Phytochemical Modulators of Mitochondria: The Search for Chemopreventive Agents and Supportive Therapeutics


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Role of Mitochondria in the Etiology and Onset of Human Diseases
3. Dietary Phytochemicals Acting on Mitochondria

4. Mitochondrial Respiration and Energy Generation


5. Reactive Oxygen Species and Oxidative Stress
6. Signaling between Mitochondria and Nucleus

7. Unfolded Protein Response and Longevity

8. Mitochondria in Cancer Stem Cells
9. Conclusions and Perspectives
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Van der Giezen, M. Hydrogenosomes and mitosomes: Conservation and evolution of functions. J. Eukaryotic. Microbiol. 2009, 56, 221–231. [Google Scholar]
- Shiflett, A.M.; Johnson, P.J. Mitochondrion-related organelles in eukaryotic protists. Annu. Rev. Microbiol. 2010, 64, 409–429. [Google Scholar] [PubMed]
- Lill, R.; Muhlenhoff, U. Iron-sulfur-protein biogenesis in eukaryotes. Trends Biochem. Sci. 2005, 30, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Van der Giezen, M.; Tovar, J. Mitosomes, Hydrogenosomes and Mitochondria: Variations on a Theme? In Organelles, Genomes and Eiukaryote Phylogeny; an Evolutionary Synthesis in the Age of Genomics; Hirt, R.P., Horner, D.S., Eds.; CRC Press: Boca Raton, FL, USA, 2004; pp. 329–351. [Google Scholar]
- Miller, S.W.; Trimmer, P.A.; Parker, W.D., Jr.; Davis, R.E. Creation and characterization of mitochondrial DNA-depleted cell lines with “neuronal-like” properties. J. Neurochem. 1996, 67, 1897–1907. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; Schumacker, P.T. Cells depleted of mitochondrial DNA (rho0) yield insight into physiological mechanisms. FEBS Lett. 1999, 454, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Baron, M.; Kudin, A.P.; Kunz, W.S. Mitochondrial dysfunction in neurodegenerative disorders. Biochem. Soc. Trans. 2007, 35, 1228–1231. [Google Scholar] [PubMed]
- Patti, M.E.; Corvera, S. The role of mitochondria in the pathogenesis of type 2 diabetes. Endocrine Rev. 2010, 31, 364–395. [Google Scholar]
- Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 2005, 6, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondrial genetics: A paradigm for aging and degenerative diseases? Science 1992, 256, 628–632. [Google Scholar] [CrossRef] [PubMed]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Bohlooly, Y.M.; Gidlof, S.; Oldfors, A.; Wibom, R.; et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004, 429, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, V.; Corao, A.I.; Alonso-Montes, C.; Sanchez-Ferrero, E.; De Mena, L.; Morales, B.; Garcia-Castro, M.; Coto, E. Mitochondrial transcription factor a (tfam) gene variation and risk of late-onset alzheimer's disease. J. Alzheimer’s Dis. 2008, 13, 275–280. [Google Scholar]
- Belin, A.C.; Bjork, B.F.; Westerlund, M.; Galter, D.; Sydow, O.; Lind, C.; Pernold, K.; Rosvall, L.; Hakansson, A.; Winblad, B.; et al. Association study of two genetic variants in mitochondrial transcription factor a (tfam) in alzheimer's and parkinson's disease. Neurosci. Lett. 2007, 420, 257–262. [Google Scholar]
- Gunther, C.; von Hadeln, K.; Muller-Thomsen, T.; Alberici, A.; Binetti, G.; Hock, C.; Nitsch, R.M.; Stoppe, G.; Reiss, J.; Gal, A.; et al. Possible association of mitochondrial transcription factor a (tfam) genotype with sporadic alzheimer disease. Neurosci. Lett. 2004, 369, 219–223. [Google Scholar]
- Gaweda-Walerych, K.; Safranow, K.; Maruszak, A.; Bialecka, M.; Klodowska-Duda, G.; Czyzewski, K.; Slawek, J.; Rudzinska, M.; Styczynska, M.; Opala, G.; et al. Mitochondrial transcription factor a variants and the risk of parkinson’s disease. Neurosci. Lett. 2010, 469, 24–29. [Google Scholar]
- Alvarez, V.; Corao, A.I.; Sanchez-Ferrero, E.; De Mena, L.; Alonso-Montes, C.; Huerta, C.; Blazquez, M.; Ribacoba, R.; Guisasola, L.M.; Salvador, C; et al. Mitochondrial transcription factor a (tfam) gene variation in parkinson’s disease. Neurosci. Lett. 2008, 432, 79–82. [Google Scholar]
- Gonzalez, J.R.; Caceres, A.; Esko, T.; Cusco, I.; Puig, M.; Esnaola, M.; Reina, J.; Siroux, V.; Bouzigon, E.; Nadif, R.; et al. A common 16p11.2 inversion underlies the joint susceptibility to asthma and obesity. Am. J. Hum. Genetics 2014, 94, 361–372. [Google Scholar]
- He, Y.; Wu, J.; Dressman, D.C.; Iacobuzio-Donahue, C.; Markowitz, S.D.; Velculescu, V.E.; Diaz, L.A., Jr.; Kinzler, K.W.; Vogelstein, B.; Papadopoulos, N. Heteroplasmic mitochondrial DNA mutations in normal and tumour cells. Nature 2010, 464, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Coller, H.A.; Khrapko, K.; Bodyak, N.D.; Nekhaeva, E.; Herrero-Jimenez, P.; Thilly, W.G. High frequency of homoplasmic mitochondrial DNA mutations in human tumors can be explained without selection. Nat. Genet. 2001, 28, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Kazi, A.; Smith, D.M.; Zhong, Q.; Dou, Q.P. Inhibition of bcl-x-l phosphorylation by tea polyphenols or epigallocatechin-3-gallate is associated with prostate cancer cell apoptosis. Mol. Pharmacol. 2002, 62, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yang, L.F.; Ye, M.; Gu, H.H.; Cao, Y. Induction of apoptosis by epigallocatechin-3-gallate via mitochondrial signal transduction pathway. Prev. Med. 2004, 39, 1172–1179. [Google Scholar] [CrossRef] [PubMed]
- Nihal, M.; Ahmad, N.; Mukhtar, H.; Wood, G.S. Anti-proliferative and proapoptotic effects of (−)-epigallocatechin-3-gallate on human melanoma: Possible implications for the chemoprevention of melanoma. Int. J. Cancer 2005, 114, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.M.; Baliga, M.S.; Katiyar, S.K. Epigallocatechin-3-gallate induces apoptosis in estrogen receptor-negative human breast carcinoma cells via modulation in protein expression of p53 and bax and caspase-3 activation. Mol. Cancer Ther. 2005, 4, 81–90. [Google Scholar] [PubMed]
- Qanungo, S.; Das, M.; Haldar, S.; Basu, A. Epigallocatechin-3-gallate induces mitochondrial membrane depolarization and caspase-dependent apoptosis in pancreatic cancer cells. Carcinogenesis 2005, 26, 958–967. [Google Scholar] [CrossRef] [PubMed]
- Shankar, S.; Suthakar, G.; Srivastava, R.K. Epigallocatechin-3-gallate inhibits cell cycle and induces apoptosis in pancreatic cancer. Front. Biosci. 2007, 12, 5039–5051. [Google Scholar] [CrossRef] [PubMed]
- Ran, Z.H.; Xu, Q.; Tong, J.L.; Xiao, S.D. Apoptotic effect of epigal locatechin-3-gallate on the human gastric cancer cell line mkn45 via activation of the mitochondrial pathway. World J. Gastroentero 2007, 13, 4255–4259. [Google Scholar]
- Singh, M.; Bhui, K.; Singh, R.; Shukla, Y. Tea polyphenols enhance cisplatin chemosensitivity in cervical cancer cells via induction of apoptosis. Life Sci. 2013, 93, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Singh, R.; Bhui, K.; Tyagi, S.; Mahmood, Z.; Shukla, Y. Tea polyphenols induce apoptosis through mitochondrial pathway and by inhibiting nuclear factor-kappab and akt activation in human cervical cancer cells. Oncol. Res. 2011, 19, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Nie, S.; Yu, Q.; Xie, M. (−)-epigallocatechin-3-gallate induces apoptosis of human hepatoma cells by mitochondrial pathways related to reactive oxygen species. J. Agric. Food Chem. 2009, 57, 6685–6691. [Google Scholar] [CrossRef] [PubMed]
- Kil, I.S.; Jung, K.H.; Nam, W.S.; Park, J.W. Attenuated mitochondrial NADP+ dependent isocitrate dehydrogenase activity enhances egcg-induced apoptosis. Biochimie 2011, 93, 1808–1815. [Google Scholar] [CrossRef] [PubMed]
- Valenti, D.; de Bari, L.; Manente, G.A.; Rossi, L.; Mutti, L.; Moro, L.; Vacca, R.A. Negative modulation of mitochondrial oxidative phosphorylation by epigallocatechin-3 gallate leads to growth arrest and apoptosis in human malignant pleural mesothelioma cells. Biochim. Biophys. Acta 2013, 1832, 2085–2096. [Google Scholar] [CrossRef]
- Meng, Q.; Velalar, C.N.; Ruan, R. Effects of epigallocatechin-3-gallate on mitochondrial integrity and antioxidative enzyme activity in the aging process of human fibroblast. Free Radic. Biol. Med. 2008, 44, 1032–1041. [Google Scholar] [CrossRef] [PubMed]
- Carrasco-Pozo, C.; Mizgier, M.L.; Speisky, H.; Gotteland, M. Differential protective effects of quercetin, resveratrol, rutin and epigallocatechin gallate against mitochondrial dysfunction induced by indomethacin in caco-2 cells. Chemico-Biol. Interact. 2012, 195, 199–205. [Google Scholar]
- Fiorani, M.; Guidarelli, A.; Blasa, M.; Azzolini, C.; Candiracci, M.; Piatti, E.; Cantoni, O. Mitochondria accumulate large amounts of quercetin: Prevention of mitochondrial damage and release upon oxidation of the extramitochondrial fraction of the flavonoid. J. Nutr. Biochem. 2010, 21, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Pham, J.; Brownlow, B.; Elbayoumi, T. Mitochondria-specific pro-apoptotic activity of genistein lipidic nanocarriers. Mol. Pharm. 2013, 10, 3789–3800. [Google Scholar] [PubMed]
- Cavallito, C.J.; Bailey, J.H. Allicin—isolation and antibacterial properties. Am. J. Chem. Soc. 1944, 66, 1950–1951. [Google Scholar] [CrossRef]
- Davis, S.R. An overview of the antifungal properties of allicin and its breakdown products—the possibility of a safe and effective antifungal prophylactic. Mycoses 2005, 48, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Cho, S.J.; Kwon, H.C.; Lee, K.R.; Rhee, D.K.; Pyo, S. Caspase-independent cell death by allicin in human epithelial carcinoma cells: Involvement of pka. Cancer Lett. 2005, 224, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Oommen, S.; Anto, R.J.; Srinivas, G.; Karunagaran, D. Allicin (from garlic) induces caspase-mediated apoptosis in cancer cells. Eur. J. Pharmacol. 2004, 485, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Miron, T.; Wilchek, M.; Sharp, A.; Nakagawa, Y.; Naoi, M.; Nozawa, Y.; Akao, Y. Allicin inhibits cell growth and induces apoptosis through the mitochondrial pathway in hl60 and u937 cells. J. Nutr. Biochem. 2008, 19, 524–535. [Google Scholar] [CrossRef] [PubMed]
- Miron, T.; Rabinkov, A.; Mirelman, D.; Wilchek, M.; Weiner, L. The mode of action of allicin: Its ready permeability through phospholipid membranes may contribute to its biological activity. Biochim. Biophys. Acta 2000, 1463, 20–30. [Google Scholar] [CrossRef]
- Rabinkov, A.; Miron, T.; Mirelman, D.; Wilchek, M.; Glozman, S.; Yavin, E.; Weiner, L. S-allylmercaptoglutathione: The reaction product of allicin with glutathione possesses SH-modifying and antioxidant properties. Biochim. Biophys. Acta 2000, 1499, 144–153. [Google Scholar] [CrossRef]
- Sareen, D.; Darjatmoko, S.R.; Albert, D.M.; Polans, A.S. Mitochondria, calcium, and calpain are key mediators of resveratrol-induced apoptosis in breast cancer. Mol. Pharmacol. 2007, 72, 1466–1475. [Google Scholar] [CrossRef] [PubMed]
- Higashida, K.; Kim, S.H.; Jung, S.R.; Asaka, M.; Holloszy, J.O.; Han, D.H. Effects of resveratrol and sirt1 on pgc-1alpha activity and mitochondrial biogenesis: A reevaluation. PLoS Biol. 2013, 11, e1001603. [Google Scholar] [CrossRef] [PubMed]
- Beher, D.; Wu, J.; Cumine, S.; Kim, K.W.; Lu, S.C.; Atangan, L.; Wang, M. Resveratrol is not a direct activator of sirt1 enzyme activity. Chem. Biol. Drug Des. 2009, 74, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Pacholec, M.; Bleasdale, J.E.; Chrunyk, B.; Cunningham, D.; Flynn, D.; Garofalo, R.S.; Griffith, D.; Griffor, M.; Loulakis, P.; Pabst, B.; et al. Srt1720, srt2183, srt1460, and resveratrol are not direct activators of sirt1. J. Biol. Chem. 2010, 285, 8340–8351. [Google Scholar] [CrossRef]
- Jager, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. Amp-activated protein kinase (ampk) action in skeletal muscle via direct phosphorylation of pgc-1alpha. Proc. Nat. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of pgc-1alpha and sirt1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating sirt1 and pgc-1alpha. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Murase, T.; Haramizu, S.; Ota, N.; Hase, T. Suppression of the aging-associated decline in physical performance by a combination of resveratrol intake and habitual exercise in senescence-accelerated mice. Biogerontology 2009, 10, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Hart, N.; Sarga, L.; Csende, Z.; Koltai, E.; Koch, L.G.; Britton, S.L.; Davies, K.J.; Kouretas, D.; Wessner, B.; Radak, Z. Resveratrol enhances exercise training responses in rats selectively bred for high running performance. Food Chem. Toxicol. 2013, 61, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Um, J.H.; Park, S.J.; Kang, H.; Yang, S.; Foretz, M.; McBurney, M.W.; Kim, M.K.; Viollet, B.; Chung, J.H. Amp-activated protein kinase-deficient mice are resistant to the metabolic effects of resveratrol. Diabetes 2010, 59, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Menzies, K.J.; Singh, K.; Saleem, A.; Hood, D.A. Sirtuin 1-mediated effects of exercise and resveratrol on mitochondrial biogenesis. J. Biol. Chem. 2013, 288, 6968–6979. [Google Scholar] [CrossRef] [PubMed]
- Hart, N.; Sarga, L.; Csende, Z.; Koch, L.G.; Britton, S.L.; Davies, K.J.; Radak, Z. Resveratrol attenuates exercise-induced adaptive responses in rats selectively bred for low running performance. Dose-Response 2014, 12, 57–71. [Google Scholar]
- Lim, H.W.; Lim, H.Y.; Wong, K.P. Uncoupling of oxidative phosphorylation by curcumin: Implication of its cellular mechanism of action. Biochem. Biophys. Res. Commun. 2009, 389, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Negrette-Guzman, M.; Huerta-Yepez, S.; Tapia, E.; Pedraza-Chaverri, J. Modulation of mitochondrial functions by the indirect antioxidant sulforaphane: A seemingly contradictory dual role and an integrative hypothesis. Free Rad. Biol. Med. 2013, 65, 1078–1089. [Google Scholar]
- Pham, T.X.; Lee, J. Dietary regulation of histone acetylases and deacetylases for the prevention of metabolic diseases. Nutrients 2012, 4, 1868–1886. [Google Scholar] [CrossRef] [PubMed]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Holmstrom, K.M.; Baird, L.; Zhang, Y.; Hargreaves, I.; Chalasani, A.; Land, J.M.; Stanyer, L.; Yamamoto, M.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 impacts cellular bioenergetics by controlling substrate availability for mitochondrial respiration. Biol. Open 2013, 2, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Brose, R.D.; Shin, G.; McGuinness, M.C.; Schneidereith, T.; Purvis, S.; Dong, G.X.; Keefer, J.; Spencer, F.; Smith, K.D. Activation of the stress proteome as a mechanism for small molecule therapeutics. Human Mol. Genet. 2012, 21, 4237–4252. [Google Scholar] [CrossRef]
- Li, Y.G.; Zhu, W.; Tao, J.P.; Xin, P.; Liu, M.Y.; Li, J.B.; Wei, M. Resveratrol protects cardiomyocytes from oxidative stress through sirt1 and mitochondrial biogenesis signaling pathways. Biochem. Biophys. Res. Commun. 2013, 438, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Leonard, S.S.; Xia, C.; Jiang, B.H.; Stinefelt, B.; Klandorf, H.; Harris, G.K.; Shi, X. Resveratrol scavenges reactive oxygen species and effects radical-induced cellular responses. Biochem. Biophys. Res. Commun. 2003, 309, 1017–1026. [Google Scholar] [CrossRef] [PubMed]
- Vina, J.; Gomez-Cabrera, M.C.; Borras, C.; Froio, T.; Sanchis-Gomar, F.; Martinez-Bello, V.E.; Pallardo, F.V. Mitochondrial biogenesis in exercise and in ageing. Adv. Drug Deliv. Rev. 2009, 61, 1369–1374. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.W.; Ho, W.Y.; Su, Y.T.; Lu, P.J.; Chen, B.Z.; Cheng, W.H.; Lu, W.H.; Sun, G.C.; Yeh, T.C.; Hsiao, M.; et al. Resveratrol decreases fructose-induced oxidative stress, mediated by nadph oxidase via an ampk-dependent mechanism. Br. J. Pharmacol. 2014, 171, 2739–2750. [Google Scholar] [CrossRef] [PubMed]
- Quincozes-Santos, A.; Bobermin, L.D.; Tramontina, A.C.; Wartchow, K.M.; Tagliari, B.; Souza, D.O.; Wyse, A.T.; Goncalves, C.A. Oxidative stress mediated by nmda, ampa/ka channels in acute hippocampal slices: Neuroprotective effect of resveratrol. Toxicol. In Vitro 2014, 28, 544–551. [Google Scholar]
- Qin, S.; Lu, Y.; Rodrigues, G.A. Resveratrol protects rpe cells from sodium iodate by modulating pparalpha and ppardelta. Exp. Eye Res. 2014, 118, 100–108. [Google Scholar]
- Mokni, M.; Hamlaoui, S.; Karkouch, I.; Amri, M.; Marzouki, L.; Limam, F.; Aouani, E. Resveratrol provides cardioprotection after ischemia/reperfusion injury via modulation of antioxidant enzyme activities. Iran. J. Pharm. Res. 2013, 12, 867–875. [Google Scholar] [PubMed]
- Shakibaei, M.; Harikumar, K.B.; Aggarwal, B.B. Resveratrol addiction: To die or not to die. Mol. Nutr. Food Res. 2009, 53, 115–128. [Google Scholar] [CrossRef]
- Morin, C.; Zini, R.; Albengres, E.; Bertelli, A.A.; Bertelli, A.; Tillement, J.P. Evidence for resveratrol-induced preservation of brain mitochondria functions after hypoxia-reoxygenation. Drugs Exp. Clin. Res. 2003, 29, 227–233. [Google Scholar] [PubMed]
- Gatson, J.W.; Liu, M.M.; Abdelfattah, K.; Wigginton, J.G.; Smith, S.; Wolf, S.; Minei, J.P. Resveratrol decreases inflammation in the brain of mice with mild traumatic brain injury. J. Trauma Acute Care Surg. 2013, 74, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Ates, O.; Cayli, S.; Altinoz, E.; Gurses, I.; Yucel, N.; Sener, M.; Kocak, A.; Yologlu, S. Neuroprotection by resveratrol against traumatic brain injury in rats. Mol. Cell. Biochem. 2007, 294, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wang, H.; Wu, Q.; Lu, Y.; Nie, J.; Xie, X.; Shi, J. Resveratrol protects cortical neurons against microglia-mediated neuroinflammation. Phytother. Res. 2013, 27, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tian, F.; Xiao, Q.; Hu, Y.; Li, J.; Jiang, F.; Liu, Y. Exploiting the role of resveratrol in rat mitochondrial permeability transition. J. Membr. Biol. 2013, 246, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Tian, X.; Huang, X.; Yan, F.; Qiao, D. Resveratrol-induced mitochondrial dysfunction and apoptosis are associated with Ca2+ and mcicr-mediated mpt activation in hepg2 cells. Mol. Cell. Biochem. 2007, 302, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.Y.; Lu, G.H.; Wu, Y.Q.; Zheng, Y.; Xu, K.; Wu, L.J.; Jiang, Z.Y.; Feng, R.; Zhou, J.Y. Curcumin induces mitochondria pathway mediated cell apoptosis in a549 lung adenocarcinoma cells. Oncol. Rep. 2010, 23, 1285–1292. [Google Scholar] [PubMed]
- Gopal, P.K.; Paul, M.; Paul, S. Curcumin induces caspase mediated apoptosis in jurkat cells by disrupting the redox balance. Asian Pac. J. Cancer Prev. 2014, 15, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Thayyullathil, F.; Chathoth, S.; Hago, A.; Patel, M.; Galadari, S. Rapid reactive oxygen species (ros) generation induced by curcumin leads to caspase-dependent and -independent apoptosis in l929 cells. Free Rad. Biol. Med. 2008, 45, 1403–1412. [Google Scholar] [CrossRef] [PubMed]
- Hirzel, E.; Lindinger, P.W.; Maseneni, S.; Giese, M.; Rhein, V.V.; Eckert, A.; Hoch, M.; Krahenbuhl, S.; Eberle, A.N. Differential modulation of ros signals and other mitochondrial parameters by the antioxidants mitoq, resveratrol and curcumin in human adipocytes. J. Recept. Signal Transduct. Res. 2013, 33, 304–312. [Google Scholar] [CrossRef]
- Chan, W.H.; Wu, H.J.; Hsuuw, Y.D. Curcumin inhibits ros formation and apoptosis in methylglyoxal-treated human hepatoma g2 cells. Ann. New York Acad. Sci. 2005, 1042, 372–378. [Google Scholar] [CrossRef]
- Sakurai, R.; Villarreal, P.; Husain, S.; Liu, J.; Sakurai, T.; Tou, E.; Torday, J.S.; Rehan, V.K. Curcumin protects the developing lung against long-term hyperoxic injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L301–L311. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.M.; Shin, D.Y.; Lee, S.J.; Joe, Y.; Zheng, M.; Yim, J.H.; Callaway, Z.; Chung, H.T. Curcumin protects retinal pigment epithelial cells against oxidative stress via induction of heme oxygenase-1 expression and reduction of reactive oxygen. Mol. Vis. 2012, 18, 901–908. [Google Scholar] [PubMed]
- Jiang, H.; Tian, X.; Guo, Y.; Duan, W.; Bu, H.; Li, C. Activation of nuclear factor erythroid 2-related factor 2 cytoprotective signaling by curcumin protect primary spinal cord astrocytes against oxidative toxicity. Biol. Pharm. Bull. 2011, 34, 1194–1197. [Google Scholar] [CrossRef] [PubMed]
- Cerny, D.; Lekic, N.; Vanova, K.; Muchova, L.; Horinek, A.; Kmonickova, E.; Zidek, Z.; Kamenikova, L.; Farghali, H. Hepatoprotective effect of curcumin in lipopolysaccharide/-galactosamine model of liver injury in rats: Relationship to ho-1/co antioxidant system. Fitoterapia 2011, 82, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Chang, Z.; Xing, J.; Yu, X. Curcumin induces osteosarcoma mg63 cells apoptosis via ros/cyto-c/caspase-3 pathway. Tumour Biol. 2014, 35, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Manach, C.; Williamson, G.; Morand, C.; Scalbert, A.; Remesy, C. Bioavailability and bioefficacy of polyphenols in humans. I. Review of 97 bioavailability studies. Am. J. Clin. Nutr. 2005, 81, 230S–242S. [Google Scholar]
- Klickovic, U.; Doberer, D.; Gouya, G.; Aschauer, S.; Weisshaar, S.; Storka, A.; Bilban, M.; Wolzt, M. Human pharmacokinetics of high dose oral curcumin and its effect on heme oxygenase-1 expression in healthy male subjects. BioMed Res. Int. 2014, 2014, 458592. [Google Scholar]
- Walle, T.; Hsieh, F.; DeLegge, M.H.; Oatis, J.E., Jr.; Walle, U.K. High absorption but very low bioavailability of oral resveratrol in humans. Drug Metabol. Disposit. 2004, 32, 1377–1382. [Google Scholar] [CrossRef]
- Shelma, R.; Sharma, C.P. In vitro and in vivo evaluation of curcumin loaded lauroyl sulphated chitosan for enhancing oral bioavailability. Carbohydr. Polym. 2013, 95, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhang, T.; Liu, L.; Wang, X.; Wu, P.; Chen, Z.; Ni, C.; Zhang, J.; Hu, F.; Huang, J. Novel micelle formulation of curcumin for enhancing antitumor activity and inhibiting colorectal cancer stem cells. Int. J. Nanomed. 2012, 7, 4487–4497. [Google Scholar]
- Verderio, P.; Bonetti, P.; Colombo, M.; Pandolfi, L.; Prosperi, D. Intracellular drug release from curcumin-loaded plga nanoparticles induces g2/m block in breast cancer cells. Biomacromolecules 2013, 14, 672–682. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Pai, R.S. Optimized plga nanoparticle platform for orally dosed trans-resveratrol with enhanced bioavailability potential. Expert Opin. Drug Deliv. 2014, 11, 647–659. [Google Scholar] [PubMed]
- Reddy, C.A.; Somepalli, V.; Golakoti, T.; Kanugula, A.K.; Karnewar, S.; Rajendiran, K.; Vasagiri, N.; Prabhakar, S.; Kuppusamy, P.; Kotamraju, S.; et al. Mitochondrial-targeted curcuminoids: A strategy to enhance bioavailability and anticancer efficacy of curcumin. PloS One 2014, 9, e89351. [Google Scholar] [CrossRef] [PubMed]
- Boddupalli, S.; Mein, J.R.; Lakkanna, S.; James, D.R. Induction of phase 2 antioxidant enzymes by broccoli sulforaphane: Perspectives in maintaining the antioxidant activity of vitamins a, c, and e. Front. Genet. 2012, 3, 7. [Google Scholar] [CrossRef] [PubMed]
- Scarpulla, R.C. Nuclear activators and coactivators in mammalian mitochondrial biogenesis. Biochim. Biophys. Acta 2002, 1576, 1–14. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Transcriptional activators and coactivators in the nuclear control of mitochondrial function in mammalian cells. Gene 2002, 286, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Scarpulla, R.C. Nuclear control of respiratory chain expression in mammalian cells. J. Bioenerg. Biomembr. 1997, 29, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator pgc-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Boss, O.; Hagen, T.; Lowell, B.B. Uncoupling proteins 2 and 3: Potential regulators of mitochondrial energy metabolism. Diabetes 2000, 49, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Jastroch, M.; Wuertz, S.; Kloas, W.; Klingenspor, M. Uncoupling protein 1 in fish uncovers an ancient evolutionary history of mammalian nonshivering thermogenesis. Physiol. Genomics 2005, 22, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Nedergaard, J.; Golozoubova, V.; Matthias, A.; Asadi, A.; Jacobsson, A.; Cannon, B. Ucp1: The only protein able to mediate adaptive non-shivering thermogenesis and metabolic inefficiency. Biochim. Biophys. Acta 2001, 1504, 82–106. [Google Scholar] [CrossRef]
- Duncan, J.G.; Fong, J.L.; Medeiros, D.M.; Finck, B.N.; Kelly, D.P. Insulin-resistant heart exhibits a mitochondrial biogenic response driven by the peroxisome proliferator-activated receptor-alpha/pgc-1alpha gene regulatory pathway. Circulation 2007, 115, 909–917. [Google Scholar] [CrossRef]
- Kelly, L.J.; Vicario, P.P.; Thompson, G.M.; Candelore, M.R.; Doebber, T.W.; Ventre, J.; Wu, M.S.; Meurer, R.; Forrest, M.J.; Conner, M.W.; et al. Peroxisome proliferator-activated receptors gamma and alpha mediate in vivo regulation of uncoupling protein (ucp-1, ucp-2, ucp-3) gene expression. Endocrinol. 1998, 139, 4920–4927. [Google Scholar]
- Brun, S.; Carmona, M.C.; Mampel, T.; Vinas, O.; Giralt, M.; Iglesias, R.; Villarroya, F. Activators of peroxisome proliferator-activated receptor-alpha induce the expression of the uncoupling protein-3 gene in skeletal muscle: A potential mechanism for the lipid intake-dependent activation of uncoupling protein-3 gene expression at birth. Diabetes 1999, 48, 1217–1222. [Google Scholar] [CrossRef] [PubMed]
- Monsalve, M.; Wu, Z.; Adelmant, G.; Puigserver, P.; Fan, M.; Spiegelman, B.M. Direct coupling of transcription and mrna processing through the thermogenic coactivator pgc-1. Mol. Cell. 2000, 6, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Guha, M.; Avadhani, N.G. Mitochondrial retrograde signaling at the crossroads of tumor bioenergetics, genetics and epigenetics. Mitochondrion 2013, 13, 577–591. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.J.; Wang, X.; Kaufman, B.A.; Butow, R.A. Aconitase couples metabolic regulation to mitochondrial DNA maintenance. Science 2005, 307, 714–717. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.W.; Yao, Z.; Vicencio, J.M.; Karkucinska-Wieckowska, A.; Szabadkai, G. Pgc-1 family coactivators and cell fate: Roles in cancer, neurodegeneration, cardiovascular disease and retrograde mitochondria-nucleus signalling. Mitochondrion 2012, 12, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Dugas, B.; Charbonnier, S.; Baarine, M.; Ragot, K.; Delmas, D.; Menetrier, F.; Lherminier, J.; Malvitte, L.; Khalfaoui, T.; Bron, A.; et al. Effects of oxysterols on cell viability, inflammatory cytokines, vegf, and reactive oxygen species production on human retinal cells: Cytoprotective effects and prevention of vegf secretion by resveratrol. Eur. J. Nutr. 2010, 49, 435–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csaki, C.; Keshishzadeh, N.; Fischer, K.; Shakibaei, M. Regulation of inflammation signalling by resveratrol in human chondrocytes in vitro. Biochem. Pharmacol. 2008, 75, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Rahman, I.; Biswas, S.K.; Kirkham, P.A. Regulation of inflammation and redox signaling by dietary polyphenols. Biochem. Pharmacol. 2006, 72, 1439–1452. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.H.; Song, I.S.; Kim, H.K.; Lee, S.R.; Song, S.; Suh, H.; Yoon, Y.G.; Yoo, Y.H.; Kim, N.; Rhee, B.D.; et al. An analogue of resveratrol hs-1793 exhibits anticancer activity against mcf-7 cells via inhibition of mitochondrial biogenesis gene expression. Mol. Cells 2012, 34, 357–365. [Google Scholar] [PubMed]
- Darvekar, S.R.; Elvenes, J.; Brenne, H.B.; Johansen, T.; Sjottem, E. Spbp is a sulforaphane induced transcriptional coactivator of nrf2 regulating expression of the autophagy receptor p62/sqstm1. PloS One 2014, 9, e85262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puissant, A.; Fenouille, N.; Auberger, P. When autophagy meets cancer through p62/sqstm1. Am. J. Cancer Res. 2012, 2, 397–413. [Google Scholar] [PubMed]
- Das, S.; Mitrovsky, G.; Vasanthi, H.R.; Das, D.K. Antiaging properties of a grape-derived antioxidant are regulated by mitochondrial balance of fusion and fission leading to mitophagy triggered by a signaling network of sirt1-sirt3-foxo3-pink1-parkin. Oxid. Med. Cell. Longev. 2014, 2014, 345105. [Google Scholar]
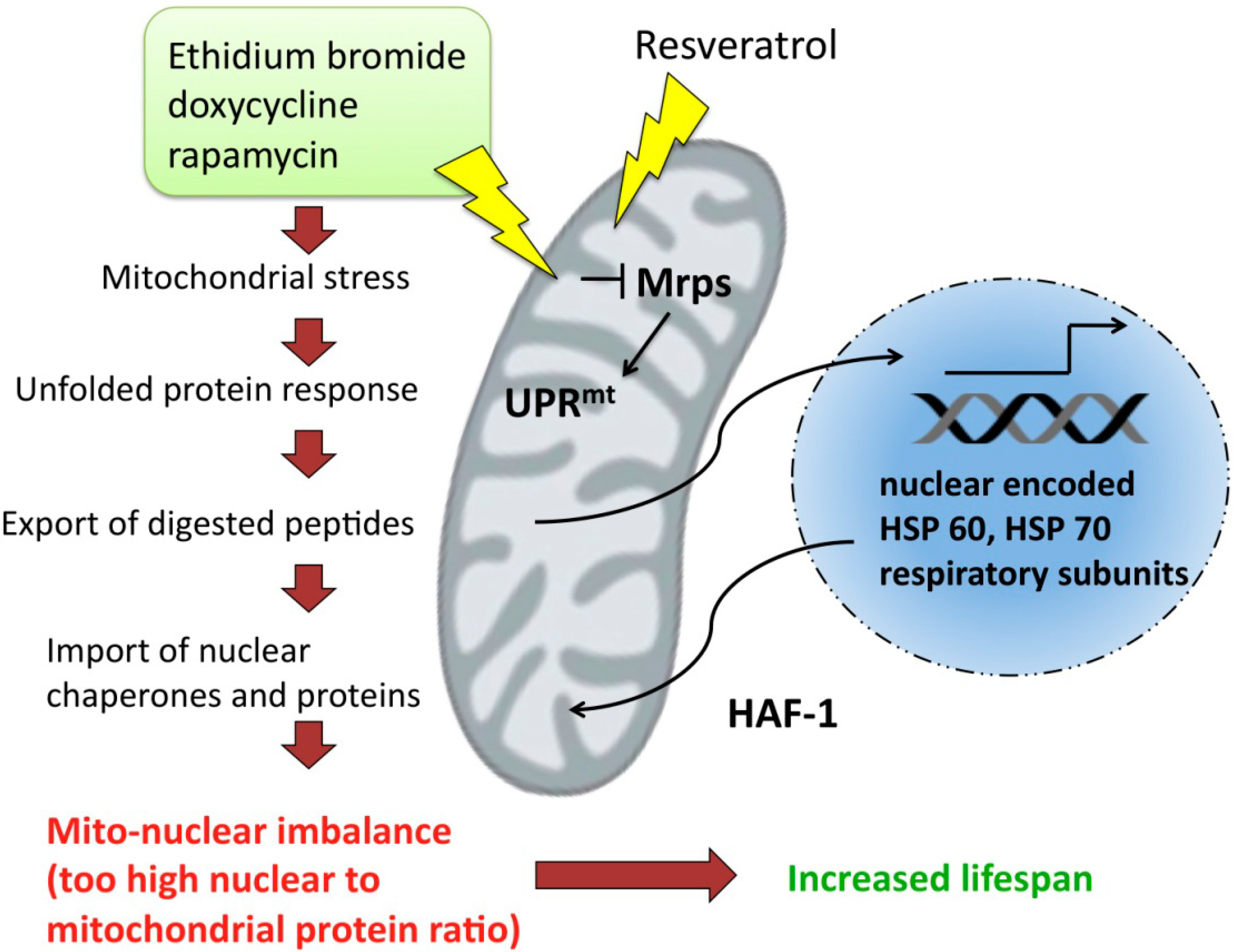
- Kirstein-Miles, J.; Morimoto, R.I. Peptides signal mitochondrial stress. Cell Metabol. 2010, 11, 177–178. [Google Scholar]
- Houtkooper, R.H.; Mouchiroud, L.; Ryu, D.; Moullan, N.; Katsyuba, E.; Knott, G.; Williams, R.W.; Auwerx, J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 2013, 497, 451–457. [Google Scholar] [CrossRef]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar] [PubMed]
- Robida-Stubbs, S.; Glover-Cutter, K.; Lamming, D.W.; Mizunuma, M.; Narasimhan, S.D.; Neumann-Haefelin, E.; Sabatini, D.M.; Blackwell, T.K. Tor signaling and rapamycin influence longevity by regulating skn-1/nrf and daf-16/foxo. Cell Metabol. 2012, 15, 713–724. [Google Scholar]
- Kirchman, P.A.; Kim, S.; Lai, C.Y.; Jazwinski, S.M. Interorganelle signaling is a determinant of longevity in saccharomyces cerevisiae. Genetics 1999, 152, 179–190. [Google Scholar] [PubMed]
- Copeland, J.M.; Cho, J.; Lo, T., Jr.; Hur, J.H.; Bahadorani, S.; Arabyan, T.; Rabie, J.; Soh, J.; Walker, D.W. Extension of drosophila life span by rnai of the mitochondrial respiratory chain. Curr. Biol. 2009, 19, 1591–1598. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Bussiere, F.; Hekimi, S. Mitochondrial electron transport is a key determinant of life span in caenorhabditis elegans. Dev. Cell 2001, 1, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Lapointe, J.; Stepanyan, Z.; Bigras, E.; Hekimi, S. Reversal of the mitochondrial phenotype and slow development of oxidative biomarkers of aging in long-lived mclk1+/− mice. J. Biol. Chem. 2009, 284, 20364–20374. [Google Scholar] [CrossRef] [PubMed]
- Van Raamsdonk, J.M.; Hekimi, S. Deletion of the mitochondrial superoxide dismutase sod-2 extends lifespan in caenorhabditis elegans. PLoS Genetics 2009, 5, e1000361. [Google Scholar]
- Yang, W.; Li, J.; Hekimi, S. A measurable increase in oxidative damage due to reduction in superoxide detoxification fails to shorten the life span of long-lived mitochondrial mutants of caenorhabditis elegans. Genetics 2007, 177, 2063–2074. [Google Scholar] [CrossRef] [PubMed]
- Durieux, J.; Wolff, S.; Dillin, A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 2011, 144, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Tamir, S.; Zuris, J.A.; Agranat, L.; Lipper, C.H.; Conlan, A.R.; Michaeli, D.; Harir, Y.; Paddock, M.L.; Mittler, R.; Cabantchik, Z.I.; et al. Nutrient-deprivation autophagy factor-1 (naf-1): Biochemical properties of a novel cellular target for anti-diabetic drugs. PloS One 2013, 8, e61202. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Pestell, T.G.; Lisanti, M.P.; Pestell, R.G. Cancer stem cells. Int. J. Biochem. Cell Biol. 2012, 44, 2144–2151. [Google Scholar] [CrossRef] [PubMed]
- Bruce, W.R.; Van Der Gaag, H. A quantitative assay for the number of murine lymphoma cells capable of proliferation in vivo. Nature 1963, 199, 79–80. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into scid mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nature Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Eramo, A.; Lotti, F.; Sette, G.; Pilozzi, E.; Biffoni, M.; Di Virgilio, A.; Conticello, C.; Ruco, L.; Peschle, C.; De Maria, R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008, 15, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.S.; Espinosa, I.; Chao, M.; Wong, D.; Ailles, L.; Diehn, M.; Gill, H.; Presti, J., Jr.; Chang, H.Y.; van de Rijn, M.; et al. Identification, molecular characterization, clinical prognosis, and therapeutic targeting of human bladder tumor-initiating cells. Proc. Nat. Acad. Sci. USA 2009, 106, 14016–14021. [Google Scholar] [CrossRef] [PubMed]
- Gu, G.; Yuan, J.; Wills, M.; Kasper, S. Prostate cancer cells with stem cell characteristics reconstitute the original human tumor in vivo. Cancer Res. 2007, 67, 4807–4815. [Google Scholar] [CrossRef]
- Zimmerer, R.M.; Korn, P.; Demougin, P.; Kampmann, A.; Kokemuller, H.; Eckardt, A.M.; Gellrich, N.C.; Tavassol, F. Functional features of cancer stem cells in melanoma cell lines. Cancer Cell Int. 2013, 13, 78. [Google Scholar] [PubMed]
- Zhang, S.; Balch, C.; Chan, M.W.; Lai, H.C.; Matei, D.; Schilder, J.M.; Yan, P.S.; Huang, T.H.; Nephew, K.P. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008, 68, 4311–4320. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, A.; Chikamatsu, K.; Sakakura, K.; Hatsushika, K.; Takahashi, G.; Masuyama, K. Expansion and characterization of cancer stem-like cells in squamous cell carcinoma of the head and neck. Oral Oncol. 2009, 45, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar]
- Dalerba, P.; Dylla, S.J.; Park, I.K.; Liu, R.; Wang, X.; Cho, R.W.; Hoey, T.; Gurney, A.; Huang, E.H.; Simeone, D.M.; et al. Phenotypic characterization of human colorectal cancer stem cells. Proc. Nat. Acad. Sci. USA 2007, 104, 10158–10163. [Google Scholar] [CrossRef]
- Sainz, B., Jr.; Heeschen, C. Standing out from the crowd: Cancer stem cells in hepatocellular carcinoma. Cancer cell 2013, 23, 431–433. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Wang, X.W. Cancer stem cells in the development of liver cancer. J. Clin. Invest. 2013, 123, 1911–1918. [Google Scholar] [CrossRef] [PubMed]
- Ponti, D.; Costa, A.; Zaffaroni, N.; Pratesi, G.; Petrangolini, G.; Coradini, D.; Pilotti, S.; Pierotti, M.A.; Daidone, M.G. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005, 65, 5506–5511. [Google Scholar] [CrossRef] [PubMed]
- Soltysova, A.; Altanerova, V.; Altaner, C. Cancer stem cells. Neoplasma 2005, 52, 435–440. [Google Scholar] [PubMed]
- Nakagawara, A.; Ohira, M. Comprehensive genomics linking between neural development and cancer: Neuroblastoma as a model. Cancer Lett. 2004, 204, 213–224. [Google Scholar] [CrossRef]
- Cozzio, A.; Passegue, E.; Ayton, P.M.; Karsunky, H.; Cleary, M.L.; Weissman, I.L. Similar mll-associated leukemias arising from self-renewing stem cells and short-lived myeloid progenitors. Genes Dev. 2003, 17, 3029–3035. [Google Scholar] [CrossRef]
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer stem cells—perspectives on current status and future directions: Aacr workshop on cancer stem cells. Cancer Res. 2006, 66, 9339–9344. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhang, Y. Cancer stem cells: Models, mechanisms and implications for improved treatment. Cell Cycle 2008, 7, 1360–1370. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Qian, F.; Tchabo, N.; Mhawech-Fauceglia, P.; Beck, A.; Qian, Z.; Wang, X.; Huss, W.J.; Lele, S.B.; Morrison, C.D.; et al. Ovarian cancer spheroid cells with stem cell-like properties contribute to tumor generation, metastasis and chemotherapy resistance through hypoxia-resistant metabolism. PloS One 2014, 9, e84941. [Google Scholar] [CrossRef] [PubMed]
- Luk, S.U.; Yap, W.N.; Chiu, Y.T.; Lee, D.T.; Ma, S.; Lee, T.K.; Vasireddy, R.S.; Wong, Y.C.; Ching, Y.P.; Nelson, C.; et al. Gamma-tocotrienol as an effective agent in targeting prostate cancer stem cell-like population. Int. J. Cancer. J. Cancer 2011, 128, 2182–2191. [Google Scholar] [CrossRef]
- Todaro, M.; Alea, M.P.; Di Stefano, A.B.; Cammareri, P.; Vermeulen, L.; Iovino, F.; Tripodo, C.; Russo, A.; Gulotta, G.; Medema, J.P.; et al. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell 2007, 1, 389–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, T.M.; McBride, W.H.; Pajonk, F. The response of cd24(-/low)/cd44+ breast cancer-initiating cells to radiation. J. Nat. Cancer Inst. 2006, 98, 1777–1785. [Google Scholar] [CrossRef] [PubMed]
- Wanet, A.; Tacheny, A.; Arnould, T.; Renard, P. Mir-212/132 expression and functions: Within and beyond the neuronal compartment. Nucleic Acids Res. 2012, 40, 4742–4753. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.Q.; Li, Q.; Wang, G.H.; Sun, F.F.; Huang, G.J.; Bian, X.W.; Yu, S.C.; Qian, G.S. Mitochondrial and energy metabolism-related properties as novel indicators of lung cancer stem cells. Int. J. Cancer. J. Int. Cancer 2011, 129, 820–831. [Google Scholar] [CrossRef]
- Tang, S.N.; Singh, C.; Nall, D.; Meeker, D.; Shankar, S.; Srivastava, R.K. The dietary bioflavonoid quercetin synergizes with epigallocathechin gallate (egcg) to inhibit prostate cancer stem cell characteristics, invasion, migration and epithelial-mesenchymal transition. J. Mol. Signaling 2010, 5, 14. [Google Scholar] [CrossRef]
- Shankar, S.; Nall, D.; Tang, S.N.; Meeker, D.; Passarini, J.; Sharma, J.; Srivastava, R.K. Resveratrol inhibits pancreatic cancer stem cell characteristics in human and krasg12d transgenic mice by inhibiting pluripotency maintaining factors and epithelial-mesenchymal transition. PloS One 2011, 6, e16530. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.K.; Tang, S.N.; Zhu, W.; Meeker, D.; Shankar, S. Sulforaphane synergizes with quercetin to inhibit self-renewal capacity of pancreatic cancer stem cells. Front. Biosci. 2011, 3, 515–528. [Google Scholar]
- Alvero, A.B.; Montagna, M.K.; Holmberg, J.C.; Craveiro, V.; Brown, D.; Mor, G. Targeting the mitochondria activates two independent cell death pathways in ovarian cancer stem cells. Mol. Cancer Ther. 2011, 10, 1385–1393. [Google Scholar] [CrossRef] [PubMed]
- Fong, D.; Yeh, A.; Naftalovich, R.; Choi, T.H.; Chan, M.M. Curcumin inhibits the side population (sp) phenotype of the rat c6 glioma cell line: Towards targeting of cancer stem cells with phytochemicals. Cancer Lett. 2010, 293, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Bleau, A.M.; Hambardzumyan, D.; Ozawa, T.; Fomchenko, E.I.; Huse, J.T.; Brennan, C.W.; Holland, E.C. Pten/pi3k/akt pathway regulates the side population phenotype and abcg2 activity in glioma tumor stem-like cells. Cell Stem Cell 2009, 4, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Resveratrol and derivatives for the prevention and treatment of cancer. Drug Discov. Today 2010, 15, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Agarwal, A.K.; Xu, T.; Feng, Q.; Baerson, S.R.; Duke, S.O.; Rimando, A.M. Identification of molecular pathways affected by pterostilbene, a natural dimethylether analog of resveratrol. BMC Med. Genom. 2008, 1, 7. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Grabacka, M.M.; Gawin, M.; Pierzchalska, M. Phytochemical Modulators of Mitochondria: The Search for Chemopreventive Agents and Supportive Therapeutics. Pharmaceuticals 2014, 7, 913-942. https://doi.org/10.3390/ph7090913
Grabacka MM, Gawin M, Pierzchalska M. Phytochemical Modulators of Mitochondria: The Search for Chemopreventive Agents and Supportive Therapeutics. Pharmaceuticals. 2014; 7(9):913-942. https://doi.org/10.3390/ph7090913
Chicago/Turabian StyleGrabacka, Maja M., Malgorzata Gawin, and Malgorzata Pierzchalska. 2014. "Phytochemical Modulators of Mitochondria: The Search for Chemopreventive Agents and Supportive Therapeutics" Pharmaceuticals 7, no. 9: 913-942. https://doi.org/10.3390/ph7090913