Personalizing Colon Cancer Therapeutics: Targeting Old and New Mechanisms of Action

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction: Personalizing Colon Cancer Therapeutics
2. Targeting Old Mechanisms of Action: Inhibitors that Target the DNA
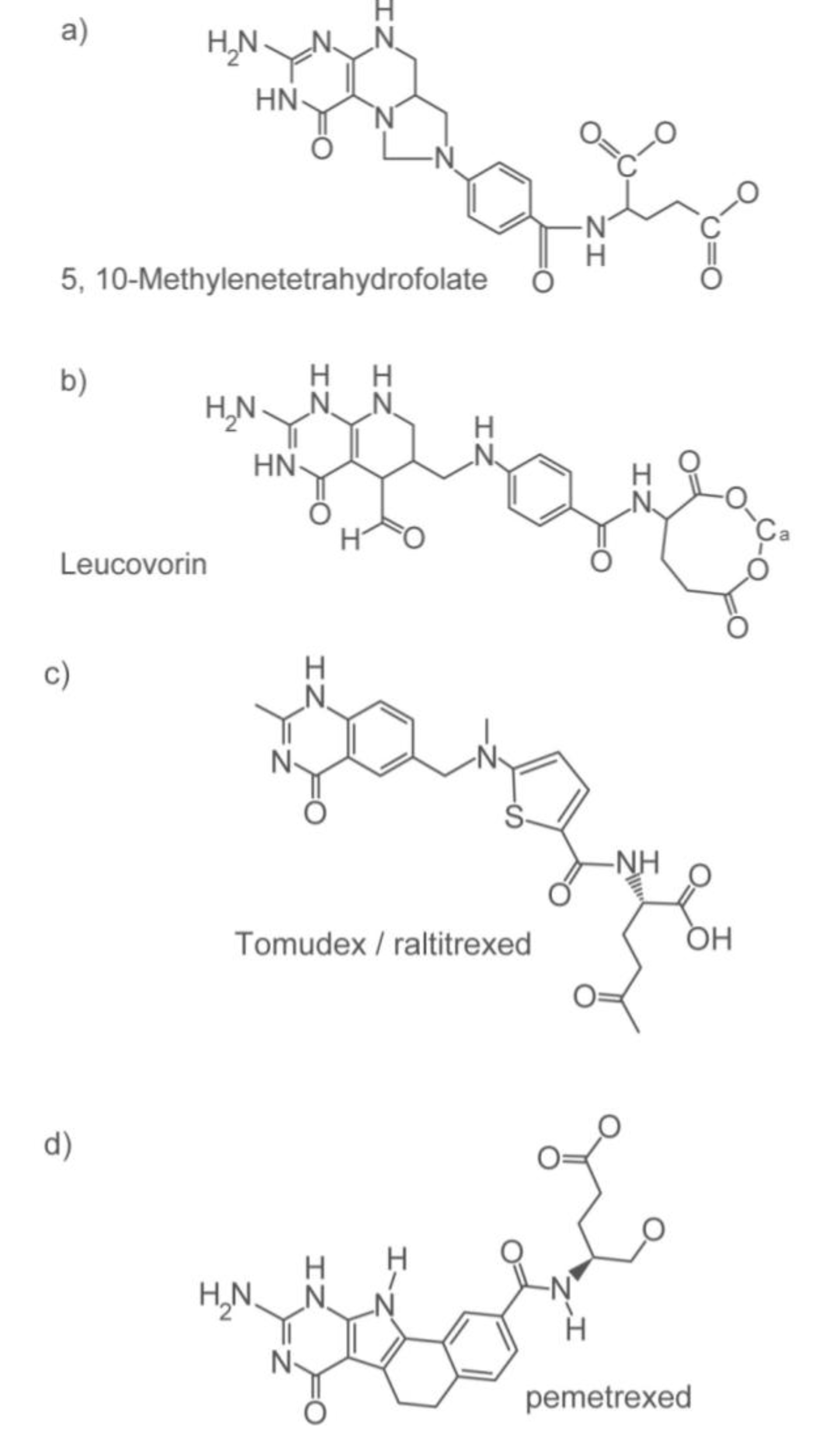
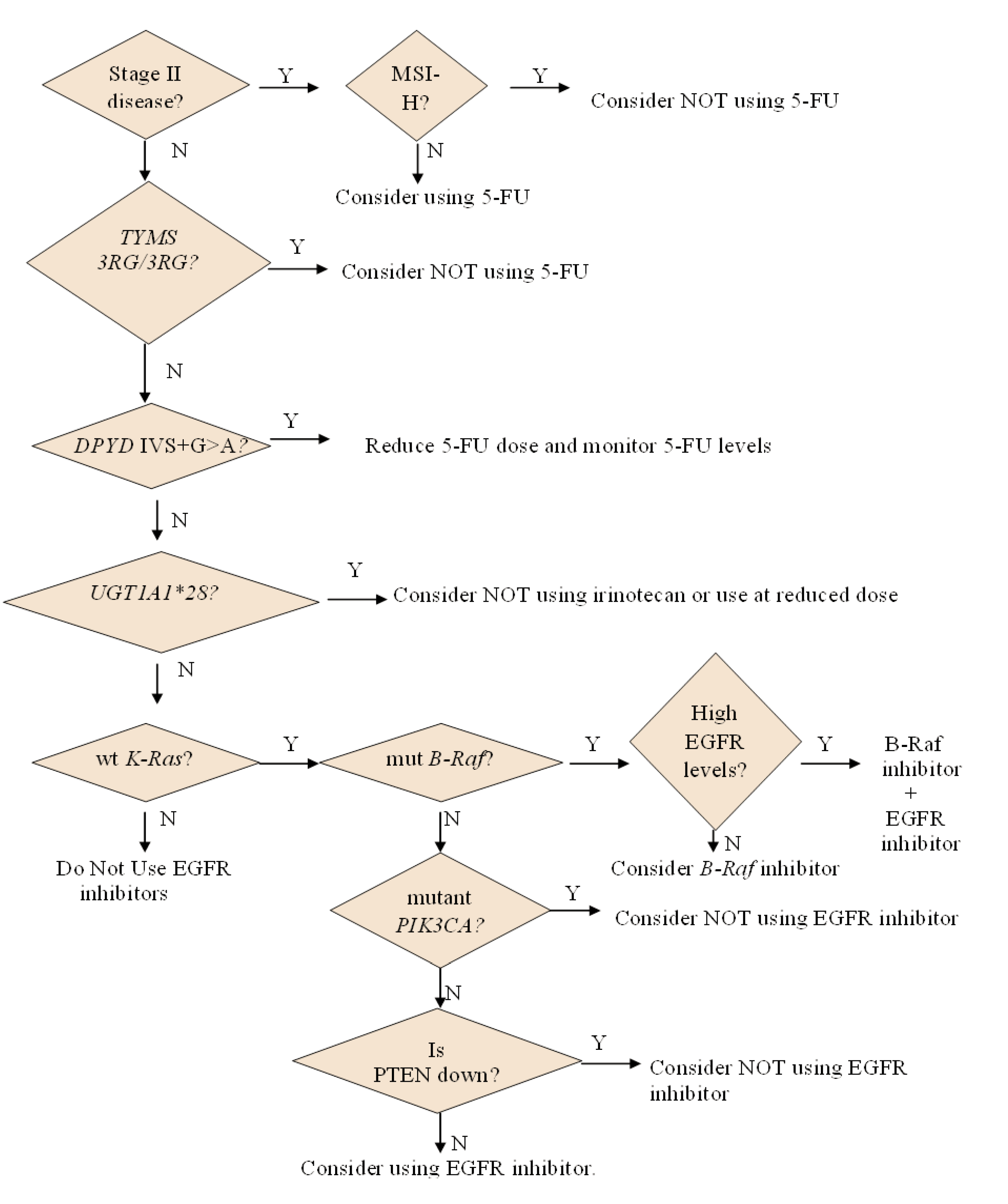
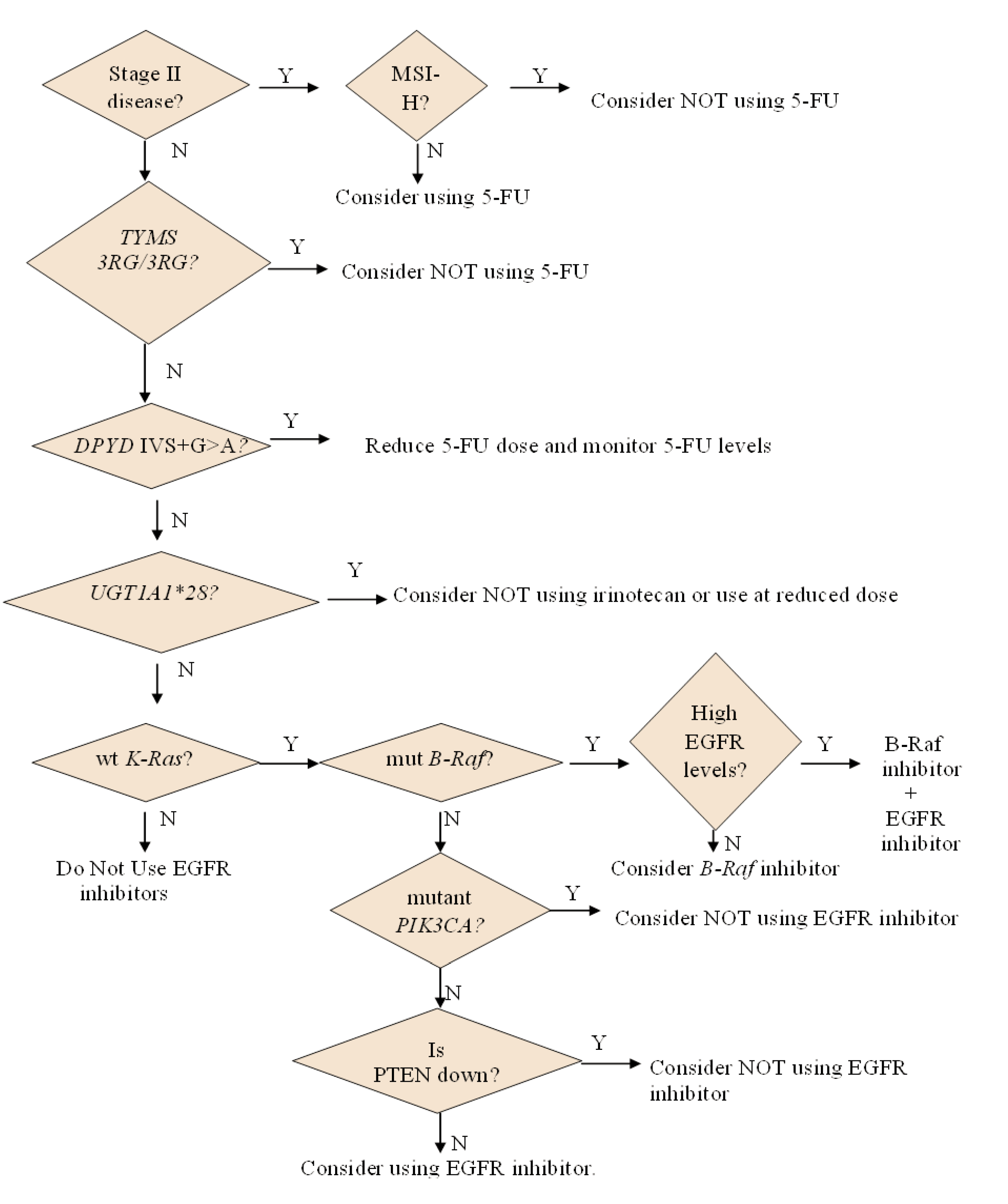
2.1. Personalizing the Use of a 55-Year Old Drug: 5-Fluorouracil (5-FU)
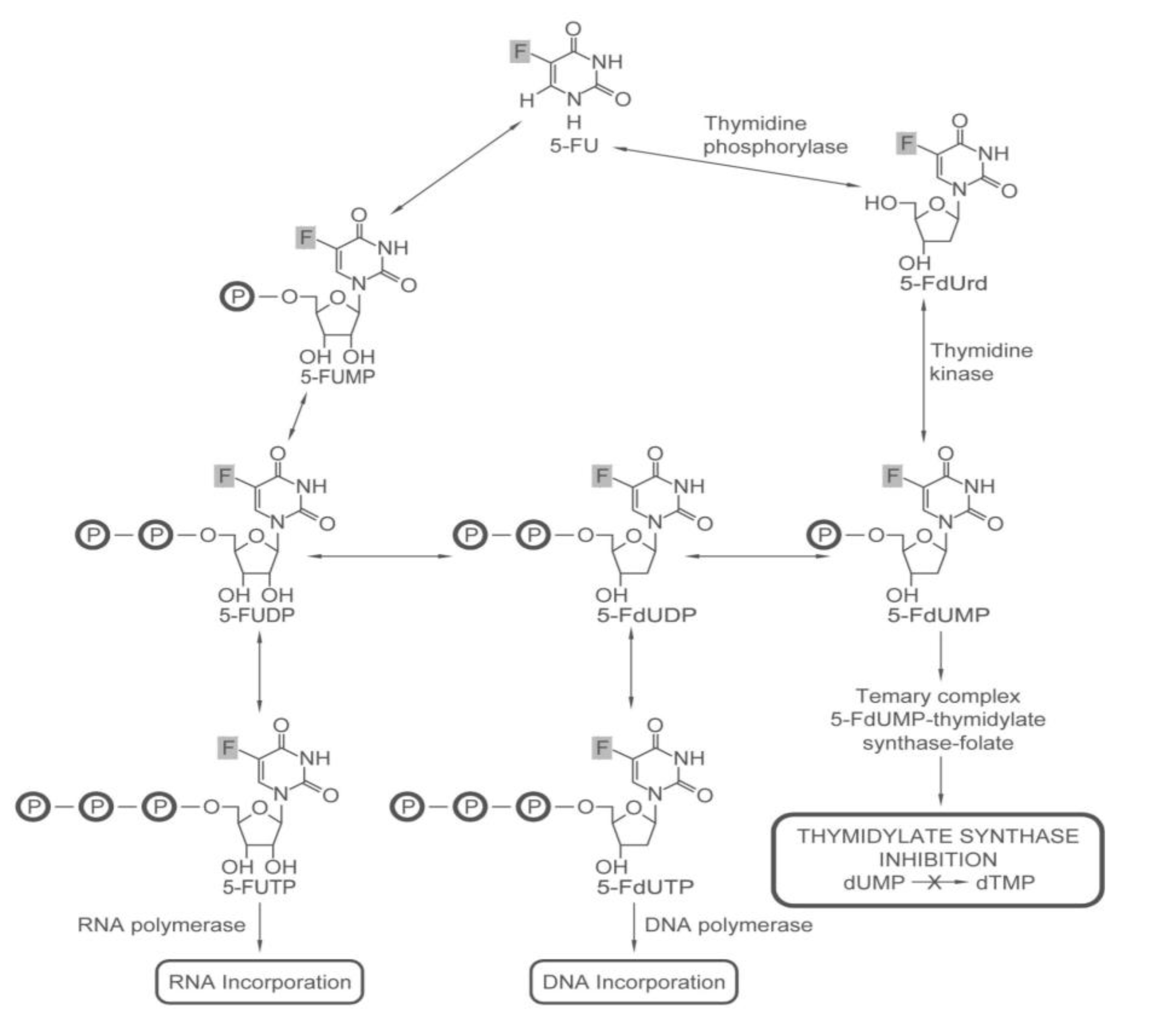
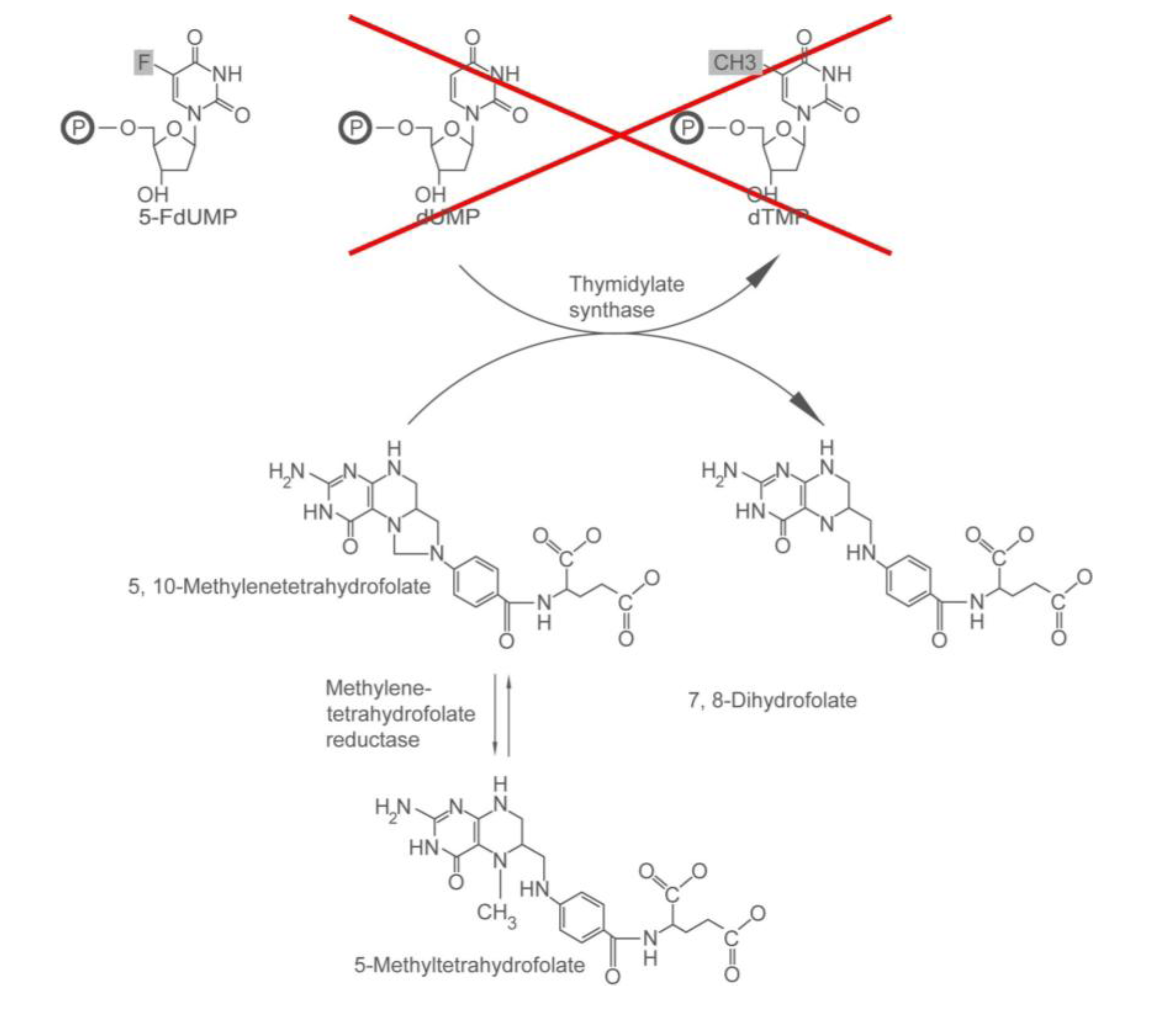
2.1.1. How 5-FU Works
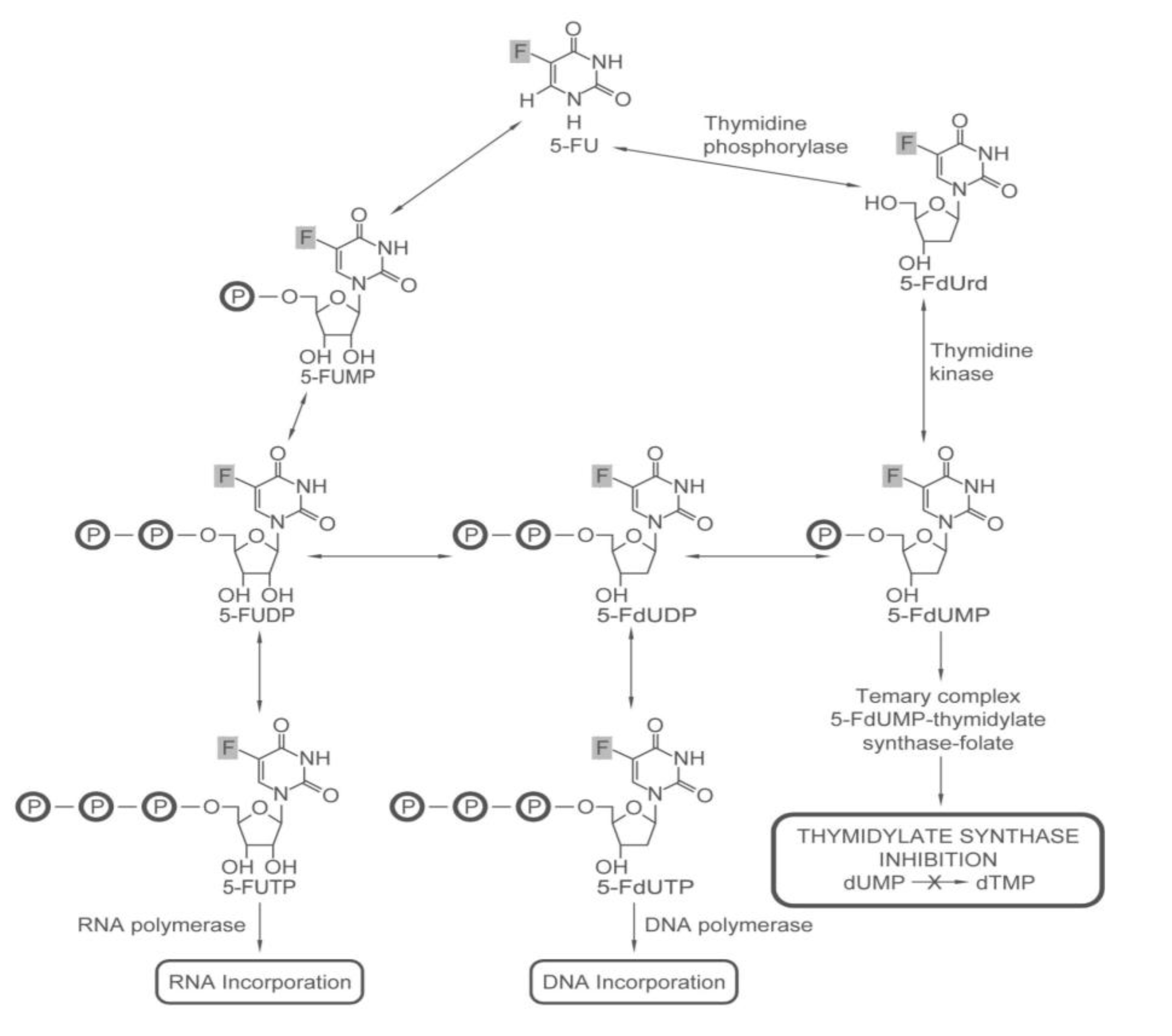
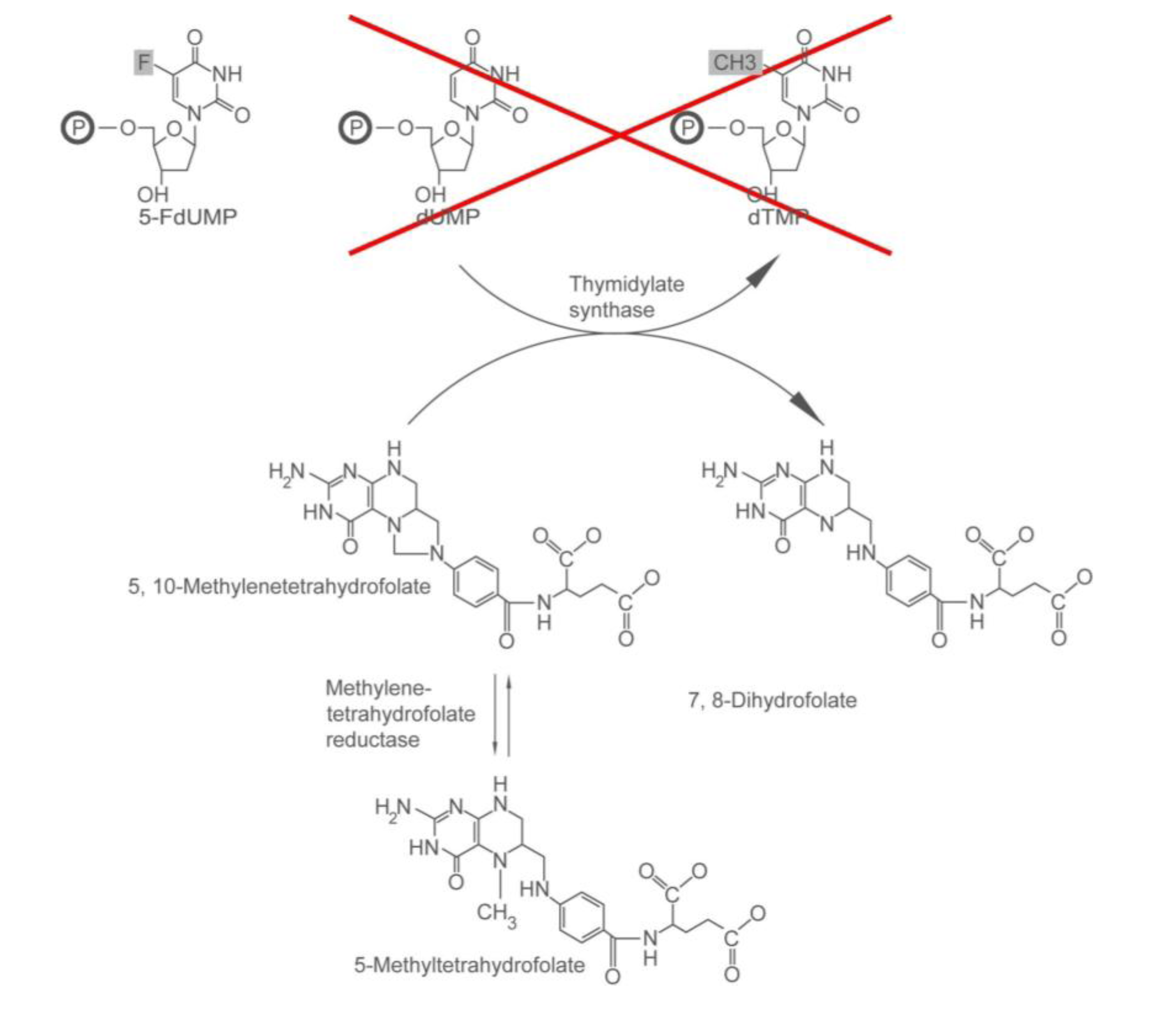
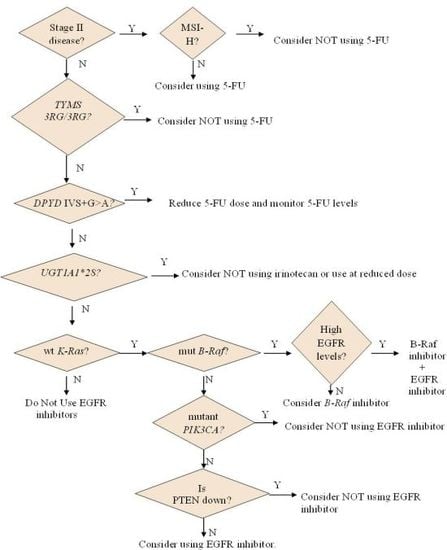
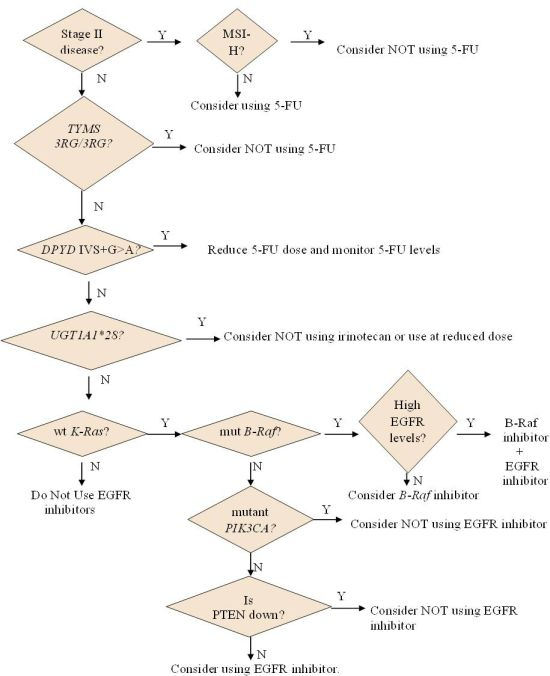
2.1.2. Who May and May not Benefit from 5-FU
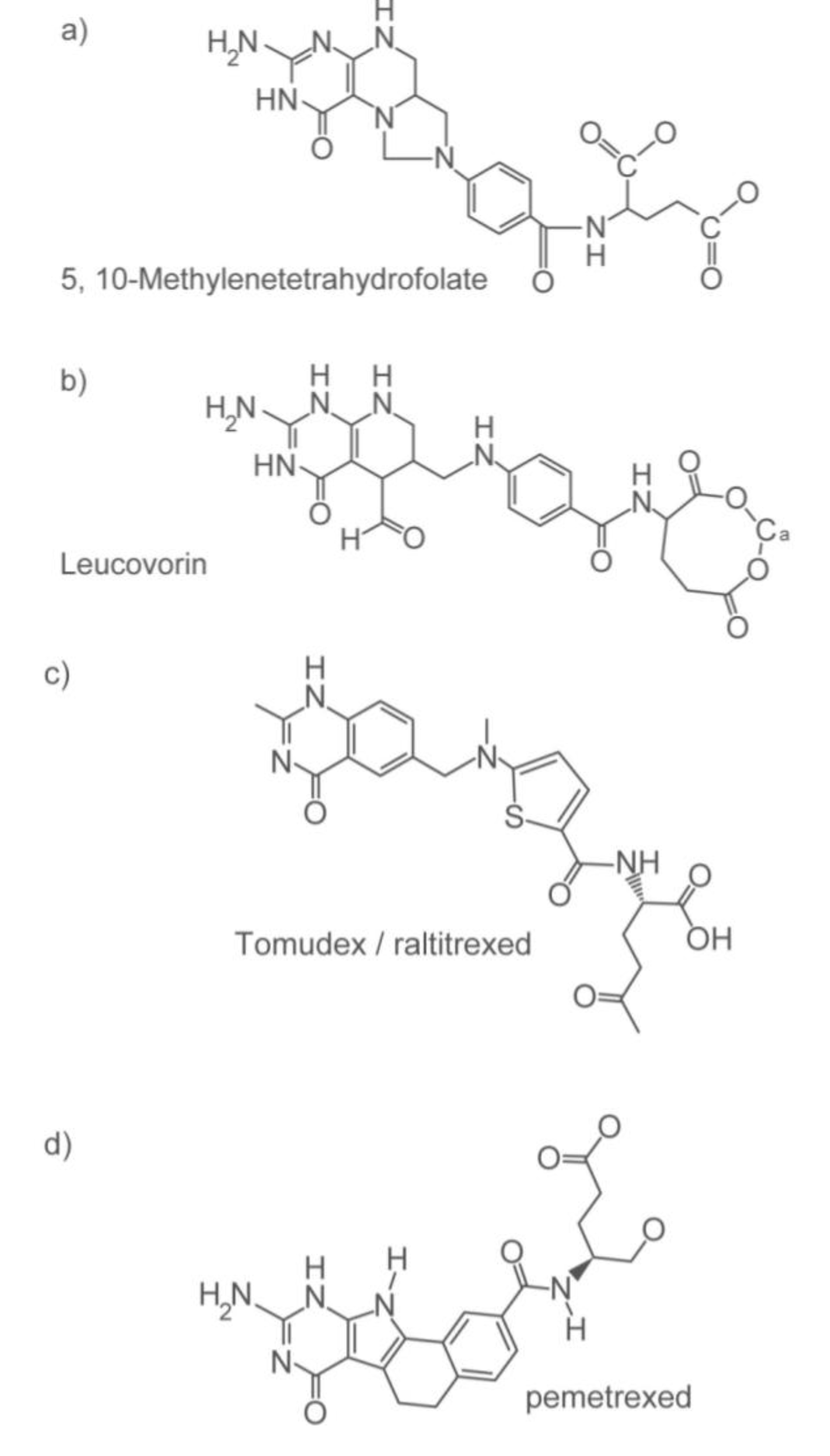
2.1.3. Options for Those Who May not Benefit from 5-FU
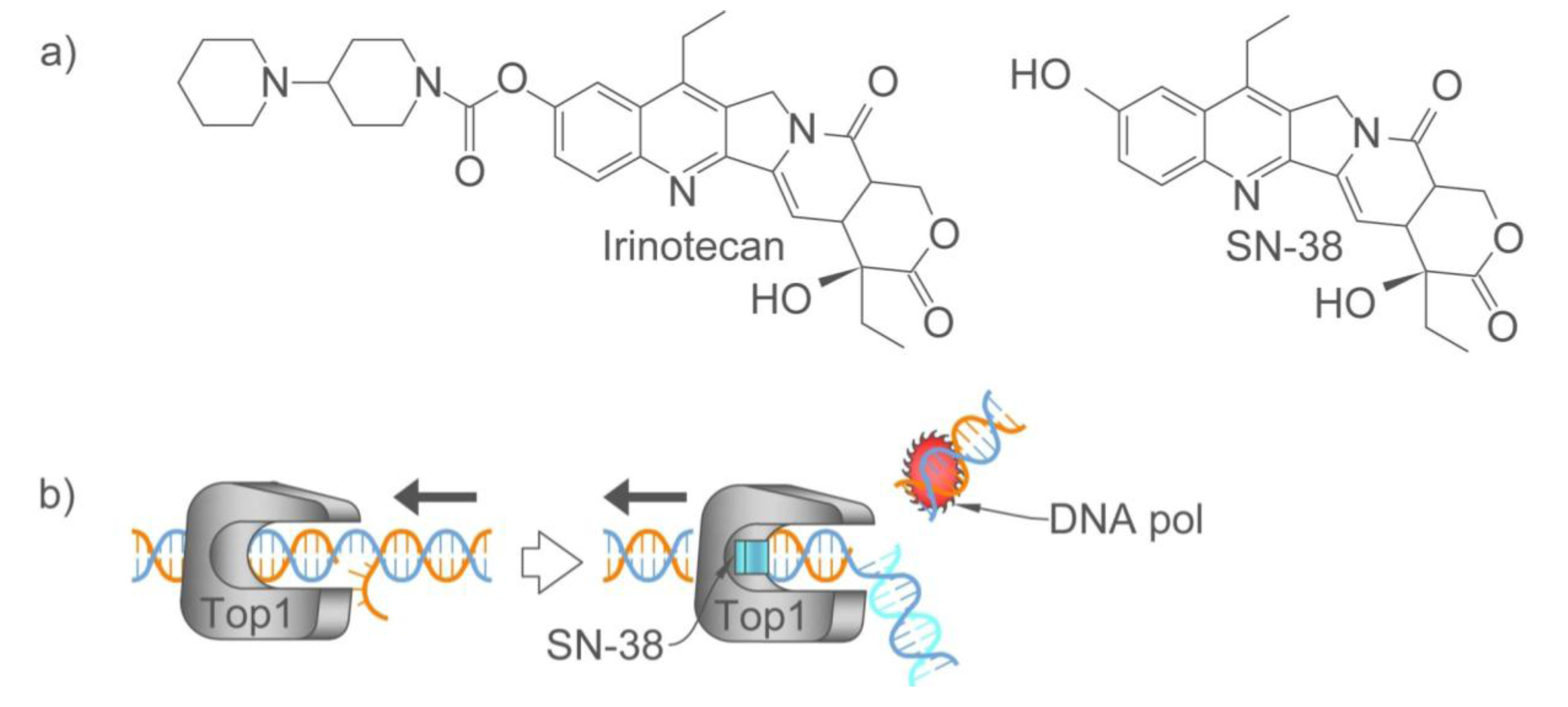
2.2. Personalizing the Choice of DNA-Damaging Agent: Oxaliplatin or Irinotecan
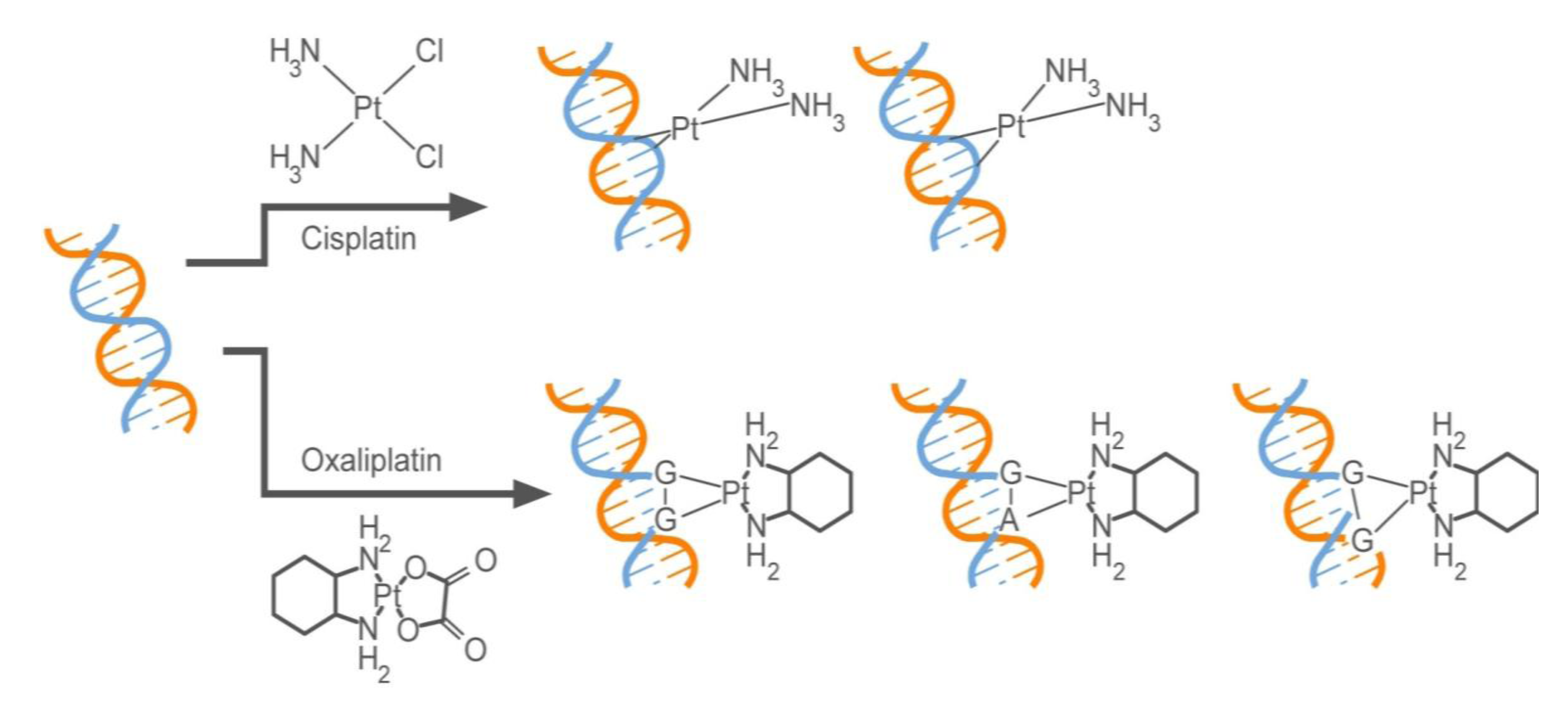
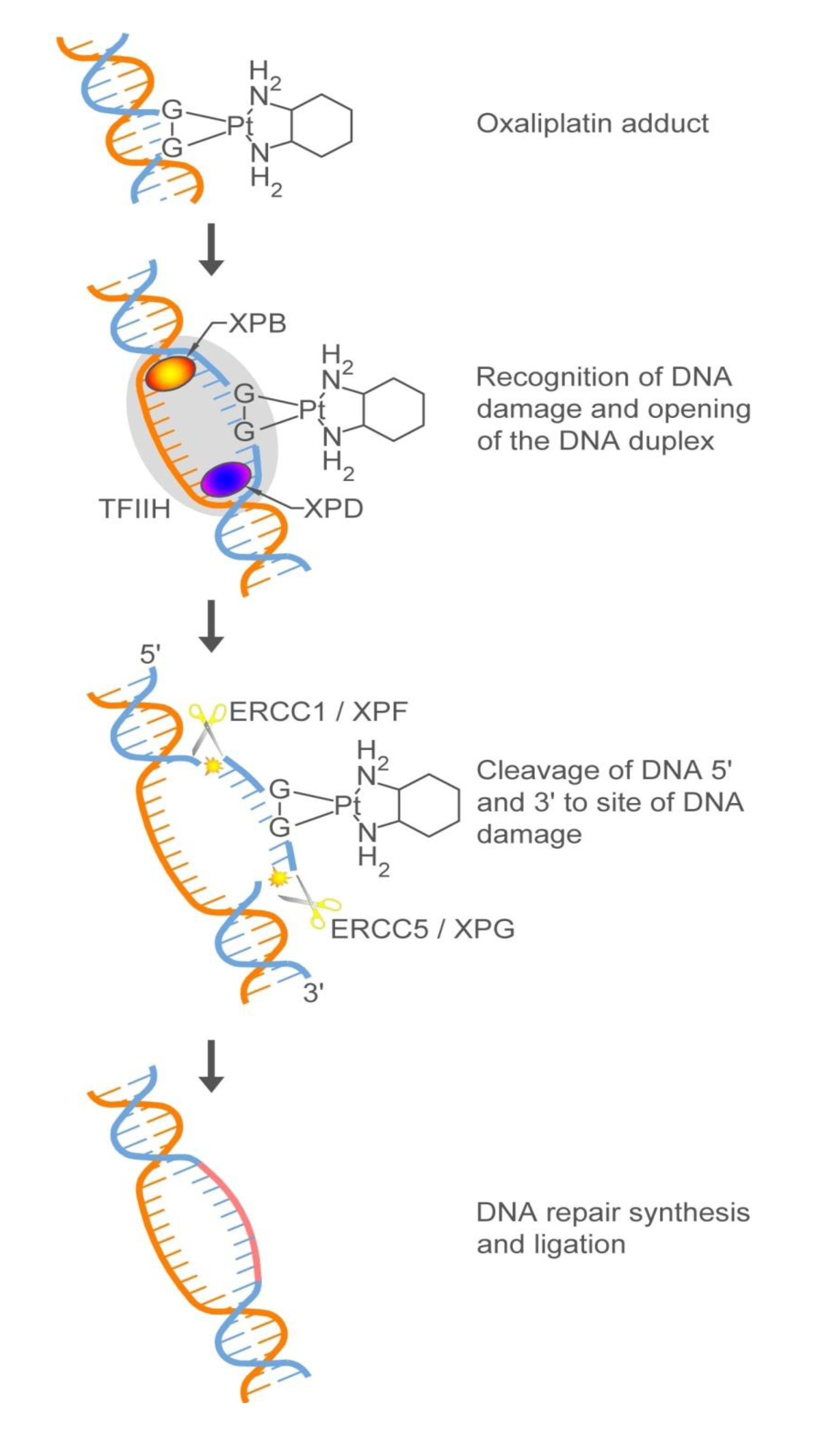
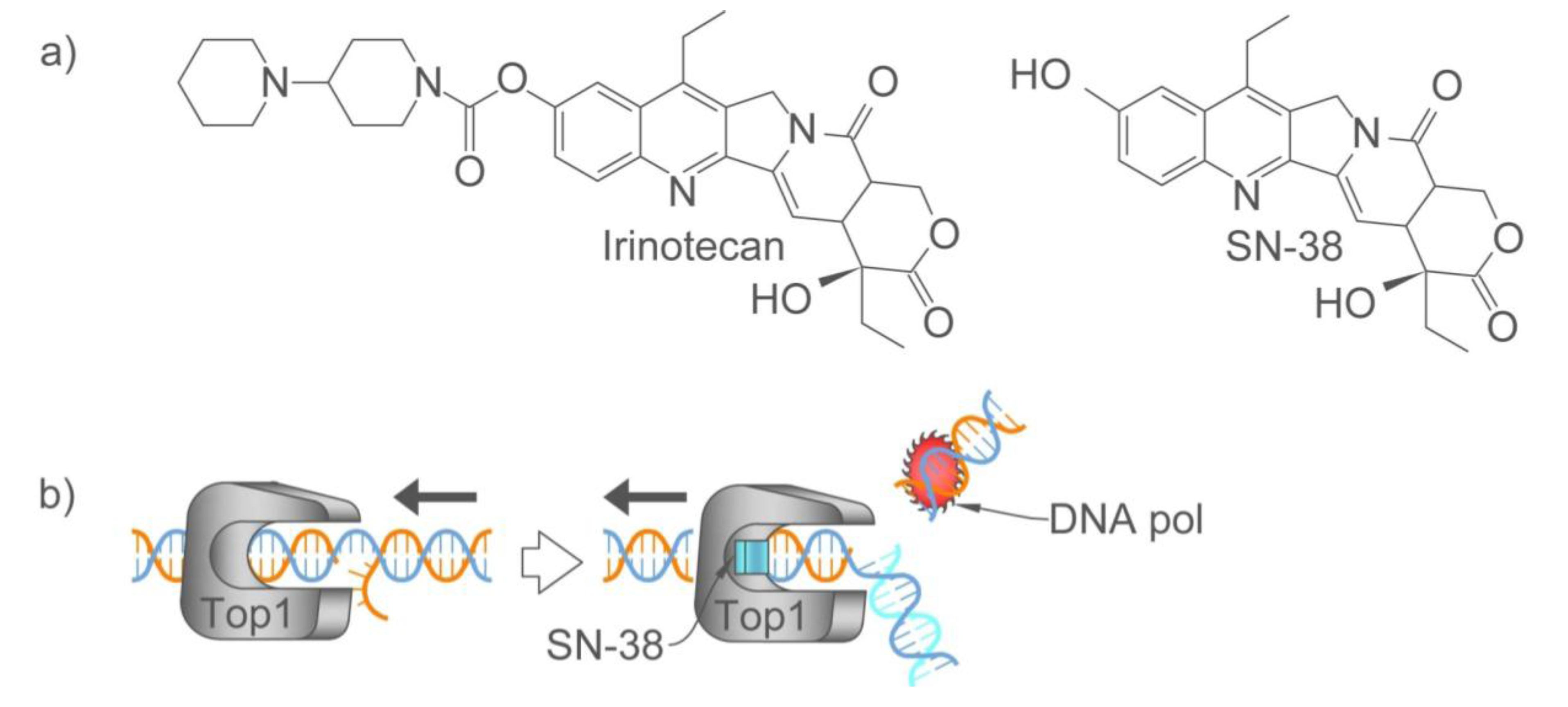
2.2.1. How Oxaliplatin and Irinotecan Work
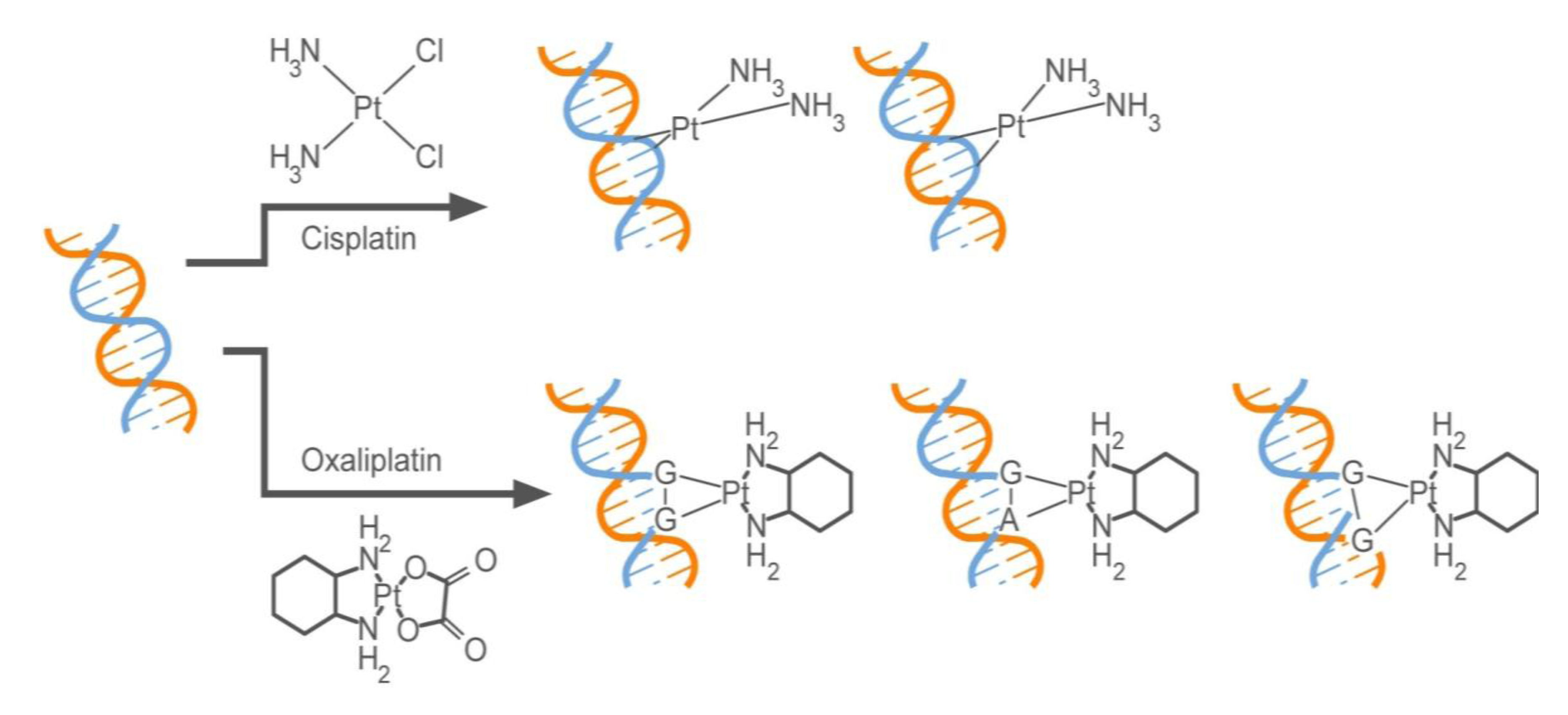
2.2.2. Who May and May not Benefit from Oxaliplatin and Irinotecan
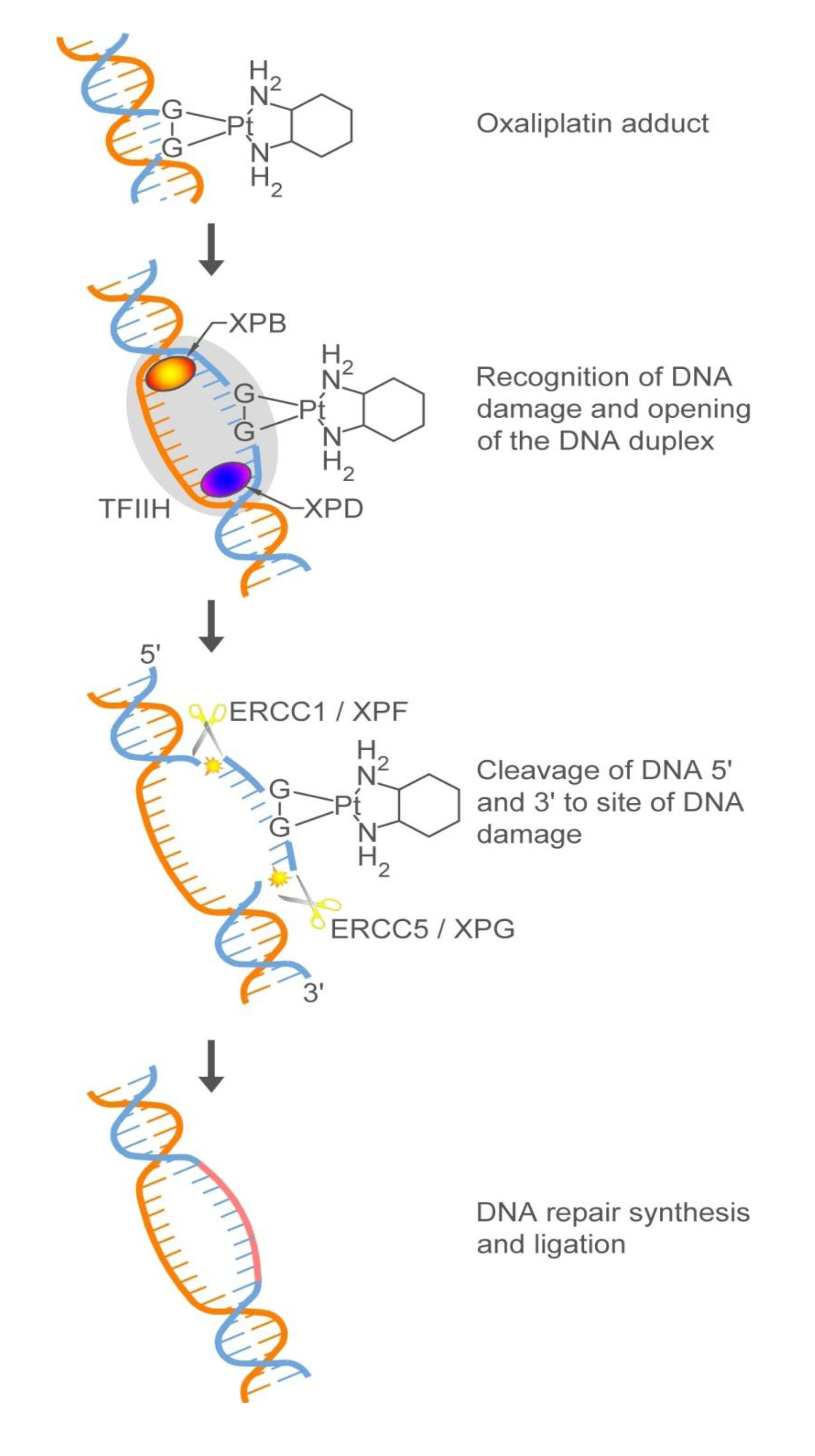
3. Targeting New Mechanisms of Action: Targeting Cancer Cell-Specific Characteristics
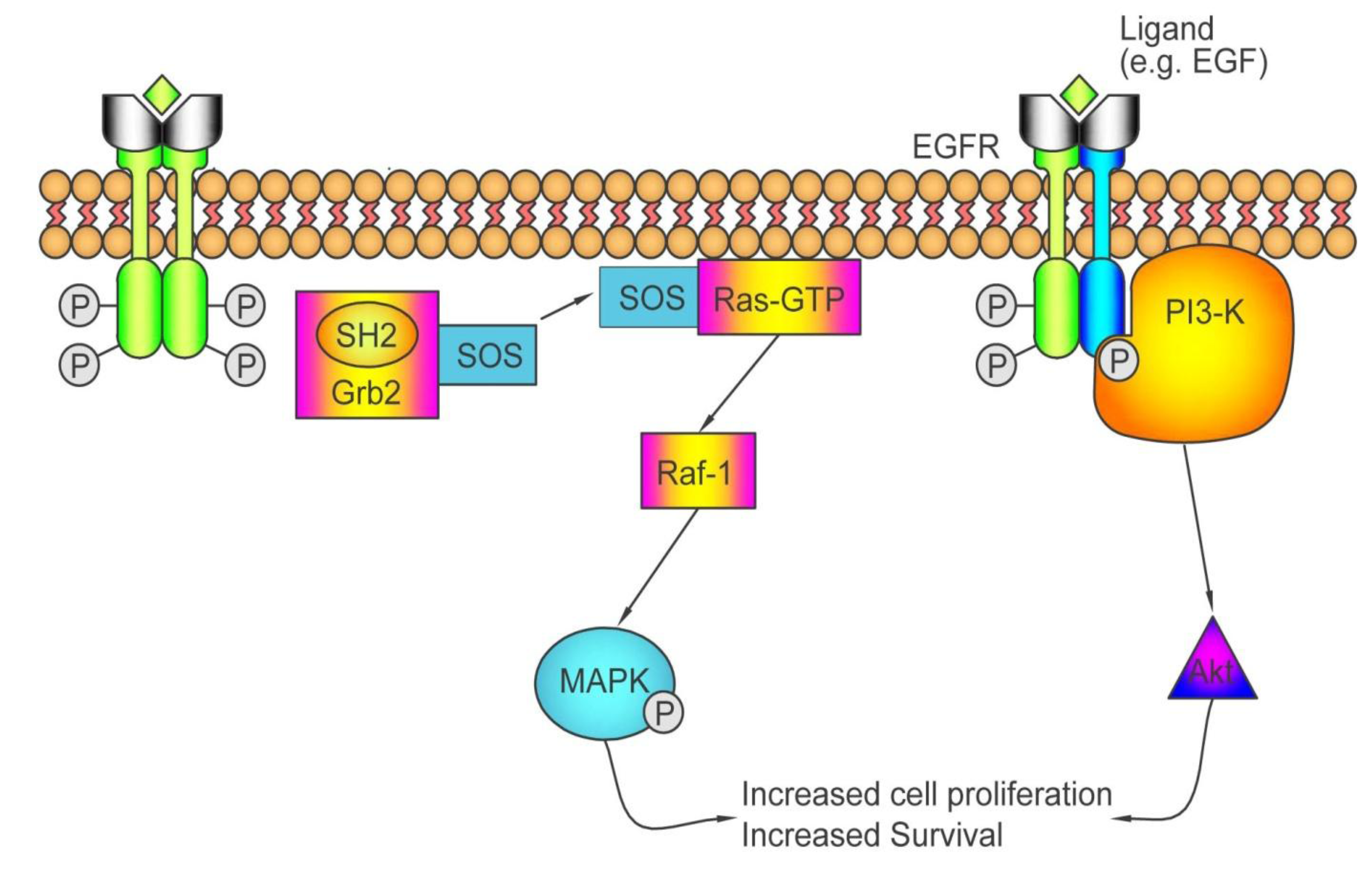
3.1. Personalizing Treatment with Antibodies against the Epidermal Growth Factor Receptor (EGFR)
3.1.1. How Antibodies against EGFR Work
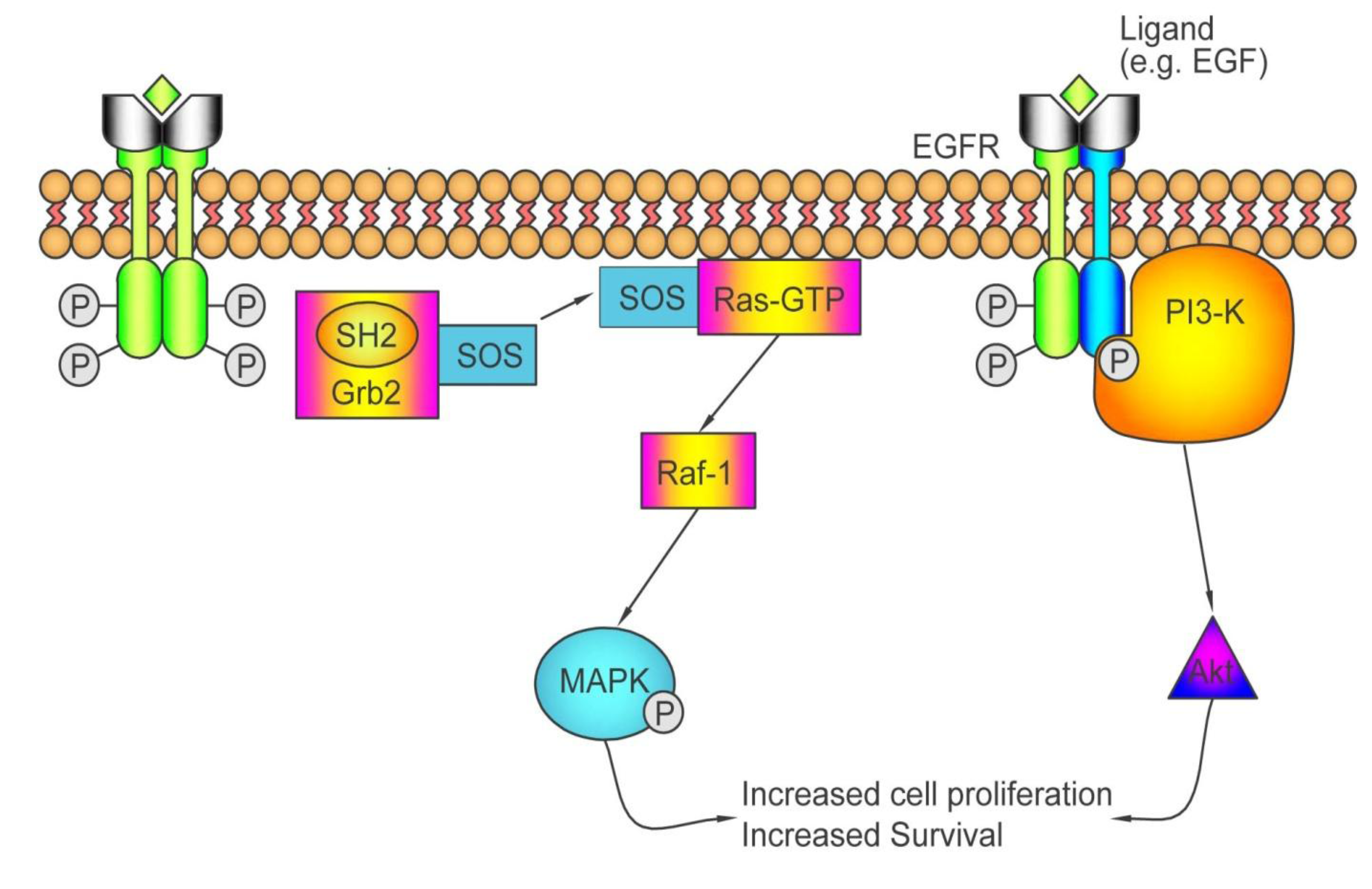
3.1.2. Who May and May not Benefit from Antibodies against EGFR
3.2. Personalizing Treatment with Inhibitors of Vascular Endothelial Growth Factor
3.2.1. Who May and May not Benefit from Inhibitors of VEGF
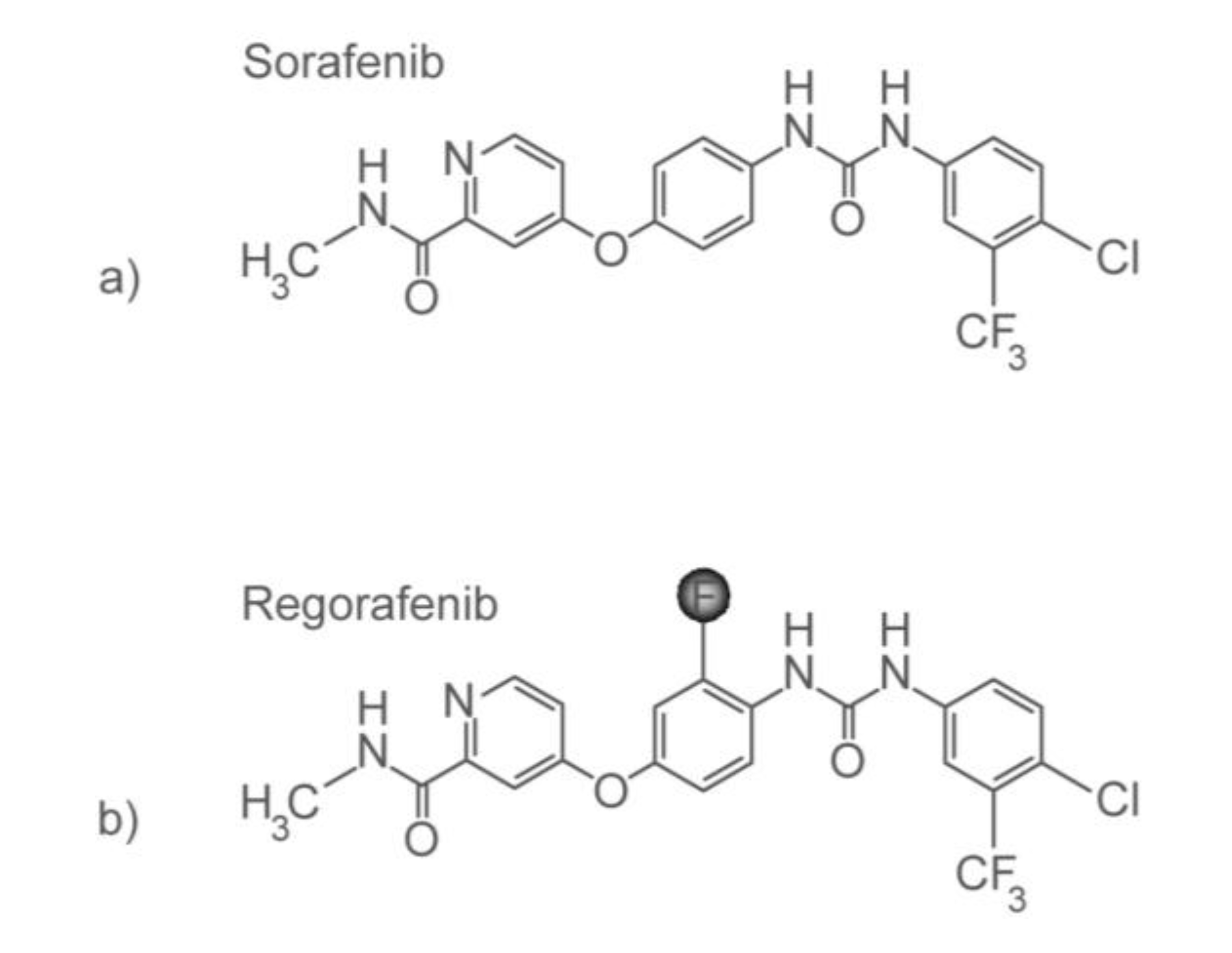
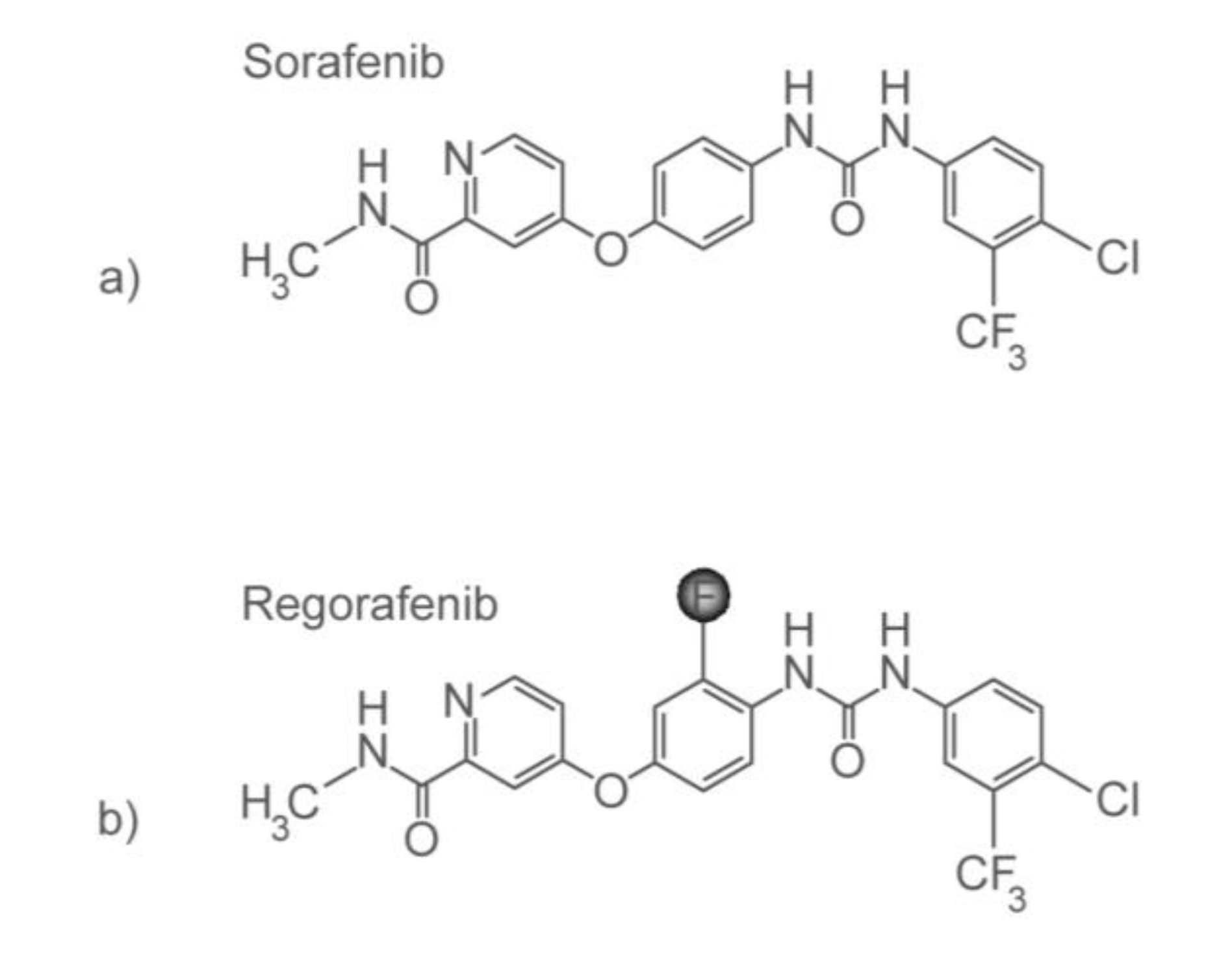
3.3. Personalizing Treatment with Multi-Kinase Inhibitors
3.4. Personalizing Treatment with an Inhibitor Specific for Mutant B-Raf
3.5. Personalizing Treatment in View of Colorectal Cancer Stem Cells (Co-CSC) and Circulating Tumor Cells (CTC)
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar] [CrossRef]
- Laurie, J.A.; Moertel, C.G.; Fleming, T.R.; Wieand, H.S.; Leigh, J.E.; Rubin, J.; McCormack, G.W.; Gerstner, J.B.; Krook, J.E.; Malliard, J.; et al. Surgical adjuvant therapy of large-bowel carcinoma: An evaluation of levamisole and the combination of levamisole and fluorouracil. The North Central Cancer Treatment Group and the Mayo Clinic. J. Clin. Oncol. 1989, 7, 1447–1456. [Google Scholar]
- Wolmark, N.; Fisher, B.; Rockette, H.; Redmond, C.; Wickerham, D.L.; Fisher, E.R.; Jones, J.; Glass, A.; Lerner, H.; Lawrence, W.; et al. Postoperative adjuvant chemotherapy or BCG for colon cancer: Results from NSABP protocol C-01. J. Natl. Cancer Inst. 1988, 80, 30–36. [Google Scholar] [CrossRef]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Neyman, N.; Aminou, R.; Waldron, W.; Altekruse, S.F.; Kosary, C.L.; Ruhl, J.; Tatalovich, Z.; et al. SEER Cancer Statistics Review, 1975–2008; National Cancer Institute: Bethesda, MD, USA, 2011. [Google Scholar]
- Schuell, B.; Gruenberger, T.; Kornek, G.V.; Dworan, N.; Depisch, D.; Lang, F.; Schneeweiss, B.; Scheithauer, W. Side effects during chemotherapy predict tumour response in advanced colorectal cancer. Br. J. Cancer 2005, 93, 744–748. [Google Scholar] [CrossRef]
- Heidelberger, C.; Chaudhuri, N.K.; Danneberg, P.; Mooren, D.; Griesbach, L.; Duschinsky, R.; Schnitzer, R.J.; Pleven, E.; Scheiner, J. Fluorinated pyrimidines, a new class of tumour-inhibitory compounds. Nature 1957, 179, 663–666. [Google Scholar] [CrossRef]
- Cudmore, J.T.; Groesbeck, H.P. Comparison of High-Dosage and Low-Dosage-Maintenance Therapy with 5-Fluorouracil in Solid Tumors. Cancer 1964, 17, 230–232. [Google Scholar] [CrossRef]
- Jacobs, E.M.; Reeves, W.J., Jr.; Wood, D.A.; Pugh, R.; Braunwald, J.; Bateman, J.R. Treatment of cancer with weekly intravenous 5-fluorouracil. Study by the Western Cooperative Cancer Chemotherapy Group (WCCCG). Cancer 1971, 27, 1302–1305. [Google Scholar] [CrossRef]
- Seifert, P.; Baker, L.H.; Reed, M.L.; Vaitkevicius, V.K. Comparison of continuously infused 5-fluorouracil with bolus injection in treatment of patients with colorectal adenocarcinoma. Cancer 1975, 36, 123–128. [Google Scholar] [CrossRef]
- Lokich, J.J.; Ahlgren, J.D.; Gullo, J.J.; Philips, J.A.; Fryer, J.G. A prospective randomized comparison of continuous infusion fluorouracil with a conventional bolus schedule in metastatic colorectal carcinoma: A Mid-Atlantic Oncology Program Study. J. Clin. Oncol. 1989, 7, 425–432. [Google Scholar]
- Brennan, M.J.; Talley, R.W.; Drake, E.H.; Vaitkevicius, V.K.; Poznanski, A.K.; Brush, B.E. 5-Fluorouracil Treatment of Liver Metastases by Continuous Hepatic Artery Infusion Via Cournand Catheter: Results and Suitability for Intensive Postsurgical Adjuvant Chemotherapy. Ann. Surg. 1963, 158, 405–419. [Google Scholar]
- Cressy, N.L.; Schell, H.W., Jr. Effectiveness and Toxicity of Prolonged Infusions of 5-Fluorouracil in the Treatment of Cancer. Am. J. Med. Sci. 1965, 249, 52–55. [Google Scholar] [CrossRef]
- Rutman, R.J.; Cantarow, A.; Paschkis, K.E. Studies in 2-acetylaminofluorene carcinogenesis. III. The utilization of uracil-2-C14 by preneoplastic rat liver and rat hepatoma. Cancer Res. 1954, 14, 119–123. [Google Scholar]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef]
- Amatori, F.; Di Paolo, A.; Del Tacca, M.; Fontanini, G.; Vannozzi, F.; Boldrini, L.; Bocci, G.; Lastella, M.; Danesi, R. Thymidylate synthase, dihydropyrimidine dehydrogenase and thymidine phosphorylase expression in colorectal cancer and normal mucosa in patients. Pharmacogenet. Genomics 2006, 16, 809–816. [Google Scholar] [CrossRef]
- Ikeguchi, M.; Makino, M.; Kaibara, N. Thymidine phosphorylase and dihydropyrimidine dehydrogenase activity in colorectal carcinoma and patients prognosis. Langenbecks Arch. Surg. 2002, 387, 240–245. [Google Scholar] [CrossRef]
- Grem, J.L.; Chabner, B.A.; Ryan, D.P.; Wadlow, R.C. 5-Fluoropyrimidines. In Cancer Chemotherapy and Biotherapy: Principles and Practice; Chabner, B.A., Longo, D.L., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2010. [Google Scholar]
- Jackson, R.C. The regulation of thymidylate biosynthesis in Novikoff hepatoma cells and the effects of amethopterin, 5-fluorodeoxyuridine, and 3-deazauridine. J. Biol. Chem. 1978, 253, 7440–7446. [Google Scholar]
- Grogan, B.C.; Parker, J.B.; Guminski, A.F.; Stivers, J.T. Effect of the thymidylate synthase inhibitors on dUTP and TTP pool levels and the activities of DNA repair glycosylases on uracil and 5-fluorouracil in DNA. Biochemistry 2011, 50, 618–627. [Google Scholar] [CrossRef]
- Goulian, M.; Bleile, B.; Tseng, B.Y. The effect of methotrexate on levels of dUTP in animal cells. J. Biol. Chem. 1980, 255, 10630–10637. [Google Scholar]
- Myers, C.E.; Young, R.C.; Chabner, B.A. Biochemical determinants of 5-fluorouracil response in vivo. The role of deoxyuridylate pool expansion. J. Clin. Invest. 1975, 56, 1231–1238. [Google Scholar] [CrossRef]
- Brynolf, K.; Eliasson, R.; Reichard, P. Formation of Okazaki fragments in polyoma DNA synthesis caused by misincorporation of uracil. Cell 1978, 13, 573–580. [Google Scholar] [CrossRef]
- Lonn, U.; Lonn, S. Interaction between 5-fluorouracil and DNA of human colon adenocarcinoma. Cancer Res. 1984, 44, 3414–3418. [Google Scholar]
- Yoshioka, A.; Tanaka, S.; Hiraoka, O.; Koyama, Y.; Hirota, Y.; Ayusawa, D.; Seno, T.; Garrett, C.; Wataya, Y. Deoxyribonucleoside triphosphate imbalance. 5-Fluorodeoxyuridine-induced DNA double strand breaks in mouse FM3A cells and the mechanism of cell death. J. Biol. Chem. 1987, 262, 8235–8241. [Google Scholar]
- Herrick, D.; Kufe, D.W. Lethality associated with incorporation of 5-fluorouracil into preribosomal RNA. Mol. Pharmacol. 1984, 26, 135–140. [Google Scholar]
- Modulation of fluorouracil by leucovorin in patients with advanced colorectal cancer: Evidence in terms of response rate. Advanced Colorectal Cancer Meta-Analysis project. J. Clin. Oncol. 1992, 10, 896–903.
- Ullman, B.; Lee, M.; Martin, D.W., Jr.; Santi, D.V. Cytotoxicity of 5-fluoro-2'-deoxyuridine: Requirement for reduced folate cofactors and antagonism by methotrexate. Proc. Natl. Acad. Sci. USA 1978, 75, 980–983. [Google Scholar] [CrossRef]
- Danenberg, P.V.; Danenberg, K.D. Effect of 5, 10-methylenetetrahydrofolate on the dissociation of 5-fluoro-2'-deoxyuridylate from thymidylate synthetase: Evidence for an ordered mechanism. Biochemistry 1978, 17, 4018–4024. [Google Scholar] [CrossRef]
- Evans, R.M.; Laskin, J.D.; Hakala, M.T. Effect of excess folates and deoxyinosine on the activity and site of action of 5-fluorouracil. Cancer Res. 1981, 41, 3288–3295. [Google Scholar]
- Erlichman, C.; Fine, S.; Wong, A.; Elhakim, T. A randomized trial of fluorouracil and folinic acid in patients with metastatic colorectal carcinoma. J. Clin. Oncol. 1988, 6, 469–475. [Google Scholar]
- Bobbio-Pallavicini, E.; Porta, C.; Moroni, M.; Spaghi, A.; Casagranda, I.; Nastasi, G. Folinic acid does improve 5-fluorouracil activity in vivo. Results of a phase III study comparing 5-fluorouracil to 5-fluorouracil and folinic acid in advanced colon cancer patients. J. Chemother. 1993, 5, 52–55. [Google Scholar]
- Peters, G.J.; van der Wilt, C.L.; van Groeningen, C.J.; Smid, K.; Meijer, S.; Pinedo, H.M. Thymidylate synthase inhibition after administration of fluorouracil with or without leucovorin in colon cancer patients: Implications for treatment with fluorouracil. J. Clin. Oncol. 1994, 12, 2035–2042. [Google Scholar]
- Donaldson, K.O.; Keresztesy, J.C. Naturally occurring forms of folic acid. I. “Prefolic A”: Preparation of concentrate and enzymatic conversion to citrovorum factor. J. Biol. Chem. 1959, 234, 3235–3240. [Google Scholar]
- Frosst, P.; Blom, H.J.; Milos, R.; Goyette, P.; Sheppard, C.A.; Matthews, R.G.; Boers, G.J.; den Heijer, M.; Kluijtmans, L.A.; van den Heuvel, L.P.; et al. A candidate genetic risk factor for vascular disease: A common mutation in methylenetetrahydrofolate reductase. Nat. Genet. 1995, 10, 111–113. [Google Scholar] [CrossRef]
- Cohen, V.; Panet-Raymond, V.; Sabbaghian, N.; Morin, I.; Batist, G.; Rozen, R. Methylenetetrahydrofolate reductase polymorphism in advanced colorectal cancer: A novel genomic predictor of clinical response to fluoropyrimidine-based chemotherapy. Clin. Cancer Res. 2003, 9, 1611–1615. [Google Scholar]
- Etienne-Grimaldi, M.C.; Milano, G.; Maindrault-Goebel, F.; Chibaudel, B.; Formento, J.L.; Francoual, M.; Lledo, G.; Andre, T.; Mabro, M.; Mineur, L.; et al. Methylenetetrahydrofolate reductase (MTHFR) gene polymorphisms and FOLFOX response in colorectal cancer patients. Br. J. Clin. Pharmacol. 2010, 69, 58–66. [Google Scholar] [CrossRef]
- Etienne, M.C.; Formento, J.L.; Chazal, M.; Francoual, M.; Magne, N.; Formento, P.; Bourgeon, A.; Seitz, J.F.; Delpero, J.R.; Letoublon, C.; et al. Methylenetetrahydrofolate reductase gene polymorphisms and response to fluorouracil-based treatment in advanced colorectal cancer patients. Pharmacogenetics 2004, 14, 785–792. [Google Scholar] [CrossRef]
- Jakobsen, A.; Nielsen, J.N.; Gyldenkerne, N.; Lindeberg, J. Thymidylate synthase and methylenetetrahydrofolate reductase gene polymorphism in normal tissue as predictors of fluorouracil sensitivity. J. Clin. Oncol. 2005, 23, 1365–1369. [Google Scholar] [CrossRef]
- Marcuello, E.; Altes, A.; Menoyo, A.; Rio, E.D.; Baiget, M. Methylenetetrahydrofolate reductase gene polymorphisms: Genomic predictors of clinical response to fluoropyrimidine-based chemotherapy? Cancer Chemother. Pharmacol. 2006, 57, 835–840. [Google Scholar]
- Suh, K.W.; Kim, J.H.; Kim do, Y.; Kim, Y.B.; Lee, C.; Choi, S. Which gene is a dominant predictor of response during FOLFOX chemotherapy for the treatment of metastatic colorectal cancer, the MTHFR or XRCC1 gene? Ann. Surg. Oncol. 2006, 13, 1379–1385. [Google Scholar] [CrossRef]
- Takeishi, K.; Kaneda, S.; Ayusawa, D.; Shimizu, K.; Gotoh, O.; Seno, T. Human thymidylate synthase gene: Isolation of phage clones which cover a functionally active gene and structural analysis of the region upstream from the translation initiation codon. J. Biochem. 1989, 106, 575–583. [Google Scholar]
- Horie, N.; Aiba, H.; Oguro, K.; Hojo, H.; Takeishi, K. Functional analysis and DNA polymorphism of the tandemly repeated sequences in the 5'-terminal regulatory region of the human gene for thymidylate synthase. Cell. Struct. Funct. 1995, 20, 191–197. [Google Scholar] [CrossRef]
- Kawakami, K.; Salonga, D.; Park, J.M.; Danenberg, K.D.; Uetake, H.; Brabender, J.; Omura, K.; Watanabe, G.; Danenberg, P.V. Different lengths of a polymorphic repeat sequence in the thymidylate synthase gene affect translational efficiency but not its gene expression. Clin. Cancer Res. 2001, 7, 4096–4101. [Google Scholar]
- Afzal, S.; Gusella, M.; Vainer, B.; Vogel, U.B.; Andersen, J.T.; Broedbaek, K.; Petersen, M.; Jimenez-Solem, E.; Bertolaso, L.; Barile, C.; et al. Combinations of polymorphisms in genes involved in the 5-Fluorouracil metabolism pathway are associated with gastrointestinal toxicity in chemotherapy-treated colorectal cancer patients. Clin. Cancer Res. 2011, 17, 3822–3829. [Google Scholar] [CrossRef]
- Kawakami, K.; Omura, K.; Kanehira, E.; Watanabe, Y. Polymorphic tandem repeats in the thymidylate synthase gene is associated with its protein expression in human gastrointestinal cancers. Anticancer Res. 1999, 19, 3249–3252. [Google Scholar]
- Pullarkat, S.T.; Stoehlmacher, J.; Ghaderi, V.; Xiong, Y.P.; Ingles, S.A.; Sherrod, A.; Warren, R.; Tsao-Wei, D.; Groshen, S.; Lenz, H.J. Thymidylate synthase gene polymorphism determines response and toxicity of 5-FU chemotherapy. Pharmacogenomics J. 2001, 1, 65–70. [Google Scholar] [CrossRef]
- Lecomte, T.; Ferraz, J.M.; Zinzindohoue, F.; Loriot, M.A.; Tregouet, D.A.; Landi, B.; Berger, A.; Cugnenc, P.H.; Jian, R.; Beaune, P.; et al. Thymidylate synthase gene polymorphism predicts toxicity in colorectal cancer patients receiving 5-fluorouracil-based chemotherapy. Clin. Cancer Res. 2004, 10, 5880–5888. [Google Scholar] [CrossRef]
- Marsh, S.; McKay, J.A.; Cassidy, J.; McLeod, H.L. Polymorphism in the thymidylate synthase promoter enhancer region in colorectal cancer. Int. J. Oncol. 2001, 19, 383–386. [Google Scholar]
- Uchida, K.; Hayashi, K.; Kawakami, K.; Schneider, S.; Yochim, J.M.; Kuramochi, H.; Takasaki, K.; Danenberg, K.D.; Danenberg, P.V. Loss of heterozygosity at the thymidylate synthase (TS) locus on chromosome 18 affects tumor response and survival in individuals heterozygous for a 28-bp polymorphism in the TS gene. Clin. Cancer Res. 2004, 10, 433–439. [Google Scholar] [CrossRef]
- Iacopetta, B.; Grieu, F.; Joseph, D.; Elsaleh, H. A polymorphism in the enhancer region of the thymidylate synthase promoter influences the survival of colorectal cancer patients treated with 5-fluorouracil. Br. J. Cancer 2001, 85, 827–830. [Google Scholar] [CrossRef]
- Mandola, M.V.; Stoehlmacher, J.; Muller-Weeks, S.; Cesarone, G.; Yu, M.C.; Lenz, H.J.; Ladner, R.D. A novel single nucleotide polymorphism within the 5' tandem repeat polymorphism of the thymidylate synthase gene abolishes USF-1 binding and alters transcriptional activity. Cancer Res. 2003, 63, 2898–2904. [Google Scholar]
- Marcuello, E.; Altes, A.; del Rio, E.; Cesar, A.; Menoyo, A.; Baiget, M. Single nucleotide polymorphism in the 5' tandem repeat sequences of thymidylate synthase gene predicts for response to fluorouracil-based chemotherapy in advanced colorectal cancer patients. Int. J. Cancer 2004, 112, 733–737. [Google Scholar] [CrossRef]
- Johnston, P.G.; Lenz, H.J.; Leichman, C.G.; Danenberg, K.D.; Allegra, C.J.; Danenberg, P.V.; Leichman, L. Thymidylate synthase gene and protein expression correlate and are associated with response to 5-fluorouracil in human colorectal and gastric tumors. Cancer Res. 1995, 55, 1407–1412. [Google Scholar]
- Leichman, C.G.; Lenz, H.J.; Leichman, L.; Danenberg, K.; Baranda, J.; Groshen, S.; Boswell, W.; Metzger, R.; Tan, M.; Danenberg, P.V. Quantitation of intratumoral thymidylate synthase expression predicts for disseminated colorectal cancer response and resistance to protracted-infusion fluorouracil and weekly leucovorin. J. Clin. Oncol. 1997, 15, 3223–3229. [Google Scholar]
- Lenz, H.J.; Hayashi, K.; Salonga, D.; Danenberg, K.D.; Danenberg, P.V.; Metzger, R.; Banerjee, D.; Bertino, J.R.; Groshen, S.; Leichman, L.P.; et al. p53 point mutations and thymidylate synthase messenger RNA levels in disseminated colorectal cancer: An analysis of response and survival. Clin. Cancer Res. 1998, 4, 1243–1250. [Google Scholar]
- Aschele, C.; Debernardis, D.; Casazza, S.; Antonelli, G.; Tunesi, G.; Baldo, C.; Lionetto, R.; Maley, F.; Sobrero, A. Immunohistochemical quantitation of thymidylate synthase expression in colorectal cancer metastases predicts for clinical outcome to fluorouracil-based chemotherapy. J. Clin. Oncol. 1999, 17, 1760–1770. [Google Scholar]
- Kumamoto, K.; Kuwabara, K.; Tajima, Y.; Amano, K.; Hatano, S.; Ohsawa, T.; Okada, N.; Ishibashi, K.; Haga, N.; Ishida, H. Thymidylate synthase and thymidine phosphorylase mRNA expression in primary lesions using laser capture microdissection is useful for prediction of the efficacy of FOLFOX treatment in colorectal cancer patients with liver metastasis. Oncol. Lett. 2012, 3, 983–989. [Google Scholar]
- Wang, T.L.; Diaz, L.A., Jr.; Romans, K.; Bardelli, A.; Saha, S.; Galizia, G.; Choti, M.; Donehower, R.; Parmigiani, G.; Shih Ie, M.; et al. Digital karyotyping identifies thymidylate synthase amplification as a mechanism of resistance to 5-fluorouracil in metastatic colorectal cancer patients. Proc. Natl. Acad. Sci. USA 2004, 101, 3089–3094. [Google Scholar] [CrossRef]
- Watson, R.G.; Muhale, F.; Thorne, L.B.; Yu, J.; O’Neil, B.H.; Hoskins, J.M.; Meyers, M.O.; Deal, A.M.; Ibrahim, J.G.; Hudson, M.L.; et al. Amplification of thymidylate synthetase in metastatic colorectal cancer patients pretreated with 5-fluorouracil-based chemotherapy. Eur. J. Cancer 2010, 46, 3358–3364. [Google Scholar] [CrossRef]
- Hillcoat, B.L.; McCulloch, P.B.; Figueredo, A.T.; Ehsan, M.H.; Rosenfeld, J.M. Clinical response and plasma levels of 5-fluorouracil in patients with colonic cancer treated by drug infusion. Br. J. Cancer 1978, 38, 719–724. [Google Scholar] [CrossRef]
- Van Groeningen, C.J.; Pinedo, H.M.; Heddes, J.; Kok, R.M.; de Jong, A.P.; Wattel, E.; Peters, G.J.; Lankelma, J. Pharmacokinetics of 5-fluorouracil assessed with a sensitive mass spectrometric method in patients on a dose escalation schedule. Cancer Res. 1988, 48, 6956–6961. [Google Scholar]
- Gamelin, E.C.; Danquechin-Dorval, E.M.; Dumesnil, Y.F.; Maillart, P.J.; Goudier, M.J.; Burtin, P.C.; Delva, R.G.; Lortholary, A.H.; Gesta, P.H.; Larra, F.G. Relationship between 5-fluorouracil (5-FU) dose intensity and therapeutic response in patients with advanced colorectal cancer receiving infusional therapy containing 5-FU. Cancer 1996, 77, 441–451. [Google Scholar] [CrossRef]
- Heggie, G.D.; Sommadossi, J.P.; Cross, D.S.; Huster, W.J.; Diasio, R.B. Clinical pharmacokineticsof 5-fluorouracil and its metabolites in plasma, urine, and bile. Cancer Res. 1987, 47, 2203–2206. [Google Scholar]
- Zhang, X.; Diasio, R.B. Regulation of human dihydropyrimidine dehydrogenase: Implications in the pharmacogenetics of 5-FU-based chemotherapy. Pharmacogenomics 2007, 8, 257–265. [Google Scholar] [CrossRef]
- Salonga, D.; Danenberg, K.D.; Johnson, M.; Metzger, R.; Groshen, S.; Tsao-Wei, D.D.; Lenz, H.J.; Leichman, C.G.; Leichman, L.; Diasio, R.B.; et al. Colorectal tumors responding to 5-fluorouracil have low gene expression levels of dihydropyrimidine dehydrogenase, thymidylate synthase, and thymidine phosphorylase. Clin. Cancer Res. 2000, 6, 1322–1327. [Google Scholar]
- Ichikawa, W.; Uetake, H.; Shirota, Y.; Yamada, H.; Nishi, N.; Nihei, Z.; Sugihara, K.; Hirayama, R. Combination of dihydropyrimidine dehydrogenase and thymidylate synthase gene expressions in primary tumors as predictive parameters for the efficacy of fluoropyrimidine-based chemotherapy for metastatic colorectal cancer. Clin. Cancer Res. 2003, 9, 786–791. [Google Scholar]
- Collie-Duguid, E.S.; Etienne, M.C.; Milano, G.; McLeod, H.L. Known variant DPYD alleles do not explain DPD deficiency in cancer patients. Pharmacogenetics 2000, 10, 217–223. [Google Scholar] [CrossRef]
- Morel, A.; Boisdron-Celle, M.; Fey, L.; Soulie, P.; Craipeau, M.C.; Traore, S.; Gamelin, E. Clinical relevance of different dihydropyrimidine dehydrogenase gene single nucleotide polymorphisms on 5-fluorouracil tolerance. Mol. Cancer Ther. 2006, 5, 2895–2904. [Google Scholar] [CrossRef]
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087.e3. [Google Scholar] [CrossRef]
- Ribic, C.M.; Sargent, D.J.; Moore, M.J.; Thibodeau, S.N.; French, A.J.; Goldberg, R.M.; Hamilton, S.R.; Laurent-Puig, P.; Gryfe, R.; Shepherd, L.E.; et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N. Engl. J. Med. 2003, 349, 247–257. [Google Scholar] [CrossRef]
- Sargent, D.J.; Marsoni, S.; Monges, G.; Thibodeau, S.N.; Labianca, R.; Hamilton, S.R.; French, A.J.; Kabat, B.; Foster, N.R.; Torri, V.; et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J. Clin. Oncol. 2010, 28, 3219–3226. [Google Scholar] [CrossRef]
- Ng, K.; Schrag, D. Microsatellite instability and adjuvant fluorouracil chemotherapy: A mismatch? J. Clin. Oncol. 2010, 28, 3207–3210. [Google Scholar] [CrossRef]
- Febbo, P.G.; Ladanyi, M.; Aldape, K.D.; de Marzo, A.M.; Hammond, M.E.; Hayes, D.F.; Iafrate, A.J.; Kelley, R.K.; Marcucci, G.; Ogino, S.; et al. NCCN Task Force report: Evaluating the clinical utility of tumor markers in oncology. J. Natl. Compr. Canc. Netw. 2011, 9, S1–S32. [Google Scholar]
- Zaanan, A.; Cuilliere-Dartigues, P.; Guilloux, A.; Parc, Y.; Louvet, C.; de Gramont, A.; Tiret, E.; Dumont, S.; Gayet, B.; Validire, P.; et al. Impact of p53 expression and microsatellite instability on stage III colon cancer disease-free survival in patients treated by 5-fluorouracil and leucovorin with or without oxaliplatin. Ann. Oncol. 2010, 21, 772–780. [Google Scholar] [CrossRef]
- Hoff, P.M.; Ansari, R.; Batist, G.; Cox, J.; Kocha, W.; Kuperminc, M.; Maroun, J.; Walde, D.; Weaver, C.; Harrison, E.; et al. Comparison of oral capecitabine versus intravenous fluorouracil plus leucovorin as first-line treatment in 605 patients with metastatic colorectal cancer: Results of a randomized phase III study. J. Clin. Oncol. 2001, 19, 2282–2292. [Google Scholar]
- Trump, D.L.; Egorin, M.J.; Forrest, A.; Willson, J.K.; Remick, S.; Tutsch, K.D. Pharmacokinetic and pharmacodynamic analysis of fluorouracil during 72-hour continuous infusion with and without dipyridamole. J. Clin. Oncol. 1991, 9, 2027–2035. [Google Scholar]
- Naguib, F.N.; el Kouni, M.H.; Cha, S. Enzymes of uracil catabolism in normal and neoplastic human tissues. Cancer Res. 1985, 45, 5405–5412. [Google Scholar]
- Diasio, R.B.; Beavers, T.L.; Carpenter, J.T. Familial deficiency of dihydropyrimidine dehydrogenase. Biochemical basis for familial pyrimidinemia and severe 5-fluorouracil-induced toxicity. J. Clin. Invest. 1988, 81, 47–51. [Google Scholar] [CrossRef]
- Di Paolo, A.; Danesi, R.; Falcone, A.; Cionini, L.; Vannozzi, F.; Masi, G.; Allegrini, G.; Mini, E.; Bocci, G.; Conte, P.F.; Del Tacca, M. Relationship between 5-fluorouracil disposition, toxicity and dihydropyrimidine dehydrogenase activity in cancer patients. Ann. Oncol. 2001, 12, 1301–1306. [Google Scholar] [CrossRef]
- Takimoto, C.H.; Lu, Z.H.; Zhang, R.; Liang, M.D.; Larson, L.V.; Cantilena, L.R., Jr.; Grem, J.L.; Allegra, C.J.; Diasio, R.B.; Chu, E. Severe neurotoxicity following 5-fluorouracil-based chemotherapy in a patient with dihydropyrimidine dehydrogenase deficiency. Clin. Cancer Res. 1996, 2, 477–481. [Google Scholar]
- Mattison, L.K.; Fourie, J.; Desmond, R.A.; Modak, A.; Saif, M.W.; Diasio, R.B. Increased prevalence of dihydropyrimidine dehydrogenase deficiency in African-Americans compared with Caucasians. Clin. Cancer Res. 2006, 12, 5491–5495. [Google Scholar] [CrossRef]
- Van Kuilenburg, A.B.; Haasjes, J.; Richel, D.J.; Zoetekouw, L.; Van Lenthe, H.; De Abreu, R.A.; Maring, J.G.; Vreken, P.; van Gennip, A.H. Clinical implications of dihydropyrimidine dehydrogenase (DPD) deficiency in patients with severe 5-fluorouracil-associated toxicity: Identification of new mutations in the DPD gene. Clin. Cancer Res. 2000, 6, 4705–4712. [Google Scholar]
- Van Kuilenburg, A.B.; Vreken, P.; Abeling, N.G.; Bakker, H.D.; Meinsma, R.; van Lenthe, H.; de Abreu, R.A.; Smeitink, J.A.; Kayserili, H.; Apak, M.Y.; et al. Genotype and phenotype in patients with dihydropyrimidine dehydrogenase deficiency. Hum. Genet. 1999, 104, 1–9. [Google Scholar] [CrossRef]
- Loganayagam, A.; Arenas-Hernandez, M.; Fairbanks, L.; Ross, P.; Sanderson, J.D.; Marinaki, A.M. The contribution of deleterious DPYD gene sequence variants to fluoropyrimidine toxicity in British cancer patients. Cancer Chemother. Pharmacol. 2010, 65, 403–406. [Google Scholar] [CrossRef]
- Capitain, O.; Boisdron-Celle, M.; Poirier, A.L.; Abadie-Lacourtoisie, S.; Morel, A.; Gamelin, E. The influence of fluorouracil outcome parameters on tolerance and efficacy in patients with advanced colorectal cancer. Pharmacogenomics J. 2008, 8, 256–267. [Google Scholar] [CrossRef]
- Mauritz, R.; van Groeningen, C.J.; Smid, K.; Jansen, G.; Pinedo, H.M.; Peters, G.J. Thymidylate synthase and dihydropyrimidine dehydrogenase mRNA expression after administration of 5-fluorouracil to patients with colorectal cancer. Int. J. Cancer 2007, 120, 2609–2612. [Google Scholar] [CrossRef]
- McLeod, H.L.; Sludden, J.; Hardy, S.C.; Lock, R.E.; Hawksworth, G.M.; Cassidy, J. Autoregulation of 5-fluorouracil metabolism. Eur. J. Cancer 1998, 34, 1623–1627. [Google Scholar] [CrossRef]
- Chu, E.; Koeller, D.M.; Johnston, P.G.; Zinn, S.; Allegra, C.J. Regulation of thymidylate synthase in human colon cancer cells treated with 5-fluorouracil and interferon-gamma. Mol. Pharmacol. 1993, 43, 527–533. [Google Scholar]
- Swain, S.M.; Lippman, M.E.; Egan, E.F.; Drake, J.C.; Steinberg, S.M.; Allegra, C.J. Fluorouracil and high-dose leucovorin in previously treated patients with metastatic breast cancer. J. Clin. Oncol. 1989, 7, 890–899. [Google Scholar]
- Kornmann, M.; Schwabe, W.; Sander, S.; Kron, M.; Strater, J.; Polat, S.; Kettner, E.; Weiser, H.F.; Baumann, W.; Schramm, H.; et al. Thymidylate synthase and dihydropyrimidine dehydrogenase mRNA expression levels: Predictors for survival in colorectal cancer patients receiving adjuvant 5-fluorouracil. Clin. Cancer Res. 2003, 9, 4116–4124. [Google Scholar]
- Takenoue, T.; Nagawa, H.; Matsuda, K.; Fujii, S.; Nita, M.E.; Hatano, K.; Kitayama, J.; Tsuruo, T.; Muto, T. Relation between thymidylate synthase expression and survival in colon carcinoma, and determination of appropriate application of 5-fluorouracil by immunohistochemical method. Ann. Surg. Oncol. 2000, 7, 193–198. [Google Scholar] [CrossRef]
- Edler, D.; Glimelius, B.; Hallstrom, M.; Jakobsen, A.; Johnston, P.G.; Magnusson, I.; Ragnhammar, P.; Blomgren, H. Thymidylate synthase expression in colorectal cancer: A prognostic and predictive marker of benefit from adjuvant fluorouracil-based chemotherapy. J. Clin. Oncol. 2002, 20, 1721–1728. [Google Scholar] [CrossRef]
- Van Kuilenburg, A.B.; Hausler, P.; Schalhorn, A.; Tanck, M.W.; Proost, J.H.; Terborg, C.; Behnke, D.; Schwabe, W.; Jabschinsky, K.; Maring, J.G. Evaluation of 5-fluorouracil pharmacokinetics in cancer patients with a c.1905+1G>A mutation in DPYD by means of a Bayesian limited sampling strategy. Clin. Pharmacokinet. 2012, 51, 163–174. [Google Scholar] [CrossRef]
- Gamelin, E.; Delva, R.; Jacob, J.; Merrouche, Y.; Raoul, J.L.; Pezet, D.; Dorval, E.; Piot, G.; Morel, A.; Boisdron-Celle, M. Individual fluorouracil dose adjustment based on pharmacokinetic follow-up compared with conventional dosage: Results of a multicenter randomized trial of patients with metastatic colorectal cancer. J. Clin. Oncol. 2008, 26, 2099–2105. [Google Scholar] [CrossRef]
- Capitain, O.; Asevoaia, A.; Boisdron-Celle, M.; Poirier, A.L.; Morel, A.; Gamelin, E. Individual Fluorouracil Dose Adjustment in FOLFOX Based on Pharmacokinetic Follow-Up Compared With Conventional Body-Area-Surface Dosing: A Phase II, Proof-of-Concept Study. Clin. Colorectal. Cancer 2012, 11, 263–267. [Google Scholar] [CrossRef] [Green Version]
- Volk, J.; Reinke, F.; van Kuilenburg, A.B.; van Gennip, A.H.; Schlichting, C.; Ganser, A.; Schoffski, P. Safe administration of irinotecan, oxaliplatin and raltitrexed in a DPD-deficient patient with metastatic colon cancer. Ann. Oncol. 2001, 12, 569–571. [Google Scholar] [CrossRef]
- Kohne, C.H.; Thuss-Patience, P.; Friedrich, M.; Daniel, P.T.; Kretzschmar, A.; Benter, T.; Bauer, B.; Dietz, R.; Dorken, B. Raltitrexed (Tomudex): An alternative drug for patients with colorectal cancer and 5-fluorouracil associated cardiotoxicity. Br. J. Cancer 1998, 77, 973–977. [Google Scholar] [CrossRef]
- Wilson, K.S.; Fitzgerald, C.A.; Barnett, J.B.; Gill, S.; Khoo, K.E. Adjuvant therapy with raltitrexed in patients with colorectal cancer intolerant of 5-fluorouracil: British Columbia Cancer Agency experience. Cancer Invest. 2007, 25, 711–714. [Google Scholar] [CrossRef]
- Gravalos, C.; Salut, A.; Garcia-Giron, C.; Garcia-Carbonero, R.; Leon, A.I.; Sevilla, I.; Maurel, J.; Esteban, B.; Garcia-Rico, E.; Murias, A.; et al. A randomized phase II study to compare oxaliplatin plus 5-fluorouracil and leucovorin (FOLFOX4) versus oxaliplatin plus raltitrexed (TOMOX) as first-line chemotherapy for advanced colorectal cancer. Clin. Transl. Oncol. 2012, 14, 606–612. [Google Scholar] [CrossRef]
- Popov, I.; Carrato, A.; Sobrero, A.; Vincent, M.; Kerr, D.; Labianca, R.; Raffaele Bianco, A.; El-Serafi, M.; Bedenne, L.; Paillot, B.; et al. Raltitrexed (Tomudex) versus standard leucovorin-modulated bolus 5-fluorouracil: Results from the randomised phase III Pan-European Trial in Adjuvant Colon Cancer 01 (PETACC-1). Eur. J. Cancer 2008, 44, 2204–2211. [Google Scholar] [CrossRef]
- Wenzel, C.; Urbauer, E.; Schwarz, C.; Funk, G.; Oehler, L.; Kornek, G.V.; Scheithauer, W. Severe enteropathy associated with raltitrexed and oxaliplatin chemotherapy: Report of two patients experiencing this rare, potentially lethal gastrointestinal adverse event. Anticancer Drugs 2006, 17, 865–868. [Google Scholar]
- Ducreux, M.; Bouche, O.; Pignon, J.P.; Mousseau, M.; Raoul, J.L.; Cassan, P.; Leduc, B.; Berger, C.; Dunant, A.; Fournet, J.; et al. Randomised trial comparing three different schedules of infusional 5FU and raltitrexed alone as first-line therapy in metastatic colorectal cancer. Final results of the Federation Francophone de Cancerologie Digestive (FFCD) 9601 trial. Oncology 2006, 70, 222–230. [Google Scholar] [CrossRef]
- Rinaldi, D.A.; Burris, H.A.; Dorr, F.A.; Woodworth, J.R.; Kuhn, J.G.; Eckardt, J.R.; Rodriguez, G.; Corso, S.W.; Fields, S.M.; Langley, C.; et al. Initial phase I evaluation of the novel thymidylate synthase inhibitor, LY231514, using the modified continual reassessment method for dose escalation. J. Clin. Oncol. 1995, 13, 2842–2850. [Google Scholar]
- Cripps, C.; Burnell, M.; Jolivet, J.; Batist, G.; Lofters, W.; Dancey, J.; Iglesias, J.; Fisher, B.; Eisenhauer, E.A. Phase II study of first-line LY231514 (multi-targeted antifolate) in patients with locally advanced or metastatic colorectal cancer: An NCIC Clinical Trials Group study. Ann. Oncol. 1999, 10, 1175–1179. [Google Scholar] [CrossRef]
- John, W.; Picus, J.; Blanke, C.D.; Clark, J.W.; Schulman, L.N.; Rowinsky, E.K.; Thornton, D.E.; Loehrer, P.J. Activity of multitargeted antifolate (pemetrexed disodium, LY231514) in patients with advanced colorectal carcinoma: Results from a phase II study. Cancer 2000, 88, 1807–1813. [Google Scholar]
- Hochster, H.; Kettner, E.; Kroning, H.; Becker, K.; Lordick, F.; Ramanathan, R.K.; Macdonald, J.; Hong, S.; John, W.; Schmoll, H.J. Phase I/II dose-escalation study of pemetrexed plus irinotecan in patients with advanced colorectal cancer. Clin. Colorectal. Cancer 2005, 5, 257–262. [Google Scholar] [CrossRef]
- Louvet, C.; Andre, T.; Gamelin, E.; Hebbar, M.; Mabro, M.; Bennamoun, M.; Rassam, H.; de Gramont, A. Phase II Study of Biweekly Pemetrexed Plus Irinotecan as Second-Line Therapy for Metastatic Colorectal Cancer. J. Oncol. 2010, 2010, 785934. [Google Scholar]
- Rosenberg, B.; VanCamp, L.; Trosko, J.E.; Mansour, V.H. Platinum compounds: A new class of potent antitumour agents. Nature 1969, 222, 385–386. [Google Scholar] [CrossRef]
- Rosenberg, B.; VanCamp, L. The successful regression of large solid sarcoma 180 tumors by platinum compounds. Cancer Res. 1970, 30, 1799–1802. [Google Scholar]
- Dentino, M.; Luft, F.C.; Yum, M.N.; Williams, S.D.; Einhorn, L.H. Long term effect of cis-diamminedichloride platinum (CDDP) on renal function and structure in man. Cancer 1978, 41, 1274–1281. [Google Scholar] [CrossRef]
- Mathe, G.; Kidani, Y.; Noji, M.; Maral, R.; Bourut, C.; Chenu, E. Antitumor activity of l-OHP in mice. Cancer Lett. 1985, 27, 135–143. [Google Scholar] [CrossRef]
- Saris, C.P.; van de Vaart, P.J.; Rietbroek, R.C.; Blommaert, F.A. In vitro formation of DNA adducts by cisplatin, lobaplatin and oxaliplatin in calf thymus DNA in solution and in cultured human cells. Carcinogenesis 1996, 17, 2763–2769. [Google Scholar] [CrossRef]
- Woynarowski, J.M.; Chapman, W.G.; Napier, C.; Herzig, M.C.; Juniewicz, P. Sequence- and region-specificity of oxaliplatin adducts in naked and cellular DNA. Mol. Pharmacol. 1998, 54, 770–777. [Google Scholar]
- Yamada, M.; O'Regan, E.; Brown, R.; Karran, P. Selective recognition of a cisplatin-DNA adduct by human mismatch repair proteins. Nucleic Acids Res. 1997, 25, 491–496. [Google Scholar] [CrossRef]
- Mello, J.A.; Acharya, S.; Fishel, R.; Essigmann, J.M. The mismatch-repair protein hMSH2 binds selectively to DNA adducts of the anticancer drug cisplatin. Chem. Biol. 1996, 3, 579–589. [Google Scholar] [CrossRef]
- Fink, D.; Nebel, S.; Aebi, S.; Zheng, H.; Cenni, B.; Nehme, A.; Christen, R.D.; Howell, S.B. The role of DNA mismatch repair in platinum drug resistance. Cancer Res. 1996, 56, 4881–4886. [Google Scholar]
- Scheeff, E.D.; Briggs, J.M.; Howell, S.B. Molecular modeling of the intrastrand guanine-guanine DNA adducts produced by cisplatin and oxaliplatin. Mol. Pharmacol. 1999, 56, 633–643. [Google Scholar]
- De Gramont, A.; Vignoud, J.; Tournigand, C.; Louvet, C.; Andre, T.; Varette, C.; Raymond, E.; Moreau, S.; Le Bail, N.; Krulik, M. Oxaliplatin with high-dose leucovorin and 5-fluorouracil 48-hour continuous infusion in pretreated metastatic colorectal cancer. Eur. J. Cancer 1997, 33, 214–219. [Google Scholar] [CrossRef]
- Levi, F.; Misset, J.L.; Brienza, S.; Adam, R.; Metzger, G.; Itzakhi, M.; Caussanel, J.P.; Kunstlinger, F.; Lecouturier, S.; Descorps-Declere, A.; et al. A chronopharmacologic phase II clinical trial with 5-fluorouracil, folinic acid, and oxaliplatin using an ambulatory multichannel programmable pump. High antitumor effectiveness against metastatic colorectal cancer. Cancer 1992, 69, 893–900. [Google Scholar]
- de Gramont, A.; Figer, A.; Seymour, M.; Homerin, M.; Hmissi, A.; Cassidy, J.; Boni, C.; Cortes-Funes, H.; Cervantes, A.; Freyer, G.; et al. Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment in advanced colorectal cancer. J. Clin. Oncol. 2000, 18, 2938–2947. [Google Scholar]
- Creemers, G.J.; Lund, B.; Verweij, J. Topoisomerase I inhibitors: Topotecan and irenotecan. Cancer Treat. Rev. 1994, 20, 73–96. [Google Scholar] [CrossRef]
- Kaneda, N.; Nagata, H.; Furuta, T.; Yokokura, T. Metabolism and pharmacokinetics of the camptothecin analogue CPT-11 in the mouse. Cancer Res. 1990, 50, 1715–1720. [Google Scholar]
- Kunimoto, T.; Nitta, K.; Tanaka, T.; Uehara, N.; Baba, H.; Takeuchi, M.; Yokokura, T.; Sawada, S.; Miyasaka, T.; Mutai, M. Antitumor activity of 7-ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxy-camptothec in, a novel water-soluble derivative of camptothecin, against murine tumors. Cancer Res. 1987, 47, 5944–5947. [Google Scholar]
- Douillard, J.Y.; Cunningham, D.; Roth, A.D.; Navarro, M.; James, R.D.; Karasek, P.; Jandik, P.; Iveson, T.; Carmichael, J.; Alakl, M.; et al. Irinotecan combined with fluorouracil compared with fluorouracil alone as first-line treatment for metastatic colorectal cancer: A multicentre randomised trial. Lancet 2000, 355, 1041–1047. [Google Scholar] [CrossRef]
- Dean, F.B.; Bullock, P.; Murakami, Y.; Wobbe, C.R.; Weissbach, L.; Hurwitz, J. Simian virus 40 (SV40) DNA replication: SV40 large T antigen unwinds DNA containing the SV40 origin of replication. Proc. Natl. Acad. Sci. USA 1987, 84, 16–20. [Google Scholar] [CrossRef]
- Garg, L.C.; DiAngelo, S.; Jacob, S.T. Role of DNA topoisomerase I in the transcription of supercoiled rRNA gene. Proc. Natl. Acad. Sci. USA 1987, 84, 3185–3188. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, J.C.; Liu, L.F. Involvement of DNA topoisomerase I in transcription of human ribosomal RNA genes. Proc. Natl. Acad. Sci. USA 1988, 85, 1060–1064. [Google Scholar] [CrossRef]
- Halligan, B.D.; Davis, J.L.; Edwards, K.A.; Liu, L.F. Intra- and intermolecular strand transfer by HeLa DNA topoisomerase I. J. Biol. Chem. 1982, 257, 3995–4000. [Google Scholar]
- Stewart, L.; Redinbo, M.R.; Qiu, X.; Hol, W.G.; Champoux, J.J. A model for the mechanism of human topoisomerase I. Science 1998, 279, 1534–1541. [Google Scholar] [CrossRef]
- Hsiang, Y.H.; Hertzberg, R.; Hecht, S.; Liu, L.F. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J. Biol. Chem. 1985, 260, 14873–14878. [Google Scholar]
- Hsiang, Y.H.; Lihou, M.G.; Liu, L.F. Arrest of replication forks by drug-stabilized topoisomerase I-DNA cleavable complexes as a mechanism of cell killing by camptothecin. Cancer Res. 1989, 49, 5077–5082. [Google Scholar]
- Tsao, Y.P.; Russo, A.; Nyamuswa, G.; Silber, R.; Liu, L.F. Interaction between replication forks and topoisomerase I-DNA cleavable complexes: Studies in a cell-free SV40 DNA replication system. Cancer Res. 1993, 53, 5908–5914. [Google Scholar]
- Tournigand, C.; Andre, T.; Achille, E.; Lledo, G.; Flesh, M.; Mery-Mignard, D.; Quinaux, E.; Couteau, C.; Buyse, M.; Ganem, G.; et al. FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: A randomized GERCOR study. J. Clin. Oncol. 2004, 22, 229–237. [Google Scholar]
- Colucci, G.; Gebbia, V.; Paoletti, G.; Giuliani, F.; Caruso, M.; Gebbia, N.; Carteni, G.; Agostara, B.; Pezzella, G.; Manzione, L.; et al. Phase III randomized trial of FOLFIRI versus FOLFOX4 in the treatment of advanced colorectal cancer: A multicenter study of the Gruppo Oncologico Dell'Italia Meridionale. J. Clin. Oncol. 2005, 23, 4866–4875. [Google Scholar] [CrossRef]
- Saif, M.W.; Reardon, J. Management of oxaliplatin-induced peripheral neuropathy. Ther. Clin. Risk Manag. 2005, 1, 249–258. [Google Scholar]
- Stein, A.; Voigt, W.; Jordan, K. Chemotherapy-induced diarrhea: Pathophysiology, frequency and guideline-based management. Ther Adv. Med. Oncol 2010, 2, 51–63. [Google Scholar] [CrossRef]
- Kamileri, I.; Karakasilioti, I.; Garinis, G.A. Nucleotide excision repair: New tricks with old bricks. Trends Genet. 2012, 28, 566–573. [Google Scholar] [CrossRef]
- You, J.S.; Wang, M.; Lee, S.H. Biochemical analysis of the damage recognition process in nucleotide excision repair. J. Biol. Chem. 2003, 278, 7476–7485. [Google Scholar] [CrossRef]
- Shen, M.R.; Jones, I.M.; Mohrenweiser, H. Nonconservative amino acid substitution variants exist at polymorphic frequency in DNA repair genes in healthy humans. Cancer Res. 1998, 58, 604–608. [Google Scholar]
- Shuck, S.C.; Short, E.A.; Turchi, J.J. Eukaryotic nucleotide excision repair: From understanding mechanisms to influencing biology. Cell. Res. 2008, 18, 64–72. [Google Scholar] [CrossRef]
- Evans, E.; Moggs, J.G.; Hwang, J.R.; Egly, J.M.; Wood, R.D. Mechanism of open complex and dual incision formation by human nucleotide excision repair factors. EMBO J. 1997, 16, 6559–6573. [Google Scholar] [CrossRef]
- Park, D.J.; Stoehlmacher, J.; Zhang, W.; Tsao-Wei, D.D.; Groshen, S.; Lenz, H.J. A Xeroderma pigmentosum group D gene polymorphism predicts clinical outcome to platinum-based chemotherapy in patients with advanced colorectal cancer. Cancer Res. 2001, 61, 8654–8658. [Google Scholar]
- Stoehlmacher, J.; Park, D.J.; Zhang, W.; Yang, D.; Groshen, S.; Zahedy, S.; Lenz, H.J. A multivariate analysis of genomic polymorphisms: Prediction of clinical outcome to 5-FU/oxaliplatin combination chemotherapy in refractory colorectal cancer. Br. J. Cancer 2004, 91, 344–354. [Google Scholar]
- Yin, M.; Yan, J.; Martinez-Balibrea, E.; Graziano, F.; Lenz, H.J.; Kim, H.J.; Robert, J.; Im, S.A.; Wang, W.S.; Etienne-Grimaldi, M.C.; et al. ERCC1 and ERCC2 polymorphisms predict clinical outcomes of oxaliplatin-based chemotherapies in gastric and colorectal cancer: A systemic review and meta-analysis. Clin. Cancer Res. 2011, 17, 1632–1640. [Google Scholar] [CrossRef]
- Lai, J.I.; Tzeng, C.H.; Chen, P.M.; Lin, J.K.; Lin, T.C.; Chen, W.S.; Jiang, J.K.; Wang, H.S.; Wang, W.S. Very low prevalence of XPD K751Q polymorphism and its association with XPD expression and outcomes of FOLFOX-4 treatment in Asian patients with colorectal carcinoma. Cancer Sci. 2009, 100, 1261–1266. [Google Scholar] [CrossRef]
- Sijbers, A.M.; de Laat, W.L.; Ariza, R.R.; Biggerstaff, M.; Wei, Y.F.; Moggs, J.G.; Carter, K.C.; Shell, B.K.; Evans, E.; de Jong, M.C.; et al. Xeroderma pigmentosum group F caused by a defect in a structure-specific DNA repair endonuclease. Cell 1996, 86, 811–822. [Google Scholar] [CrossRef]
- O'Donovan, A.; Davies, A.A.; Moggs, J.G.; West, S.C.; Wood, R.D. XPG endonuclease makes the 3' incision in human DNA nucleotide excision repair. Nature 1994, 371, 432–435. [Google Scholar] [CrossRef]
- Shirota, Y.; Stoehlmacher, J.; Brabender, J.; Xiong, Y.P.; Uetake, H.; Danenberg, K.D.; Groshen, S.; Tsao-Wei, D.D.; Danenberg, P.V.; Lenz, H.J. ERCC1 and thymidylate synthase mRNA levels predict survival for colorectal cancer patients receiving combination oxaliplatin and fluorouracil chemotherapy. J. Clin. Oncol. 2001, 19, 4298–4304. [Google Scholar]
- Yu, J.J.; Mu, C.; Lee, K.B.; Okamoto, A.; Reed, E.L.; Bostick-Bruton, F.; Mitchell, K.C.; Reed, E. A nucleotide polymorphism in ERCC1 in human ovarian cancer cell lines and tumor tissues. Mutat Res. 1997, 382, 13–20. [Google Scholar] [CrossRef]
- Viguier, J.; Boige, V.; Miquel, C.; Pocard, M.; Giraudeau, B.; Sabourin, J.C.; Ducreux, M.; Sarasin, A.; Praz, F. ERCC1 codon 118 polymorphism is a predictive factor for the tumor response to oxaliplatin/5-fluorouracil combination chemotherapy in patients with advanced colorectal cancer. Clin. Cancer Res. 2005, 11, 6212–6217. [Google Scholar] [CrossRef]
- Pare, L.; Marcuello, E.; Altes, A.; del Rio, E.; Sedano, L.; Salazar, J.; Cortes, A.; Barnadas, A.; Baiget, M. Pharmacogenetic prediction of clinical outcome in advanced colorectal cancer patients receiving oxaliplatin/5-fluorouracil as first-line chemotherapy. Br. J. Cancer 2008, 99, 1050–1055. [Google Scholar] [CrossRef]
- Chen, J.; Xie, F.; Chen, K.; Wang, D.; Jiang, H.; Li, J.; Pan, F.; Chen, S.; Zhang, Y.; Ruan, Z.; Huang, H.; Zou, L.; Liang, H. ERCC5 promoter polymorphisms at −763 and +25 predict the response to oxaliplatin-based chemotherapy in patients with advanced colorectal cancer. Cancer Biol. Ther. 2009, 8, 1424–1430. [Google Scholar] [CrossRef]
- McWhinney, S.R.; Goldberg, R.M.; McLeod, H.L. Platinum neurotoxicity pharmacogenetics. Mol. Cancer Ther. 2009, 8, 10–16. [Google Scholar] [CrossRef]
- Goto, S.; Iida, T.; Cho, S.; Oka, M.; Kohno, S.; Kondo, T. Overexpression of glutathione S-transferase pi enhances the adduct formation of cisplatin with glutathione in human cancer cells. Free Radic Res. 1999, 31, 549–558. [Google Scholar] [CrossRef]
- Mannervik, B.; Awasthi, Y.C.; Board, P.G.; Hayes, J.D.; Di Ilio, C.; Ketterer, B.; Listowsky, I.; Morgenstern, R.; Muramatsu, M.; Pearson, W.R.; et al. Nomenclature for human glutathione transferases. Biochem. J. 1992, 282((Pt. 1)), 305–306. [Google Scholar]
- Watson, M.A.; Stewart, R.K.; Smith, G.B.; Massey, T.E.; Bell, D.A. Human glutathione S-transferase P1 polymorphisms: Relationship to lung tissue enzyme activity and population frequency distribution. Carcinogenesis 1998, 19, 275–280. [Google Scholar] [CrossRef]
- Ralat, L.A.; Colman, R.F. Glutathione S-transferase Pi has at least three distinguishable xenobiotic substrate sites close to its glutathione-binding site. J. Biol. Chem. 2004, 279, 50204–50213. [Google Scholar] [CrossRef]
- Ranganathan, S.; Tew, K.D. Immunohistochemical localization of glutathione S-transferases alpha, mu, and pi in normal tissue and carcinomas from human colon. Carcinogenesis 1991, 12, 2383–2387. [Google Scholar] [CrossRef]
- Moorghen, M.; Cairns, J.; Forrester, L.M.; Hayes, J.D.; Hall, A.; Cattan, A.R.; Wolf, C.R.; Harris, A.L. Enhanced expression of glutathione S-transferases in colorectal carcinoma compared to non-neoplastic mucosa. Carcinogenesis 1991, 12, 13–17. [Google Scholar] [CrossRef]
- Peters, W.H.; Boon, C.E.; Roelofs, H.M.; Wobbes, T.; Nagengast, F.M.; Kremers, P.G. Expression of drug-metabolizing enzymes and P-170 glycoprotein in colorectal carcinoma and normal mucosa. Gastroenterology 1992, 103, 448–455. [Google Scholar]
- Miyazaki, M.; Kohno, K.; Saburi, Y.; Matsuo, K.; Ono, M.; Kuwano, M.; Tsuchida, S.; Sato, K.; Sakai, M.; Muramatsu, M. Drug resistance to cis-diamminedichloroplatinum (II) in Chinese hamster ovary cell lines transfected with glutathione S-transferase pi gene. Biochem. Biophys. Res. Commun. 1990, 166, 1358–1364. [Google Scholar] [CrossRef]
- Ban, N.; Takahashi, Y.; Takayama, T.; Kura, T.; Katahira, T.; Sakamaki, S.; Niitsu, Y. Transfection of glutathione S-transferase (GST)-pi antisense complementary DNA increases the sensitivity of a colon cancer cell line to adriamycin, cisplatin, melphalan, and etoposide. Cancer Res. 1996, 56, 3577–3582. [Google Scholar]
- Board, P.G.; Webb, G.C.; Coggan, M. Isolation of a cDNA clone and localization of the human glutathione S-transferase 3 genes to chromosome bands 11q13 and 12q13–14. Ann. Hum. Genet. 1989, 53, 205–213. [Google Scholar] [CrossRef]
- Inada, M.; Sato, M.; Morita, S.; Kitagawa, K.; Kawada, K.; Mitsuma, A.; Sawaki, M.; Fujita, K.; Ando, Y. Associations between oxaliplatin-induced peripheral neuropathy and polymorphisms of the ERCC1 and GSTP1 genes. Int. J. Clin. Pharmacol. Ther. 2010, 48, 729. [Google Scholar]
- Chen, Y.C.; Tzeng, C.H.; Chen, P.M.; Lin, J.K.; Lin, T.C.; Chen, W.S.; Jiang, J.K.; Wang, H.S.; Wang, W.S. Influence of GSTP1 I105V polymorphism on cumulative neuropathy and outcome of FOLFOX-4 treatment in Asian patients with colorectal carcinoma. Cancer Sci. 2010, 101, 530–535. [Google Scholar] [CrossRef]
- Lecomte, T.; Landi, B.; Beaune, P.; Laurent-Puig, P.; Loriot, M.A. Glutathione S-transferase P1 polymorphism (Ile105Val) predicts cumulative neuropathy in patients receiving oxaliplatin-based chemotherapy. Clin. Cancer Res. 2006, 12, 3050–3056. [Google Scholar]
- Peng, Z.; Wang, Q.; Gao, J.; Ji, Z.; Yuan, J.; Tian, Y.; Shen, L. Association between GSTP1 Ile105Val polymorphism and oxaliplatin-induced neuropathy: A systematic review and meta-analysis. Cancer Chemother. Pharmacol. 2013, 72, 305–314. [Google Scholar] [CrossRef]
- Ruzzo, A.; Graziano, F.; Loupakis, F.; Rulli, E.; Canestrari, E.; Santini, D.; Catalano, V.; Ficarelli, R.; Maltese, P.; Bisonni, R.; et al. Pharmacogenetic profiling in patients with advanced colorectal cancer treated with first-line FOLFOX-4 chemotherapy. J. Clin. Oncol. 2007, 25, 1247–1254. [Google Scholar] [CrossRef]
- Iyer, L.; King, C.D.; Whitington, P.F.; Green, M.D.; Roy, S.K.; Tephly, T.R.; Coffman, B.L.; Ratain, M.J. Genetic predisposition to the metabolism of irinotecan (CPT-11). Role of uridine diphosphate glucuronosyltransferase isoform 1A1 in the glucuronidation of its active metabolite (SN-38) in human liver microsomes. J. Clin Invest. 1998, 101, 847–854. [Google Scholar] [CrossRef]
- Gagne, J.F.; Montminy, V.; Belanger, P.; Journault, K.; Gaucher, G.; Guillemette, C. Common human UGT1A polymorphisms and the altered metabolism of irinotecan active metabolite 7-ethyl-10-hydroxycamptothecin (SN-38). Mol. Pharmacol. 2002, 62, 608–617. [Google Scholar] [CrossRef]
- Gupta, E.; Lestingi, T.M.; Mick, R.; Ramirez, J.; Vokes, E.E.; Ratain, M.J. Metabolic fate of irinotecan in humans: Correlation of glucuronidation with diarrhea. Cancer Res. 1994, 54, 3723–3725. [Google Scholar]
- Guillemette, C. Pharmacogenomics of human UDP-glucuronosyltransferase enzymes. Pharmacogenomics J. 2003, 3, 136–158. [Google Scholar] [CrossRef]
- Guillemette, C.; Ritter, J.K.; Auyeung, D.J.; Kessler, F.K.; Housman, D.E. Structural heterogeneity at the UDP-glucuronosyltransferase 1 locus: Functional consequences of three novel missense mutations in the human UGT1A7 gene. Pharmacogenetics 2000, 10, 629–644. [Google Scholar] [CrossRef]
- Cecchin, E.; Innocenti, F.; D’Andrea, M.; Corona, G.; De Mattia, E.; Biason, P.; Buonadonna, A.; Toffoli, G. Predictive role of the UGT1A1, UGT1A7, and UGT1A9 genetic variants and their haplotypes on the outcome of metastatic colorectal cancer patients treated with fluorouracil, leucovorin, and irinotecan. J. Clin. Oncol. 2009, 27, 2457–2465. [Google Scholar] [CrossRef]
- Beutler, E.; Gelbart, T.; Demina, A. Racial variability in the UDP-glucuronosyltransferase 1 (UGT1A1) promoter: A balanced polymorphism for regulation of bilirubin metabolism? Proc. Natl. Acad. Sci. USA 1998, 95, 8170–8174. [Google Scholar] [CrossRef]
- Bosma, P.J.; Chowdhury, J.R.; Bakker, C.; Gantla, S.; de Boer, A.; Oostra, B.A.; Lindhout, D.; Tytgat, G.N.; Jansen, P.L.; Oude Elferink, R.P.; et al. The genetic basis of the reduced expression of bilirubin UDP-glucuronosyltransferase 1 in Gilbert’s syndrome. N. Engl. J. Med. 1995, 333, 1171–1175. [Google Scholar] [CrossRef]
- Monaghan, G.; Ryan, M.; Seddon, R.; Hume, R.; Burchell, B. Genetic variation in bilirubin UPD-glucuronosyltransferase gene promoter and Gilbert’s syndrome. Lancet 1996, 347, 578–581. [Google Scholar] [CrossRef]
- Iyer, L.; Das, S.; Janisch, L.; Wen, M.; Ramirez, J.; Karrison, T.; Fleming, G.F.; Vokes, E.E.; Schilsky, R.L.; Ratain, M.J. UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharmacogenomics J. 2002, 2, 43–47. [Google Scholar] [CrossRef]
- Innocenti, F.; Undevia, S.D.; Iyer, L.; Chen, P.X.; Das, S.; Kocherginsky, M.; Karrison, T.; Janisch, L.; Ramirez, J.; Rudin, C.M.; et al. Genetic variants in the UDP-glucuronosyltransferase 1A1 gene predict the risk of severe neutropenia of irinotecan. J. Clin. Oncol. 2004, 22, 1382–1388. [Google Scholar] [CrossRef]
- Shulman, K.; Cohen, I.; Barnett-Griness, O.; Kuten, A.; Gruber, S.B.; Lejbkowicz, F.; Rennert, G. Clinical implications of UGT1A1*28 genotype testing in colorectal cancer patients. Cancer 2011, 117, 3156–3162. [Google Scholar] [CrossRef]
- Marcuello, E.; Paez, D.; Pare, L.; Salazar, J.; Sebio, A.; del Rio, E.; Baiget, M. A genotype-directed phase I-IV dose-finding study of irinotecan in combination with fluorouracil/leucovorin as first-line treatment in advanced colorectal cancer. Br. J. Cancer 2011, 105, 53–57. [Google Scholar] [CrossRef]
- Ychou, M.; Raoul, J.L.; Desseigne, F.; Borel, C.; Caroli-Bosc, F.X.; Jacob, J.H.; Seitz, J.F.; Kramar, A.; Hua, A.; Lefebvre, P.; et al. High-dose, single-agent irinotecan as first-line therapy in the treatment of metastatic colorectal cancer. Cancer Chemother. Pharmacol. 2002, 50, 383–391. [Google Scholar] [CrossRef]
- Toffoli, G.; Cecchin, E.; Gasparini, G.; D’Andrea, M.; Azzarello, G.; Basso, U.; Mini, E.; Pessa, S.; de Mattia, E.; Lo Re, G.; et al. Genotype-driven phase I study of irinotecan administered in combination with fluorouracil/leucovorin in patients with metastatic colorectal cancer. J. Clin. Oncol. 2010, 28, 866–871. [Google Scholar] [CrossRef]
- Sawyers, C. Targeted cancer therapy. Nature 2004, 432, 294–297. [Google Scholar] [CrossRef]
- Kluftinger, A.M.; Robinson, B.W.; Quenville, N.F.; Finley, R.J.; Davis, N.L. Correlation of epidermal growth factor receptor and c-erbB2 oncogene product to known prognostic indicators of colorectal cancer. Surg. Oncol. 1992, 1, 97–105. [Google Scholar] [CrossRef]
- Radinsky, R.; Risin, S.; Fan, D.; Dong, Z.; Bielenberg, D.; Bucana, C.D.; Fidler, I.J. Level and function of epidermal growth factor receptor predict the metastatic potential of human colon carcinoma cells. Clin. Cancer Res. 1995, 1, 19–31. [Google Scholar]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell. 2010, 141, 1117–1134. [Google Scholar] [CrossRef]
- Marmor, M.D.; Skaria, K.B.; Yarden, Y. Signal transduction and oncogenesis by ErbB/HER receptors. Int. J. Radiat. Oncol. Biol. Phys. 2004, 58, 903–913. [Google Scholar] [CrossRef]
- Sato, J.D.; Kawamoto, T.; Le, A.D.; Mendelsohn, J.; Polikoff, J.; Sato, G.H. Biological effects in vitro of monoclonal antibodies to human epidermal growth factor receptors. Mol. Biol. Med. 1983, 1, 511–529. [Google Scholar]
- Kawamoto, T.; Sato, J.D.; Le, A.; Polikoff, J.; Sato, G.H.; Mendelsohn, J. Growth stimulation of A431 cells by epidermal growth factor: Identification of high-affinity receptors for epidermal growth factor by an anti-receptor monoclonal antibody. Proc. Natl. Acad. Sci. USA 1983, 80, 1337–1341. [Google Scholar] [CrossRef]
- Masui, H.; Kawamoto, T.; Sato, J.D.; Wolf, B.; Sato, G.; Mendelsohn, J. Growth inhibition of human tumor cells in athymic mice by anti-epidermal growth factor receptor monoclonal antibodies. Cancer Res. 1984, 44, 1002–1007. [Google Scholar]
- Fan, Z.; Masui, H.; Altas, I.; Mendelsohn, J. Blockade of epidermal growth factor receptor function by bivalent and monovalent fragments of 225 anti-epidermal growth factor receptor monoclonal antibodies. Cancer Res. 1993, 53, 4322–4328. [Google Scholar]
- Masui, H.; Moroyama, T.; Mendelsohn, J. Mechanism of antitumor activity in mice for anti-epidermal growth factor receptor monoclonal antibodies with different isotypes. Cancer Res. 1986, 46, 5592–5598. [Google Scholar]
- Gill, G.N.; Kawamoto, T.; Cochet, C.; Le, A.; Sato, J.D.; Masui, H.; McLeod, C.; Mendelsohn, J. Monoclonal anti-epidermal growth factor receptor antibodies which are inhibitors of epidermal growth factor binding and antagonists of epidermal growth factor binding and antagonists of epidermal growth factor-stimulated tyrosine protein kinase activity. J. Biol. Chem. 1984, 259, 7755–7760. [Google Scholar]
- Fan, Z.; Lu, Y.; Wu, X.; Mendelsohn, J. Antibody-induced epidermal growth factor receptor dimerization mediates inhibition of autocrine proliferation of A431 squamous carcinoma cells. J. Biol. Chem. 1994, 269, 27595–27602. [Google Scholar]
- Naramura, M.; Gillies, S.D.; Mendelsohn, J.; Reisfeld, R.A.; Mueller, B.M. Therapeutic potential of chimeric and murine anti-(epidermal growth factor receptor) antibodies in a metastasis model for human melanoma. Cancer Immunol. Immunother. 1993, 37, 343–349. [Google Scholar] [CrossRef]
- Goldstein, N.I.; Prewett, M.; Zuklys, K.; Rockwell, P.; Mendelsohn, J. Biological efficacy of a chimeric antibody to the epidermal growth factor receptor in a human tumor xenograft model. Clin. Cancer Res. 1995, 1, 1311–1318. [Google Scholar]
- Baselga, J.; Pfister, D.; Cooper, M.R.; Cohen, R.; Burtness, B.; Bos, M.; D’Andrea, G.; Seidman, A.; Norton, L.; Gunnett, K.; et al. Phase I studies of anti-epidermal growth factor receptor chimeric antibody C225 alone and in combination with cisplatin. J. Clin. Oncol. 2000, 18, 904–914. [Google Scholar]
- Robert, F.; Ezekiel, M.P.; Spencer, S.A.; Meredith, R.F.; Bonner, J.A.; Khazaeli, M.B.; Saleh, M.N.; Carey, D.; LoBuglio, A.F.; Wheeler, R.H.; et al. Phase I study of anti-epidermal growth factor receptor antibody cetuximab in combination with radiation therapy in patients with advanced head and neck cancer. J. Clin. Oncol. 2001, 19, 3234–3243. [Google Scholar]
- Foon, K.A.; Yang, X.D.; Weiner, L.M.; Belldegrun, A.S.; Figlin, R.A.; Crawford, J.; Rowinsky, E.K.; Dutcher, J.P.; Vogelzang, N.J.; Gollub, J.; et al. Preclinical and clinical evaluations of ABX-EGF, a fully human anti-epidermal growth factor receptor antibody. Int. J. Radiat. Oncol. Biol. Phys. 2004, 58, 984–990. [Google Scholar] [CrossRef]
- Baselga, J.; Rischin, D.; Ranson, M.; Calvert, H.; Raymond, E.; Kieback, D.G.; Kaye, S.B.; Gianni, L.; Harris, A.; Bjork, T.; et al. Phase I safety, pharmacokinetic, and pharmacodynamic trial of ZD1839, a selective oral epidermal growth factor receptor tyrosine kinase inhibitor, in patients with five selected solid tumor types. J. Clin. Oncol. 2002, 20, 4292–4302. [Google Scholar] [CrossRef]
- Rothenberg, M.L.; LaFleur, B.; Levy, D.E.; Washington, M.K.; Morgan-Meadows, S.L.; Ramanathan, R.K.; Berlin, J.D.; Benson, A.B., 3rd; Coffey, R.J. Randomized phase II trial of the clinical and biological effects of two dose levels of gefitinib in patients with recurrent colorectal adenocarcinoma. J. Clin. Oncol. 2005, 23, 9265–9274. [Google Scholar]
- Jimeno, A.; Gravalos, C.; Escudero, P.; Sevilla, I.; Vega-Villegas, M.E.; Alonso, V.; Juez, I.; Garcia-Carbonero, R.; Bovio, H.; Colomer, R.; et al. Phase I/II study of gefitinib and capecitabine in patients with colorectal cancer. Clin.Transl. Oncol. 2008, 10, 52–57. [Google Scholar] [CrossRef]
- Trarbach, T.; Reinacher-Schick, A.; Hegewisch-Becker, S.; Vanhoefer, U.; Frieling, T.; Lehnert, L.; Schmiegel, W.; Graeven, U. Gefitinib in combination with capecitabine as second-line therapy in patients with advanced colorectal cancer (aCRC): A phase I/II study of the Arbeitsgemeinschaft Internistische Onkologie (AIO). Onkologie 2010, 33, 89–93. [Google Scholar] [CrossRef]
- Chau, I.; Cunningham, D.; Hickish, T.; Massey, A.; Higgins, L.; Osborne, R.; Botwood, N.; Swaisland, A. Gefitinib and irinotecan in patients with fluoropyrimidine-refractory, irinotecan-naive advanced colorectal cancer: A phase I-II study. Ann. Oncol. 2007, 18, 730–737. [Google Scholar]
- Weickhardt, A.J.; Price, T.J.; Chong, G.; Gebski, V.; Pavlakis, N.; Johns, T.G.; Azad, A.; Skrinos, E.; Fluck, K.; Dobrovic, A.; et al. Dual targeting of the epidermal growth factor receptor using the combination of cetuximab and erlotinib: Preclinical evaluation and results of the phase II DUX study in chemotherapy-refractory, advanced colorectal cancer. J. Clin. Oncol. 2012, 30, 1505–1512. [Google Scholar] [CrossRef]
- Sweet, R.W.; Yokoyama, S.; Kamata, T.; Feramisco, J.R.; Rosenberg, M.; Gross, M. The product of ras is a GTPase and the T24 oncogenic mutant is deficient in this activity. Nature 1984, 311, 273–275. [Google Scholar] [CrossRef]
- Bourne, H.R.; Sanders, D.A.; McCormick, F. The GTPase superfamily: A conserved switch for diverse cell functions. Nature 1990, 348, 125–132. [Google Scholar] [CrossRef]
- Schubbert, S.; Shannon, K.; Bollag, G. Hyperactive Ras in developmental disorders and cancer. Nat. Rev. Cancer 2007, 7, 295–308. [Google Scholar] [CrossRef]
- Buday, L.; Downward, J. Many faces of Ras activation. Biochim. Biophys. Acta 2008, 1786, 178–187. [Google Scholar]
- Krasinskas, A.M. EGFR Signaling in Colorectal Carcinoma. Patholog. Res. Int. 2011, 2011, 932932. [Google Scholar]
- Bos, J.L.; Fearon, E.R.; Hamilton, S.R.; Verlaan-de Vries, M.; van Boom, J.H.; van der Eb, A.J.; Vogelstein, B. Prevalence of ras gene mutations in human colorectal cancers. Nature 1987, 327, 293–297. [Google Scholar] [CrossRef]
- Normanno, N.; Tejpar, S.; Morgillo, F.; de Luca, A.; Van Cutsem, E.; Ciardiello, F. Implications for KRAS status and EGFR-targeted therapies in metastatic CRC. Nat. Rev. Clin. Oncol. 2009, 6, 519–527. [Google Scholar] [CrossRef]
- Forbes, S.; Clements, J.; Dawson, E.; Bamford, S.; Webb, T.; Dogan, A.; Flanagan, A.; Teague, J.; Wooster, R.; Futreal, P.A.; Stratton, M.R. Cosmic 2005. Br. J. Cancer 2006, 94, 318–322. [Google Scholar] [CrossRef]
- Kimura, T.; Okamoto, K.; Miyamoto, H.; Kimura, M.; Kitamura, S.; Takenaka, H.; Muguruma, N.; Okahisa, T.; Aoyagi, E.; Kajimoto, M.; et al. Clinical benefit of high-sensitivity KRAS mutation testing in metastatic colorectal cancer treated with anti-EGFR antibody therapy. Oncology 2012, 82, 298–304. [Google Scholar] [CrossRef]
- Allegra, C.J.; Jessup, J.M.; Somerfield, M.R.; Hamilton, S.R.; Hammond, E.H.; Hayes, D.F.; McAllister, P.K.; Morton, R.F.; Schilsky, R.L. American Society of Clinical Oncology provisional clinical opinion: Testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J. Clin. Oncol. 2009, 27, 2091–2096. [Google Scholar] [CrossRef]
- Lievre, A.; Bachet, J.B.; le Corre, D.; Boige, V.; Landi, B.; Emile, J.F.; Cote, J.F.; Tomasic, G.; Penna, C.; Ducreux, M.; et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006, 66, 3992–3995. [Google Scholar] [CrossRef]
- Laurent-Puig, P.; Cayre, A.; Manceau, G.; Buc, E.; Bachet, J.B.; Lecomte, T.; Rougier, P.; Lievre, A.; Landi, B.; Boige, V.; et al. Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer. J. Clin. Oncol. 2009, 27, 5924–5930. [Google Scholar] [CrossRef]
- Khambata-Ford, S.; Garrett, C.R.; Meropol, N.J.; Basik, M.; Harbison, C.T.; Wu, S.; Wong, T.W.; Huang, X.; Takimoto, C.H.; Godwin, A.K.; et al. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J. Clin. Oncol. 2007, 25, 3230–3237. [Google Scholar] [CrossRef]
- Bollag, G.; McCormick, F. Regulators and effectors of ras proteins. Annu. Rev. Cell. Biol. 1991, 7, 601–632. [Google Scholar] [CrossRef]
- De Roock, W.; Jonker, D.J.; di Nicolantonio, F.; Sartore-Bianchi, A.; Tu, D.; Siena, S.; Lamba, S.; Arena, S.; Frattini, M.; Piessevaux, H.; et al. Association of KRAS p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. JAMA 2010, 304, 1812–1820. [Google Scholar] [CrossRef]
- Tejpar, S.; Celik, I.; Schlichting, M.; Sartorius, U.; Bokemeyer, C.; Van Cutsem, E. Association of KRAS G13D tumor mutations with outcome in patients with metastatic colorectal cancer treated with first-line chemotherapy with or without cetuximab. J. Clin. Oncol. 2012, 30, 3570–3577. [Google Scholar] [CrossRef]
- Horsch, M.; Recktenwald, C.V.; Schadler, S.; Hrabe de Angelis, M.; Seliger, B.; Beckers, J. Overexpressed vs mutated Kras in murine fibroblasts: A molecular phenotyping study. Br. J. Cancer 2009, 100, 656–662. [Google Scholar] [CrossRef]
- Recktenwald, C.V.; Mendler, S.; Lichtenfels, R.; Kellner, R.; Seliger, B. Influence of Ki-ras-driven oncogenic transformation on the protein network of murine fibroblasts. Proteomics 2007, 7, 385–398. [Google Scholar] [CrossRef]
- Van Houdt, W.J.; Hoogwater, F.J.; de Bruijn, M.T.; Emmink, B.L.; Nijkamp, M.W.; Raats, D.A.; van der Groep, P.; van Diest, P.; Borel Rinkes, I.H.; Kranenburg, O. Oncogenic KRAS desensitizes colorectal tumor cells to epidermal growth factor receptor inhibition and activation. Neoplasia 2010, 12, 443–452. [Google Scholar]
- Dunn, E.F.; Iida, M.; Myers, R.A.; Campbell, D.A.; Hintz, K.A.; Armstrong, E.A.; Li, C.; Wheeler, D.L. Dasatinib sensitizes KRAS mutant colorectal tumors to cetuximab. Oncogene 2011, 30, 561–574. [Google Scholar] [CrossRef]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravegna, G.; et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012, 486, 532–536. [Google Scholar]
- Peeters, M.; Douillard, J.Y.; Van Cutsem, E.; Siena, S.; Zhang, K.; Williams, R.; Wiezorek, J. Mutant KRAS codon 12 and 13 alleles in patients with metastatic colorectal cancer: Assessment as prognostic and predictive biomarkers of response to panitumumab. J. Clin Oncol 2013, 31, 759–765. [Google Scholar] [CrossRef]
- Patel, D.; Guo, X.; Ng, S.; Melchior, M.; Balderes, P.; Burtrum, D.; Persaud, K.; Luna, X.; Ludwig, D.L.; Kang, X. IgG isotype, glycosylation, and EGFR expression determine the induction of antibody-dependent cellular cytotoxicity in vitro by cetuximab. Hum. Antibodies 2010, 19, 89–99. [Google Scholar]
- Desjarlais, J.R.; Lazar, G.A.; Zhukovsky, E.A.; Chu, S.Y. Optimizing engagement of the immune system by anti-tumor antibodies: An engineer’s perspective. Drug Discov. Today 2007, 12, 898–910. [Google Scholar] [CrossRef]
- Modest, D.P.; Reinacher-Schick, A.; Stintzing, S.; Giessen, C.; Tannapfel, A.; Laubender, R.P.; Brodowicz, T.; Knittelfelder, R.; Vrbanec, D.; Schmiegel, W.; et al. Cetuximab-based or bevacizumab-based first-line treatment in patients with KRAS p.G13D-mutated metastatic colorectal cancer: A pooled analysis. Anticancer Drugs 2012, 23, 666–673. [Google Scholar] [CrossRef]
- De Roock, W.; Claes, B.; Bernasconi, D.; De Schutter, J.; Biesmans, B.; Fountzilas, G.; Kalogeras, K.T.; Kotoula, V.; Papamichael, D.; Laurent-Puig, P.; et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: A retrospective consortium analysis. Lancet Oncol. 2010, 11, 753–762. [Google Scholar] [CrossRef]
- Pratilas, C.A.; Xing, F.; Solit, D.B. Targeting oncogenic BRAF in human cancer. Curr. Top. Microbiol. Immunol. 2012, 355, 83–98. [Google Scholar] [CrossRef]
- Di Nicolantonio, F.; Martini, M.; Molinari, F.; Sartore-Bianchi, A.; Arena, S.; Saletti, P.; de Dosso, S.; Mazzucchelli, L.; Frattini, M.; Siena, S.; et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J. Clin. Oncol. 2008, 26, 5705–5712. [Google Scholar] [CrossRef]
- Al-Marrawi, M.; Saroya, B.; Brennan, M.; Yang, Z.; Dykes, T.; El-Deiry, W. Off-label use of cetuximab plus sorafenib and panitumumab plus regorafenib to personalize therapy for a patient with V600E BRAF-mutant metastatic colon cancer. Cancer Biol. Ther. 2013, in press. [Google Scholar]
- Galal, K.M.; Khaled, Z.; Mourad, A.M. Role of cetuximab and sorafenib in treatment of metastatic colorectal cancer. Indian J. Cancer 2011, 48, 47–54. [Google Scholar] [CrossRef]
- W.T.S Institute. Catalogue of Somatic Mutations in Cancer. Available online: http://www.sanger.ac.uk/genetics/CGP/cosmic/ (accessed on 14 August 2013).
- Yu, J.; Zhang, Y.; McIlroy, J.; Rordorf-Nikolic, T.; Orr, G.A.; Backer, J.M. Regulation of the p85/p110 phosphatidylinositol 3'-kinase: Stabilization and inhibition of the p110alpha catalytic subunit by the p85 regulatory subunit. Mol. Cell. Biol. 1998, 18, 1379–1387. [Google Scholar]
- Miyaki, M.; Iijima, T.; Yamaguchi, T.; Takahashi, K.; Matsumoto, H.; Yasutome, M.; Funata, N.; Mori, T. Mutations of the PIK3CA gene in hereditary colorectal cancers. Int. J. Cancer 2007, 121, 1627–1630. [Google Scholar] [CrossRef]
- Velho, S.; Oliveira, C.; Ferreira, A.; Ferreira, A.C.; Suriano, G.; Schwartz, S., Jr.; Duval, A.; Carneiro, F.; Machado, J.C.; Hamelin, R.; et al. The prevalence of PIK3CA mutations in gastric and colon cancer. Eur. J. Cancer 2005, 41, 1649–1654. [Google Scholar] [CrossRef]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef]
- Kang, S.; Bader, A.G.; Vogt, P.K. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc. Natl.Acad. Sci.USA 2005, 102, 802–807. [Google Scholar] [CrossRef]
- Ikenoue, T.; Kanai, F.; Hikiba, Y.; Obata, T.; Tanaka, Y.; Imamura, J.; Ohta, M.; Jazag, A.; Guleng, B.; Tateishi, K.; et al. Functional analysis of PIK3CA gene mutations in human colorectal cancer. Cancer Res. 2005, 65, 4562–4567. [Google Scholar] [CrossRef]
- Sartore-Bianchi, A.; Martini, M.; Molinari, F.; Veronese, S.; Nichelatti, M.; Artale, S.; di Nicolantonio, F.; Saletti, P.; de Dosso, S.; Mazzucchelli, L.; et al. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res. 2009, 69, 1851–1857. [Google Scholar] [CrossRef]
- Frattini, M.; Saletti, P.; Romagnani, E.; Martin, V.; Molinari, F.; Ghisletta, M.; Camponovo, A.; Etienne, L.L.; Cavalli, F.; Mazzucchelli, L. PTEN loss of expression predicts cetuximab efficacy in metastatic colorectal cancer patients. Br. J. Cancer 2007, 97, 1139–1145. [Google Scholar] [CrossRef]
- Sartore-Bianchi, A.; Moroni, M.; Veronese, S.; Carnaghi, C.; Bajetta, E.; Luppi, G.; Sobrero, A.; Barone, C.; Cascinu, S.; Colucci, G.; et al. Epidermal growth factor receptor gene copy number and clinical outcome of metastatic colorectal cancer treated with panitumumab. J. Clin. Oncol. 2007, 25, 3238–3245. [Google Scholar] [CrossRef]
- Cappuzzo, F.; Finocchiaro, G.; Rossi, E.; Janne, P.A.; Carnaghi, C.; Calandri, C.; Bencardino, K.; Ligorio, C.; Ciardiello, F.; Pressiani, T.; et al. FGFR FISH assay predicts for response to cetuximab in chemotherapy refractory colorectal cancer patients. Ann. Oncol. 2008, 19, 717–723. [Google Scholar]
- Li, Y.H.; Wang, F.; Shen, L.; Deng, Y.M.; Shao, Q.; Feng, F.; An, X.; Wang, F.H.; Wang, Z.Q.; Xu, R.H.; Shao, J.Y. EGFR fluorescence in situ hybridization pattern of chromosome 7 disomy predicts resistance to cetuximab in KRAS wild-type metastatic colorectal cancer patients. Clin. Cancer Res. 2011, 17, 382–390. [Google Scholar] [CrossRef]
- Scartozzi, M.; Bearzi, I.; Mandolesi, A.; Pierantoni, C.; Loupakis, F.; Zaniboni, A.; Negri, F.; Quadri, A.; Zorzi, F.; Galizia, E.; Berardi, R.; et al. Epidermal Growth Factor Receptor (EGFR) gene copy number (GCN) correlates with clinical activity of irinotecan-cetuximab in K-RAS wild-type colorectal cancer: A fluorescence in situ (FISH) and chromogenic in situ hybridization (CISH) analysis. BMC Cancer 2009, 9, 303. [Google Scholar] [CrossRef]
- Lenz, H.J.; van Cutsem, E.; Khambata-Ford, S.; Mayer, R.J.; Gold, P.; Stella, P.; Mirtsching, B.; Cohn, A.L.; Pippas, A.W.; Azarnia, N.; et al. Multicenter phase II and translational study of cetuximab in metastatic colorectal carcinoma refractory to irinotecan, oxaliplatin, and fluoropyrimidines. J. Clin. Oncol. 2006, 24, 4914–4921. [Google Scholar]
- Sartore-Bianchi, A.; Fieuws, S.; Veronese, S.; Moroni, M.; Personeni, N.; Frattini, M.; Torri, V.; Cappuzzo, F.; Vander Borght, S.; Martin, V.; et al. Standardisation of EGFR FISH in colorectal cancer: Results of an international interlaboratory reproducibility ring study. J. Clin. Pathol. 2012, 65, 218–223. [Google Scholar] [CrossRef]
- Jacobs, B.; de Roock, W.; Piessevaux, H.; van Oirbeek, R.; Biesmans, B.; de Schutter, J.; Fieuws, S.; Vandesompele, J.; Peeters, M.; van Laethem, J.L.; et al. Amphiregulin and epiregulin mRNA expression in primary tumors predicts outcome in metastatic colorectal cancer treated with cetuximab. J. Clin. Oncol. 2009, 27, 5068–5074. [Google Scholar] [CrossRef]
- Oliveras-Ferraros, C.; Cufi, S.; Queralt, B.; Vazquez-Martin, A.; Martin-Castillo, B.; de Llorens, R.; Bosch-Barrera, J.; Brunet, J.; Menendez, J.A. Cross-suppression of EGFR ligands amphiregulin and epiregulin and de-repression of FGFR3 signalling contribute to cetuximab resistance in wild-type KRAS tumour cells. Br. J. Cancer 2012, 106, 1406–1414. [Google Scholar] [CrossRef]
- Sonoda, H.; Mekata, E.; Shimizu, T.; Endo, Y.; Tani, T. Safety and efficacy of panitumumab therapy after metastatic colorectal cancer progression with cetuximab: Experience at a single Japanese institution. Oncol. Lett. 2013, 5, 1331–1334. [Google Scholar]
- Montagut, C.; Dalmases, A.; Bellosillo, B.; Crespo, M.; Pairet, S.; Iglesias, M.; Salido, M.; Gallen, M.; Marsters, S.; Tsai, S.P.; et al. Identification of a mutation in the extracellular domain of the Epidermal Growth Factor Receptor conferring cetuximab resistance in colorectal cancer. Nat. Med. 2012, 18, 221–223. [Google Scholar] [CrossRef]
- Voigt, M.; Braig, F.; Gothel, M.; Schulte, A.; Lamszus, K.; Bokemeyer, C.; Binder, M. Functional dissection of the epidermal growth factor receptor epitopes targeted by panitumumab and cetuximab. Neoplasia 2012, 14, 1023–1031. [Google Scholar]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef]
- Folkman, J. What is the evidence that tumors are angiogenesis dependent? J. Natl. Cancer Inst. 1990, 82, 4–6. [Google Scholar] [CrossRef]
- Takahashi, Y.; Kitadai, Y.; Bucana, C.D.; Cleary, K.R.; Ellis, L.M. Expression of vascular endothelial growth factor and its receptor, KDR, correlates with vascularity, metastasis, and proliferation of human colon cancer. Cancer Res. 1995, 55, 3964–3968. [Google Scholar]
- Takebayashi, Y.; Aklyama, S.; Yamada, K.; Akiba, S.; Aikou, T. Angiogenesis as an unfavorable prognostic factor in human colorectal carcinoma. Cancer 1996, 78, 226–231. [Google Scholar] [CrossRef]
- Takahashi, Y.; Tucker, S.L.; Kitadai, Y.; Koura, A.N.; Bucana, C.D.; Cleary, K.R.; Ellis, L.M. Vessel counts and expression of vascular endothelial growth factor as prognostic factors in node-negative colon cancer. Arch. Surg. 1997, 132, 541–546. [Google Scholar] [CrossRef]
- Ishigami, S.I.; Arii, S.; Furutani, M.; Niwano, M.; Harada, T.; Mizumoto, M.; Mori, A.; Onodera, H.; Imamura, M. Predictive value of vascular endothelial growth factor (VEGF) in metastasis and prognosis of human colorectal cancer. Br. J. Cancer 1998, 78, 1379–1384. [Google Scholar] [CrossRef]
- Li, X.; Eriksson, U. Novel VEGF family members: VEGF-B, VEGF-C and VEGF-D. Int J. Biochem. Cell. Biol. 2001, 33, 421–426. [Google Scholar] [CrossRef]
- Maglione, D.; Guerriero, V.; Viglietto, G.; Delli-Bovi, P.; Persico, M.G. Isolation of a human placenta cDNA coding for a protein related to the vascular permeability factor. Proc. Natl. Acad. Sci. USA 1991, 88, 9267–9271. [Google Scholar]
- Escudero-Esparza, A.; Martin, T.A.; Davies, M.L.; Jiang, W.G. PGF isoforms, PLGF-1 and PGF-2, in colorectal cancer and the prognostic significance. Cancer Genomics Proteomics 2009, 6, 239–246. [Google Scholar]
- Wei, S.C.; Tsao, P.N.; Yu, S.C.; Shun, C.T.; Tsai-Wu, J.J.; Wu, C.H.; Su, Y.N.; Hsieh, F.J.; Wong, J.M. Placenta growth factor expression is correlated with survival of patients with colorectal cancer. Gut 2005, 54, 666–672. [Google Scholar] [CrossRef]
- Ferrara, N.; Hillan, K.J.; Novotny, W. Bevacizumab (Avastin), a humanized anti-VEGF monoclonal antibody for cancer therapy. Biochem. Biophys. Res. Commun. 2005, 333, 328–335. [Google Scholar] [CrossRef]
- Cao, Y.; Chen, H.; Zhou, L.; Chiang, M.K.; Anand-Apte, B.; Weatherbee, J.A.; Wang, Y.; Fang, F.; Flanagan, J.G.; Tsang, M.L. Heterodimers of placenta growth factor/vascular endothelial growth factor. Endothelial activity, tumor cell expression, and high affinity binding to Flk-1/KDR. J. Biol. Chem. 1996, 271, 3154–3162. [Google Scholar] [CrossRef]
- Presta, L.G.; Chen, H.; O’Connor, S.J.; Chisholm, V.; Meng, Y.G.; Krummen, L.; Winkler, M.; Ferrara, N. Humanization of an anti-vascular endothelial growth factor monoclonal antibody for the therapy of solid tumors and other disorders. Cancer Res. 1997, 57, 4593–4599. [Google Scholar]
- Kim, K.J.; Li, B.; Winer, J.; Armanini, M.; Gillett, N.; Phillips, H.S.; Ferrara, N. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature 1993, 362, 841–844. [Google Scholar] [CrossRef]
- O'Connor, J.P.; Carano, R.A.; Clamp, A.R.; Ross, J.; Ho, C.C.; Jackson, A.; Parker, G.J.; Rose, C.J.; Peale, F.V.; Friesenhahn, M.; et al. Quantifying antivascular effects of monoclonal antibodies to vascular endothelial growth factor: Insights from imaging. Clin. Cancer Res. 2009, 15, 6674–6682. [Google Scholar] [CrossRef]
- Koukourakis, M.I.; Mavanis, I.; Kouklakis, G.; Pitiakoudis, M.; Minopoulos, G.; Manolas, C.; Simopoulos, C. Early antivascular effects of bevacizumab anti-VEGF monoclonal antibody on colorectal carcinomas assessed with functional CT imaging. Am. J. Clin. Oncol. 2007, 30, 315–318. [Google Scholar] [CrossRef]
- Saltz, L.B.; Clarke, S.; Diaz-Rubio, E.; Scheithauer, W.; Figer, A.; Wong, R.; Koski, S.; Lichinitser, M.; Yang, T.S.; Rivera, F.; et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: A randomized phase III study. J. Clin. Oncol. 2008, 26, 2013–2019. [Google Scholar] [CrossRef]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; Ferrara, N.; Fyfe, G.; Rogers, B.; Ross, R.; Kabbinavar, F. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef]
- Fuchs, C.S.; Marshall, J.; Barrueco, J. Randomized, controlled trial of irinotecan plus infusional, bolus, or oral fluoropyrimidines in first-line treatment of metastatic colorectal cancer: Updated results from the BICC-C study. J. Clin. Oncol. 2008, 26, 689–690. [Google Scholar] [CrossRef]
- Giantonio, B.J.; Catalano, P.J.; Meropol, N.J.; O’Dwyer, P.J.; Mitchell, E.P.; Alberts, S.R.; Schwartz, M.A.; Benson, A.B., 3rd. Bevacizumab in combination with oxaliplatin, fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer: Results from the Eastern Cooperative Oncology Group Study E3200. J. Clin. Oncol. 2007, 25, 1539–1544. [Google Scholar] [CrossRef]
- Grothey, A.; Sugrue, M.M.; Purdie, D.M.; Dong, W.; Sargent, D.; Hedrick, E.; Kozloff, M. Bevacizumab beyond first progression is associated with prolonged overall survival in metastatic colorectal cancer: Results from a large observational cohort study (BRiTE). J. Clin. Oncol. 2008, 26, 5326–5334. [Google Scholar] [CrossRef]
- Grothey, A.; Allegra, C. Antiangiogenesis therapy in the treatment of metastatic colorectal cancer. Ther. Adv. Med. Oncol. 2012, 4, 301–319. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Tabernero, J.; Lakomy, R.; Prenen, H.; Prausova, J.; Macarulla, T.; Ruff, P.; van Hazel, G.A.; Moiseyenko, V.; Ferry, D.; et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J. Clin. Oncol. 2012, 30, 3499–3506. [Google Scholar] [CrossRef]
- Kim, J.G.; Chae, Y.S.; Sohn, S.K.; Cho, Y.Y.; Moon, J.H.; Park, J.Y.; Jeon, S.W.; Lee, I.T.; Choi, G.S.; Jun, S.H. Vascular endothelial growth factor gene polymorphisms associated with prognosis for patients with colorectal cancer. Clin. Cancer Res. 2008, 14, 62–66. [Google Scholar] [CrossRef]
- Formica, V.; Palmirotta, R.; del Monte, G.; Savonarola, A.; Ludovici, G.; de Marchis, M.L.; Grenga, I.; Schirru, M.; Guadagni, F.; Roselli, M. Predictive value of VEGF gene polymorphisms for metastatic colorectal cancer patients receiving first-line treatment including fluorouracil, irinotecan, and bevacizumab. Int. J. Colorectal. Dis. 2011, 26, 143–151. [Google Scholar]
- Watson, C.J.; Webb, N.J.; Bottomley, M.J.; Brenchley, P.E. Identification of polymorphisms within the vascular endothelial growth factor (VEGF) gene: Correlation with variation in VEGF protein production. Cytokine 2000, 12, 1232–1235. [Google Scholar] [CrossRef]
- Awata, T.; Inoue, K.; Kurihara, S.; Ohkubo, T.; Watanabe, M.; Inukai, K.; Inoue, I.; Katayama, S. A common polymorphism in the 5'-untranslated region of the VEGF gene is associated with diabetic retinopathy in type 2 diabetes. Diabetes 2002, 51, 1635–1639. [Google Scholar] [CrossRef]
- Koutras, A.K.; Antonacopoulou, A.G.; Eleftheraki, A.G.; Dimitrakopoulos, F.I.; Koumarianou, A.; Varthalitis, I.; Fostira, F.; Sgouros, J.; Briasoulis, E.; Bournakis, E.; et al. Vascular endothelial growth factor polymorphisms and clinical outcome in colorectal cancer patients treated with irinotecan-based chemotherapy and bevacizumab. Pharmacogenomics J. 2012, 12, 468–475. [Google Scholar]
- Shahbazi, M.; Fryer, A.A.; Pravica, V.; Brogan, I.J.; Ramsay, H.M.; Hutchinson, I.V.; Harden, P.N. Vascular endothelial growth factor gene polymorphisms are associated with acute renal allograft rejection. J. Am. Soc. Nephrol. 2002, 13, 260–264. [Google Scholar]
- Mohammadi, M.; Ollier, W.E.; Hutchinson, I.V. A functional association study of VEGF gene promoter polymorphisms with VEGF expression by stimulated PBM cells. Hum. Immunol. 2003, 64, S125. [Google Scholar]
- Loupakis, F.; Cremolini, C.; Fioravanti, A.; Orlandi, P.; Salvatore, L.; Masi, G.; di Desidero, T.; Canu, B.; Schirripa, M.; Frumento, P.; et al. Pharmacodynamic and pharmacogenetic angiogenesis-related markers of first-line FOLFOXIRI plus bevacizumab schedule in metastatic colorectal cancer. Br. J. Cancer 2011, 104, 1262–1269. [Google Scholar] [CrossRef]
- Pander, J.; Wessels, J.A.; Gelderblom, H.; van der Straaten, T.; Punt, C.J.; Guchelaar, H.J. Pharmacogenetic interaction analysis for the efficacy of systemic treatment in metastatic colorectal cancer. Ann. Oncol. 2011, 22, 1147–1153. [Google Scholar] [CrossRef]
- Cacev, T.; Loncar, B.; Seiwerth, S.; Spaventi, S.; Kapitanovic, S. Vascular endothelial growth factor polymorphisms -1154 G/A and -460 C/T are not associated with VEGF mRNA expression and susceptibility to sporadic colon cancer. DNA Cell Biol. 2008, 27, 569–574. [Google Scholar] [CrossRef]
- Manzoni, M.; Mariucci, S.; Delfanti, S.; Rovati, B.; Ronzoni, M.; Loupakis, F.; Brugnatelli, S.; Tinelli, C.; Villa, E.; Falcone, A.; et al. Circulating endothelial cells and their apoptotic fraction are mutually independent predictive biomarkers in Bevacizumab-based treatment for advanced colorectal cancer. J. Cancer Res. Clin. Oncol. 2012, 138, 1187–1196. [Google Scholar] [CrossRef]
- Ronzoni, M.; Manzoni, M.; Mariucci, S.; Loupakis, F.; Brugnatelli, S.; Bencardino, K.; Rovati, B.; Tinelli, C.; Falcone, A.; Villa, E.; et al. Circulating endothelial cells and endothelial progenitors as predictive markers of clinical response to bevacizumab-based first-line treatment in advanced colorectal cancer patients. Ann. Oncol. 2010, 21, 2382–2389. [Google Scholar] [CrossRef]
- Matsusaka, S.; Mishima, Y.; Suenaga, M.; Terui, Y.; Kuniyoshi, R.; Mizunuma, N.; Hatake, K. Circulating endothelial progenitors and CXCR4-positive circulating endothelial cells are predictive markers for bevacizumab. Cancer 2011, 117, 4026–4032. [Google Scholar] [CrossRef]
- Guleng, B.; Tateishi, K.; Ohta, M.; Kanai, F.; Jazag, A.; Ijichi, H.; Tanaka, Y.; Washida, M.; Morikane, K.; Fukushima, Y.; et al. Blockade of the stromal cell-derived factor-1/CXCR4 axis attenuates in vivo tumor growth by inhibiting angiogenesis in a vascular endothelial growth factor-independent manner. Cancer Res. 2005, 65, 5864–5871. [Google Scholar] [CrossRef]
- Simkens, L.H.; Tol, J.; Terstappen, L.W.; Teerenstra, S.; Punt, C.J.; Nagtegaal, I.D. The predictive and prognostic value of circulating endothelial cells in advanced colorectal cancer patients receiving first-line chemotherapy and bevacizumab. Ann. Oncol. 2010, 21, 2447–2448. [Google Scholar] [CrossRef]
- Bertolini, F.; Marighetti, P.; Shaked, Y. Cellular and soluble markers of tumor angiogenesis: From patient selection to the identification of the most appropriate postresistance therapy. Biochim. Biophys. Acta 2010, 1806, 131–137. [Google Scholar]
- Bertolini, F.; Shaked, Y.; Mancuso, P.; Kerbel, R.S. The multifaceted circulating endothelial cell in cancer: Towards marker and target identification. Nat. Rev. Cancer 2006, 6, 835–845. [Google Scholar] [CrossRef]
- Gordon, M.S.; Margolin, K.; Talpaz, M.; Sledge, G.W., Jr.; Holmgren, E.; Benjamin, R.; Stalter, S.; Shak, S.; Adelman, D. Phase I safety and pharmacokinetic study of recombinant human anti-vascular endothelial growth factor in patients with advanced cancer. J. Clin. Oncol. 2001, 19, 843–850. [Google Scholar]
- Willett, C.G.; Boucher, Y.; Duda, D.G.; di Tomaso, E.; Munn, L.L.; Tong, R.T.; Kozin, S.V.; Petit, L.; Jain, R.K.; Chung, D.C.; et al. Surrogate markers for antiangiogenic therapy and dose-limiting toxicities for bevacizumab with radiation and chemotherapy: Continued experience of a phase I trial in rectal cancer patients. J. Clin. Oncol. 2005, 23, 8136–8139. [Google Scholar] [CrossRef]
- Yang, J.C.; Haworth, L.; Sherry, R.M.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Steinberg, S.M.; Chen, H.X.; Rosenberg, S.A. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N. Engl. J. Med. 2003, 349, 427–434. [Google Scholar] [CrossRef]
- Loupakis, F.; Falcone, A.; Masi, G.; Fioravanti, A.; Kerbel, R.S.; del Tacca, M.; Bocci, G. Vascular endothelial growth factor levels in immunodepleted plasma of cancer patients as a possible pharmacodynamic marker for bevacizumab activity. J. Clin. Oncol. 2007, 25, 1816–1818. [Google Scholar] [CrossRef]
- Jubb, A.M.; Hurwitz, H.I.; Bai, W.; Holmgren, E.B.; Tobin, P.; Guerrero, A.S.; Kabbinavar, F.; Holden, S.N.; Novotny, W.F.; Frantz, G.D.; et al. Impact of vascular endothelial growth factor-A expression, thrombospondin-2 expression, and microvessel density on the treatment effect of bevacizumab in metastatic colorectal cancer. J. Clin. Oncol. 2006, 24, 217–227. [Google Scholar]
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar]
- Lyons, J.F.; Wilhelm, S.; Hibner, B.; Bollag, G. Discovery of a novel Raf kinase inhibitor. Endocr. Relat. Cancer 2001, 8, 219–225. [Google Scholar] [CrossRef]
- Smith, R.A.; Barbosa, J.; Blum, C.L.; Bobko, M.A.; Caringal, Y.V.; Dally, R.; Johnson, J.S.; Katz, M.E.; Kennure, N.; Kingery-Wood, J.; et al. Discovery of heterocyclic ureas as a new class of raf kinase inhibitors: Identification of a second generation lead by a combinatorial chemistry approach. Bioorg. Med. Chem. Lett. 2001, 11, 2775–2778. [Google Scholar] [CrossRef]
- Khire, U.R.; Bankston, D.; Barbosa, J.; Brittelli, D.R.; Caringal, Y.; Carlson, R.; Dumas, J.; Gane, T.; Heald, S.L.; Hibner, B.; et al. Omega-carboxypyridyl substituted ureas as Raf kinase inhibitors: SAR of the amide substituent. Bioorg. Med. Chem. Lett. 2004, 14, 783–786. [Google Scholar]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43–9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef]
- Alavi, A.; Hood, J.D.; Frausto, R.; Stupack, D.G.; Cheresh, D.A. Role of Raf in vascular protection from distinct apoptotic stimuli. Science 2003, 301, 94–96. [Google Scholar] [CrossRef]
- Hood, J.D.; Bednarski, M.; Frausto, R.; Guccione, S.; Reisfeld, R.A.; Xiang, R.; Cheresh, D.A. Tumor regression by targeted gene delivery to the neovasculature. Science 2002, 296, 2404–2407. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Adnane, L.; Newell, P.; Villanueva, A.; Llovet, J.M.; Lynch, M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol. Cancer Ther. 2008, 7, 3129–3140. [Google Scholar] [CrossRef]
- Activity of the Raf kinase inhibitor BAY 43–9006 in patients with advanced solid tumors. Clin. Colorectal. Cancer 2003, 3, 16–18. [CrossRef]
- Martinelli, E.; Troiani, T.; Morgillo, F.; Rodolico, G.; Vitagliano, D.; Morelli, M.P.; Tuccillo, C.; Vecchione, L.; Capasso, A.; Orditura, M.; et al. Synergistic antitumor activity of sorafenib in combination with epidermal growth factor receptor inhibitors in colorectal and lung cancer cells. Clin. Cancer Res. 2010, 16, 4990–5001. [Google Scholar] [CrossRef]
- Azad, N.; Dasari, A.; Arcaroli, J.; Taylor, G.E.; Laheru, D.A.; Carducci, M.A.; McManus, M.; Quackenbush, K.; Wright, J.J.; Hidalgo, M.; et al. Phase I pharmacokinetic and pharmacodynamic study of cetuximab, irinotecan and sorafenib in advanced colorectal cancer. Invest. New Drugs 2013, 31, 345–354. [Google Scholar] [CrossRef]
- Wehler, T.C.; Hamdi, S.; Maderer, A.; Graf, C.; Gockel, I.; Schmidtmann, I.; Hainz, M.; Berger, M.R.; Theobald, M.; Galle, P.R.; et al. Single-agent therapy with sorafenib or 5-FU is equally effective in human colorectal cancer xenograft-no benefit of combination therapy. Int. J. Colorectal Dis. 2013, 28, 385–398. [Google Scholar]
- Mross, K.; Steinbild, S.; Baas, F.; Gmehling, D.; Radtke, M.; Voliotis, D.; Brendel, E.; Christensen, O.; Unger, C. Results from an in vitro and a clinical/pharmacological phase I study with the combination irinotecan and sorafenib. Eur. J. Cancer 2007, 43, 55–63. [Google Scholar] [CrossRef]
- Kupsch, P.; Henning, B.F.; Passarge, K.; Richly, H.; Wiesemann, K.; Hilger, R.A.; Scheulen, M.E.; Christensen, O.; Brendel, E.; Schwartz, B.; et al. Results of a phase I trial of sorafenib (BAY 43–9006) in combination with oxaliplatin in patients with refractory solid tumors, including colorectal cancer. Clin. Colorectal. Cancer 2005, 5, 188–196. [Google Scholar]
- Wilhelm, S.M.; Dumas, J.; Adnane, L.; Lynch, M.; Carter, C.A.; Schutz, G.; Thierauch, K.H.; Zopf, D. Regorafenib (BAY 73–4506): A new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int. J. Cancer 2011, 129, 245–255. [Google Scholar] [CrossRef]
- Strumberg, D.; Scheulen, M.E.; Schultheis, B.; Richly, H.; Frost, A.; Buchert, M.; Christensen, O.; Jeffers, M.; Heinig, R.; Boix, O.; et al. Regorafenib (BAY 73–4506) in advanced colorectal cancer: A phase I study. Br. J. Cancer 2012, 106, 1722–1727. [Google Scholar] [CrossRef]
- Grothey, A. Regorafenib in metastatic colorectal cancer. Clin. Adv. Hematol. Oncol. 2012, 10, 324–325. [Google Scholar]
- Yokota, T.; Ura, T.; Shibata, N.; Takahari, D.; Shitara, K.; Nomura, M.; Kondo, C.; Mizota, A.; Utsunomiya, S.; Muro, K.; et al. BRAF mutation is a powerful prognostic factor in advanced and recurrent colorectal cancer. Br. J. Cancer 2011, 104, 856–862. [Google Scholar] [CrossRef]
- Ogino, S.; Nosho, K.; Kirkner, G.J.; Kawasaki, T.; Meyerhardt, J.A.; Loda, M.; Giovannucci, E.L.; Fuchs, C.S. CpG island methylator phenotype, microsatellite instability, BRAF mutation and clinical outcome in colon cancer. Gut 2009, 58, 90–96. [Google Scholar] [CrossRef]
- Farina-Sarasqueta, A.; van Lijnschoten, G.; Moerland, E.; Creemers, G.J.; Lemmens, V.E.; Rutten, H.J.; van den Brule, A.J. The BRAF V600E mutation is an independent prognostic factor for survival in stage II and stage III colon cancer patients. Ann. Oncol. 2010, 21, 2396–2402. [Google Scholar] [CrossRef]
- Sala, E.; Mologni, L.; Truffa, S.; Gaetano, C.; Bollag, G.E.; Gambacorti-Passerini, C. BRAF silencing by short hairpin RNA or chemical blockade by PLX4032 leads to different responses in melanoma and thyroid carcinoma cells. Mol. Cancer Res. 2008, 6, 751–759. [Google Scholar] [CrossRef]
- Yang, H.; Higgins, B.; Kolinsky, K.; Packman, K.; Bradley, W.D.; Lee, R.J.; Schostack, K.; Simcox, M.E.; Kopetz, S.; Heimbrook, D.; Lestini, B.; Bollag, G.; Su, F. Antitumor activity of BRAF inhibitor vemurafenib in preclinical models of BRAF-mutant colorectal cancer. Cancer Res. 2012, 72, 779–789. [Google Scholar] [CrossRef]
- Kopetz, S.; Desai, J.; Chan, E.; Hecht, J.R.; O’Dwyer, P.; Lee, R.; Nolop, K.; Saltz, L. PLX4032 in metastatic colorectal cancer patients with mutant BRaf tumors. J. Clin. Oncol. 2010, 28, 15s. [Google Scholar]
- Mao, M.; Tian, F.; Mariadason, J.M.; Tsao, C.C.; Lemos, R., Jr.; Dayyani, F.; Gopal, Y.N.; Jiang, Z.Q.; Wistuba, II; Tang, X.M.; et al. Resistance to BRAF inhibition in BRAF-mutant colon cancer can be overcome with PI3K inhibition or demethylating agents. Clin. Cancer Res. 2013, 19, 657–667. [Google Scholar] [CrossRef]
- Prahallad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Ebi, H.; Turke, A.B.; Coffee, E.M.; Nishino, M.; Cogdill, A.P.; Brown, R.D.; Della Pelle, P.; Dias-Santagata, D.; Hung, K.E.; et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012, 2, 227–235. [Google Scholar] [CrossRef]
- Coffee, E.M.; Faber, A.C.; Roper, J.; Sinnamon, M.J.; Goel, G.; Keung, L.; Wang, W.V.; Vecchione, L.; de Vriendt, V.; Weinstein, B.J.; et al. Concomitant BRAF and PI3K/mTOR Blockade Is Required for Effective Treatment of BRAFV600E Colorectal Cancer. Clin. Cancer Res. 2013, 19, 2688–2698. [Google Scholar] [CrossRef]
- Pang, R.; Law, W.L.; Chu, A.C.; Poon, J.T.; Lam, C.S.; Chow, A.K.; Ng, L.; Cheung, L.W.; Lan, X.R.; Lan, H.Y.; et al. A subpopulation of CD26+ cancer stem cells with metastatic capacity in human colorectal cancer. Cell Stem Cell 2010, 6, 603–615. [Google Scholar] [CrossRef]
- Dylla, S.J.; Beviglia, L.; Park, I.K.; Chartier, C.; Raval, J.; Ngan, L.; Pickell, K.; Aguilar, J.; Lazetic, S.; Smith-Berdan, S.; et al. Colorectal cancer stem cells are enriched in xenogeneic tumors following chemotherapy. PLoS One 2008, 3, e2428. [Google Scholar] [CrossRef]
- Wilson, B.J.; Schatton, T.; Frank, M.H.; Frank, N.Y. Colorectal Cancer Stem Cells: Biology and Therapeutic Implications. Curr. Colorectal. Cancer Rep. 2011, 7, 128–135. [Google Scholar] [CrossRef]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef]
- Todaro, M.; Alea, M.P.; di Stefano, A.B.; Cammareri, P.; Vermeulen, L.; Iovino, F.; Tripodo, C.; Russo, A.; Gulotta, G.; Medema, J.P.; et al. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell 2007, 1, 389–402. [Google Scholar]
- Dalerba, P.; Dylla, S.J.; Park, I.K.; Liu, R.; Wang, X.; Cho, R.W.; Hoey, T.; Gurney, A.; Huang, E.H.; Simeone, D.M.; et al. Phenotypic characterization of human colorectal cancer stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 10158–10163. [Google Scholar] [CrossRef]
- Huang, E.H.; Hynes, M.J.; Zhang, T.; Ginestier, C.; Dontu, G.; Appelman, H.; Fields, J.Z.; Wicha, M.S.; Boman, B.M. Aldehyde dehydrogenase 1 is a marker for normal and malignant human colonic stem cells (SC) and tracks SC overpopulation during colon tumorigenesis. Cancer Res. 2009, 69, 3382–3389. [Google Scholar] [CrossRef]
- Langan, R.C.; Mullinax, J.E.; Raiji, M.T.; Upham, T.; Summers, T.; Stojadinovic, A.; Avital, I. Colorectal cancer biomarkers and the potential role of cancer stem cells. J. Cancer 2013, 4, 241–250. [Google Scholar] [CrossRef]
- Ong, C.W.; Kim, L.G.; Kong, H.H.; Low, L.Y.; Iacopetta, B.; Soong, R.; Salto-Tellez, M. CD133 expression predicts for non-response to chemotherapy in colorectal cancer. Mod. Pathol. 2010, 23, 450–457. [Google Scholar] [CrossRef]
- Mia-Jan, K.; Jung, S.Y.; Kim, I.Y.; Oh, S.S.; Choi, E.; Chang, S.J.; Kang, T.Y.; Cho, M.Y. CD133 expression is not an independent prognostic factor in stage II and III colorectal cancer but may predict the better outcome in patients with adjuvant therapy. BMC Cancer 2013, 13, 166. [Google Scholar]
- Shmelkov, S.V.; Butler, J.M.; Hooper, A.T.; Hormigo, A.; Kushner, J.; Milde, T.; St Clair, R.; Baljevic, M.; White, I.; Jin, D.K.; et al. CD133 expression is not restricted to stem cells, and both CD133+ and CD133- metastatic colon cancer cells initiate tumors. J. Clin. Invest. 2008, 118, 2111–2120. [Google Scholar]
- Allard, W.J.; Matera, J.; Miller, M.C.; Repollet, M.; Connelly, M.C.; Rao, C.; Tibbe, A.G.; Uhr, J.W.; Terstappen, L.W. Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin. Cancer Res. 2004, 10, 6897–6904. [Google Scholar] [CrossRef]
- Cohen, S.J.; Punt, C.J.; Iannotti, N.; Saidman, B.H.; Sabbath, K.D.; Gabrail, N.Y.; Picus, J.; Morse, M.; Mitchell, E.; Miller, M.C.; et al. Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. J. Clin. Oncol. 2008, 26, 3213–3221. [Google Scholar] [CrossRef]
- Otsuka, K.; Imai, H.; Soeda, H.; Komine, K.; Ishioka, C.; Shibata, H. Practical utility of circulating tumour cells as biomarkers in cancer chemotherapy for advanced colorectal cancer. Anticancer Res. 2013, 33, 625–629. [Google Scholar]
- Gazzaniga, P.; Raimondi, C.; Gradilone, A.; Biondi Zoccai, G.; Nicolazzo, C.; Gandini, O.; Longo, F.; Tomao, S.; Lo Russo, G.; Seminara, P.; et al. Circulating tumor cells in metastatic colorectal cancer: Do we need an alternative cutoff? J. Cancer Res. Clin. Oncol. 2013, 139, 1411–1416. [Google Scholar] [CrossRef]
- Sastre, J.; Maestro, M.L.; Gomez-Espana, A.; Rivera, F.; Valladares, M.; Massuti, B.; Benavides, M.; Gallen, M.; Marcuello, E.; Abad, A.; et al. Circulating tumor cell count is a prognostic factor in metastatic colorectal cancer patients receiving first-line chemotherapy plus bevacizumab: A Spanish Cooperative Group for the Treatment of Digestive Tumors study. Oncologist 2012, 17, 947–955. [Google Scholar] [CrossRef]
- De Albuquerque, A.; Kubisch, I.; Stolzel, U.; Ernst, D.; Boese-Landgraf, J.; Breier, G.; Stamminger, G.; Fersis, N.; Kaul, S. Prognostic and predictive value of circulating tumor cell analysis in colorectal cancer patients. J. Transl. Med. 2012, 10, 222. [Google Scholar] [CrossRef]
- Roychowdhury, S.; Iyer, M.K.; Robinson, D.R.; Lonigro, R.J.; Wu, Y.M.; Cao, X.; Kalyana-Sundaram, S.; Sam, L.; Balbin, O.A.; Quist, M.J.; et al. Personalized oncology through integrative high-throughput sequencing: A pilot study. Sci. Transl. Med. 2011, 3, 111ra121. [Google Scholar] [CrossRef]
- Lamparella, N.; Saroya, B.; Yang, Z.; Sarwani, N.; El-Deiry, W. Contradictory KRAS mutation test results in a patient with metastatic colon cancer: A clinical dilemma in the era of personalized medicine. Cancer Biol. Ther. 2013, in press. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kline, C.L.B.; El-Deiry, W.S. Personalizing Colon Cancer Therapeutics: Targeting Old and New Mechanisms of Action. Pharmaceuticals 2013, 6, 988-1038. https://doi.org/10.3390/ph6080988
Kline CLB, El-Deiry WS. Personalizing Colon Cancer Therapeutics: Targeting Old and New Mechanisms of Action. Pharmaceuticals. 2013; 6(8):988-1038. https://doi.org/10.3390/ph6080988
Chicago/Turabian StyleKline, Christina Leah B., and Wafik S. El-Deiry. 2013. "Personalizing Colon Cancer Therapeutics: Targeting Old and New Mechanisms of Action" Pharmaceuticals 6, no. 8: 988-1038. https://doi.org/10.3390/ph6080988