 inhibitors. Cancer Res. 2004, 64, 4309–4318. [Google Scholar]
- Thompson, W.J.; Piazza, G.A.; Li, H.; Liu, L.; Fetter, J.; Zhu, B.; Sperl, G.; Ahnen, D.; Pamukcu, R. Exisulind induction of apoptosis involves guanosine 3',5'-cyclic monophosphate phosphodiesterase inhibition, protein kinase G activation, and attenuated beta-catenin. Cancer Res. 2000, 60, 3338–3342. [Google Scholar] [PubMed]
- Zerbini, L.F.; Czibere, A.; Wang, Y.; Correa, R.G.; Otu, H.; Joseph, M.; Takayasu, Y.; Silver, M.; Gu, X.; Ruchusatsawat, K.; Li, L.; Sarkar, D.; Zhou, J.R.; Fisher, P.B.; Libermann, T.A. A novel pathway involving melanoma differentiation associated gene-7/interleukin-24 mediates nonsteroidal anti-inflammatory drug-induced apoptosis and growth arrest of cancer cells. Cancer Res. 2006, 66, 11922–11931. [Google Scholar]
- Eling, T.E.; Baek, S.J.; Shim, M.; Lee, C.H. NSAID activated gene (NAG-1), a modulator of tumorigenesis. J. Biochem. Mol. Biol. 2006, 39, 649–655. [Google Scholar]
- Soh, J.W.; Weinstein, I.B. Role of COX-independent targets of NSAIDs and related compounds in cancer prevention and treatment. Prog. Exp. Tumor Res. 2003, 37, 261–285. [Google Scholar]
- Tinsley, H.N.; Gary, B.D.; Keeton, A.B.; Zhang, W.; Abadi, A.H.; Reynolds, R.C.; Piazza, G.A. Sulindac sulfide selectively inhibits growth and induces apoptosis of human breast tumor cells by phosphodiesterase 5 inhibition, elevation of cyclic GMP, and activation of protein kinase G. Mol. Cancer Ther. 2009, 8, 3331–3340. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, L.; David, M.L.; Whitehead, C.M.; Chen, M.; Fetter, J.R.; Sperl, G.J.; Pamukcu, R.; Thompson, W.J. Pro-apoptotic actions of exisulind and CP461 in SW480 colon tumor cells involve beta-catenin and cyclin D1 down-regulation. Biochem. Pharmacol. 2002, 64, 1325–1336. [Google Scholar]
- Liu, L.; Li, H.; Underwood, T.; Lloyd, M.; David, M.; Sperl, G.; Pamukcu, R.; Thompson, W.J. Cyclic GMP-dependent protein kinase activation and induction by exisulind and CP461 in colon tumor cells. J. Pharmacol. Exp. Ther. 2001, 299, 583–592. [Google Scholar]
- Rice, P.L.; Kelloff, J.; Sullivan, H.; Driggers, L.J.; Beard, K.S.; Kuwada, S.; Piazza, G.; Ahnen, D.J. Sulindac metabolites induce caspase- and proteasome-dependent degradation of beta-catenin protein in human colon cancer cells. Mol. Cancer Ther. 2003, 2, 885–892. [Google Scholar]
- Vogelstein, B.; Kinzler, K.W. Cancer genes and the pathways they control. Nat. Med. 2004, 10, 789–799. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Piazza, G.A.; Keeton, A.B.; Tinsley, H.N.; Whitt, J.D.; Gary, B.D.; Mathew, B.; Singh, R.; Grizzle, W.E.; Reynolds, R.C. NSAIDs: Old Drugs Reveal New Anticancer Targets. Pharmaceuticals 2010, 3, 1652-1667. https://doi.org/10.3390/ph3051652
Piazza GA, Keeton AB, Tinsley HN, Whitt JD, Gary BD, Mathew B, Singh R, Grizzle WE, Reynolds RC. NSAIDs: Old Drugs Reveal New Anticancer Targets. Pharmaceuticals. 2010; 3(5):1652-1667. https://doi.org/10.3390/ph3051652
Chicago/Turabian StylePiazza, Gary A., Adam B. Keeton, Heather N. Tinsley, Jason D. Whitt, Bernard D. Gary, Bini Mathew, Raj Singh, William E. Grizzle, and Robert C. Reynolds. 2010. "NSAIDs: Old Drugs Reveal New Anticancer Targets" Pharmaceuticals 3, no. 5: 1652-1667. https://doi.org/10.3390/ph3051652
APA StylePiazza, G. A., Keeton, A. B., Tinsley, H. N., Whitt, J. D., Gary, B. D., Mathew, B., Singh, R., Grizzle, W. E., & Reynolds, R. C. (2010). NSAIDs: Old Drugs Reveal New Anticancer Targets. Pharmaceuticals, 3(5), 1652-1667. https://doi.org/10.3390/ph3051652