Hsp90 Inhibitors and the Reduction of Anti-Cancer Drug Resistance by Non-Genetic and Genetic Mechanisms


{kind=link}
Abstract
:1. Introduction
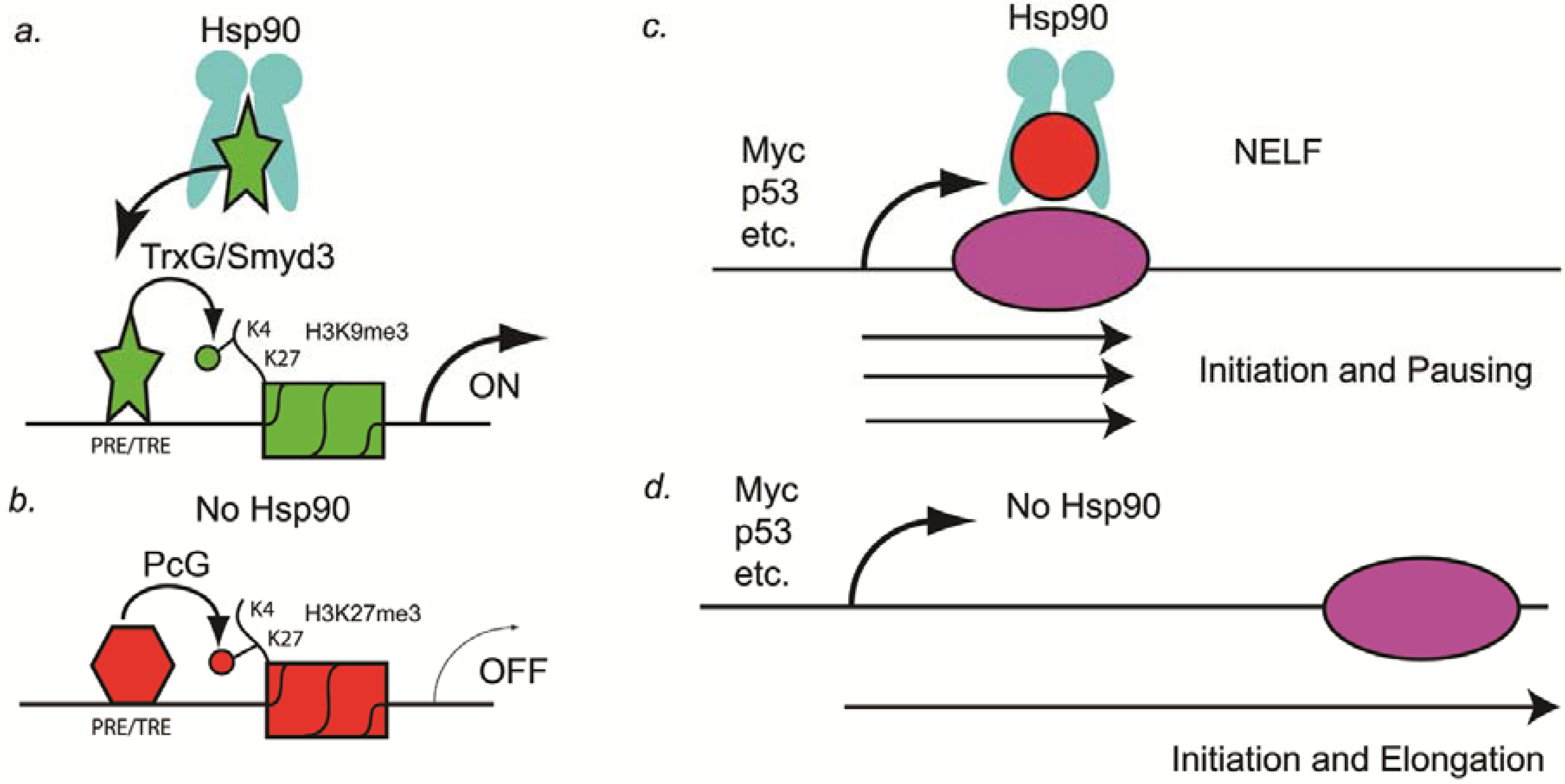
2. Hsp90 as a Capacitor for Phenotypic Variation and Cancer Cell Survival
3. Hsp90 and the Generation of Chromosomal Aneuploidies
4. Hsp90 and the Initiation of Polymerase Pausing

5. Conclusions
Acknowledgements
References
- Darwin, C. Variation in Animals and Plants under Domestication; Appleton and Co.: New York, NY, USA, 1883. [Google Scholar]
- Darwin, C. The Origin of the Species; Suriano, G., Ed.; Random House: London, UK, 1859. [Google Scholar]
- Lamarck, J.B.P. Zoological Philosophy; Chicago Press: Chicago, IL, USA, 1809. [Google Scholar]
- Ruden, D.M.; Jamison, D.C.; Zeeberg, B.R.; Garfinkel, M.D.; Weinstein, J.N.; Rasouli, P.; Lu, X. The EDGE hypothesis: Epigenetically directed genetic errors in repeat-containing proteins (RCPs) involved in evolution, neuroendocrine signaling, and cancer. Front. Neuroendocrinol. 2008, 29, 428–444. [Google Scholar] [CrossRef]
- Ruden, D.M.; Xiao, L.; Garfinkel, M.D.; Lu, X. Hsp90 and environmental impacts on epigenetic states: A model for the trans-generational effects of diethylstibesterol on uterine development and cancer. Hum. Mol. Genet. 2005, 14, R149–R155. [Google Scholar] [CrossRef]
- Taipale, M.; Jarosz, D.F.; Lindquist, S. HSP90 at the hub of protein homeostasis: Emerging mechanistic insights. Nat. Rev. Mol. Cell Biol. 2010, 11, 515–528. [Google Scholar]
- Dezwaan, D.C.; Freeman, B.C. HSP90: The Rosetta stone for cellular protein dynamics? Cell Cycle 2008, 7, 1006–1012. [Google Scholar] [CrossRef]
- Whitesell, L.; Lindquist, S.L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef]
- Rutherford, S.; Hirate, Y.; Swalla, B.J. The Hsp90 capacitor, developmental remodeling, and evolution: The robustness of gene networks and the curious evolvability of metamorphosis. Crit. Rev. Biochem. Mol. Biol. 2007, 42, 355–372. [Google Scholar] [CrossRef]
- Queitsch, C.; Sangster, T.A.; Lindquist, S. Hsp90 as a capacitor of phenotypic variation. Nature 2002, 417, 618–624. [Google Scholar] [CrossRef]
- Ruden, D.M.; Garfinkel, M.D.; Sollars, V.E.; Lu, X. Waddington’s widget: Hsp90 and the inheritance of acquired characters. Semin. Cell Dev. Biol. 2003, 14, 301–310. [Google Scholar]
- Sollars, V.; Lu, X.; Xiao, L.; Wang, X.; Garfinkel, M.D.; Ruden, D.M. Evidence for an epigenetic mechanism by which Hsp90 acts as a capacitor for morphological evolution. Nat. Genet. 2003, 33, 70–74. [Google Scholar] [CrossRef]
- Rutherford, S.L.; Lindquist, S. Hsp90 as a capacitor for morphological evolution. Nature 1998, 396, 336–342. [Google Scholar] [CrossRef]
- Meiklejohn, C.D.; Hartl, D.L. A single mode of canalization. Trends Ecol. Evol. 2002, 17, 468–473. [Google Scholar] [CrossRef]
- Lu, X.; Xiao, L.; Wang, L.; Ruden, D.M. Hsp90 inhibitors and drug resistance in cancer: The potential benefits of combination therapies of Hsp90 inhibitors and other anti-cancer drugs. Biochem. Pharmacol. 2011, 83, 995–1004. [Google Scholar]
- McLaren, A. Too late for the midwife toad: Stress, variability and Hsp90. Trends Genet. 1999, 15, 169–171. [Google Scholar]
- Waddington, C.H. Genetic Assimilation of an acquired character. Evolution 1953, 7, 118–126. [Google Scholar] [CrossRef]
- Waddington, C.H. Canalization of development and the inheritance of acquired characters. Nature 1942, 150, 563–565. [Google Scholar] [CrossRef]
- Jablonka, E.; Lamb, M.J. Evolution in Four Dimensions: Genetic, Epigenetic, Behavioral, and Symbolic Variation in the History of Life, Lif and Mind; The MIT Press: Cambridge, MA, USA, 2005; p. X, 462. [Google Scholar]
- Kamal, A.; Thao, L.; Sensintaffar, J.; Zhang, L.; Boehm, M.F.; Fritz, L.C.; Burrows, F.J. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 2003, 425, 407–410. [Google Scholar] [CrossRef]
- Chen, G.; Bradford, W.D.; Seidel, C.W.; Li, R. Hsp90 stress potentiates rapid cellular adaptation through induction of aneuploidy. Nature 2012, 482, 246–250. [Google Scholar]
- Gerson, S.L. MGMT: Its role in cancer aetiology and cancer therapeutics. Nat. Rev. Cancer 2004, 4, 296–307. [Google Scholar] [CrossRef]
- Solar, P.; Horváth, V.; Kleban, J.; Koval', J.; Solárová, Z.; Kozubík, A.; Fedorocko, P. Hsp90 inhibitor geldanamycin increases the sensitivity of resistant ovarian adenocarcinoma cell line A2780cis to cisplatin. Neoplasma 2007, 54, 127–130. [Google Scholar]
- Vasilevskaya, I.A.; Rakitina, T.V.; O’Dwyer, P.J. Geldanamycin and its 17-allylamino-17-demethoxy analogue antagonize the action of Cisplatin in human colon adenocarcinoma cells: Differential caspase activation as a basis for interaction. Cancer Res. 2003, 63, 3241–3246. [Google Scholar]
- Kwon, H.M.; Kim, Y.; Yang, S.I.; Kim, Y.J.; Lee, S.H.; Yoon, B.W. Geldanamycin protects rat brain through overexpression of HSP70 and reducing brain edema after cerebral focal ischemia. Neurol. Res. 2008, 30, 740–745. [Google Scholar] [CrossRef]
- McLean, P.J.; Klucken, J.; Shin, Y.; Hyman, B.T. Geldanamycin induces Hsp70 and prevents alpha-synuclein aggregation and toxicity in vitro. Biochem. Biophys. Res. Commun. 2004, 321, 665–669. [Google Scholar]
- McCollum, A.K.; Lukasiewicz, K.B.; Teneyck, C.J.; Lingle, W.L.; Toft, D.O.; Erlichman, C. Cisplatin abrogates the geldanamycin-induced heat shock response. Mol. Cancer Ther. 2008, 7, 3256–3264. [Google Scholar]
- Paraiso, K.H.; Haarberg, H.E.; Wood, E.; Rebecca, V.W.; Chen, Y.A.; Xiang, Y.; Ribas, A.; Lo, R.S.; Weber, J.S.; Sondak, V.K.; et al. The HSP90 inhibitor XL888 overcomes BRAF inhibitor resistance mediated through diverse mechanisms. Clin. Cancer Res. 2012, 18, 2502–2514. [Google Scholar]
- Weigert, O.; Lane, A.A.; Bird, L.; Kopp, N.; Chapuy, B.; van Bodegom, D.; Toms, A.V.; Marubayashi, S.; Christie, A.L.; McKeown, M.; et al. Genetic resistance to JAK2 enzymatic inhibitors is overcome by HSP90 inhibition. J. Exp. Med. 2012, 209, 259–273. [Google Scholar] [CrossRef]
- Reddy, M.M.; Deshpande, A.; Sattler, M. Targeting JAK2 in the therapy of myeloproliferative neoplasms. Expert Opin. Ther. Targets 2012, 16, 313–324. [Google Scholar] [CrossRef]
- Best, O.G.; Che, Y.; Singh, N.; Forsyth, C.; Christopherson, R.I.; Mulligan, S.P. The Hsp90 inhibitor SNX-7081 synergizes with and restores sensitivity to fludarabine in chronic lymphocytic leukemia cells with lesions in the TP53 pathway: A potential treatment strategy for fludarabine refractory disease. Leuk. Lymphoma 2012, 53, 1367–1375. [Google Scholar] [CrossRef]
- Tariq, M.; Nussbaumer, U.; Chen, Y.; Beisel, C.; Paro, R. Trithorax requires Hsp90 for maintenance of active chromatin at sites of gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 1157–1162. [Google Scholar]
- Sawarkar, R.; Sievers, C.; Paro, R. Hsp90 globally targets paused RNA polymerase to regulate gene expression in response to environmental stimuli. Cell 2012, 149, 807–818. [Google Scholar]
- Nechaev, S.; Adelman, K. Pol II waiting in the starting gates: Regulating the transition from transcription initiation into productive elongation. Biochim. Biophys. Acta 2011, 1809, 34–45. [Google Scholar]
- Pigliucci, M.; Müller, G. Konrad Lorenz Institute for Evolution and Cognition Research, Evolution, the Extended Synthesis 2010, VIII; The MIT Press: Cambridge, MA, USA, 2010; p. 495. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lu, X.; Wang, L.; Ruden, D.M. Hsp90 Inhibitors and the Reduction of Anti-Cancer Drug Resistance by Non-Genetic and Genetic Mechanisms. Pharmaceuticals 2012, 5, 890-898. https://doi.org/10.3390/ph5090890
Lu X, Wang L, Ruden DM. Hsp90 Inhibitors and the Reduction of Anti-Cancer Drug Resistance by Non-Genetic and Genetic Mechanisms. Pharmaceuticals. 2012; 5(9):890-898. https://doi.org/10.3390/ph5090890
Chicago/Turabian StyleLu, Xiangyi, Luan Wang, and Douglas M. Ruden. 2012. "Hsp90 Inhibitors and the Reduction of Anti-Cancer Drug Resistance by Non-Genetic and Genetic Mechanisms" Pharmaceuticals 5, no. 9: 890-898. https://doi.org/10.3390/ph5090890
APA StyleLu, X., Wang, L., & Ruden, D. M. (2012). Hsp90 Inhibitors and the Reduction of Anti-Cancer Drug Resistance by Non-Genetic and Genetic Mechanisms. Pharmaceuticals, 5(9), 890-898. https://doi.org/10.3390/ph5090890