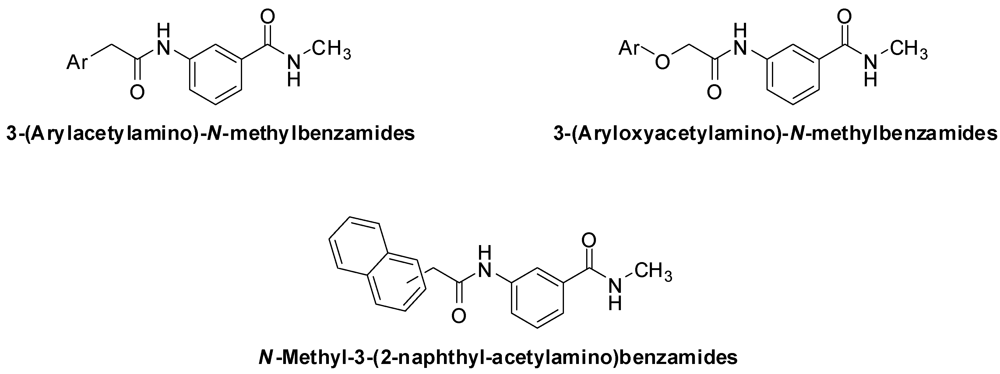
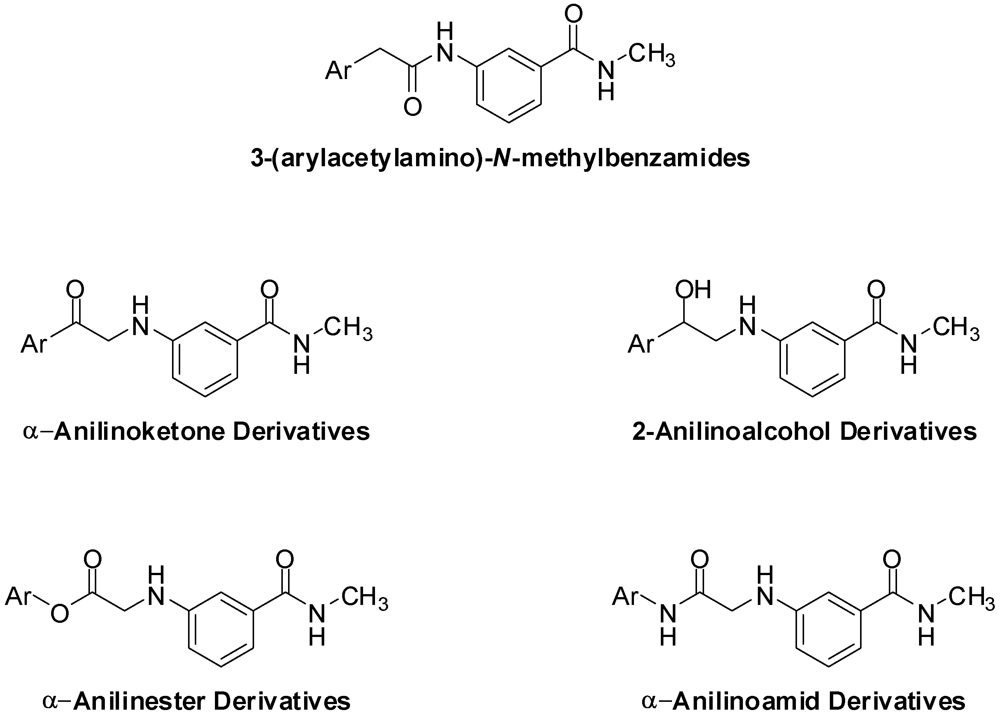
3.2. Synthesis
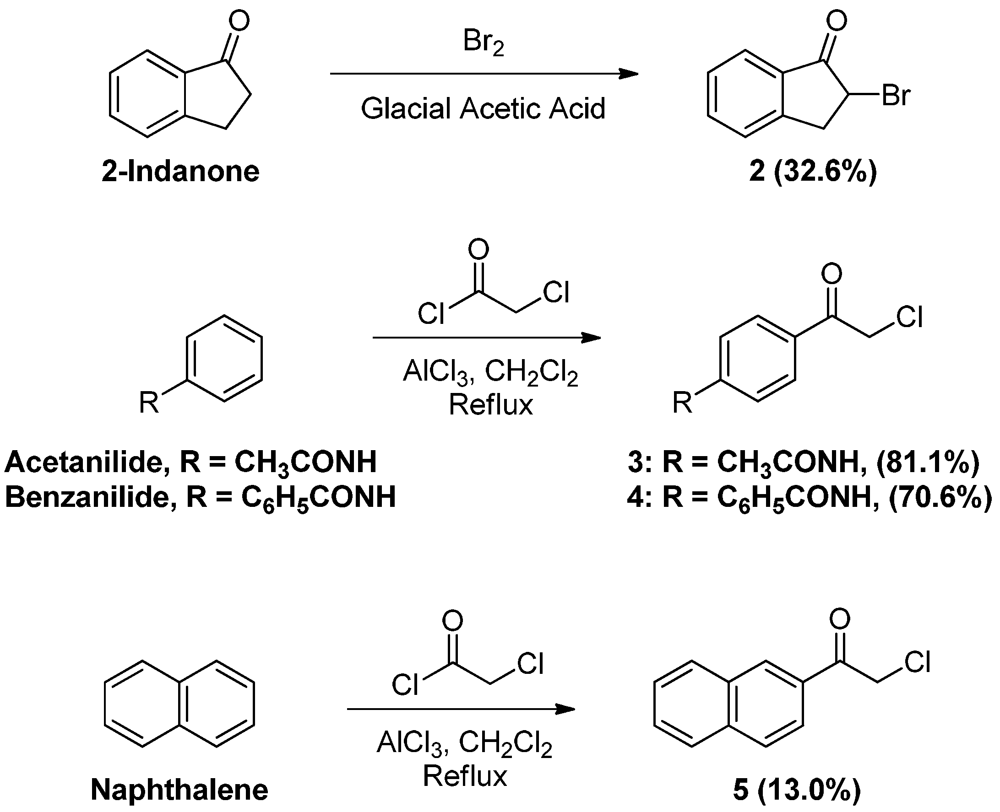
2-Bromoindan-1-one (
2). To a solution of 1-indanone (2.0 g, 15.1 mmol) dissolved in glacial acetic acid (10.0 mL), bromine (1.00 mL, 19.50 mmol) was added dropwise and the mixture was stirred for 30 min. The reaction mixture was then dissolved in ether (100 mL) and washed with water (100 mL × 2) and saturated sodium bicarbonate solution (100 mL). The ether layer was then dried (MgSO
4), filtered and evaporated. The residue was crystallized from ethyl acetate-hexane to yield 1.0 g of the title compound (32.6 %) as white crystals, mp: 41–44 °C, (published: 39–44 °C [
26]);
1H-NMR (CDCl
3): δ 3.43 (dd, 1H,
J = 18.1, 3.2 Hz); 3.84 (dd, 2H,
J = 18.1, 7.5 Hz); 4.66 (dd, 2H, CO-CH-Br,
J = 7.6, 3.2 Hz); 7.49 (m, Ar-H, 2H); 7.72 (t, 2H, Ar-H,
J = 7.7 Hz); 7.84 (d, 2H, Ar-H,
J = 7.7 Hz).
p-Chloroacetyl acetanilide (
3). To a solution of acetanilide (20.0 g, 148.0 mmol) and α-chloroacetyl chloride (27.0 mL, 339.5 mmol) in CH
2Cl
2 (100 mL), aluminum chloride (59.2 g, 444.0 mmol) was added in portions while stirring, turning the color to dark brown. After completion of addition, the mixture was heated at reflux for 30 min. The reaction mixture was then allowed to cool to room temperature and added slowly with stirring to crushed ice (300 g) containing conc. HCl (10 mL). The precipitate was vacuum filtered, washed thoroughly with water, then left to dry. The solid residue was crystallized from absolute ethanol to yield 25.4 g of
3 (81.1%) as brownish crystals, mp: 219–221 °C (published: 213–214 °C [
14]);
1H-NMR (DMSO-
d6): δ 2.09 (s, COCH
3, 3H); 5.09 (s, 2H, CO-CH
2-Cl,
J = 18.1, 3.2 Hz) 7.73 (d, 2H, Ar-H,
J = 8.6 Hz); 7.93 (d, 2H, Ar-H,
J = 8.5 Hz); 10.34 (s, NH, 1H).
p-Chloroacetyl benzanilide (4). To a suspension of benzanilide (20.0 g, 101.4 mmol) and α-chloroacetyl chloride (16.0 mL, 201.2 mmol) in CH2Cl2 (100 mL), aluminum chloride (40.6 g, 304.5 mmol) was added in portions while stirring, turning the color to dark brown. After completion of addition, the mixture was heated at reflux for 2 h. The reaction mixture was then allowed to cool to room temperature and added slowly with stirring to crushed ice (300 g) containing conc. HCl (10 mL). The pale yellow precipitate was vacuum filtered, washed thoroughly with water, then left to dry. The solid residue was crystallized from absolute ethanol to yield 19.6 g of 4 (70.6%) as yellowish-white crystals, mp: 188–191 °C; IR (KBr): 3,353, 1,660, 1,519 cm−1; 1H-NMR (DMSO-d6): δ 5.15 (s, 2H, CO-CH2-Cl); 7.56 (t, 2H, Ar-H, J = 7.4 Hz); 7.63 (t, 1H, Ar-H, J = 7.2 Hz); 7.98 (m, 7H, Ar-H); 10.64 (s, 1H, Ar-H, CO-NH). 13C-NMR (CDCl3): δ 189.81, 165.80, 143.65, 134.27, 132.46, 130.17, 130.02, 129.01, 127.09, 11.92, 54.82, 29.71; MS (EI) m/z: 273 (M+, 60%); 105 (C7H5O, 100%).
2-Naphthacyl chloride (
5). To a solution of naphthalene (5.0 g, 39.0 mmol) and α-chloroacetyl chloride (6.2 mL, 78.0 mmol) in CH
2Cl
2 (30 mL), aluminum chloride (10.4 g, 78.0 mmol) was added in portions while stirring, turning the color to dark brown. After completion of addition, the mixture was heated at reflux for 30 min. The reaction mixture was then allowed to cool to room temperature and added slowly with stirring to crushed ice (300 g) containing conc. HCl (10 mL). The organic layer was taken, dried (MgSO
4) and evaporated. The residue was crystallized from ethyl acetate-hexane to yield 2.0 g of
5 (13.0%) as yellow crystals, mp: 115–118 °C, (published: 119–120 °C [
27]); IR (KBr): 3,360, 1,675, 1,600 cm
−1;
1H-NMR (CDCl
3): δ 4.46 (s, 2H, CO-CH
2-Cl); 7.60 (t, 1H, Ar-H,
J = 8 Hz); 7.66 (t, 1H, Ar-H,
J = 8 Hz); 7.91 (d, 1H, Ar-H,
J = 8 Hz); 7.95 (d, 1H, Ar-H,
J = 8 Hz); 8.00 (d, 1H, Ar-H,
J = 8 Hz); 8.01 (dd, 1H, Ar-H,
J = 2, 8 Hz); 8.49 (s, 1H, Ar-H). MS (EI)
m/z: 204 (M
+, 12%), 63 (COCl, 100%).
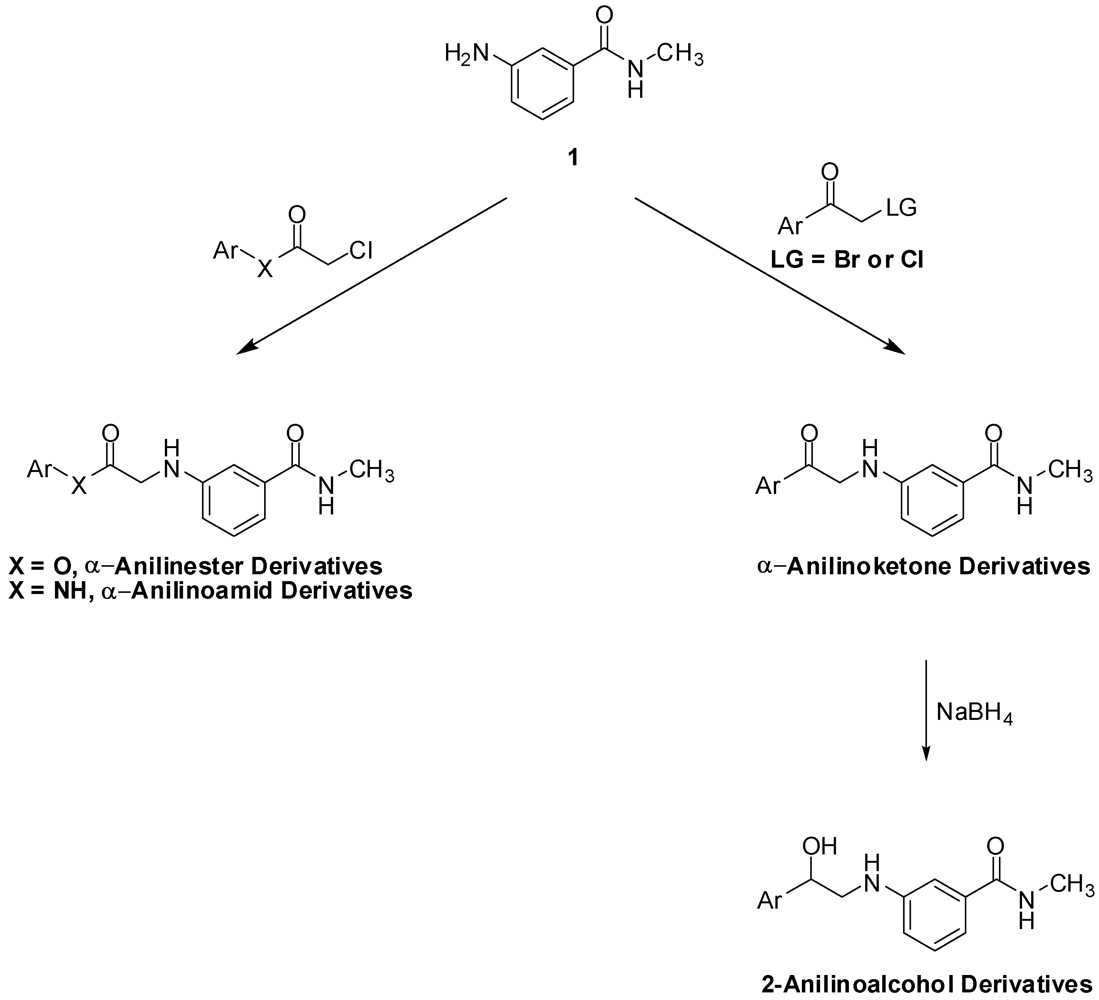
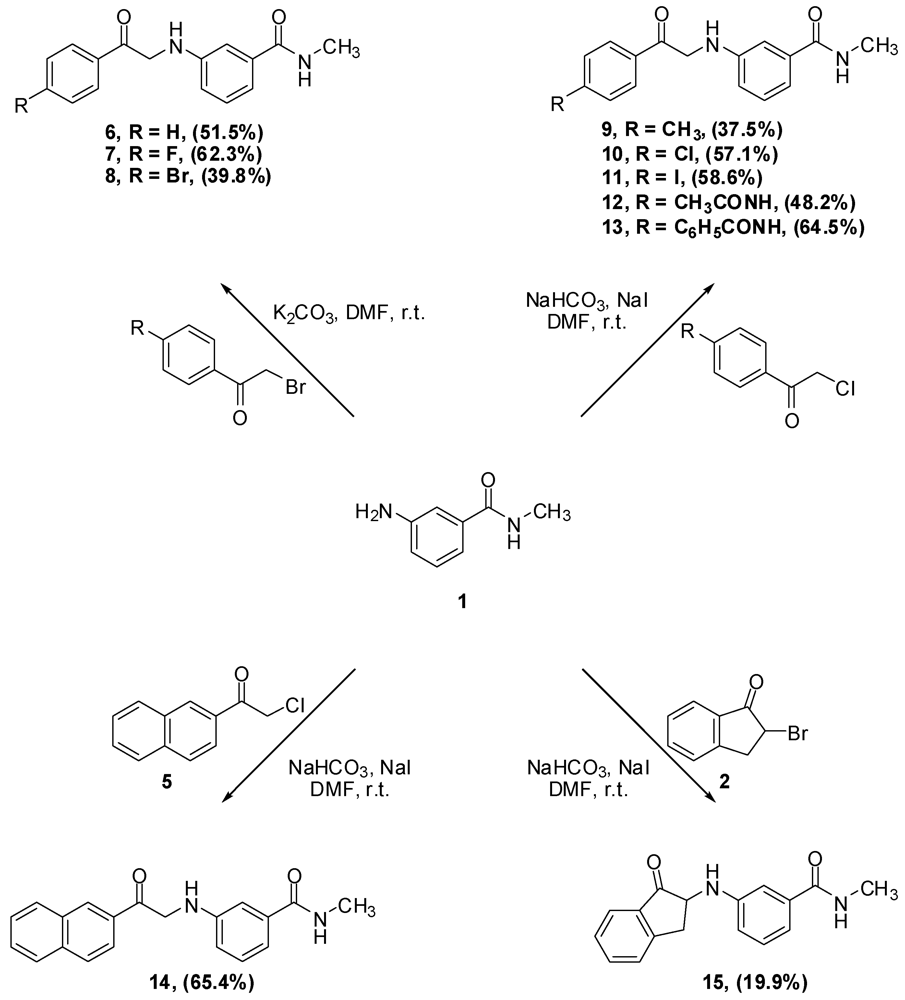
N-Methyl-N-3-(2-Phenyl-2-oxo-ethylamino)benzamide (6). Aniline 1 (10.0 g, 66.6 mmol) and phenacyl bromide (13.3 g, 66.6 mmol) were dissolved in DMF (330 mL). To that solution potassium carbonate (9.2 g, 66.6 mmol) was added and the mixture was stirred overnight. The reaction mixture was then added to water (1.2 L) and the precipitate was vacuum filtered and left to dry. The solid residue was crystallized from ethyl acetate to yield 9.2 g of 6 (51.5%) as yellow crystals, mp: 177–179 °C; IR: 3,371, 1,691, 1,515, 1,358 cm−1; 1H-NMR (DMSO-d6): δ 2.73 (d, 3H, N-CH3, J = 4.8 Hz); 4.71 (d, 2H, CO-CH2-N-Ar, J = 4 Hz); 6.01 (t, 1H, Ar-NH, J = 5.2 Hz); 6.81 (dd, 1H, Ar-H, J = 8, 2 Hz); 7.00 (d, 1H, Ar-H, J = 7.6 Hz); 7.07 (s, 1H, Ar-H); 7.13 (t, 1H, Ar-H, J = 7.8 Hz); 7.56 (t, 2H, Ar-H, J = 7.6 Hz); 7.66 (t, 1H, Ar-H, J = 7 Hz); 8.06 (d, 2H, Ar-H, J = 8 Hz); 8.19 (d, 1H, CO-NH, J = 4.4 Hz). 13C-NMR (DMSO-d6): δ 196.96, 167.76, 18.60, 135.83, 135.51, 134.04, 29.28, 129.11, 128.31, 115; MS (EI) m/z: 268 (M+, 42%); 163 (C9H11N2O, 100%).
N-3-[2-(4-Flourophenyl)-2-oxo-ethylamino]-N-methylbenzamide (7). Aniline 1 (1.0 g, 6.7 mmol) and 4-fluorophenacyl bromide (1.4 g, 6.7 mmol) were dissolved in DMF (15 mL). To that solution, potassium carbonate (0.9 g, 6.5 mmol) was added and the mixture was stirred for 4 h. The reaction mixture was then added to water (200 mL) and the precipitate was vacuum filtered and left to dry. The solid residue was crystallized from absolute ethanol to yield 1.2 g of 7 (62.3%) as yellow crystals, mp: 194–197 °C; IR: 3,381, 3,319, 1,685, 1,600 cm−1; 1H-NMR (DMSO-d6): δ 2.73 (d, 3H, N-CH3, J = 4.5 Hz); 4.70 (d, 2H, CO-CH2-N-Ar, J = 5.5 Hz); 6.02 (t, 1H, Ar-NH, J = 5.4 Hz); 6.81 (dd, 1H, Ar-H, J = 8, 2.3 Hz); 7.00 (d, 1H, Ar-H, J = 7.5 Hz); 7.07 (s, 1H, Ar-H); 7.12 (t, 1H, Ar-H, J = 7.8 Hz); 7.39 (t, 2H, Ar-H, J = 7.9 Hz); 8.15 (m, 3H, Ar-H and CO-NH); 13C-NMR (DMSO-d6): δ 195.28, 167.36, 165.16 (C-F, J = 250 Hz), 148.18, 135.45, 131.89, 130.98 (C-F, J = 9 Hz), 128.70, 116.90 (C-F, J = 21 Hz), 115.54 (C-F, J = 45 Hz), 110.86, 49.79, 26.26; MS (EI) m/z: 286 (M+, 42%); 163 (C9H11N2O, 100%).
N-3-[2-(4-Bromophenyl)-2-oxo-ethylamino]-N-methylbenzamide (8). Aniline 1 (5.0 g, 33.3 mmol) and 4-bromophenacyl bromide (9.3 g, 33.3 mmol) were dissolved in DMF (100 mL). To that solution, potassium carbonate (4.6 g, 33.3 mmol) was added and the mixture was stirred for 2 h. The reaction mixture was then added to water (300 mL) and the precipitate was vacuum filtered and left to dry. The solid residue was crystallized from absolute ethanol to yield 4.6 g of 8 (39.8%) as yellow crystals, mp: 196–199 °C; IR: 3,385, 3,311, 1,684, 1,586, 995 cm−1; 1H-NMR (DMSO-d6): δ 2.73 (d, 3H, N-CH3, J = 4.4 Hz); 4.69 (d, 2H, CO-CH2-N-Ar, J = 5.2 Hz); 6.02 (t, 1H, Ar-NH, J = 5.2 Hz); 6.80 (d, 1H, Ar-H, J = 8 Hz); 7.00 (d, 1H, Ar-H, J = 8 Hz); 7.06 (s, 1H, Ar-H); 7.12 (t, 1H, Ar-H, J = 7.8 Hz); 7.77 (d, 2H, Ar-H, J = 7.8 Hz); 7.99 (d, 2H, Ar-H, J = 7.6 Hz); 8.18 (d, 1H, CO-NH, J = 4.4 Hz); 13C-NMR (DMSO-d6): δ 196.33, 167.76, 148.51, 135.83, 134.50, 132.31, 130.34, 129.11, 128.11, 115.71, 115.32, 111.26, 56.51, 26.67; MS (EI) m/z: 346 (M+, 23%); 163 (C9H11N2O, 100%).
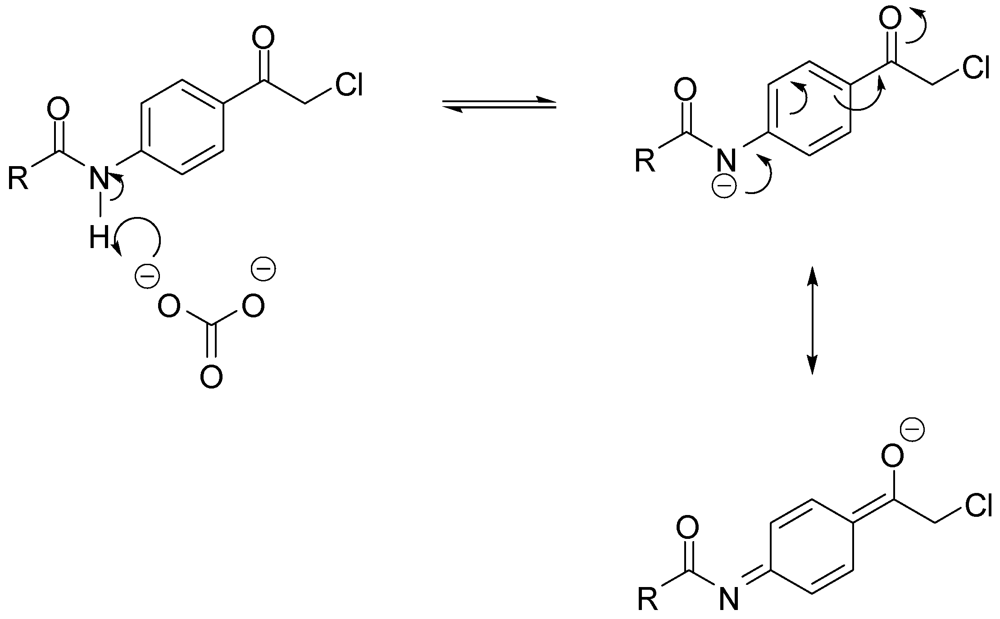
N-Methyl-N-3-(2-p-tolyl-2-oxo-ethylamino)benzamide (9). To a solution of aniline 1 (3.6 g, 23.6 mmol) and 4-methylphenacy chloride (4.0 g, 23.6 mmol) in DMF (30 mL), sodium bicarbonate (2.0 g, 23.6 mmol) and sodium iodide (5.1 g, 23.6 mmol) were added and the mixture was stirred overnight. The reaction mixture was then added to water (500 mL) and the precipitate was vacuum filtered and left to dry. The solid residue was crystallized from absolute ethanol to yield 2.5 g of 9 (37.5%) as a white powder, mp: 161–163 °C; IR: 3,386, 3,318, 1,688, 1,604, 1,539, 811 cm−1; 1H-NMR (DMSO-d6): δ 2.39 (s, 3H, Ar-CH3); 2.74 (d, 3H, N-CH3, J = 4.4 Hz); 4.67 (d, 2H, CO-CH2-N-Ar, J = 5.2 Hz); 5.98 (t, 1H, Ar-NH, J = 5.4 Hz); 6.81 (d, 1H, Ar-H, J = 7.6 Hz); 7.00 (d, 1H, Ar-H, J = 7.6 Hz); 7.07 (s, 1H, Ar-H); 7.12 (t, 1H, Ar-H, J = 7.8 Hz); 7.36 (d, 2H, Ar-H, J = 8 Hz); 7.96 (d, 2H, Ar-H, J = 8 Hz); 8.19 (d, 1H, CO-NH, J = 4.4 Hz); 13C-NMR (DMSO-d6): δ 196.39, 167.76, 148.59, 144.50, 135.83, 133.02, 129.80, 129.09, 128.41, 115.74, 115.23, 111.20, 50.08, 26.66, 21.67; MS (EI) m/z: 282 (M+, 42%); 163 (C9H11N2O, 100%).
N-3-[2-(4-Chlorophenyl)-2-oxo-ethylamino]-N-methylbenzamide (10). To a solution of aniline 1 (2.0 g, 13.3 mmol) and 4-chlorophenacyl chloride (2.5 g, 13.4 mmol) in DMF (30 mL), sodium bicarbonate (2.9 g, 20.9 mmol) and sodium iodide (1.1 g, 51.4 mmol) were added and the mixture was stirred overnight. The reaction mixture was then added to water (250 mL) and the precipitate was vacuum filtered and left to dry. The solid residue was crystallized from absolute ethanol to yield 2.4 g of 10 (57.1%) as a white powder, mp: 188–190 °C; IR: 3,386, 3,319, 1,685, 1,612, 997 cm−1; 1H-NMR (DMSO-d6): δ 2.73 (d, 3H, N-CH3, J = 4.4 Hz); 4.70 (d, 2H, CO-CH2-N-Ar, J = 5.6 Hz); 6.03 (t, 1H, Ar-NH, J = 5.4 Hz); 6.81 (dd, 1H, Ar-H, J = 2, 8 Hz); 7.00 (d, 1H, Ar-H, J = 7.6 Hz); 7.06 (s, 1H, Ar-H); 7.12 (t, 1H, Ar-H, J = 7.8 Hz); 7.63 (d, 2H, Ar-H, J = 8.4 Hz); 8.07 (d, 2H, Ar-H, J = 8.8 Hz); 8.18 (d, 1H, CO-NH, J = 4.8 Hz); 13C-NMR (DMSO-d6): δ 200.85, 172.52, 135.27, 143.68, 140.61, 140.57, 138.92, 134.90, 134.10, 135.85, 120.47, 120.08, 116.02, 55.04, 31.28; MS (EI) m/z: 302 (M+, 37%); 163 (C9H11N2O, 100%).
N-3-[2-(4-Iodophenyl)-2-oxo-ethylamino]-N-methylbenzamide (11). To a solution of aniline 1 (1.2 g, 8.2 mmol) and 4-iodophenacyl chloride (2.3 g, 8.2 mmol) in DMF (30 mL), sodium bicarbonate (688.8 mg, 8.2 mmol) and sodium iodide (1.8 g, 8.2 mmol) were added and the mixture was stirred overnight. The reaction mixture was then added to water (200 mL) and the precipitate was vacuum filtered and left to dry. The solid residue was crystallized from absolute ethanol to yield 1.9 g of 11 (58.8%) as yellowish crystals, mp: 210–213 °C; IR: 3,388, 1,686, 993 cm−1; 1H-NMR (DMSO-d6): δ 2.73 (d, 3H, N-CH3, J = 4.4 Hz); 4.67 (d, 2H, CO-CH2-N-Ar, J = 5.6 Hz); 6.01 (t, 1H, Ar-NH, J = 5.4 Hz); 6.80 (dd, 1H, Ar-H, J = 2, 8 Hz); 7.00 (d, 1H, Ar-H, J = 7.6 Hz); 7.06 (s, 1H, Ar-H); 7.12 (t, 1H, Ar-H, J = 7.8 Hz); 7.81 (d, 2H, Ar-H, J = 8.4 Hz); 7.95 (d, 2H, Ar-H, J = 8.4 Hz); 8.18 (d, 1H, CO-NH, J = 4.4 Hz); 13C-NMR (DMSO-d6): δ 196.66, 167.73, 167.66, 148.50, 138.19, 135.81, 134.79, 130.04, 129.10, 115.70, 115.30, 111.24, 102.65, 50.19, 26.54; MS (EI) m/z: 394 (M+, 38%); 163 (C9H11N2O, 100%).
N-3-[2-(4-Acetylaminophenyl)-2-oxo-ethylamino]-N-methylbenzamide (12). To a solution of aniline 1 (4.5 g, 30.0 mmol) and p-chloroacetyl acetanilide (3) (6.4 g, 30.2 mmol) in DMF (100 mL), sodium bicarbonate (2.5 g, 29.4 mmol) and sodium iodide (6.5 g, 43.4 mmol) were added and the mixture was stirred overnight. The reaction mixture was then added to water (300 mL) and the precipitate was vacuum filtered and left to dry. The solid residue was crystallized from DMF-water to yield 4.7 g of 12 (48.2%) as yellowish brown crystals, mp: 226–230 °C; IR: 3,375, 3,330, 1,682, 1,598, 1,538 cm−1; 1H-NMR (DMSO-d6): δ 2.08 (s, 3H, CH3-CO-N); 2.74 (d, 3H, N-CH3, J = 4.4 Hz); 4.64 (d, 2H, CO-CH2-N-Ar, J = 5.6 Hz); 5.96 (t, 1H, Ar-NH, J = 5.4 Hz); 6.80 (d, 1H, Ar-H, J = 7.6 Hz); 6.99 (d, 1H, Ar-H, J = 7.6 Hz); 7.06 (s, 1H, Ar-H); 7.12 (t, 1H, Ar-H, J = 7.8 Hz); 7.74 (d, 2H, Ar-H, J = 8.4 Hz); 8.02 (d, 2H, Ar-H, J = 8.8 Hz); 8.19 (d, 1H, CO-NH, J = 4.4 Hz); 10.30 (s, 1H, CO-NH-Ar); 13C-NMR (DMSO-d6): δ 195.32, 169.51, 167.80, 148.61, 144.43, 135.84, 130.05, 129.63, 129.09, 118.70, 115.74, 115.20, 111.20, 49.86, 26.67, 24.64; MS (EI) m/z: 325 (M+, 55%); 163 (C9H11N2O, 100%).
N-Methyl-N-3-[2-(4-phenacylaminophenyl)-2-oxo-ethylamino]benzamide (13). To a solution of aniline 1 (3.0g, 20.0 mmol) and 4-benzoylaminophenacyl chloride (4) (5.5g, 20.0 mmol) in DMF (75 mL), sodium bicarbonate (1.7 g, 20.0 mmol) and sodium iodide (4.3 g, 28. mmol) were added and the mixture was stirred over 72 h. The reaction mixture was then added to water (800 mL) and the precipitate was vacuum filtered and left to dry. The solid residue was crystallized from DMF-water to yield 5.0 g of 13 (64.6%) as yellowish brown crystals; mp: 252–255 °C; IR: 3,406, 3,377, 3,312, 1,676, 1,600, 1,538 cm−1; 1H-NMR (DMSO-d6): δ 2.74 (d, 3H, N-CH3, J = 4.4 Hz); 4.68 (d, 2H, CO-CH2-N-Ar, J = 5.2 Hz); 5.99 (t, 1H, Ar-NH, J = 5.4 Hz); 6.82 (dd, 1H, Ar-H, J = 2, 8 Hz); 7.00 (d, 1H, Ar-H, J = 7.6 Hz); 7.08 (s, 1H, Ar-H); 7.13 (t, 1H, Ar-H, J = 7.8 Hz); 7.55 (t, 2H, Ar-H, J = 7.4 Hz); 7.61 (t, 1H, Ar-H, J = 7.2 Hz); 7.98 (m, 4H, Ar-H); 8.09 (d, 2H, Ar-H, J = 8.8 Hz); 8.20 (d, 1H, CO-NH, J = 4.4 Hz); 10.58 (s, 1H, CO-NH-Ar); 13C-NMR (DMSO-d6): δ 195.52, 180.59, 167.81, 167.22, 148.60, 144.41, 135.86, 134.95, 132.42, 130.60, 129.47, 129.11, 128.94, 128.27, 120.01, 119.92, 115.74, 115.23, 111.22, 49.97, 26.67; MS (EI) m/z: 387 (M+, 8%); 163 (C9H11N2O, 100%).
N-3-(2-Naphth-2-yl-2-oxo-ethylamino)-N-methylbenzamide (14). Aniline 1 (350.0 mg, 2.3 mmol) and 2-naphthacyl chloride (5) (0.5 g, 2.4 mmol) were dissolved in DMF (10 mL). To that solution, sodium bicarbonate (193.2 g, 2.3 mmol) of and sodium iodide (480.0 mg, 3.3 mmol) were added and the reaction mixture was stirred overnight. The reaction mixture was then added to water (100 mL) and the precipitate was vacuum filtered and left to dry. The solid residue was crystallized from ethyl acetate to yield 0.5 g of 14 (65.4%) as yellow crystals; mp: 173–175 °C; IR: 3,362, 3,304, 1,683, 1,579 cm−1; 1H-NMR (DMSO-d6): δ 2.74 (d, 3H, N-CH3, J = 4.4 Hz); 4.85 (d, 2H, CO-CH2-N-Ar, J = 2.7 Hz); 6.08 (s, 1H, Ar-NH); 6.85 (dd, 1H, Ar-H, J = 1.6, 8 Hz); 7.01 (d, 1H, Ar-H, J = 7.5 Hz); 7.14 (m, 2H, Ar-H); 7.66 (m, 2H, Ar-H); 8.01 (m, 3H, Ar-H); 8.14 (d, 1H, Ar-H, J = 7.4 Hz); 8.21 (d, 1H, CO-NH, J = 4.4 Hz); 13C-NMR (DMSO-d6): δ 196.92, 167.76, 148.64, 135.89, 135.68, 132.81, 132.64, 130.19, 130.05, 129.24, 129.11, 128.87, 128.20, 127.51, 123.91, 115.75, 115.22, 111.28, 50.27, 26.67; MS (EI) m/z: 318 (M+, 30%); 163 (C9H11N2O, 100%).
N-Methyl-N-3-(1-oxo-indan-2-ylamino]benzamide (15). To a solution of aniline 1 (1.0 g, 6.7 mmol) and 2-bromo-indan-1-one (2) (1.4 g, 6.7 mmol) in DMF (25 mL), sodium bicarbonate (1.1 g, 13.3 mmol) was added and the reaction mixture was stirred overnight. The reaction mixture was then added to water (300 mL) and the precipitate was vacuum filtered and left to dry. The aqueous layer was extracted with CH2Cl2 (100 mL × 2) and the organic layer was dried (MgSO4), filtered then evaporated. The solid residue combined with the residue from CH2Cl2 extract was crystallized from ethyl acetate to yield 0.4 g of 15 (19.9 %) as violet crystals, mp: 149–152 °C; IR: 3,368, 3,297, 1,729, 1,638 cm−1; 1H-NMR (DMSO-d6): δ 2.73 (d, 3H, N-CH3, J = 4.4 Hz); 2.89 (dd, 1H, Ar-CH2, J = 5.2, 16.8 Hz); 3.70 (dd, 1H, Ar-CH2, J = 8, 16.8 Hz); 4.50 (m, 1H, CO-CH-N-Ar); 6.20 (d, 1H, Ar-NH, J = 7.2 Hz); 6.77 (dd, 1H, Ar-H, J = 2, 8 Hz); 7.01 (d, 1H, Ar-H, J = 7.6 Hz); 7.13 (m, 2H, Ar-H, J = 8 Hz); 7.47 (t, 1H, Ar-H, J = 7.4 Hz); 7.59 (d, 1H, Ar-H, J = 7.6 Hz); 7.71 (t, 2H, Ar-H, J = 7 Hz); 8.19 (d, 1H, CO-NH, J = 4 Hz); 13C-NMR (DMSO-d6): δ 205.10, 167.73, 151.95, 148.47, 135.94, 135.88, 135.37, 129.22, 128.27, 123.90, 115.64, 115.37, 111.65, 58.79, 34.38, 26.69; MS (EI) m/z: 280 (M+, 91%); 161 (C9H9N2O, 100%).
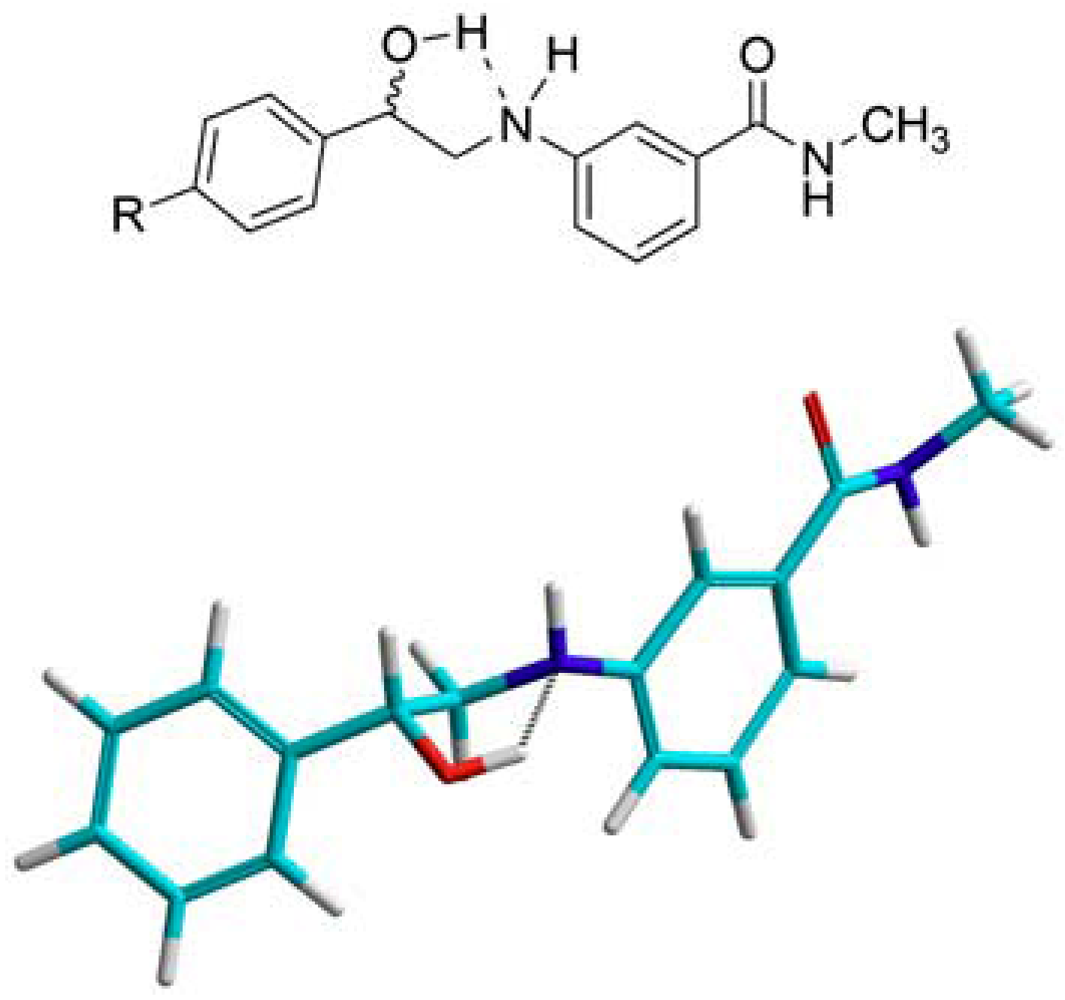
N-3-(2-Hydroxy-2-phenylethylamino)-N-methylbenzamide (16). To a suspension of aminoketone 6 (0.5 g, 1.9 mmol) in methanol (10 mL) at 15 °C, sodium borohydride (71.2 mg, 1.9 mmol) was added, then the mixture was allowed to stir at room temperature till complete dissolution. CH2Cl2 (20 mL) was then added and the resultant mixture was washed with water (30 mL × 2) followed by 1 M NaOH solution (30 mL × 2). The combined aqueous layers were extracted with CH2Cl2 (30 mL), and the combined organic layers were dried (MgSO4), filtered and evaporated. The residue was crystallized from ethyl acetate-ether to yield 380.0 mg of 16 (75.4%) as white crystals; mp: 116–118 °C; IR: 3,409, 3,328, 1,613, 1,543, 1,333, 694 cm−1; 1H-NMR (DMSO-d6): δ 2.75 (d, 3H, N-CH3, J = 4.5 Hz); 3.15 (m, 1H, CH2-N-Ar); 3.26 (m, 1H, CH2-N-Ar); 4.76 (m, 1H, Ar-CH); 5.49 (d, 1H, OH, J = 4.3 Hz); 5.71 (t, 1H, Ar-NH, J = 5.7 Hz); 6.77 (dd, 1H, Ar-H, J = 2.2, 8 Hz); 6.99 (d, 1H, Ar-H, J = 6.9 Hz); 7.05 (d, 1H, Ar-H, J = 1.4 Hz); 7.12 (t, 1H, Ar-H, J = 7.2 Hz); 7.26 (t, 1H, Ar-H, J = 7.2 Hz); 7.35 (t, 2H, Ar-H, J = 7 Hz); 7.42 (d, 2H, Ar-H, J = 7.6 Hz); 8.21 (d, 1H, CO-NH, J = 4.2 Hz); 13C-NMR (DMSO-d6): δ 167.84, 149.04, 144.58, 135.85, 129.17, 128.63, 127.48, 126.50, 115.29, 114.89, 110.85, 170.24, 51.79, 26.69; MS (EI) m/z: 270 (M+, 19%); 163 (C9H11N2O, 100%).
N-3-[2-(4-Flourophenyl)-2-hydroxyethylamino]-N-methylbenzamide (17). To a suspension of the aminoketone 7 (0.3 g, 1.0 mmol) in methanol (8 mL) at 15 °C, sodium borohydride (37.8 mg, 1.0 mmol) was added, then the mixture was allowed to stir at room temperature till complete dissolution. CH2Cl2 (30 mL) was then added and the resultant mixture was washed with water (30 mL) followed by 1 M NaOH solution (30 mL × 2). The combined aqueous layers were extracted with CH2Cl2 (30 mL), and the combined organic layers were dried (MgSO4), filtered and evaporated. The residue was crystallized from ethyl acetate-ether to yield 0.2 g of 17 (51.3%) as white crystals; mp: 100–102 °C; IR: 3,345, 1,604, 1,340, 1,225, 829 cm−1; 1H-NMR (DMSO-d6): δ 2.73 (d, 3H, N-CH3, J = 4.4 Hz); 3.13 (m, 1H, CH2-N-Ar); 3.21 (m, 1H, CH2-N-Ar); 4.73 (m, 1H, Ar-CH); 5.52 (d, 1H, OH, J = 4 Hz); 5.68 (t, 1H, Ar-NH, J = 5.8 Hz); 6.74 (dd, 1H, Ar-H, J = 2.4, 8 Hz); 6.96 (d, 1H, Ar-H, J = 7.6 Hz); 7.02 (s, 1H, Ar-H); 7.11 (t, 1H, Ar-H, J = 7.8 Hz); 7.16 (d, 2H, Ar-H, J = 8.6 Hz); 7.42 (m, 2H, Ar-H); 8.19 (d, 1H, CO-NH, J = 4.5 Hz); 13C-NMR (DMSO-d6): δ 167.77, 161.95 (C-F, J = 240 Hz), 149.04, 140.74, 135.84, 129.13, 128.41 (C-F, J = 8 Hz), 115.38 (C-F, J = 25 Hz), 114.95 (C-F, J = 19 Hz), 110.85, 70.57, 51.65, 26.66; MS (EI) m/z: 288 (M+, 16%); 163 (C9H11N2O, 100%).
N-3-[2-(4-Bromophenyl)-2-hydroxyethylamino]-N-methylbenzamide (18). To a suspension of aminoketone 8 (380.0 mg, 1.0 mmol) in methanol (10 mL) at 15 °C, sodium borohydride (45.2 g, 1.2 mmol) was added then the mixture was allowed to stir at room temperature till complete dissolution. CH2Cl2(20 mL) was then added and the resultant mixture was washed with water (30 mL × 2) followed by 1 M NaOH solution (30 mL × 2). The combined aqueous layers were extracted with CH2Cl2 (60 mL), and the combined organic layers were dried (MgSO4), filtered and evaporated. The residue was crystallized from ethyl acetate-ether to yield 0.2 g of 18 (56.8%) as white crystals, mp: 127–129 °C; IR: 3,443, 3,357, 1,637, 1,604 cm−1; 1H-NMR (DMSO-d6): δ 2.73 (d, 3H, N-CH3, J = 4.4 Hz); 3.15 (m, 1H, CH2-N-Ar); 3.20 (m, 1H, CH2-N-Ar); 4.73 (m, 1H, Ar-CH); 5.57 (d, 1H, OH, J = 4.2 Hz); 5.70 (t, 1H, Ar-NH, J = 5.7 Hz); 6.74 (dd, 1H, Ar-H, J = 2, 8 Hz); 6.97 (dd, 1H, Ar-H, J = 7 Hz); 7.02 (s, 1H, Ar-H); 7.10 (t, 1H, Ar-H, J = 7.8 Hz); 7.35 (d, 2H, Ar-H, J = 8.8 Hz); 7.52 (d, 2H, Ar-H, J = 9 Hz); 8.19 (d, 1H, CO-NH, J = 4.4 Hz); 13C-NMR (DMSO-d6): δ 172.55, 153.76, 148.30, 140.61, 140.57, 136.68, 135.89, 133.16, 120.25, 119.65, 115.66, 99.98, 75.28, 56.19, 31.29; MS (EI) m/z: 348 (M+, 7%); 163 (C9H11N2O, 100%).
N-3-(2-Hydroxy-2-p-tolylethylamino)-N-methylbenzamide (19). To a suspension of aminoketone 9(0.5 g, 1.8 mmol) in methanol (10 mL) of at 15 °C, sodium borohydride (68.1 mg, 1.8 mmol) was added, then the mixture was allowed to stir at room temperature till complete dissolution. CH2Cl2 (30 mL) was then added and the resultant mixture was washed with 2 M NaOH solution (30 mL × 3). The combined aqueous layers were extracted with CH2Cl2 (30 mL), then the combined organic layers were dried (MgSO4), filtered and evaporated. The residue was crystallized from ethanol to yield 270.0 mg of 19 (59.6%) as white crystals, mp: 137–139 °C; IR: 3,349, 3,299, 1,603, 1,583, 1,341, 1,060 cm−1; 1H-NMR (DMSO-d6): δ 2.28 (s, 3H, Ar-CH3); 2.73 (d, 3H, N-CH3, J = 4.4 Hz); 3.10 (m, 1H, CH2-N-Ar); 3.20 (m, 1H, CH2-N-Ar); 4.70 (m, 1H, Ar-CH); 5.39 (d, 1H, OH, J = 4 Hz); 5.65 (t, 1H, Ar-NH, J = 4.8, 6.8 Hz); 6.74 (dd, 1H, Ar-H, J = 1.6, 8 Hz); 6.96 (d, 1H, Ar-H, J = 7.8 Hz); 7.02 (s, 1H, Ar-H); 7.09 (d, 1H, Ar-H, J = 7.6 Hz); 7.14 (d, 2H, Ar-H, J = 8 Hz); 8.19 (d, 1H, CO-NH, J = 4.4 Hz); 13C-NMR (DMSO-d6): δ 167.79, 149.09, 141.59, 136.47, 135.84, 129.13, 129.04, 126.42, 115.53, 114.81, 110.82, 71.02, 51.80, 26.68, 21.18; MS (EI) m/z: 284 (M+, 11%); 163 (C9H11N2O, 100%).
N-3-[2-(4-Chlorophenyl)-2-hydroxyethylamino]-N-methylbenzamide (20). To a suspension of aminoketone 10(0.5 g, 1.7 mmol) in methanol (10 mL) at 15 °C, sodium borohydride (64.3 mg, 1.7 mmol) was added, and then the mixture was allowed to stir at room temperature till complete dissolution. CH2Cl2 (30 mL) was then added and the resultant mixture was then washed with water (30 mL × 2) followed by 1 M NaOH solution (30 mL). The combined aqueous layers were extracted with CH2Cl2 (60 mL), and the combined organic layers were dried (MgSO4), filtered and evaporated. The residue was crystallized from ethyl acetate-ether to yield 0.3 g of 20 (53.6%) as white crystals, mp: 120–122 °C; IR: 3,445, 3,355, 2,904, 1,638, 1,342, 825 cm−1; 1H-NMR (DMSO-d6): δ 2.73 (d, 3H, N-CH3, J = 4.5 Hz); 3.14 (m, 1H, CH2-N-Ar); 3.22 (m, 1H, CH2-N-Ar); 4.74 (m, 1H, Ar-CH); 5.57 (d, 1H, OH, J = 4.4 Hz); 5.70 (t, 1H, Ar-NH, J = 5.8 Hz); 6.74 (dd, 1H, Ar-H, J = 1.7, 8 Hz); 6.96 (d, 1H, Ar-H, J = 7.8 Hz); 7.02 (s, 1H, Ar-H); 7.10 (t, 1H, Ar-H, J = 7.8 Hz); 7.40 (m, 4H, Ar-H); 8.19 (d, 1H, CO-NH, J = 4.5 Hz); 13C-NMR (DMSO-d6): δ 167.76, 148.99, 144.00, 135.85, 131.32, 129.14, 128.80, 120.44, 115.49, 114.87, 110.88, 70.55, 51.45, 26.68; MS (IR) m/z: 304 (M+, 11%); 163 (C9H11N2O, 100%).
N-3-[2-Hydroxy-2-(4-iodophenyl)ethylamino]-N-methylbenzamide (21). To a suspension of the aminoketone 11 (0.5 g, 1.3 mmol) in methanol (10 mL) at 15 °C, sodium borohydride (49.2 mg, 1.3 mmol) was added, then the mixture was allowed to stir at room temperature till complete dissolution. CH2Cl2 (50 mL) was then added and the resultant mixture washed with 0.5M NaOH solution (50 mL × 3). The combined aqueous layers were extracted with CH2Cl2 (50 mL), and the combined organic layers were dried (MgSO4), filtered and evaporated. The residue was crystallized from ethyl acetate-ether to yield 360.0 g of 21 (71.6 %) as white crystals; mp: 132–134 °C; IR: 3,352, 3,300, 1,603, 1,584, 1,342, 1,062 cm−1; 1H-NMR (DMSO-d6): δ 2.73 (d, 3H, N-CH3, J = 4.6 Hz); 3.12 (m, 1H, CH2-N-Ar); 3.21 (m, 1H, CH2-N-Ar); 4.70 (m, 1H, Ar-CH); 5.55 (d, 1H, OH, J = 4.4 Hz); 5.69 (t, 1H, Ar-NH, J = 5.9 Hz); 6.74 (dd, 1H, Ar-H, J = 1.6, 8.1 Hz); 6.96 (d, 1H, Ar-H, J = 7.6 Hz); 7.02 (s, 1H, Ar-H); 7.10 (t, 1H, Ar-H, J = 7.8 Hz); 7.21 (d, 2H, Ar-H, J = 8.2 Hz); 7.69 (d, 2H, Ar-H, J = 8.3 Hz); 8.19 (d, 1H, CO-NH, J = 4.4 Hz); 13C-NMR (DMSO-d6): δ 167.80, 148.99, 144.39, 137.19, 135.86, 135.82, 129.16, 128.98, 128.41, 115.50, 115.91, 110.91, 93.23, 70.66, 51.41, 26.70; MS (EI) m/z: 396 (M+, 11%); 163 (C9H11N2O, 100%).
N-3-[2-(4-Acetylaminophenyl)-2-hydroxyethylamino]-N-methylbenzamide (22). To a suspension of aminoketone 12 (2.5 g, 7.7 mmol) in CH2Cl2 (40 mL) and isopropanol (40 mL), sodium borohydride (351.8 mg, 9.3 mmol) was added and the reaction mixture was stirred overnight. Solvents were then evaporated and the residue taken up in ethyl acetate (50 mL) and washed with water (50 mL × 3). The organic layer was dried (MgSO4), filtered and evaporated. The residue was crystallized from absolute ethanol to yield 1.4 g of 22 (55.7%) as a white solid, mp: 177–179 °C; IR (KBr); 3,398, 3,307, 1,603, 1,545 cm−1; 1H-NMR (DMSO-d6): δ 2.02 (s, 3H, CH3-CO); 2.73 (d, 3H, N-CH3, J = 4.9 Hz); 3.11 (m, 1H, CH2-N-Ar); 3.21 (m, 1H, CH2-N-Ar); 4.68 (m, 1H, Ar-CH); 5.39 (d, 1H, OH, J = 4.2 Hz); 5.65 (t, 1H, Ar-NH, J = 5.7 Hz); 6.74 (dd, 1H, Ar-H, J = 1.3, 8.1 Hz); 6.96 (d, 1H, Ar-H, J = 7.6 Hz); 7.02 (s, 1H, Ar-H); 7.10 (t, 1H, Ar-H, J = 7.8 Hz); 7.30 (d, 2H, Ar-H, J = 8.4 Hz); 7.52 (d, 2H, Ar-H, J = 8.4 Hz); 8.19 (d, 1H, CO-NH, J = 4.6 Hz); 13C-NMR (DMSO-d6): δ 168.68, 167.83, 149.09, 139.15, 138.65, 135.83, 132.83, 129.15, 126.80, 119.20, 115.52, 114.82, 110.83, 70.90, 51.87, 26.68, 24.41; MS (EI) m/z: 327 (M+, 8%); 163 (C9H11N2O, 100%).
N-3-[2-Hydroxy-2-(4-benzamidoaminophenyl)-ethylamino]-N-methylbenzamide (23). To a suspension of aminoketone 13 (3.0 g, 7.5 mmol) in CH2Cl2 (80 mL) and isopropanol (80 mL), sodium borohydride (438.8 mg, 11.6 mmol) was added and the reaction mixture was stirred overnight. The suspension was then vacuum filtered, washed thoroughly with water then left to dry. The solid residue was crystallized from MeOH to yield 280.0 mg of 23 (9.3%) as a yellowish solid, mp: 189–191 °C; IR: 3,327, 1,651, 1,528, 1,322, 691 cm−1; 1H-NMR (DMSO-d6): δ 2.73 (d, 3H, N-CH3, J = 4.4 Hz); 3.14 (m, 1H, CH2-N-Ar); 3.24 (m, 1H, CH2-N-Ar); 4.73 (m, 1H, Ar-CH); 5.54 (bs, 1H, OH); 5.68 (t, 1H, Ar-NH, J = 5.8 Hz); 6.76 (dd, 1H, Ar-H, J = 2, 7.6 Hz); 6.97 (d, 1H, Ar-H, J = 7.6 Hz); 7.04 (s, 1H, Ar-H); 7.11 (t, 1H, Ar-H, J = 7.8 Hz); 7.37 (d, 2H, Ar-H, J = 8.4 Hz); 7.51 (m, 3H, Ar-H); 7.74 (d, 2H, Ar-H, J = 8.4 Hz); 7.95 (d, 2H, Ar-H, J = 7.2 Hz); 8.19 (d, 1H, CO-NH, J = 4.8 Hz); 10.22 (s, 1H, CO-NH-Ar); 13C-NMR (DMSO-d6): δ 176.82, 165.88, 149.11, 139.86, 138.44, 135.87, 135.38, 131.99, 129.15, 128.84, 128.08, 126.72, 120.52, 115.52, 114.85, 110.86, 70.87, 51.63, 26.68; MS (EI) m/z: 371 (M+-water, 2%); 227 (C14H14NO2, 34%).
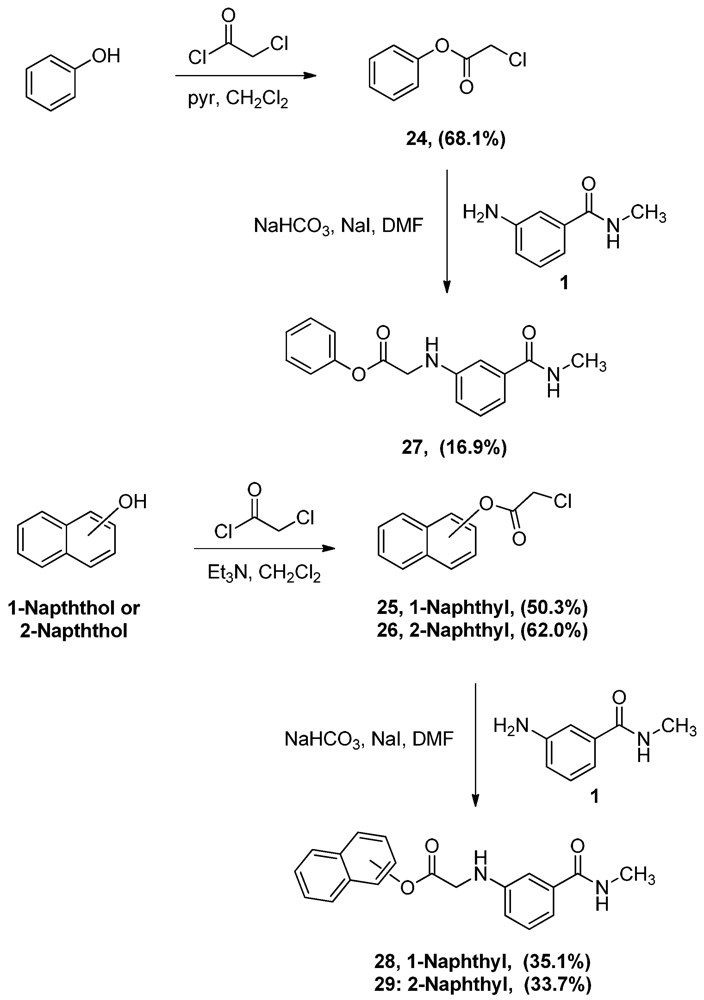
Phenyl α-chloroacetate (24). A solution of phenol (5.0 g, 53.1 mmol) and pyridine (4.3 mL, 53.1 mmol) in CH2Cl2 (80 mL) was added dropwise to α-chloroacetyl chloride (8.5 mL, 106.9 mmol) and the mixture was allowed to stir vigorously for 2 h. The reaction mixture was then washed with water (200 mL × 2), 0.1 M NaOH solution (200 mL × 2) and 1 M HCl solution (100 mL). The organic layer was then dried (MgSO4), filtered then evaporated. The residue was crystallized from hexane to yield 6.2 g of 24 (68.1%) as white crystals, mp: 47–50 °C; IR: 3,068, 2,951, 1,773 cm−1; 1H-NMR (CDCl3): δ 4.47 (s, 2H, CO-CH2-Cl); 7.16 (d, 2H, Ar-H, J = 7.6 Hz); 7.30 (t, 1H, Ar-H, J = 7.4 Hz); 7.43 (t, 2H, Ar-H, J = 7.9 Hz); MS (EI) m/z: 170 (M+, 1%); 63 (COCl, 100%).
Naphthalen-1-yl α-chloroacetate (25). To a solution of 1-naphthol (10.0 g, 69.4 mmol) and α-chloroacetyl chloride (6.6 mL, 82.8 mmol) in CH2Cl2 (150 mL), triethyl amine (9.6 mL, 76.5 mmol) was added dropwise and the mixture was stirred for one hour. The reaction mixture was then washed with 1 M HCl solution (100 mL) followed by 1 M NaOH solution (100 mL × 3). The CH2Cl2 layer was then dried (MgSO4), filtered and evaporated. The residue was crystallized from ethyl acetate-hexane to yield 7.7 g of 25 (50.3%) as white crystals, mp: 113–115 °C; IR: 1,761, 1,162, 770 cm−1; 1H-NMR (CDCl3): δ 4.50 (s, 2H, CO-CH2-Cl); 7.32 (d, 1H, Ar-H, J = 7.6 Hz); 7.49 (t, 2H, Ar-H, J = 8 Hz); 7.55 (t, 1H, Ar-H, J = 4.6 Hz); 7.79 (d, 1H, Ar-H, J = 8 Hz); 7.91 (m, 2H, Ar-H); MS (EI) m/z: 220 (M+, 62%); 63 (COCl, 100%).
Naphthalen-2-yl α-chloroacetate (26). To a solution of 2-naphthol (10.0 g, 69.4 mmol) and α-chloroacetyl chloride (7.0 mL, 88.0 mmol) in CH2Cl2 (150 mL), triethyl amine (9.6 mL, 76.5 mmol) was added dropwise and the mixture was stirred for one hour. The reaction mixture was then washed with 1 M HCl solution (100 mL) and 1 M NaOH solution (100 mL × 3). The CH2Cl2 layer was then dried (MgSO4), filtered and evaporated. The residue was crystallized from ethyl acetate-hexane to yield 9.5 g of 26 (62.0%) as white crystals, mp: 99–102 °C; IR: 1,173, 1,163, 810 cm−1; 1H-NMR (CDCl3): δ 4.37 (s, 2H, CO-CH2-Cl); 7.27 (dd, 1H, Ar-H, J = 2, 8 Hz); 7.51 (m, 2H, Ar-H); 7.63 (d, 1H, Ar-H, J = 2 Hz); 7.86 (m, 3H, Ar-H); MS (EI) m/z: 220 (M+, 61%); 63 (COCl, 100%).
N-Methyl-N-3-(1-phenoxyacetylamino)benzamide (27). To a solution of aniline 1 (1.0 g, 6.7 mmol) and acetate 24 (1.1 g, 6.7 mmol) in DMF (17 mL), sodium bicarbonate (562.9 mg, 6.7 mmol) of and sodium iodide (1.4 g, 9.3 mmol) were added and the mixture was stirred overnight. The reaction mixture was then added to water (150 mL) and the precipitate was vacuum filtered and left to dry. The solid residue was crystallized from ethyl acetate-hexane to yield 320.0 mg of 27 (16.9%) as white crystals, mp: 137–139 °C; IR: 3,405, 3,320, 1,752 cm−1; 1H-NMR (DMSO-d6): δ 2.75 (d, 3H, N-CH3, J = 4.5 Hz); 4.23 (d, 2H, CO-CH2-N-Ar, J = 6.4 Hz); 6.34 (t, 1H, Ar-NH, J = 6.4 Hz); 6.78 (dd, 1H, Ar-H, J = 2.2, 7.8 Hz); 7.05 (d, 1H, Ar-H, J = 7.6 Hz); 7.11 (d, 3H, Ar-H, J = 8.4 Hz); 7.17 (t, 1H, Ar-H, J = 7.8 Hz); 7.25 (t, 1H, Ar-H, J = 7.4 Hz); 7.42 (t, 2H, Ar-H, J = 7.8 Hz); 8.25 (d, 1H, CO-NH, J = 4.6 Hz); 13C-NMR (DMSO-d6): δ 170.75, 167.63, 150.82, 148.42, 135.88, 130.04, 129.25, 126.39, 122.11, 115.59, 115.38, 111.22, 45.22, 26.69; MS (EI) m/z: 284 (M+, 62%); 163 (C9H11N2O, 100%).
N-Methyl-N-3-[1-(1-naphthoxy)acetylamino]-benzamide (28). To a solution of the aniline 1 (1.0 g, 6.7 mmol) and acetate 25 (1.5 g, 6.7 mmol) in DMF (25 mL), sodium bicarbonate (0.6 g, 6.7 mmol) of and sodium iodide (1.5 g, 1.0 mmol) were added and the mixture was stirred for 4 h. The reaction mixture was then added to water (100 mL) and the precipitate was vacuum filtered and left to dry. The solid residue was crystallized from ethyl acetate-hexane to yield 783.0 mg (35.1%). IR (KBr): 3,397, 3,301, 1,758, cm−1; 1H-NMR (400 MHz, DMSO-d6): δ 2.8 (d, 3H, N-CH3, J = 3.8 Hz); 4.49 (d, 2H, CO-CH2-N-Ar, J = 6.6 Hz); 6.48 (t, 1H, Ar-NH, J = 5.4. 6.6 Hz); 6.89 (d, 1H, Ar-H, J = 8 Hz); 7.10 (d, 1H, Ar-H, J = 6.6 Hz); 7.22 (m, 2H, Ar-H); 7.32 (d, 1H, Ar-H, J = 7.5 Hz); 7.55 (m, 3H, Ar-H); 7.87 (t, 2H, Ar-H, J = 8.8, 10.9 Hz); 7.99 (d, 1H, Ar-H, J = 8.1 Hz); 8.31 (s, 1H, CO-NH). 13C-NMR (100 MHz, DMSO-d6): δ 171.12, 167.62, 148.51, 136.67, 135.92, 135.59, 129.32, 128.38, 127.12, 126.78, 126.46, 126.18, 121.73, 118.81, 115.70, 111.14, 45.20, 26.74. MS (EI) m/z: 334 (M+, 42%); 163 (C9H11N2O, 100%). mp: 164–166 °C.
N-Methyl-N-3-[1-(2-naphthoxy)acetylamino]benzamide (29). To a solution of the aniline 1(1.0 g, 6.7 mmol) and acetate 26 (1.5 g, 6.7 mmol) in DMF (25 mL), sodium bicarbonate (562.9 mg, 6.7 mmol) of and sodium iodide (1.5 g, 1.0 mmol) were added and the mixture was stirred for 4 h. The reaction mixture was then added to water (100 mL) and the precipitate was vacuum filtered and left to dry. The solid residue was crystallized from ethyl acetate-hexane to yield 750.0 mg (33.7%) of 29, mp: 150–152 °C; IR: 3,397, 3,312, 1,753, 1,644 cm−1; 1H-NMR (DMSO-d6): δ 2.77 (d, 3H, N-CH3, J = 4.4 Hz); 4.31 (d, 2H, CO-CH2-N-Ar, J = 6.6 Hz); 6.39 (t, 1H, Ar-NH, J = 6 Hz); 6.82 (dd, 1H, Ar-H, J = 1.5, 6.9 Hz); 7.07 (d, 1H, Ar-H, J = 5 Hz); 7.14 (s, 1H, Ar-H); 7.19 (t, 1H, Ar-H, J = 7.8 Hz); 7.30 (dd, 1H, Ar-H, J = 2.3, 8.8 Hz); 7.52 (dp, 2H, Ar-H, J = 1.6, 6.9 Hz); 7.68 (d, 1H, Ar-H, J = 2.3 Hz); 7.92 (m, 1H, Ar-H); 7.97 (d, 2H, Ar-H, J = 8.8 Hz); 8.27 (q, 1H, CO-NH, J = 4.4 Hz); 13C-NMR (DMSO-d6): δ 170.99, 167.65, 148.50, 135.90, 133.73, 131.47, 129.91, 129.30, 128.17, 127.92, 127.23, 126.35, 121.86, 118.89, 115.60, 111.28, 42.26, 26.72; MS (EI) m/z: 334 (M+, 63%); 144 (C10H8O, 100%).
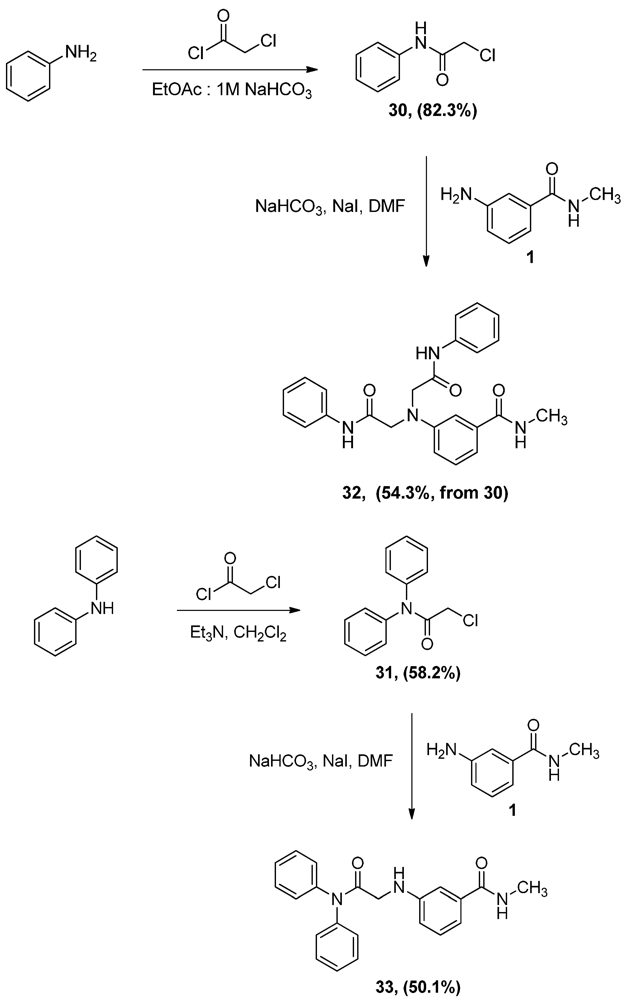
N-Phenyl α-chloroacetamide (30). Aniline 1 (9.8 mL, 107.5 mmol) was dissolved in a biphasic mixture of ethyl acetate (220 mL) of and 1 M aqueous solution of sodium bicarbonate (129 mL). α-Chloroacetyl chloride (12.8 mL, 160.9 mmol) was then added dropwise to the biphasic mixture and the resultant mixture was stirred for one hour. The two layers were then separated and the aqueous layer extracted with ethyl acetate (100 mL). The combined organic layers were dried, filtered and evaporated to half the volume of ethyl acetate then allowed to cool to yield 15.0 g (82.3%) of grayish white crystals, mp: 140–143°C; 1H-NMR (CDCl3): δ 4.19 (s, 2H, CO-CH2-Cl); 7.18 (t, 1H, Ar-H, J = 7.4 Hz); 7.37 (t, 2H, Ar-H, J = 8 Hz); 7.55 (d, 2H, Ar-H, J = 7.6 Hz); 8.24 (bs, 1H, Ar-NH).
N,N-Diphenyl α-chloroacetamide (31). A solution of diphenylamine (5.0 g, 29.5 mmol) and triethyl-amine (4.1 mL, 29.5 mmol) in CH2Cl2 (70 mL) was added dropwise to a 2 M solution of α-chloroacetyl chloride (4.7 mL, 59.1 mmol) in CH2Cl2 and the mixture was stirred vigorously overnight. The reaction mixture was then washed with water (100 mL × 2) and 0.1N HCl solution (100 mL), dried (MgSO4), filtered then evaporated. The residue was crystallized from ethyl acetate to yield 4.2 g (58.2%) of 31, mp: 124–127 °C; IR: 2,945, 1,682, 1,491, 1,265 cm−1; 1H-NMR (CDCl3): δ 4.03 (s, 2H, CO-CH2-Cl); 7.34 (m, 10H, Ar-H); 13C-NMR (CDCl3): δ 166.20, 141.81, 130.07, 129.118, 128.57, 126.66, 126.06; MS (EI) m/z: 245 (M+, 71%); 63 (COCl, 100%).
N-Methyl 3-N,N-Di(phenylamidomethylamino)benzamide (32). α-Chloroacetamide 30 (3.0 g, 17.7 mmol) and the aniline 1 (2.9 g, 19.3 mmol) were dissolved in DMF (40 mL). To that solution, sodium bicarbonate (1.5 g, 17.7 mmol) and sodium iodide (3.8 g, 25.4 mmol) were added and the mixture was stirred overnight. The reaction mixture was then added to water (300 mL) and the precipitate was vacuum filtered and left to dry. The solid residue was crystallized from ethanol to yield 2.0 g of 32 (54.3%, from 30 as the limiting reagent) as white crystals, mp: 248–250 °C; IR: 3,365, 3,300, 1,682, 1,660 cm−1; 1H-NMR (DMSO-d6): δ 2.72 (d, 3H, N-CH3, J = 4.4 Hz); δ 4.38 (s, 4H, CO-CH2-N-Ar); δ 6.67 (dd, 1H, Ar-H, J = 2.4, 8 Hz); δ 7.07 (m, 3H, Ar-H); δ 7.14 (d, 1H, Ar-H, J = 7.6 Hz); δ 7.27 (t, 2H, Ar-H, J = 8 Hz); δ 7.33 (t, 1H, Ar-H, J = 7.8 Hz); δ 7.65 (d, 4H, Ar-H, J = 8 Hz); δ 8.30 (d, 1H, CO-NH, J = 4.4 Hz), δ 10.88 (s, 2H, Ar-H); 13C-NMR (DMSO-d6): δ 170.36, 167.37, 147.42, 139.06, 136.09, 129.66, 129.40, 124.18, 119.67, 116.11, 114.12, 110.81, 57.30, 26.71; MS (EI) m/z: 296 (C17H18N3O2, 29%); 119 (C7H6NO, 9%).
3-(Diphenylamidomethylamino)-N-methylbenzamide (33). α-Chloroacetamide 31 (1.6 g, 6.7 mmol) and the aniline 1 (1.0 g, 6.7 mmol) were dissolved in DMF (40 mL). To that solution, sodium carbonate (562.9 mg, 6.7 mmol) and sodium iodide (1.5 g, 1.0 mmol) were added and the mixture was stirred overnight. The reaction mixture was then added to water (200 mL) and the precipitate was vacuum filtered and left to dry. The solid residue was crystallized from ethyl acetate to yield 1.2 g (50.1%) of 33 as white crystals, mp: 171–173 °C; IR): 3,449, 3,373, 1,684, 1,505, 1,294 cm−1; 1H-NMR (DMSO-d6): δ 2.74 (d, 3H, N-CH3, J = 4.5 Hz); δ 3.72 (d, 2H, CO-CH2-N-Ar, J = 5.7 Hz); δ 5.94 (t, 1H, Ar-NH, J = 5.8 Hz); δ 6.64 (dd, 1H, Ar-H, J = 1.8, 8 Hz); δ 6.93 (s, 1H, Ar-H); δ 6.99 (d, 1H, Ar-H, J = 7.9 Hz); δ 7.11 (t, 1H, Ar-H, J = 7.8 Hz); δ 7.43 (m, 10H, Ar-H); δ 8.2 (d, 1H, CO-NH, J = 4.5 Hz); 13C-NMR (DMSO-d6): δ 169.93, 167.76, 148.50, 142.65, 135.81, 129.81, 129.13, 127.24, 118.77, 115.40, 115.11, 111.33, 46.22, 26.72 MS (EI) m/z: 359 (M+, 73%); 163 (C9H11N2O, 100%).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}












