
The Role of Alarmins in the Pathogenesis of Rheumatoid Arthritis, Osteoarthritis, and Psoriasis

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
3. Brief Overview of Alarmins
4. Alarmins and Rheumatoid Arthritis
4.1. High-Mobility Group Box 1
4.2. S100 Proteins
4.3. Interleukin-33
5. Alarmins and Osteoarthritis
5.1. High-Mobility Group Box 1
5.2. S100 Proteins
5.3. Interleukin-33
6. Alarmins and Psoriasis
6.1. Typical Features and Epidemiology of Psoriasis
6.2. Pathogenesis of Psoriasis
6.3. High-Mobility Group Box 1
6.4. S100 Proteins
6.5. Cathelicidin
6.6. Heat Shock Proteins
6.7. Defensins
6.8. Thymic Stromal Lymphopoietin
6.9. Interleukin-33
6.10. Interleukin-1α
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, D.; Han, Z.; Oppenheim, J.J. Alarmins and immunity. Immunol. Rev. 2017, 280, 41–56. [Google Scholar] [CrossRef]
- Oppenheim, J.J.; Yang, D. Alarmins: Chemotactic activators of immune responses. Curr. Opin. Immunol. 2005, 17, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Chaly, Y.V.; Paleolog, E.M.; Kolesnikova, T.S.; Tikhonov, I.I.; Petratchenko, E.V.; Voitenok, N.N. Neutrophil alpha-defensin human neutrophil peptide modulates cytokine production in human monocytes and adhesion molecule expression in endothelial cells. Eur. Cytokine Netw. 2000, 11, 257–266. [Google Scholar]
- Andersson, U.; Wang, H.; Palmblad, K.; Aveberger, A.C.; Bloom, O.; Erlandsson-Harris, H.; Janson, A.; Kokkola, R.; Zhang, M.; Yang, H.; et al. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J. Exp. Med. 2000, 192, 565–570. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef]
- De Almeida, D.E.; Ling, S.; Pi, X.; Hartmann-Scruggs, A.M.; Pumpens, P.; Holoshitz, J. Immune dysregulation by the rheumatoid arthritis shared epitope. J. Immunol. 2010, 185, 1927–1934. [Google Scholar] [CrossRef] [PubMed]
- Barik, R.R.; Bhatt, L.K. Emerging epigenetic targets in rheumatoid arthritis. Rheumatol. Int. 2021, 41, 2047–2067. [Google Scholar] [CrossRef]
- Klein, K.; Gay, S. Epigenetics in rheumatoid arthritis. Curr. Opin. Rheumatol. 2015, 27, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Kang, R.; Tang, D. The mechanism of HMGB1 secretion and release. Exp. Mol. Med. 2022, 54, 91–102. [Google Scholar] [CrossRef]
- Yang, H.; Wang, H.; Andersson, U. Targeting Inflammation Driven by HMGB1. Front. Immunol. 2020, 11, 484. [Google Scholar] [CrossRef]
- Kawasaki, H.; Nakayama, S.; Kretsinger, R.H. Classification and evolution of EF-hand proteins. Biometals 1998, 11, 277–295. [Google Scholar] [CrossRef] [PubMed]
- Marenholz, I.; Heizmann, C.W.; Fritz, G. S100 proteins in mouse and man: From evolution to function and pathology (including an update of the nomenclature). Biochem. Biophys. Res. Commun. 2004, 322, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Ali, S.A. Multifunctional Role of S100 Protein Family in the Immune System: An Update. Cells 2022, 11, 2274. [Google Scholar] [CrossRef] [PubMed]
- Gilston, B.A.; Skaar, E.P.; Chazin, W.J. Binding of transition metals to S100 proteins. Sci. China Life Sci. 2016, 59, 792–801. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, L.C.; Donor, M.T.; Prell, J.S.; Harms, M.J. Multiple Evolutionary Origins of Ubiquitous Cu2+ and Zn2+ Binding in the S100 Protein Family. PLoS ONE 2016, 11, e0164740. [Google Scholar] [CrossRef]
- Fritz, G.; Botelho, H.M.; Morozova-Roche, L.A.; Gomes, C.M. Natural and amyloid self-assembly of S100 proteins: Structural basis of functional diversity. FEBS J. 2010, 277, 4578–4590. [Google Scholar] [CrossRef] [PubMed]
- Donato, R.; Cannon, B.R.; Sorci, G.; Riuzzi, F.; Hsu, K.; Weber, D.J.; Geczy, C.L. Functions of S100 proteins. Curr. Mol. Med. 2013, 13, 24–57. [Google Scholar] [CrossRef] [PubMed]
- Bou-Dargham, M.J.; Khamis, Z.I.; Cognetta, A.B.; Sang, Q.A. The Role of Interleukin-1 in Inflammatory and Malignant Human Skin Diseases and the Rationale for Targeting Interleukin-1 Alpha. Med. Res. Rev. 2017, 37, 180–216. [Google Scholar] [CrossRef] [PubMed]
- Perez, F.; Ruera, C.N.; Miculan, E.; Carasi, P.; Dubois-Camacho, K.; Garbi, L.; Guzman, L.; Hermoso, M.A.; Chirdo, F.G. IL-33 Alarmin and Its Active Proinflammatory Fragments Are Released in Small Intestine in Celiac Disease. Front. Immunol. 2020, 11, 581445. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, G.; Yamamura, K.; Kawamura, K.; Kido-Nakahara, M.; Ito, T.; Nakahara, T. Regulatory Mechanism of the IL-33-IL-37 Axis via Aryl Hydrocarbon Receptor in Atopic Dermatitis and Psoriasis. Int. J. Mol. Sci. 2023, 24, 14633. [Google Scholar] [CrossRef]
- Borgia, F.; Custurone, P.; Peterle, L.; Pioggia, G.; Gangemi, S. Role of Epithelium-Derived Cytokines in Atopic Dermatitis and Psoriasis: Evidence and Therapeutic Perspectives. Biomolecules 2021, 11, 1843. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.; Ozaki, K.; Baumann, H.; Levin, S.D.; Puel, A.; Farr, A.G.; Ziegler, S.F.; Leonard, W.J.; Lodish, H.F. Cloning of a receptor subunit required for signaling by thymic stromal lymphopoietin. Nat. Immunol. 2000, 1, 59–64. [Google Scholar] [CrossRef]
- He, R.; Geha, R.S. Thymic stromal lymphopoietin. Ann. N. Y. Acad. Sci. 2010, 1183, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Aletaha, D.; McInnes, I.B. Rheumatoid arthritis. Lancet 2016, 388, 2023–2038. [Google Scholar] [CrossRef] [PubMed]
- Safiri, S.; Kolahi, A.A.; Hoy, D.; Smith, E.; Bettampadi, D.; Mansournia, M.A.; Almasi-Hashiani, A.; Ashrafi-Asgarabad, A.; Moradi-Lakeh, M.; Qorbani, M.; et al. Global, regional and national burden of rheumatoid arthritis 1990–2017: A systematic analysis of the Global Burden of Disease study 2017. Ann. Rheum. Dis. 2019, 78, 1463–1471. [Google Scholar] [CrossRef] [PubMed]
- Deane, K.D.; Demoruelle, M.K.; Kelmenson, L.B.; Kuhn, K.A.; Norris, J.M.; Holers, V.M. Genetic and environmental risk factors for rheumatoid arthritis. Best Pract. Res. Clin. Rheumatol. 2017, 31, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, R.S.; Bruchfeld, A.; Yang, L.; Qureshi, A.R.; Gallowitsch-Puerta, M.; Patel, N.B.; Huston, B.J.; Chavan, S.; Rosas-Ballina, M.; Gregersen, P.K.; et al. Cholinergic anti-inflammatory pathway activity and High Mobility Group Box-1 (HMGB1) serum levels in patients with rheumatoid arthritis. Mol. Med. 2007, 13, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Sandoghchian Shotorbani, S.; Su, Z.; Liu, Y.; Tong, J.; Zheng, D.; Chen, J.; Xu, Y.; Jiao, Z.; Wang, S.; et al. Enhanced HMGB1 expression may contribute to Th17 cells activation in rheumatoid arthritis. Clin. Dev. Immunol. 2012, 2012, 295081. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zeng, T.; Zhang, X.; Tian, Y.; Wu, Y.; Yu, J.; Pei, Z.; Liu, Y.; Hu, T.; Tan, L. Clinical diagnostic significance of 14-3-3η protein, high-mobility group box-1, anti-cyclic citrullinated peptide antibodies, anti-mutated citrullinated vimentin antibodies and rheumatoid factor in rheumatoid arthritis. Br. J. Biomed. Sci. 2020, 77, 19–23. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, Y.; Jiang, S.; Fan, Y.; Huang, J.; Xiao, B.; Rao, H.; Huang, L. Effects of metformin therapy on HMGB1 levels in rheumatoid arthritis patients. Eur. J. Med. Res. 2023, 28, 512. [Google Scholar] [CrossRef]
- Taniguchi, N.; Kawahara, K.; Yone, K.; Hashiguchi, T.; Yamakuchi, M.; Goto, M.; Inoue, K.; Yamada, S.; Ijiri, K.; Matsunaga, S.; et al. High mobility group box chromosomal protein 1 plays a role in the pathogenesis of rheumatoid arthritis as a novel cytokine. Arthritis Rheumatol. 2003, 48, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.B.; Xu, P.; Xu, K.; Cai, Y.S.; Sun, M.Y.; Yang, L.; Sun, J.; Lu, S.M. Methotrexate affects HMGB1 expression in rheumatoid arthritis, and the downregulation of HMGB1 prevents rheumatoid arthritis progression. Mol. Cell. Biochem. 2016, 420, 161–170. [Google Scholar] [CrossRef] [PubMed]
- He, Z.W.; Qin, Y.H.; Wang, Z.W.; Chen, Y.; Shen, Q.; Dai, S.M. HMGB1 acts in synergy with lipopolysaccharide in activating rheumatoid synovial fibroblasts via p38 MAPK and NF-κB signaling pathways. Mediat. Inflamm. 2013, 2013, 596716. [Google Scholar] [CrossRef] [PubMed]
- Elshabrawy, H.A.; Chen, Z.; Volin, M.V.; Ravella, S.; Virupannavar, S.; Shahrara, S. The pathogenic role of angiogenesis in rheumatoid arthritis. Angiogenesis 2015, 18, 433–448. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.F.; Liu, Z.L.; Pan, C.; Yang, X.Q.; Ning, S.L.; Liu, H.D.; Guo, S.; Yu, J.M.; Zhang, Z.L. HMGB1 correlates with angiogenesis and poor prognosis of perihilar cholangiocarcinoma via elevating VEGFR2 of vessel endothelium. Oncogene 2019, 38, 868–880. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Luo, H.; Wu, R.; Wang, J.; Zhou, B.; Zhang, Y.; Jiang, Y.; Xu, J. Internalization of HMGB1 (High Mobility Group Box 1) Promotes Angiogenesis in Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2922–2940. [Google Scholar] [CrossRef]
- Biscetti, F.; Flex, A.; Pecorini, G.; Angelini, F.; Arena, V.; Stigliano, E.; Gremese, E.; Tolusso, B.; Ferraccioli, G. The role of high-mobility group box protein 1 in collagen antibody-induced arthritis is dependent on vascular endothelial growth factor. Clin. Exp. Immunol. 2016, 184, 62–72. [Google Scholar] [CrossRef]
- Park, S.Y.; Lee, S.W.; Kim, H.Y.; Lee, W.S.; Hong, K.W.; Kim, C.D. HMGB1 induces angiogenesis in rheumatoid arthritis via HIF-1α activation. Eur. J. Immunol. 2015, 45, 1216–1227. [Google Scholar] [CrossRef]
- Siouti, E.; Andreakos, E. The many facets of macrophages in rheumatoid arthritis. Biochem. Pharmacol. 2019, 165, 152–169. [Google Scholar] [CrossRef]
- Peng, L.; Zhu, N.; Mao, J.; Huang, L.; Yang, Y.; Zhou, Z.; Wang, L.; Wu, B. Expression levels of CXCR4 and CXCL12 in patients with rheumatoid arthritis and its correlation with disease activity. Exp. Ther. Med. 2020, 20, 1925–1934. [Google Scholar] [CrossRef]
- Cecchinato, V.; D’Agostino, G.; Raeli, L.; Nerviani, A.; Schiraldi, M.; Danelon, G.; Manzo, A.; Thelen, M.; Ciurea, A.; Bianchi, M.E.; et al. Redox-Mediated Mechanisms Fuel Monocyte Responses to CXCL12/HMGB1 in Active Rheumatoid Arthritis. Front. Immunol. 2018, 9, 2118. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.Y.; Li, K.T.; Yang, H.X.; Yang, B.; Lu, X.; Zhao, L.D.; Fei, Y.Y.; Chen, H.; Wang, L.; Li, J.; et al. Complement C1q synergizes with PTX3 in promoting NLRP3 inflammasome over-activation and pyroptosis in rheumatoid arthritis. J. Autoimmun. 2020, 106, 102336. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Zhao, F.; Cao, Y.; Ping, F.; Wang, W.; Li, Y. HMGB1 mediates lipopolysaccharide-induced macrophage autophagy and pyroptosis. BMC Mol. Cell Biol. 2023, 24, 2. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Zhang, Q.; Tan, J.; Xiong, Y.; Liang, Y.; Yan, J.; Gu, F.; Xu, Y. HMGB1 induces macrophage pyroptosis in chronic endometritis. Int. Immunopharmacol. 2023, 123, 110706. [Google Scholar] [CrossRef]
- Salo, H.; Qu, H.; Mitsiou, D.; Aucott, H.; Han, J.; Zhang, X.M.; Aulin, C.; Erlandsson Harris, H. Disulfide and Fully Reduced HMGB1 Induce Different Macrophage Polarization and Migration Patterns. Biomolecules 2021, 11, 800. [Google Scholar] [CrossRef]
- Zhu, C.; Chen, T.; Liu, B. Inhibitory effects of miR-25 targeting HMGB1 on macrophage secretion of inflammatory cytokines in sepsis. Oncol. Lett. 2018, 16, 5027–5033. [Google Scholar] [CrossRef] [PubMed]
- Paradowska-Gorycka, A.; Wajda, A.; Romanowska-Próchnicka, K.; Walczuk, E.; Kuca-Warnawin, E.; Kmiolek, T.; Stypinska, B.; Rzeszotarska, E.; Majewski, D.; Jagodzinski, P.P.; et al. Th17/Treg-Related Transcriptional Factor Expression and Cytokine Profile in Patients with Rheumatoid Arthritis. Front. Immunol. 2020, 11, 572858. [Google Scholar] [CrossRef] [PubMed]
- Roeleveld, D.M.; Koenders, M.I. The role of the Th17 cytokines IL-17 and IL-22 in Rheumatoid Arthritis pathogenesis and developments in cytokine immunotherapy. Cytokine 2015, 74, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Sun, C.; Zhou, C.; Liu, Y.; Zhu, H.; Sandoghchian, S.; Zheng, D.; Peng, T.; Zhang, Y.; Jiao, Z.; et al. HMGB1 blockade attenuates experimental autoimmune myocarditis and suppresses Th17-cell expansion. Eur. J. Immunol. 2011, 41, 3586–3595. [Google Scholar] [CrossRef]
- Xu, K.; Ren, X.; Ju, B.; Aihaiti, Y.; Cai, Y.; Zhang, Y.; He, L.; Wang, J. Clinical markers combined with HMGB1 polymorphisms to predict efficacy of conventional DMARDs in rheumatoid arthritis patients. Clin. Immunol. 2020, 221, 108592. [Google Scholar] [CrossRef]
- Schierbeck, H.; Wähämaa, H.; Andersson, U.; Harris, H.E. Immunomodulatory drugs regulate HMGB1 release from activated human monocytes. Mol. Med. 2010, 16, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Kuroiwa, Y.; Takakusagi, Y.; Kusayanagi, T.; Kuramochi, K.; Imai, T.; Hirayama, T.; Ito, I.; Yoshida, M.; Sakaguchi, K.; Sugawara, F. Identification and characterization of the direct interaction between methotrexate (MTX) and high-mobility group box 1 (HMGB1) protein. PLoS ONE 2013, 8, e63073. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Cai, Y.S.; Lu, S.M.; Li, X.L.; Liu, L.; Li, Z.; Liu, H.; Xu, P. Autophagy induction contributes to the resistance to methotrexate treatment in rheumatoid arthritis fibroblast-like synovial cells through high mobility group box chromosomal protein 1. Arthritis Res. Ther. 2015, 17, 374. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Leng, M.; Ding, C.; Zhu, X.; Zhang, Y.; Sun, C.; Lou, P. Ferritinophagy-mediated ferroptosis facilitates methotrexate-induced hepatotoxicity by high-mobility group box 1 (HMGB1). Liver Int. 2024, 44, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Sundberg, E.; Grundtman, C.; Af Klint, E.; Lindberg, J.; Ernestam, S.; Ulfgren, A.K.; Harris, H.E.; Andersson, U. Systemic TNF blockade does not modulate synovial expression of the pro-inflammatory mediator HMGB1 in rheumatoid arthritis patients--a prospective clinical study. Arthritis Res. Ther. 2008, 10, R33. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, T.; Sakata, N.; Narumi, Y.; Kimura, T.; Hayashi, T.; Nagano, K.; Liu, K.; Nishibori, M.; Tsukita, S.; Yamada, T.; et al. Metformin directly binds the alarmin HMGB1 and inhibits its proinflammatory activity. J. Biol. Chem. 2017, 292, 8436–8446. [Google Scholar] [CrossRef] [PubMed]
- Jarlborg, M.; Courvoisier, D.S.; Lamacchia, C.; Martinez Prat, L.; Mahler, M.; Bentow, C.; Finckh, A.; Gabay, C.; Nissen, M.J. Serum calprotectin: A promising biomarker in rheumatoid arthritis and axial spondyloarthritis. Arthritis Res. Ther. 2020, 22, 105. [Google Scholar] [CrossRef]
- Mansour, H.E.; Abdullrhman, M.A.; Mobasher, S.A.; El Mallah, R.; Abaza, N.; Hamed, F.; Khalil, A.A.F. Serum Calprotectin in Rheumatoid Arthritis: A Promising Diagnostic Marker, How Far Is It Related to Activity and Sonographic Findings? J. Med. Ultrasound. 2017, 25, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liang, Y. Clinical significance of serum calprotectin level for the disease activity in active rheumatoid arthritis with normal C-reactive protein. Int. J. Clin. Exp. Pathol. 2019, 12, 1009–1014. [Google Scholar] [PubMed]
- Bettner, L.F.; Peterson, R.A.; Bergstedt, D.T.; Kelmenson, L.B.; Demoruelle, M.K.; Mikuls, T.R.; Edison, J.D.; Parish, M.C.; Feser, M.L.; Frazer-Abel, A.A.; et al. Combinations of Anticyclic Citrullinated Protein Antibody, Rheumatoid Factor, and Serum Calprotectin Positivity Are Associated with the Diagnosis of Rheumatoid Arthritis within 3 Years. ACR Open Rheumatol. 2021, 3, 684–689. [Google Scholar] [CrossRef]
- Bae, S.C.; Lee, Y.H. Calprotectin levels in rheumatoid arthritis and their correlation with disease activity: A meta-analysis. Postgrad. Med. 2017, 129, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Roszkowski, L.; Jaszczyk, B.; Plebańczyk, M.; Ciechomska, M. S100A8 and S100A12 Proteins as Biomarkers of High Disease Activity in Patients with Rheumatoid Arthritis That Can Be Regulated by Epigenetic Drugs. Int. J. Mol. Sci. 2022, 24, 710. [Google Scholar] [CrossRef] [PubMed]
- Hurnakova, J.; Hulejova, H.; Zavada, J.; Komarc, M.; Cerezo, L.A.; Mann, H.; Vencovsky, J.; Pavelka, K.; Senolt, L. Serum calprotectin may reflect inflammatory activity in patients with active rheumatoid arthritis despite normal to low C-reactive protein. Clin. Rheumatol. 2018, 37, 2055–2062. [Google Scholar] [CrossRef]
- Sejersen, K.; Weitoft, T.; Knight, A.; Lysholm, J.; Larsson, A.; Rönnelid, J. Serum calprotectin correlates more strongly with inflammation and disease activity in ACPA positive than ACPA negative rheumatoid arthritis. Rheumatology, 2023; ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Gernert, M.; Schmalzing, M.; Tony, H.P.; Strunz, P.P.; Schwaneck, E.C.; Fröhlich, M. Calprotectin (S100A8/S100A9) detects inflammatory activity in rheumatoid arthritis patients receiving tocilizumab therapy. Arthritis Res. Ther. 2022, 24, 200. [Google Scholar] [CrossRef] [PubMed]
- Inciarte-Mundo, J.; Ramirez, J.; Hernández, M.V.; Ruiz-Esquide, V.; Cuervo, A.; Cabrera-Villalba, S.R.; Pascal, M.; Yagüe, J.; Cañete, J.D.; Sanmarti, R. Calprotectin and TNF trough serum levels identify power Doppler ultrasound synovitis in rheumatoid arthritis and psoriatic arthritis patients in remission or with low disease activity. Arthritis Res. Ther. 2016, 18, 160. [Google Scholar] [CrossRef] [PubMed]
- Hurnakova, J.; Zavada, J.; Hanova, P.; Hulejova, H.; Klein, M.; Mann, H.; Sleglova, O.; Olejarova, M.; Forejtova, S.; Ruzickova, O.; et al. Serum calprotectin (S100A8/9): An independent predictor of ultrasound synovitis in patients with rheumatoid arthritis. Arthritis Res. Ther. 2015, 17, 252. [Google Scholar] [CrossRef] [PubMed]
- Andrés Cerezo, L.; Mann, H.; Pecha, O.; Pleštilová, L.; Pavelka, K.; Vencovský, J.; Senolt, L. Decreases in serum levels of S100A8/9 (calprotectin) correlate with improvements in total swollen joint count in patients with recent-onset rheumatoid arthritis. Arthritis Res. Ther. 2011, 13, R122. [Google Scholar] [CrossRef] [PubMed]
- de Moel, E.C.; Rech, J.; Mahler, M.; Roth, J.; Vogl, T.; Schouffoer, A.; Goekoop, R.J.; Huizinga, T.W.J.; Allaart, C.F.; Toes, R.E.M.; et al. Circulating calprotectin (S100A8/A9) is higher in rheumatoid arthritis patients that relapse within 12 months of tapering anti-rheumatic drugs. Arthritis Res. Ther. 2019, 21, 268. [Google Scholar] [CrossRef]
- Romand, X.; Clapasson, M.; Chuong, M.V.; Paclet, M.H.; Fautrel, B.; Baillet, A. Serum calprotectin levels do not predict subsequent relapse in rheumatoid arthritis in remission: A post-hoc analysis of STRASS study. RMD Open 2023, 9, e003198. [Google Scholar] [CrossRef]
- Chen, W.; Wang, Q.; Ke, Y.; Lin, J. Neutrophil Function in an Inflammatory Milieu of Rheumatoid Arthritis. J. Immunol. Res. 2018, 2018, 8549329. [Google Scholar] [CrossRef] [PubMed]
- Sprenkeler, E.G.G.; Zandstra, J.; van Kleef, N.D.; Goetschalckx, I.; Verstegen, B.; Aarts, C.E.M.; Janssen, H.; Tool, A.T.J.; van Mierlo, G.; van Bruggen, R.; et al. S100A8/A9 Is a Marker for the Release of Neutrophil Extracellular Traps and Induces Neutrophil Activation. Cells 2022, 11, 236. [Google Scholar] [CrossRef] [PubMed]
- Schenten, V.; Plançon, S.; Jung, N.; Hann, J.; Bueb, J.L.; Bréchard, S.; Tschirhart, E.J.; Tolle, F. Secretion of the Phosphorylated Form of S100A9 from Neutrophils Is Essential for the Proinflammatory Functions of Extracellular S100A8/A9. Front. Immunol. 2018, 9, 447. [Google Scholar] [CrossRef] [PubMed]
- Jung, N.; Schenten, V.; Bueb, J.L.; Tolle, F.; Bréchard, S. miRNAs Regulate Cytokine Secretion Induced by Phosphorylated S100A8/A9 in Neutrophils. Int. J. Mol. Sci. 2019, 20, 5699. [Google Scholar] [CrossRef] [PubMed]
- Sunahori, K.; Yamamura, M.; Yamana, J.; Takasugi, K.; Kawashima, M.; Yamamoto, H.; Chazin, W.J.; Nakatani, Y.; Yui, S.; Makino, H. The S100A8/A9 heterodimer amplifies proinflammatory cytokine production by macrophages via activation of nuclear factor kappa B and p38 mitogen-activated protein kinase in rheumatoid arthritis. Arthritis Res. Ther. 2006, 8, R69. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Kong, X.; You, Y.; Xiang, L.; Zhang, Y.; Wu, R.; Zhou, L.; Duan, L. S100A8-Mediated NLRP3 Inflammasome-Dependent Pyroptosis in Macrophages Facilitates Liver Fibrosis Progression. Cells 2022, 11, 3579. [Google Scholar] [CrossRef] [PubMed]
- Carrión, M.; Juarranz, Y.; Martínez, C.; González-Álvaro, I.; Pablos, J.L.; Gutiérrez-Cañas, I.; Gomariz, R.P. IL-22/IL-22R1 axis and S100A8/A9 alarmins in human osteoarthritic and rheumatoid arthritis synovial fibroblasts. Rheumatology 2013, 52, 2177–2186. [Google Scholar] [CrossRef] [PubMed]
- Zreiqat, H.; Howlett, C.R.; Gronthos, S.; Hume, D.; Geczy, C.L. S100A8/S100A9 and their association with cartilage and bone. J. Mol. Histol. 2007, 38, 381–391. [Google Scholar] [CrossRef]
- Grevers, L.C.; de Vries, T.J.; Vogl, T.; Abdollahi-Roodsaz, S.; Sloetjes, A.W.; Leenen, P.J.; Roth, J.; Everts, V.; van den Berg, W.B.; van Lent, P.L. S100A8 enhances osteoclastic bone resorption in vitro through activation of Toll-like receptor 4: Implications for bone destruction in murine antigen-induced arthritis. Arthritis Rheum. 2011, 63, 1365–1375. [Google Scholar] [CrossRef]
- Di Ceglie, I.; Blom, A.B.; Davar, R.; Logie, C.; Martens, J.H.A.; Habibi, E.; Böttcher, L.M.; Roth, J.; Vogl, T.; Goodyear, C.S.; et al. The alarmin S100A9 hampers osteoclast differentiation from human circulating precursors by reducing the expression of RANK. FASEB J. 2019, 33, 10104–10115. [Google Scholar] [CrossRef]
- Cesaro, A.; Anceriz, N.; Plante, A.; Pagé, N.; Tardif, M.R.; Tessier, P.A. An inflammation loop orchestrated by S100A9 and calprotectin is critical for development of arthritis. PLoS ONE 2012, 7, e45478. [Google Scholar] [CrossRef]
- Yasuda, K.; Shimodan, S.; Maehara, N.; Hirota, A.; Iijima, R.; Nishijima, A.; Mori, H.; Toyama, R.; Ito, A.; Yoshikawa, Y.; et al. AIM/CD5L ameliorates autoimmune arthritis by promoting removal of inflammatory DAMPs at the lesions. J. Autoimmun. 2023, 142, 103149. [Google Scholar] [CrossRef] [PubMed]
- Obry, A.; Lequerré, T.; Hardouin, J.; Boyer, O.; Fardellone, P.; Philippe, P.; Le Loët, X.; Cosette, P.; Vittecoq, O. Identification of S100A9 as biomarker of responsiveness to the methotrexate/etanercept combination in rheumatoid arthritis using a proteomic approach. PLoS ONE 2014, 9, e115800. [Google Scholar] [CrossRef]
- Tweehuysen, L.; den Broeder, N.; van Herwaarden, N.; Joosten, L.A.B.; van Lent, P.L.; Vogl, T.; van den Hoogen, F.H.J.; Thurlings, R.M.; den Broeder, A.A. Predictive value of serum calprotectin (S100A8/A9) for clinical response after starting or tapering anti-TNF treatment in patients with rheumatoid arthritis. RMD Open 2018, 4, e000654. [Google Scholar] [CrossRef]
- Andrés Cerezo, L.; Šumová, B.; Prajzlerová, K.; Veigl, D.; Damgaard, D.; Nielsen, C.H.; Pavelka, K.; Vencovský, J.; Šenolt, L. Calgizzarin (S100A11): A novel inflammatory mediator associated with disease activity of rheumatoid arthritis. Arthritis Res. Ther. 2017, 19, 79. [Google Scholar] [CrossRef]
- Navrátilová, A.; Bečvář, V.; Baloun, J.; Damgaard, D.; Nielsen, C.H.; Veigl, D.; Pavelka, K.; Vencovský, J.; Šenolt, L.; Andrés Cerezo, L. S100A11 (calgizzarin) is released via NETosis in rheumatoid arthritis (RA) and stimulates IL-6 and TNF secretion by neutrophils. Sci. Rep. 2021, 11, 6063. [Google Scholar] [CrossRef] [PubMed]
- Foell, D.; Kane, D.; Bresnihan, B.; Vogl, T.; Nacken, W.; Sorg, C.; Fitzgerald, O.; Roth, J. Expression of the pro-inflammatory protein S100A12 (EN-RAGE) in rheumatoid and psoriatic arthritis. Rheumatology 2003, 42, 1383–1389. [Google Scholar] [CrossRef]
- Rouleau, P.; Vandal, K.; Ryckman, C.; Poubelle, P.E.; Boivin, A.; Talbot, M.; Tessier, P.A. The calcium-binding protein S100A12 induces neutrophil adhesion, migration, and release from bone marrow in mouse at concentrations similar to those found in human inflammatory arthritis. Clin. Immunol. 2003, 107, 46–54. [Google Scholar] [CrossRef]
- Nordal, H.H.; Brun, J.G.; Halse, A.K.; Jonsson, R.; Fagerhol, M.K.; Hammer, H.B. The neutrophil protein S100A12 is associated with a comprehensive ultrasonographic synovitis score in a longitudinal study of patients with rheumatoid arthritis treated with adalimumab. BMC Musculoskelet. Disord. 2014, 15, 335. [Google Scholar] [CrossRef]
- Nishida, M.; Saegusa, J.; Tanaka, S.; Morinobu, A. S100A12 facilitates osteoclast differentiation from human monocytes. PLoS ONE 2018, 13, e0204140. [Google Scholar] [CrossRef] [PubMed]
- Liew, F.Y.; Girard, J.P.; Turnquist, H.R. Interleukin-33 in health and disease. Nat. Rev. Immunol. 2016, 16, 676–689. [Google Scholar] [CrossRef] [PubMed]
- Kakkar, R.; Lee, R.T. The IL-33/ST2 pathway: Therapeutic target and novel biomarker. Nat. Rev. Drug Discov. 2008, 7, 827–840. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.S.; Moon, S.J.; Joo, Y.B.; Jeon, C.H.; Cho, M.L.; Ju, J.H.; Oh, H.J.; Heo, Y.J.; Park, S.H.; Kim, H.Y.; et al. Measurement of interleukin-33 (IL-33) and IL-33 receptors (sST2 and ST2L) in patients with rheumatoid arthritis. J. Korean Med. Sci. 2011, 26, 1132–1139. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Huang, H.; Hu, F.; Zhou, W.; Guo, J.; Jiang, H.; Mu, R.; Li, Z. Increased IL-33 in synovial fluid and paired serum is associated with disease activity and autoantibodies in rheumatoid arthritis. Clin. Dev. Immunol. 2013, 2013, 985301. [Google Scholar] [CrossRef] [PubMed]
- Talabot-Ayer, D.; McKee, T.; Gindre, P.; Bas, S.; Baeten, D.L.; Gabay, C.; Palmer, G. Distinct serum and synovial fluid interleukin (IL)-33 levels in rheumatoid arthritis, psoriatic arthritis and osteoarthritis. Joint Bone Spine 2012, 79, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Poole, J.A.; England, B.R.; Sayles, H.; Johnson, T.M.; Duryee, M.J.; Hunter, C.D.; Baker, J.F.; Kerr, G.S.; Kunkel, G.; Cannon, G.W.; et al. Serum alarmins and the risk of incident interstitial lung disease in rheumatoid arthritis. Rheumatology, 2023; ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Iwaszko, M.; Wielińska, J.; Świerkot, J.; Kolossa, K.; Sokolik, R.; Bugaj, B.; Chaszczewska-Markowska, M.; Jeka, S.; Bogunia-Kubik, K. Gene Polymorphisms as Potential Biomarkers of Disease Susceptibility and Response to TNF Inhibitors in Rheumatoid Arthritis, Ankylosing Spondylitis, and Psoriatic Arthritis Patients. Front. Immunol. 2021, 12, 631603. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Jiang, H.R.; Li, Y.; Pushparaj, P.N.; Kurowska-Stolarska, M.; Leung, B.P.; Mu, R.; Tay, H.K.; McKenzie, A.N.; McInnes, I.B.; et al. IL-33 exacerbates autoantibody-induced arthritis. J. Immunol. 2010, 184, 2620–2626. [Google Scholar] [CrossRef]
- Wu, J.; Li, Q.; Deng, J.; Zhao, J.J.; Yu, Q.H. Association between IL-33 and other inflammatory factors in patients with rheumatoid arthritis and in fibroblast-like synoviocytes. Exp. Ther. Med. 2021, 21, 161. [Google Scholar] [CrossRef]
- Lee, E.J.; So, M.W.; Hong, S.; Kim, Y.G.; Yoo, B.; Lee, C.K. Interleukin-33 acts as a transcriptional repressor and extracellular cytokine in fibroblast-like synoviocytes in patients with rheumatoid arthritis. Cytokine 2016, 77, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Fu, W.; Zhao, S.; Jiao, T.; Wu, D.; Wang, Y. miR-483-3p Promotes IL-33 Production from Fibroblast-Like Synoviocytes by Regulating ERK Signaling in Rheumatoid Arthritis. Inflammation 2021, 44, 2302–2308. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Chen, B.; Wen, Z.; Huang, Z.; Ye, L. IL-33/ST2-mediated inflammation in macrophages is directly abrogated by IL-10 during rheumatoid arthritis. Oncotarget 2017, 8, 32407–32418. [Google Scholar] [CrossRef] [PubMed]
- Onodera, S.; Tanji, H.; Suzuki, K.; Kaneda, K.; Mizue, Y.; Sagawa, A.; Nishihira, J. High expression of macrophage migration inhibitory factor in the synovial tissues of rheumatoid joints. Cytokine 1999, 11, 163–167. [Google Scholar] [CrossRef] [PubMed]
- García-Arellano, S.; Hernández-Palma, L.A.; Cerpa-Cruz, S.; Sánchez-Zuno, G.A.; Herrera-Godina, M.G.; Muñoz-Valle, J.F. The Novel Role of MIF in the Secretion of IL-25, IL-31, and IL-33 from PBMC of Patients with Rheumatoid Arthritis. Molecules 2021, 26, 4968. [Google Scholar] [CrossRef] [PubMed]
- Lutzky, V.; Hannawi, S.; Thomas, R. Cells of the synovium in rheumatoid arthritis. Dendritic cells. Arthritis Res. Ther. 2007, 9, 219. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Kim, M.S.; Lim, H.X.; Cho, D.; Kim, T.S. IL-33-matured dendritic cells promote Th17 cell responses via IL-1β and IL-6. Cytokine 2017, 99, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Verri, W.A.; Souto, F.O.; Vieira, S.M.; Almeida, S.C.; Fukada, S.Y.; Xu, D.; Alves-Filho, J.C.; Cunha, T.M.; Guerrero, A.T.; Mattos-Guimaraes, R.B.; et al. IL-33 induces neutrophil migration in rheumatoid arthritis and is a target of anti-TNF therapy. Ann. Rheum. Dis. 2010, 69, 1697–1703. [Google Scholar] [CrossRef] [PubMed]
- Leung, B.P.; Xu, D.; Culshaw, S.; McInnes, I.B.; Liew, F.Y. A novel therapy of murine collagen-induced arthritis with soluble T1/ST2. J. Immunol. 2004, 173, 145–150. [Google Scholar] [CrossRef]
- Kageyama, Y.; Torikai, E.; Tsujimura, K.; Kobayashi, M. Involvement of IL-33 in the pathogenesis of rheumatoid arthritis: The effect of etanercept on the serum levels of IL-33. Mod. Rheumatol. 2012, 22, 89–93. [Google Scholar] [CrossRef]
- Matsuyama, Y.; Okazaki, H.; Hoshino, M.; Onishi, S.; Kamata, Y.; Nagatani, K.; Nagashima, T.; Iwamoto, M.; Yoshio, T.; Ohto-Ozaki, H.; et al. Sustained elevation of interleukin-33 in sera and synovial fluids from patients with rheumatoid arthritis non-responsive to anti-tumor necrosis factor: Possible association with persistent IL-1β signaling and a poor clinical response. Rheumatol. Int. 2012, 32, 1397–1401. [Google Scholar] [CrossRef] [PubMed]
- Sellam, J.; Rivière, E.; Courties, A.; Rouzaire, P.O.; Tolusso, B.; Vital, E.M.; Emery, P.; Ferraccioli, G.; Soubrier, M.; Ly, B.; et al. Serum IL-33, a new marker predicting response to rituximab in rheumatoid arthritis. Arthritis Res. Ther. 2016, 18, 294. [Google Scholar] [CrossRef] [PubMed]
- Das, S.K.; Farooqi, A. Osteoarthritis. Best Pract. Res. Clin. Rheumatol. 2008, 22, 657–675. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, R.C.; Felson, D.T.; Helmick, C.G.; Arnold, L.M.; Choi, H.; Deyo, R.A.; Gabriel, S.; Hirsch, R.; Hochberg, M.C.; Hunder, G.G.; et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part II. Arthritis Rheum. 2008, 58, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Ashford, S.; Williard, J. Osteoarthritis: A review. Nurse Pract. 2014, 39, 1–8. [Google Scholar] [CrossRef]
- Coaccioli, S.; Sarzi-Puttini, P.; Zis, P.; Rinonapoli, G.; Varrassi, G. Osteoarthritis: New Insight on Its Pathophysiology. J. Clin. Med. 2022, 11, 6013. [Google Scholar] [CrossRef] [PubMed]
- Maccagnano, G.; Pesce, V.; Noia, G.; Coviello, M.; Vicenti, G.; Vitiello, R.; Ziranu, A.; Spinarelli, A.; Moretti, B. The effects of a new protocol on blood loss in total knee arthroplasty. Orthop. Rev. 2022, 14, 37625. [Google Scholar] [CrossRef] [PubMed]
- Glauser, T.A.; Salinas, G.D.; Roepke, N.L.; Williamson, J.C.; Reese, A.; Gutierrez, G.; Abdolrasulnia, M. Management of mild-to-moderate osteoarthritis: A study of the primary care perspective. Postgrad. Med. 2011, 123, 126–134. [Google Scholar] [CrossRef]
- Shelton, L.R. A closer look at osteoarthritis. Nurse Pract. 2013, 38, 30–36, quiz 36–37. [Google Scholar] [CrossRef]
- Sun, X.H.; Liu, Y.; Han, Y.; Wang, J. Expression and Significance of High-Mobility Group Protein B1 (HMGB1) and the Receptor for Advanced Glycation End-Product (RAGE) in Knee Osteoarthritis. Med. Sci. Monit. 2016, 22, 2105–2112. [Google Scholar] [CrossRef]
- Wagner, G.; Lehmann, C.; Bode, C.; Miosge, N.; Schubert, A. High Mobility Group Box 1 Protein in Osteoarthritic Knee Tissue and Chondrogenic Progenitor Cells: An ex vivo and in vitro study. Cartilage 2021, 12, 484–495. [Google Scholar] [CrossRef]
- Li, Z.-C.; Cheng, G.-Q.; Hu, K.-Z.; Li, M.-Q.; Zang, W.-P.; Dong, Y.-Q.; Wang, W.-L.; Liu, Z.-D. Correlation of Synovial Fluid HMGB-1 Levels with Radiographic Severity of Knee Osteoarthritis. Clin. Investig. Med. 2011, 34, 298. [Google Scholar] [CrossRef] [PubMed]
- Aulin, C.; Lassacher, T.; Palmblad, K.; Erlandsson Harris, H. Early stage blockade of the alarmin HMGB1 reduces cartilage destruction in experimental OA. Osteoarthr. Cartil. 2020, 28, 698–707. [Google Scholar] [CrossRef]
- Zhou, S.; Liu, G.; Si, Z.; Yu, L.; Hou, L. Glycyrrhizin, an HMGB1 inhibitor, Suppresses Interleukin-1β-Induced Inflammatory Responses in Chondrocytes from Patients with Osteoarthritis. Cartilage 2021, 13, 947S–955S. [Google Scholar] [CrossRef]
- Pan, H.; Dai, H.; Wang, L.; Lin, S.; Tao, Y.; Zheng, Y.; Jiang, R.; Fang, F.; Wu, Y. MicroRNA-410-3p modulates chondrocyte apoptosis and inflammation by targeting high mobility group box 1 (HMGB1) in an osteoarthritis mouse model. BMC Musculoskelet. Disord. 2020, 21, 486. [Google Scholar] [CrossRef]
- Qiu, M.; Liu, D.; Fu, Q. MiR-129-5p shuttled by human synovial mesenchymal stem cell-derived exosomes relieves IL-1β induced osteoarthritis via targeting HMGB1. Life Sci. 2021, 269, 118987. [Google Scholar] [CrossRef]
- Wang, X.; Guo, Y.; Wang, C.; Yu, H.; Yu, X. MicroRNA-142-3p Inhibits Chondrocyte Apoptosis and Inflammation in Osteoarthritis by Targeting HMGB1. Inflammation 2016, 39, 1718–1728. [Google Scholar] [CrossRef] [PubMed]
- Shu, Z.; Miao, X.; Tang, T.; Zhan, P.; Zeng, L.; Jiang, Y. The GSK-3β/β-catenin signaling pathway is involved in HMGB1-induced chondrocyte apoptosis and cartilage matrix degradation. Int. J. Mol. Med. 2020, 45, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Zhang, Q.; Pu, F.; Zhu, Z.; Lu, K.; Lu, W.W.; Tong, L.; Yu, H.; Chen, D. Signalling interaction between β-catenin and other signalling molecules during osteoarthritis development. Cell Prolif. 2024, e13600. [Google Scholar] [CrossRef]
- Foell, D.; Wittkowski, H.; Vogl, T.; Roth, J. S100 proteins expressed in phagocytes: A novel group of damage-associated molecular pattern molecules. J. Leukoc. Biol. 2007, 81, 28–37. [Google Scholar] [CrossRef]
- van Lent, P.L.; Blom, A.B.; Schelbergen, R.F.; Slöetjes, A.; Lafeber, F.P.; Lems, W.F.; Cats, H.; Vogl, T.; Roth, J.; van den Berg, W.B. Active involvement of alarmins S100A8 and S100A9 in the regulation of synovial activation and joint destruction during mouse and human osteoarthritis. Arthritis Rheum. 2012, 64, 1466–1476. [Google Scholar] [CrossRef] [PubMed]
- Zreiqat, H.; Belluoccio, D.; Smith, M.M.; Wilson, R.; Rowley, L.A.; Jones, K.; Ramaswamy, Y.; Vogl, T.; Roth, J.; Bateman, J.F.; et al. S100A8 and S100A9 in experimental osteoarthritis. Arthritis Res. Ther. 2010, 12, R16. [Google Scholar] [CrossRef] [PubMed]
- Ruan, G.; Xu, J.; Wang, K.; Zheng, S.; Wu, J.; Ren, J.; Bian, F.; Chang, B.; Zhu, Z.; Han, W.; et al. Associations between serum S100A8/S100A9 and knee symptoms, joint structures and cartilage enzymes in patients with knee osteoarthritis. Osteoarthr. Cartil. 2019, 27, 99–105. [Google Scholar] [CrossRef]
- Huang, X.; Liu, J.; Huang, W. Identification of S100A8 as a common diagnostic biomarkers and exploring potential pathogenesis for osteoarthritis and metabolic syndrome. Front. Immunol. 2023, 14, 1185275. [Google Scholar] [CrossRef]
- Lourido, L.; Balboa-Barreiro, V.; Ruiz-Romero, C.; Rego-Pérez, I.; Camacho-Encina, M.; Paz-González, R.; Calamia, V.; Oreiro, N.; Nilsson, P.; Blanco, F.J. A clinical model including protein biomarkers predicts radiographic knee osteoarthritis: A prospective study using data from the Osteoarthritis Initiative. Osteoarthr. Cartil. 2021, 29, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Lourido, L.; Ayoglu, B.; Fernández-Tajes, J.; Oreiro, N.; Henjes, F.; Hellström, C.; Schwenk, J.M.; Ruiz-Romero, C.; Nilsson, P.; Blanco, F.J. Discovery of circulating proteins associated to knee radiographic osteoarthritis. Sci. Rep. 2017, 7, 137. [Google Scholar] [CrossRef]
- Mahler, E.A.; Zweers, M.C.; van Lent, P.L.; Blom, A.B.; van den Hoogen, F.H.; van den Berg, W.B.; Roth, J.; Vogl, T.; Bijlsma, J.W.; van den Ende, C.H.; et al. Association between serum levels of the proinflammatory protein S100A8/A9 and clinical and structural characteristics of patients with established knee, hip, and hand osteoarthritis. Scand. J. Rheumatol. 2015, 44, 56–60. [Google Scholar] [CrossRef]
- van den Bosch, M.H.; Blom, A.B.; Schelbergen, R.F.; Koenders, M.I.; van de Loo, F.A.; van den Berg, W.B.; Vogl, T.; Roth, J.; van der Kraan, P.M.; van Lent, P.L. Alarmin S100A9 Induces Proinflammatory and Catabolic Effects Predominantly in the M1 Macrophages of Human Osteoarthritic Synovium. J. Rheumatol. 2016, 43, 1874–1884. [Google Scholar] [CrossRef]
- van Kooten, N.J.T.; Blom, A.B.; Teunissen van Manen, I.J.; Theeuwes, W.F.; Roth, J.; Gorris, M.A.J.; Walgreen, B.; Sloetjes, A.W.; Helsen, M.M.; Vitters, E.L.; et al. S100A8/A9 drives monocytes towards M2-like macrophage differentiation and associates with M2-like macrophages in osteoarthritic synovium. Rheumatology, 2024; ahead of pint. [Google Scholar] [CrossRef]
- Schelbergen, R.F.; de Munter, W.; van den Bosch, M.H.; Lafeber, F.P.; Sloetjes, A.; Vogl, T.; Roth, J.; van den Berg, W.B.; van der Kraan, P.M.; Blom, A.B.; et al. Alarmins S100A8/S100A9 aggravate osteophyte formation in experimental osteoarthritis and predict osteophyte progression in early human symptomatic osteoarthritis. Ann. Rheum. Dis. 2016, 75, 218–225. [Google Scholar] [CrossRef]
- van den Bosch, M.H.; Blom, A.B.; Schelbergen, R.F.; Vogl, T.; Roth, J.P.; Slöetjes, A.W.; van den Berg, W.B.; van der Kraan, P.M.; van Lent, P.L. Induction of Canonical Wnt Signaling by the Alarmins S100A8/A9 in Murine Knee Joints: Implications for Osteoarthritis. Arthritis Rheumatol. 2016, 68, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Schelbergen, R.F.; van Dalen, S.; ter Huurne, M.; Roth, J.; Vogl, T.; Noël, D.; Jorgensen, C.; van den Berg, W.B.; van de Loo, F.A.; Blom, A.B.; et al. Treatment efficacy of adipose-derived stem cells in experimental osteoarthritis is driven by high synovial activation and reflected by S100A8/A9 serum levels. Osteoarthr. Cartil. 2014, 22, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Schelbergen, R.F.; Blom, A.B.; van den Bosch, M.H.; Slöetjes, A.; Abdollahi-Roodsaz, S.; Schreurs, B.W.; Mort, J.S.; Vogl, T.; Roth, J.; van den Berg, W.B.; et al. Alarmins S100A8 and S100A9 elicit a catabolic effect in human osteoarthritic chondrocytes that is dependent on Toll-like receptor 4. Arthritis Rheum. 2012, 64, 1477–1487. [Google Scholar] [CrossRef] [PubMed]
- Schelbergen, R.F.; Geven, E.J.; van den Bosch, M.H.; Eriksson, H.; Leanderson, T.; Vogl, T.; Roth, J.; van de Loo, F.A.; Koenders, M.I.; van der Kraan, P.M.; et al. Prophylactic treatment with S100A9 inhibitor paquinimod reduces pathology in experimental collagenase-induced osteoarthritis. Ann. Rheum. Dis. 2015, 74, 2254–2258. [Google Scholar] [CrossRef] [PubMed]
- Corr, E.M.; Cunningham, C.C.; Helbert, L.; McCarthy, G.M.; Dunne, A. Osteoarthritis-associated basic calcium phosphate crystals activate membrane proximal kinases in human innate immune cells. Arthritis Res. Ther. 2017, 19, 23. [Google Scholar] [CrossRef]
- Stack, J.; McCarthy, G. Basic calcium phosphate crystals and osteoarthritis pathogenesis: Novel pathways and potential targets. Curr. Opin. Rheumatol. 2016, 28, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Yi, C.; Yi, Y.; Wei, J.; Jin, Q.; Li, J.; Sacitharan, P.K. Targeting IL-22 and IL-22R protects against experimental osteoarthritis. Cell. Mol. Immunol. 2021, 18, 1329–1331. [Google Scholar] [CrossRef]
- Li, C.; Chen, K.; Kang, H.; Yan, Y.; Liu, K.; Guo, C.; Qi, J.; Yang, K.; Wang, F.; Guo, L.; et al. Double-stranded RNA released from damaged articular chondrocytes promotes cartilage degeneration via Toll-like receptor 3-interleukin-33 pathway. Cell Death Dis. 2017, 8, e3165. [Google Scholar] [CrossRef]
- Rai, V.; Radwan, M.M.; Agrawal, D.K. IL-33, IL-37, and Vitamin D Interaction Mediate Immunomodulation of Inflammation in Degenerating Cartilage. Antibodies 2021, 10, 41. [Google Scholar] [CrossRef]
- He, Z.; Song, Y.; Yi, Y.; Qiu, F.; Wang, J.; Li, J.; Jin, Q.; Sacitharan, P.K. Blockade of IL-33 signalling attenuates osteoarthritis. Clin. Transl. Immunol. 2020, 9, e1185. [Google Scholar] [CrossRef]
- Rai, V.; Dilisio, M.F.; Samadi, F.; Agrawal, D.K. Counteractive Effects of IL-33 and IL-37 on Inflammation in Osteoarthritis. Int. J. Environ. Res. Public Health 2022, 19, 5690. [Google Scholar] [CrossRef] [PubMed]
- van Geffen, E.W.; van Caam, A.P.M.; Schreurs, W.; van de Loo, F.A.; van Lent, P.L.E.M.; Koenders, M.I.; Thudium, C.S.; Bay-Jensen, A.C.; Blaney Davidson, E.N.; van der Kraan, P.M. IL-37 diminishes proteoglycan loss in human OA cartilage: Donor-specific link between IL-37 and MMP-3. Osteoarthr. Cartil. 2019, 27, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Nicols, B.F.; Robledo-Pulido, J.J.; Alvarado-Navarro, A. Etiopathogenesis of Psoriasis: Integration of Proposed Theories. Immunol. Investig. 2024, 53, 348–415. [Google Scholar] [CrossRef]
- Oliveira, M.e.F.; Rocha, B.e.O.; Duarte, G.V. Psoriasis: Classical and emerging comorbidities. An. Bras. Dermatol. 2015, 90, 9–20. [Google Scholar] [CrossRef]
- Yen, H.; Chi, C.C. Association between Psoriasis and Vitiligo: A Systematic Review and Meta-Analysis. Am. J. Clin. Dermatol. 2019, 20, 31–40. [Google Scholar] [CrossRef]
- Wu, J.J.; Nguyen, T.U.; Poon, K.Y.; Herrinton, L.J. The association of psoriasis with autoimmune diseases. J. Am. Acad. Dermatol. 2012, 67, 924–930. [Google Scholar] [CrossRef]
- Najarian, D.J.; Gottlieb, A.B. Connections between psoriasis and Crohn’s disease. J. Am. Acad. Dermatol. 2003, 48, 805–821, quiz 822–804. [Google Scholar] [CrossRef]
- Ayala-Fontánez, N.; Soler, D.C.; McCormick, T.S. Current knowledge on psoriasis and autoimmune diseases. Psoriasis 2016, 6, 7–32. [Google Scholar] [CrossRef] [PubMed]
- Makredes, M.; Robinson, D.; Bala, M.; Kimball, A.B. The burden of autoimmune disease: A comparison of prevalence ratios in patients with psoriatic arthritis and psoriasis. J. Am. Acad. Dermatol. 2009, 61, 405–410. [Google Scholar] [CrossRef]
- Nussbaum, L.; Chen, Y.L.; Ogg, G.S. Role of regulatory T cells in psoriasis pathogenesis and treatment. Br. J. Dermatol. 2021, 184, 14–24. [Google Scholar] [CrossRef]
- Borsky, P.; Fiala, Z.; Andrys, C.; Beranek, M.; Hamakova, K.; Malkova, A.; Svadlakova, T.; Krejsek, J.; Palicka, V.; Borska, L.; et al. Alarmins HMGB1, IL-33, S100A7, and S100A12 in Psoriasis Vulgaris. Mediat. Inflamm. 2020, 2020, 8465083. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Guo, Z.-P.; Li, L.; Wang, L.; Jia, R.-Z.; Cao, N.; Qin, S.; Li, M.-M. Increased HMGB1 serum levels and altered HMGB1 expression in patients with psoriasis vulgaris. Arch. Dermatol. Res. 2013, 305, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, C.; Strohbuecker, L.; Lotfi, R.; Sucker, A.; Joosten, I.; Koenen, H.; Korber, A. High mobility group box 1 is increased in the sera of psoriatic patients with disease progression. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Yamaguchi, Y.; Watanabe, Y.; Takamura, N.; Aihara, M. Increased level of high mobility group box 1 in the serum and skin in patients with generalized pustular psoriasis. J. Dermatol. 2020, 47, 1033–1036. [Google Scholar] [CrossRef] [PubMed]
- Mohamad, N.E.; Hashem, N.; Eltahlawy, S.; Gomaa, E. Involvement of high-mobility group box-1 and Dermoscopy in diagnosis of psoriasis severity. J. Cosmet. Dermatol. 2022, 21, 6336–6342. [Google Scholar] [CrossRef] [PubMed]
- Maurelli, M.; Gisondi, P.; Danese, E.; Gelati, M.; Papagrigoraki, A.; Del Giglio, M.; Lippi, G.; Girolomoni, G. Psoriasin (S100A7) is increased in the serum of patients with moderate-to-severe psoriasis. Br. J. Dermatol. 2020, 182, 1502–1503. [Google Scholar] [CrossRef]
- Chen, J.; Fu, Y.; Xiong, S. Keratinocyte derived HMGB1 aggravates psoriasis dermatitis via facilitating inflammatory polarization of macrophages and hyperproliferation of keratinocyte. Mol. Immunol. 2023, 163, 1–12. [Google Scholar] [CrossRef]
- Jeon, S.; Song, J.; Lee, D.; Kim, G.T.; Park, S.H.; Shin, D.Y.; Shin, K.O.; Park, K.; Shim, S.M.; Park, T.S. Inhibition of sphingosine 1-phosphate lyase activates human keratinocyte differentiation and attenuates psoriasis in mice. J. Lipid Res. 2020, 61, 20–32. [Google Scholar] [CrossRef]
- Wang, Z.; Zhou, H.; Zheng, H.; Zhou, X.; Shen, G.; Teng, X.; Liu, X.; Zhang, J.; Wei, X.; Hu, Z.; et al. Autophagy-based unconventional secretion of HMGB1 by keratinocytes plays a pivotal role in psoriatic skin inflammation. Autophagy 2021, 17, 529–552. [Google Scholar] [CrossRef]
- Li, S.; Li, G.; Li, X.; Wu, F.; Li, L. Etanercept ameliorates psoriasis progression through regulating high mobility group box 1 pathway. Ski. Res. Technol. 2023, 29, e13329. [Google Scholar] [CrossRef] [PubMed]
- Holmannová, D.; Císařová, B.; Borský, P.; Fiala, Z.; Andrýs, C.; Hamáková, K.; Švadláková, T.; Krejsek, J.; Palička, V.; Kotingová, L.; et al. Goeckerman Regimen Reduces Alarmin Levels and PASI Score in Paediatric Patients with Psoriasis. Acta Med. 2021, 64, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Christmann, C.; Zenker, S.; Martens, L.; Hubner, J.; Loser, K.; Vogl, T.; Roth, J. Interleukin 17 Promotes Expression of Alarmins S100A8 and S100A9 During the Inflammatory Response of Keratinocytes. Front. Immunol. 2020, 11, 599947. [Google Scholar] [CrossRef] [PubMed]
- Dolivo, D.; Rodrigues, A.; Sun, L.; Galiano, R.; Mustoe, T.; Hong, S.J. Reduced hydration regulates pro-inflammatory cytokines via CD14 in barrier function-impaired skin. Biochim. Biophys. Acta Mol. Basis Dis. 2022, 1868, 166482. [Google Scholar] [CrossRef] [PubMed]
- Wilsmann-Theis, D.; Wagenpfeil, J.; Holzinger, D.; Roth, J.; Koch, S.; Schnautz, S.; Bieber, T.; Wenzel, J. Among the S100 proteins, S100A12 is the most significant marker for psoriasis disease activity. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 1165–1170. [Google Scholar] [CrossRef] [PubMed]
- Schonthaler, H.B.; Guinea-Viniegra, J.; Wculek, S.K.; Ruppen, I.; Ximénez-Embún, P.; Guío-Carrión, A.; Navarro, R.; Hogg, N.; Ashman, K.; Wagner, E.F. S100A8-S100A9 protein complex mediates psoriasis by regulating the expression of complement factor C3. Immunity 2013, 39, 1171–1181. [Google Scholar] [CrossRef] [PubMed]
- Berg, A.R.; Hong, C.G.; Svirydava, M.; Li, H.; Parel, P.M.; Florida, E.; O’Hagan, R.; Pantoja, C.J.; Lateef, S.S.; Anzenberg, P.; et al. Association of S100A8/A9 with Lipid-Rich Necrotic Core and Treatment with Biologic Therapy in Patients with Psoriasis: Results from an Observational Cohort Study. J. Investig. Dermatol. 2022, 142, 2909–2919. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, M.; Sawada, Y.; Saito-Sasaki, N.; Yoshioka, H.; Hama, K.; Omoto, D.; Ohmori, S.; Okada, E.; Nakamura, M. High S100A2 expression in keratinocytes in patients with drug eruption. Sci. Rep. 2021, 11, 5493. [Google Scholar] [CrossRef]
- da Costa, L.C.O.; Gardinassi, L.G.; Veras, F.P.; Milanezi, C.; Ramalho, L.N.Z.; Benevides, L.; Alves-Filho, J.C.; da Silva, J.S.; da Silva Souza, C. Expression of B lymphocyte-induced maturation protein 1 (Blimp-1) in keratinocyte and cytokine signalling drives human Th17 response in psoriasis. Arch. Dermatol. Res. 2023, 315, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Fritz, Y.; Klenotic, P.A.; Swindell, W.R.; Yin, Z.Q.; Groft, S.G.; Zhang, L.; Baliwag, J.; Camhi, M.I.; Diaconu, D.; Young, A.B.; et al. Induction of Alternative Proinflammatory Cytokines Accounts for Sustained Psoriasiform Skin Inflammation in IL-17C+IL-6KO Mice. J. Investig. Dermatol. 2017, 137, 696–705. [Google Scholar] [CrossRef]
- Kim, M.J.; Im, M.A.; Lee, J.S.; Mun, J.Y.; Kim, D.H.; Gu, A.; Kim, I.S. Effect of S100A8 and S100A9 on expressions of cytokine and skin barrier protein in human keratinocytes. Mol. Med. Rep. 2019, 20, 2476–2483. [Google Scholar] [CrossRef]
- Benedyk, M.; Sopalla, C.; Nacken, W.; Bode, G.; Melkonyan, H.; Banfi, B.; Kerkhoff, C. HaCaT keratinocytes overexpressing the S100 proteins S100A8 and S100A9 show increased NADPH oxidase and NF-kappaB activities. J. Investig. Dermatol. 2007, 127, 2001–2011. [Google Scholar] [CrossRef]
- Benezeder, T.; Painsi, C.; Patra, V.; Dey, S.; Holcmann, M.; Lange-Asschenfeldt, B.; Sibilia, M.; Wolf, P. Dithranol targets keratinocytes, their crosstalk with neutrophils and inhibits the IL-36 inflammatory loop in psoriasis. Elife 2020, 9, e56991. [Google Scholar] [CrossRef]
- Krueger, J.G.; Wharton, K.A., Jr.; Schlitt, T.; Suprun, M.; Torene, R.I.; Jiang, X.; Wang, C.Q.; Fuentes-Duculan, J.; Hartmann, N.; Peters, T.; et al. IL-17A inhibition by secukinumab induces early clinical, histopathologic, and molecular resolution of psoriasis. J. Allergy Clin. Immunol. 2019, 144, 750–763. [Google Scholar] [CrossRef]
- Visvanathan, S.; Baum, P.; Vinisko, R.; Schmid, R.; Flack, M.; Lalovic, B.; Kleiner, O.; Fuentes-Duculan, J.; Garcet, S.; Davis, J.W.; et al. Psoriatic skin molecular and histopathologic profiles after treatment with risankizumab versus ustekinumab. J. Allergy Clin. Immunol. 2019, 143, 2158–2169. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Guo, J.; Yin, L.; Tu, J.; Yin, Z. Tacrolimus Inhibits TNF-alpha/IL-17A-Produced pro-Inflammatory Effect on Human Keratinocytes by Regulating IkappaBzeta. Inflammation 2020, 43, 692–700. [Google Scholar] [CrossRef] [PubMed]
- Catlett, I.M.; Hu, Y.; Gao, L.; Banerjee, S.; Gordon, K.; Krueger, J.G. Molecular and clinical effects of selective tyrosine kinase 2 inhibition with deucravacitinib in psoriasis. J. Allergy Clin. Immunol. 2022, 149, 2010–2020.e2018. [Google Scholar] [CrossRef] [PubMed]
- Ten Bergen, L.L.; Petrovic, A.; Aarebrot, A.K.; Appel, S. Current knowledge on autoantigens and autoantibodies in psoriasis. Scand. J. Immunol. 2020, 92, e12945. [Google Scholar] [CrossRef]
- Lande, R.; Gregorio, J.; Facchinetti, V.; Chatterjee, B.; Wang, Y.H.; Homey, B.; Cao, W.; Wang, Y.H.; Su, B.; Nestle, F.O.; et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature 2007, 449, 564–569. [Google Scholar] [CrossRef]
- Yuan, Y.; Qiu, J.; Lin, Z.T.; Li, W.; Haley, C.; Mui, U.N.; Ning, J.; Tyring, S.K.; Wu, T. Identification of Novel Autoantibodies Associated with Psoriatic Arthritis. Arthritis Rheumatol. 2019, 71, 941–951. [Google Scholar] [CrossRef]
- Emmungil, H.; Ilgen, U.; Direskeneli, R.H. Autoimmunity in psoriatic arthritis: Pathophysiological and clinical aspects. Turk. J. Med. Sci. 2021, 51, 1601–1614. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, K.; Schauber, J.; Coda, A.; Lin, H.; Dorschner, R.A.; Schechter, N.M.; Bonnart, C.; Descargues, P.; Hovnanian, A.; Gallo, R.L. Kallikrein-mediated proteolysis regulates the antimicrobial effects of cathelicidins in skin. FASEB J. 2006, 20, 2068–2080. [Google Scholar] [CrossRef] [PubMed]
- Ong, P.Y.; Ohtake, T.; Brandt, C.; Strickland, I.; Boguniewicz, M.; Ganz, T.; Gallo, R.L.; Leung, D.Y. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N. Engl. J. Med. 2002, 347, 1151–1160. [Google Scholar] [CrossRef] [PubMed]
- Herster, F.; Bittner, Z.; Archer, N.K.; Dickhofer, S.; Eisel, D.; Eigenbrod, T.; Knorpp, T.; Schneiderhan-Marra, N.; Loffler, M.W.; Kalbacher, H.; et al. Neutrophil extracellular trap-associated RNA and LL37 enable self-amplifying inflammation in psoriasis. Nat. Commun. 2020, 11, 105. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, D.; Chamilos, G.; Lande, R.; Gregorio, J.; Meller, S.; Facchinetti, V.; Homey, B.; Barrat, F.J.; Zal, T.; Gilliet, M. Self-RNA-antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J. Exp. Med. 2009, 206, 1983–1994. [Google Scholar] [CrossRef] [PubMed]
- Nestle, F.O.; Conrad, C.; Tun-Kyi, A.; Homey, B.; Gombert, M.; Boyman, O.; Burg, G.; Liu, Y.J.; Gilliet, M. Plasmacytoid predendritic cells initiate psoriasis through interferon-alpha production. J. Exp. Med. 2005, 202, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Di Meglio, P.; Perera, G.K.; Nestle, F.O. The multitasking organ: Recent insights into skin immune function. Immunity 2011, 35, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Alwan, W.; Nestle, F.O. Pathogenesis and treatment of psoriasis: Exploiting pathophysiological pathways for precision medicine. Clin. Exp. Rheumatol. 2015, 33, S2–S6. [Google Scholar]
- Furue, M.; Kadono, T. The contribution of IL-17 to the development of autoimmunity in psoriasis. Innate Immun. 2019, 25, 337–343. [Google Scholar] [CrossRef]
- Lande, R.; Botti, E.; Jandus, C.; Dojcinovic, D.; Fanelli, G.; Conrad, C.; Chamilos, G.; Feldmeyer, L.; Marinari, B.; Chon, S.; et al. The antimicrobial peptide LL37 is a T-cell autoantigen in psoriasis. Nat. Commun. 2014, 5, 5621. [Google Scholar] [CrossRef]
- Wang, W.M.; Jin, H.Z. Heat shock proteins and psoriasis. Eur. J. Dermatol. 2019, 29, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Curry, J.L.; Qin, J.Z.; Bonish, B.; Carrick, R.; Bacon, P.; Panella, J.; Robinson, J.; Nickoloff, B.J. Innate immune-related receptors in normal and psoriatic skin. Arch. Pathol. Lab. Med. 2003, 127, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz, S.; Korkmaz, H. Effect of alterations in apoptotic pathway on development of metabolic syndrome in patients with psoriasis vulgaris. Br. J. Dermatol. 2017, 176, 1549–1557. [Google Scholar] [CrossRef] [PubMed]
- Damasiewicz-Bodzek, A.; Szumska, M.; Tyrpien-Golder, K. Antibodies to Heat Shock Proteins 90alpha and 90beta in Psoriasis. Arch. Immunol. Ther. Exp. 2020, 68, 9. [Google Scholar] [CrossRef]
- Besgen, P.; Trommler, P.; Vollmer, S.; Prinz, J.C. Ezrin, maspin, peroxiredoxin 2, and heat shock protein 27: Potential targets of a streptococcal-induced autoimmune response in psoriasis. J. Immunol. 2010, 184, 5392–5402. [Google Scholar] [CrossRef] [PubMed]
- Puig, L.; Fernandez-Figueras, M.T.; Ferrandiz, C.; Ribera, M.; de Moragas, J.M. Epidermal expression of 65 and 72 kd heat shock proteins in psoriasis and AIDS-associated psoriasiform dermatitis. J. Am. Acad. Dermatol. 1995, 33, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wu, L.; Bulek, K.; Martin, B.N.; Zepp, J.A.; Kang, Z.; Liu, C.; Herjan, T.; Misra, S.; Carman, J.A.; et al. The psoriasis-associated D10N variant of the adaptor Act1 with impaired regulation by the molecular chaperone hsp90. Nat. Immunol. 2013, 14, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Boyman, O.; Conrad, C.; Dudli, C.; Kielhorn, E.; Nickoloff, B.J.; Nestle, F.O. Activation of dendritic antigen-presenting cells expressing common heat shock protein receptor CD91 during induction of psoriasis. Br. J. Dermatol. 2005, 152, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Bayramgurler, D.; Ozkara, S.K.; Apaydin, R.; Ercin, C.; Bilen, N. Heat shock proteins 60 and 70 expression of cutaneous lichen planus: Comparison with normal skin and psoriasis vulgaris. J. Cutan. Pathol. 2004, 31, 586–594. [Google Scholar] [CrossRef]
- Baroni, A.; Paoletti, I.; Ruocco, E.; Agozzino, M.; Tufano, M.A.; Donnarumma, G. Possible role of Malassezia furfur in psoriasis: Modulation of TGF-beta1, integrin, and HSP70 expression in human keratinocytes and in the skin of psoriasis-affected patients. J. Cutan. Pathol. 2004, 31, 35–42. [Google Scholar] [CrossRef]
- Cancino-Diaz, M.E.; Ruiz-Gonzalez, V.; Ramirez-Resendiz, L.; Ortiz, B.; Dominguez-Lopez, M.L.; Paredes-Cabrera, G.C.; Leon-Dorantes, G.; Blancas-Gonzalez, F.; Jimenez-Zamudio, L.; Garcia-Latorre, E. IgG class antibodies from psoriasis patients recognize the 60-KDa heat-shock protein of Streptococcus pyogenes. Int. J. Dermatol. 2004, 43, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Seifarth, F.G.; Lax, J.E.; Harvey, J.; DiCorleto, P.E.; Husni, M.E.; Chandrasekharan, U.M.; Tytell, M. Topical heat shock protein 70 prevents imiquimod-induced psoriasis-like inflammation in mice. Cell Stress Chaperones 2018, 23, 1129–1135. [Google Scholar] [CrossRef] [PubMed]
- Tukaj, S.; Sitko, K. Heat Shock Protein 90 (Hsp90) and Hsp70 as Potential Therapeutic Targets in Autoimmune Skin Diseases. Biomolecules 2022, 12, 1153. [Google Scholar] [CrossRef] [PubMed]
- Ben Abdallah, H.; Seeler, S.; Bregnhoj, A.; Ghatnekar, G.; Kristensen, L.S.; Iversen, L.; Johansen, C. Heat shock protein 90 inhibitor RGRN-305 potently attenuates skin inflammation. Front. Immunol. 2023, 14, 1128897. [Google Scholar] [CrossRef] [PubMed]
- Kakeda, M.; Arock, M.; Schlapbach, C.; Yawalkar, N. Increased expression of heat shock protein 90 in keratinocytes and mast cells in patients with psoriasis. J. Am. Acad. Dermatol. 2014, 70, 683–690.e681. [Google Scholar] [CrossRef] [PubMed]
- Stenderup, K.; Rosada, C.; Gavillet, B.; Vuagniaux, G.; Dam, T.N. Debio 0932, a new oral Hsp90 inhibitor, alleviates psoriasis in a xenograft transplantation model. Acta Derm. Venereol. 2014, 94, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Qiao, P.; Guo, W.; Ke, Y.; Fang, H.; Zhuang, Y.; Jiang, M.; Zhang, J.; Shen, S.; Qiao, H.; Dang, E.; et al. Mechanical Stretch Exacerbates Psoriasis by Stimulating Keratinocyte Proliferation and Cytokine Production. J. Investig. Dermatol. 2019, 139, 1470–1479. [Google Scholar] [CrossRef] [PubMed]
- Tseng, P.Y.; Hoon, M.A. Specific beta-Defensins Stimulate Pruritus through Activation of Sensory Neurons. J. Investig. Dermatol. 2022, 142, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Harder, J.; Schroder, J.M. Psoriatic scales: A promising source for the isolation of human skin-derived antimicrobial proteins. J. Leukoc. Biol. 2005, 77, 476–486. [Google Scholar] [CrossRef]
- Harder, J.; Bartels, J.; Christophers, E.; Schroder, J.M. A peptide antibiotic from human skin. Nature 1997, 387, 861. [Google Scholar] [CrossRef]
- Harder, J.; Bartels, J.; Christophers, E.; Schroder, J.M. Isolation and characterization of human beta -defensin-3, a novel human inducible peptide antibiotic. J. Biol. Chem. 2001, 276, 5707–5713. [Google Scholar] [CrossRef] [PubMed]
- Tampa, M.; Sarbu, M.I.; Mitran, M.I.; Mitran, C.I.; Matei, C.; Georgescu, S.R. The Pathophysiological Mechanisms and the Quest for Biomarkers in Psoriasis, a Stress-Related Skin Disease. Dis. Markers 2018, 2018, 5823684. [Google Scholar] [CrossRef] [PubMed]
- Jansen, P.A.; Rodijk-Olthuis, D.; Hollox, E.J.; Kamsteeg, M.; Tjabringa, G.S.; de Jongh, G.J.; van Vlijmen-Willems, I.M.; Bergboer, J.G.; van Rossum, M.M.; de Jong, E.M.; et al. Beta-defensin-2 protein is a serum biomarker for disease activity in psoriasis and reaches biologically relevant concentrations in lesional skin. PLoS ONE 2009, 4, e4725. [Google Scholar] [CrossRef] [PubMed]
- Kolbinger, F.; Loesche, C.; Valentin, M.A.; Jiang, X.; Cheng, Y.; Jarvis, P.; Peters, T.; Calonder, C.; Bruin, G.; Polus, F.; et al. β-Defensin 2 is a responsive biomarker of IL-17A-driven skin pathology in patients with psoriasis. J. Allergy Clin. Immunol. 2017, 139, 923–932.e928. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Yamasaki, K. Psoriasis and Antimicrobial Peptides. Int. J. Mol. Sci. 2020, 21, 6791. [Google Scholar] [CrossRef] [PubMed]
- Johansen, C.; Bertelsen, T.; Ljungberg, C.; Mose, M.; Iversen, L. Characterization of TNF-alpha- and IL-17A-Mediated Synergistic Induction of DEFB4 Gene Expression in Human Keratinocytes through IkappaBzeta. J. Investig. Dermatol. 2016, 136, 1608–1616. [Google Scholar] [CrossRef] [PubMed]
- Pourani, M.R.; Abdollahimajd, F.; Zargari, O.; Shahidi Dadras, M. Soluble biomarkers for diagnosis, monitoring, and therapeutic response assessment in psoriasis. J. Dermatolog. Treat. 2022, 33, 1967–1974. [Google Scholar] [CrossRef] [PubMed]
- Schakel, K.; Reich, K.; Asadullah, K.; Pinter, A.; Jullien, D.; Weisenseel, P.; Paul, C.; Gomez, M.; Wegner, S.; Personke, Y.; et al. Early disease intervention with guselkumab in psoriasis leads to a higher rate of stable complete skin clearance (‘clinical super response’): Week 28 results from the ongoing phase IIIb randomized, double-blind, parallel-group, GUIDE study. J. Eur. Acad. Dermatol. Venereol. 2023, 37, 2016–2027. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.T.; Qiao, Z.H.; Tian, J.B.; Lin, J.L.; Hou, S.C.; Liu, X.M. The effect of secukinumab treatment for psoriasis on serum cytokines and correlation with disease severity. Skin Res. Technol. 2023, 29, e13405. [Google Scholar] [CrossRef]
- Cardner, M.; Tuckwell, D.; Kostikova, A.; Forrer, P.; Siegel, R.M.; Marti, A.; Vandemeulebroecke, M.; Ferrero, E. Analysis of serum proteomics data identifies a quantitative association between beta-defensin 2 at baseline and clinical response to IL-17 blockade in psoriatic arthritis. RMD Open 2023, 9, e003042. [Google Scholar] [CrossRef]
- Uzuncakmak, T.K.; Karadag, A.S.; Ozkanli, S.; Akbulak, O.; Ozlu, E.; Akdeniz, N.; Oguztuzun, S. Alteration of tissue expression of human beta defensin-1 and human beta defensin-2 in psoriasis vulgaris following phototherapy. Biotech. Histochem. 2020, 95, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Ham, J.; Shin, J.W.; Ko, B.C.; Kim, H.Y. Targeting the Epithelium-Derived Innate Cytokines: From Bench to Bedside. Immune Netw. 2022, 22, e11. [Google Scholar] [CrossRef] [PubMed]
- El-Ghareeb, M.I.; Helmy, A.; Al Kazzaz, S.; Samir, H. Serum TSLP is a potential biomarker of psoriasis vulgaris activity. Psoriasis 2019, 9, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Suwarsa, O.; Dharmadji, H.P.; Sutedja, E.; Herlina, L.; Sori, P.R.; Hindritiani, R.; Dwiyana, R.F.; Gunawan, H. Skin tissue expression and serum level of thymic stromal lymphopoietin in patients with psoriasis vulgaris. Dermatol. Rep. 2019, 11, 8006. [Google Scholar] [CrossRef] [PubMed]
- Fromm, S.; Cunningham, C.C.; Dunne, M.R.; Veale, D.J.; Fearon, U.; Wade, S.M. Enhanced angiogenic function in response to fibroblasts from psoriatic arthritis synovium compared to rheumatoid arthritis. Arthritis Res. Ther. 2019, 21, 297. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.H.; Zuo, Y.G. Thymic Stromal Lymphopoietin in Cutaneous Immune-Mediated Diseases. Front. Immunol. 2021, 12, 698522. [Google Scholar] [CrossRef] [PubMed]
- Gago-Lopez, N.; Mellor, L.F.; Megias, D.; Martin-Serrano, G.; Izeta, A.; Jimenez, F.; Wagner, E.F. Role of bulge epidermal stem cells and TSLP signaling in psoriasis. EMBO Mol. Med. 2019, 11, e10697. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.; Chong, H.J.; So, J.; Jo, Y.; Yune, T.Y.; Ju, B.G. Ghrelin Represses Thymic Stromal Lymphopoietin Gene Expression through Activation of Glucocorticoid Receptor and Protein Kinase C Delta in Inflamed Skin Keratinocytes. Int. J. Mol. Sci. 2022, 23, 3977. [Google Scholar] [CrossRef] [PubMed]
- Tashiro, N.; Segawa, R.; Tobita, R.; Asakawa, S.; Mizuno, N.; Hiratsuka, M.; Hirasawa, N. Hypoxia inhibits TNF-alpha-induced TSLP expression in keratinocytes. PLoS ONE 2019, 14, e0224705. [Google Scholar] [CrossRef]
- Segawa, R.; Shigeeda, K.; Hatayama, T.; Dong, J.; Mizuno, N.; Moriya, T.; Hiratsuka, M.; Hirasawa, N. EGFR transactivation is involved in TNF-alpha-induced expression of thymic stromal lymphopoietin in human keratinocyte cell line. J. Dermatol. Sci. 2018, 89, 290–298. [Google Scholar] [CrossRef]
- Meephansan, J.; Komine, M.; Tsuda, H.; Karakawa, M.; Tominaga, S.; Ohtsuki, M. Expression of IL-33 in the epidermis: The mechanism of induction by IL-17. J. Dermatol. Sci. 2013, 71, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Hu, Y.; Gong, Y.; Zhang, X.; Cui, L.; Chen, R.; Yu, Y.; Yu, Q.; Chen, Y.; Diao, H.; et al. Interleukin-33 alleviates psoriatic inflammation by suppressing the T helper type 17 immune response. Immunology 2020, 160, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.; Chen, H.; Chen, L.; Mao, J.; Cai, S.; Xiao, Y.; Li, J.; Shi, J.; Li, B.; Xu, Y.; et al. An Autocrine Circuit of IL-33 in Keratinocytes Is Involved in the Progression of Psoriasis. J. Investig. Dermatol. 2021, 141, 596–606.e597. [Google Scholar] [CrossRef] [PubMed]
- Ampawong, S.; Kengkoom, K.; Sukphopetch, P.; Aramwit, P.; Muangkaew, W.; Kanjanapruthipong, T.; Buaban, T. Evaluating the effect of rice (Oryza sativa L.: SRNC05053-6-2) crude extract on psoriasis using in vitro and in vivo models. Sci. Rep. 2020, 10, 17618. [Google Scholar] [CrossRef] [PubMed]
- Barr, T.P.; Garzia, C.; Guha, S.; Fletcher, E.K.; Nguyen, N.; Wieschhaus, A.J.; Ferrer, L.; Covic, L.; Kuliopulos, A. PAR2 Pepducin-Based Suppression of Inflammation and Itch in Atopic Dermatitis Models. J. Investig. Dermatol. 2019, 139, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Meephansan, J.; Tsuda, H.; Komine, M.; Tominaga, S.; Ohtsuki, M. Regulation of IL-33 expression by IFN-gamma and tumor necrosis factor-alpha in normal human epidermal keratinocytes. J. Investig. Dermatol. 2012, 132, 2593–2600. [Google Scholar] [CrossRef] [PubMed]
- Du, H.Y.; Fu, H.Y.; Li, D.N.; Qiao, Y.; Wang, Q.W.; Liu, W. The Expression and Regulation of Interleukin-33 in Human Epidermal Keratinocytes: A New Mediator of Atopic Dermatitis and Its Possible Signaling Pathway. J. Interferon Cytokine Res. 2016, 36, 552–562. [Google Scholar] [CrossRef]
- Tsuji, G.; Hashimoto-Hachiya, A.; Yen, V.H.; Miake, S.; Takemura, M.; Mitamura, Y.; Ito, T.; Murata, M.; Furue, M.; Nakahara, T. Aryl Hydrocarbon Receptor Activation Downregulates IL-33 Expression in Keratinocytes via Ovo-Like 1. J. Clin. Med. 2020, 9, 891. [Google Scholar] [CrossRef] [PubMed]
- Cannavo, S.P.; Bertino, L.; Di Salvo, E.; Papaianni, V.; Ventura-Spagnolo, E.; Gangemi, S. Possible Roles of IL-33 in the Innate-Adaptive Immune Crosstalk of Psoriasis Pathogenesis. Mediat. Inflamm. 2019, 2019, 7158014. [Google Scholar] [CrossRef]
- Imai, Y. ILC2s in skin disorders. Allergol. Int. 2023, 72, 201–206. [Google Scholar] [CrossRef]
- Raimondo, A.; Lembo, S.; Di Caprio, R.; Donnarumma, G.; Monfrecola, G.; Balato, N.; Ayala, F.; Balato, A. Psoriatic cutaneous inflammation promotes human monocyte differentiation into active osteoclasts, facilitating bone damage. Eur. J. Immunol. 2017, 47, 1062–1074. [Google Scholar] [CrossRef] [PubMed]
- Meephansan, J.; Subpayasarn, U.; Ponnikorn, S.; Chakkavittumrong, P.; Juntongjin, P.; Komine, M.; Ohtsuki, M.; Poovorawan, Y. Methotrexate, but not narrowband ultraviolet B radiation, suppresses interleukin-33 mRNA levels in psoriatic plaques and protein levels in serum of patients with psoriasis. J. Dermatol. 2018, 45, 322–325. [Google Scholar] [CrossRef] [PubMed]
- Guilloteau, K.; Paris, I.; Pedretti, N.; Boniface, K.; Juchaux, F.; Huguier, V.; Guillet, G.; Bernard, F.X.; Lecron, J.C.; Morel, F. Skin Inflammation Induced by the Synergistic Action of IL-17A, IL-22, Oncostatin M, IL-1{alpha}, and TNF-{alpha} Recapitulates Some Features of Psoriasis. J. Immunol. 2010, 184, 5263–5270. [Google Scholar] [CrossRef] [PubMed]
- Conti, P.; Pregliasco, F.E.; Bellomo, R.G.; Gallenga, C.E.; Caraffa, A.; Kritas, S.K.; Lauritano, D.; Ronconi, G. Mast Cell Cytokines IL-1, IL-33, and IL-36 Mediate Skin Inflammation in Psoriasis: A Novel Therapeutic Approach with the Anti-Inflammatory Cytokines IL-37, IL-38, and IL-1Ra. Int. J. Mol. Sci. 2021, 22, 8076. [Google Scholar] [CrossRef] [PubMed]
- Iznardo, H.; Puig, L. IL-1 Family Cytokines in Inflammatory Dermatoses: Pathogenetic Role and Potential Therapeutic Implications. Int. J. Mol. Sci. 2022, 23, 9479. [Google Scholar] [CrossRef] [PubMed]
- Rabeony, H.; Petit-Paris, I.; Garnier, J.; Barrault, C.; Pedretti, N.; Guilloteau, K.; Jegou, J.F.; Guillet, G.; Huguier, V.; Lecron, J.C.; et al. Inhibition of keratinocyte differentiation by the synergistic effect of IL-17A, IL-22, IL-1α, TNFα and oncostatin M. PLoS ONE 2014, 9, e101937. [Google Scholar] [CrossRef] [PubMed]
- Romero, L.I.; Ikejima, T.; Pincus, S.H. In Situ Localization of Interleukin-1 in Normal and Psoriatic Skin. J. Investig. Dermatol. 1989, 93, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Portugal-Cohen, M.; Horev, L.; Ruffer, C.; Schlippe, G.; Voss, W.; Ma’or, Z.; Oron, M.; Soroka, Y.; Frušić-Zlotkin, M.; Milner, Y.; et al. Non-invasive skin biomarkers quantification of psoriasis and atopic dermatitis: Cytokines, antioxidants and psoriatic skin auto-fluorescence. Biomed. Pharmacother. 2012, 66, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Schaap, M.J.; Bruins, F.M.; He, X.; Orro, K.; Peppelman, M.; van Erp, P.E.J.; de Jong, E.M.G.J.; Koenen, H.J.P.M.; van den Bogaard, E.H.; Seyger, M.M.B. Skin Surface Protein Detection by Transdermal Analysis Patches in Pediatric Psoriasis. Skin Pharmacol. Physiol. 2021, 34, 271–280. [Google Scholar] [CrossRef]
- Orro, K.; Salk, K.; Merkulova, A.; Abram, K.; Karelson, M.; Traks, T.; Neuman, T.; Spee, P.; Kingo, K. Non-Invasive Assessment of Skin Surface Proteins of Psoriasis Vulgaris Patients in Response to Biological Therapy. Int. J. Mol. Sci. 2023, 24, 16248. [Google Scholar] [CrossRef]
- Orro, K.; Salk, K.; Abram, K.; Arshavskaja, J.; Meikas, A.; Karelson, M.; Neuman, T.; Kingo, K.; Spee, P. Assessment of soluble skin surface protein levels for monitoring. Front. Med. 2023, 10, 1072160. [Google Scholar] [CrossRef]
- Tamilselvi, E.; Haripriya, D.; Hemamalini, M.; Pushpa, G.; Swapna, S. Association of disease severity with IL-1 levels in methotrexate-treated psoriasis patients. Scand. J. Immunol. 2013, 78, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Mee, J.B.; Cork, M.J.; di Giovine, F.S.; Duff, G.W.; Groves, R.W. Interleukin-1: A key inflammatory mediator in psoriasis? Cytokine 2006, 33, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Krzesicki, R.F.; Hatfield, C.A.; Bienkowski, M.J.; McGuire, J.C.; Winterrowd, G.E.; Chapman, D.L.; Berger, A.E.; McEwan, R.N.; Carter, D.B.; Chosay, J.G. Regulation of expression of IL-1 receptor antagonist protein in human synovial and dermal fibroblasts. J. Immunol. 1993, 150, 4008–4018. [Google Scholar] [CrossRef] [PubMed]
- McColl, S.R.; Paquin, R.; Ménard, C.; Beaulieu, A.D. Human neutrophils produce high levels of the interleukin 1 receptor antagonist in response to granulocyte/macrophage colony-stimulating factor and tumor necrosis factor alpha. J. Exp. Med. 1992, 176, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Janson, R.W.; Hance, K.R.; Arend, W.P. Production of IL-1 receptor antagonist by human in vitro-derived macrophages. Effects of lipopolysaccharide and granulocyte-macrophage colony-stimulating factor. J. Immunol. 1991, 147, 4218–4223. [Google Scholar] [CrossRef] [PubMed]
- Arend, W.P. Interleukin 1 receptor antagonist. A new member of the interleukin 1 family. J. Clin. Investig. 1991, 88, 1445–1451. [Google Scholar] [CrossRef] [PubMed]
- Yegorov, S.; Babenko, D.; Kozhakhmetov, S.; Akhmaltdinova, L.; Kadyrova, I.; Nurgozhina, A.; Nurgaziyev, M.; Good, S.V.; Hortelano, G.H.; Yermekbayeva, B.; et al. Psoriasis Is Associated with Elevated Gut IL-1α and Intestinal Microbiome Alterations. Front. Immunol. 2020, 11, 571319. [Google Scholar] [CrossRef]
- Boraschi, D.; Tagliabue, A. The interleukin-1 receptor family. Semin. Immunol. 2013, 25, 394–407. [Google Scholar] [CrossRef]
- Lücke, G. Interplay of a well-known medication and newly discovered transporters driving bacteria-induced inflammation in psoriasis. Allergy, 2024; online ahead of print. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alarmin | Blood/Synovial Fluid Level/PBMC Expression | Synovial Expression | Potential Role in the Pathogenesis | References |
|---|---|---|---|---|
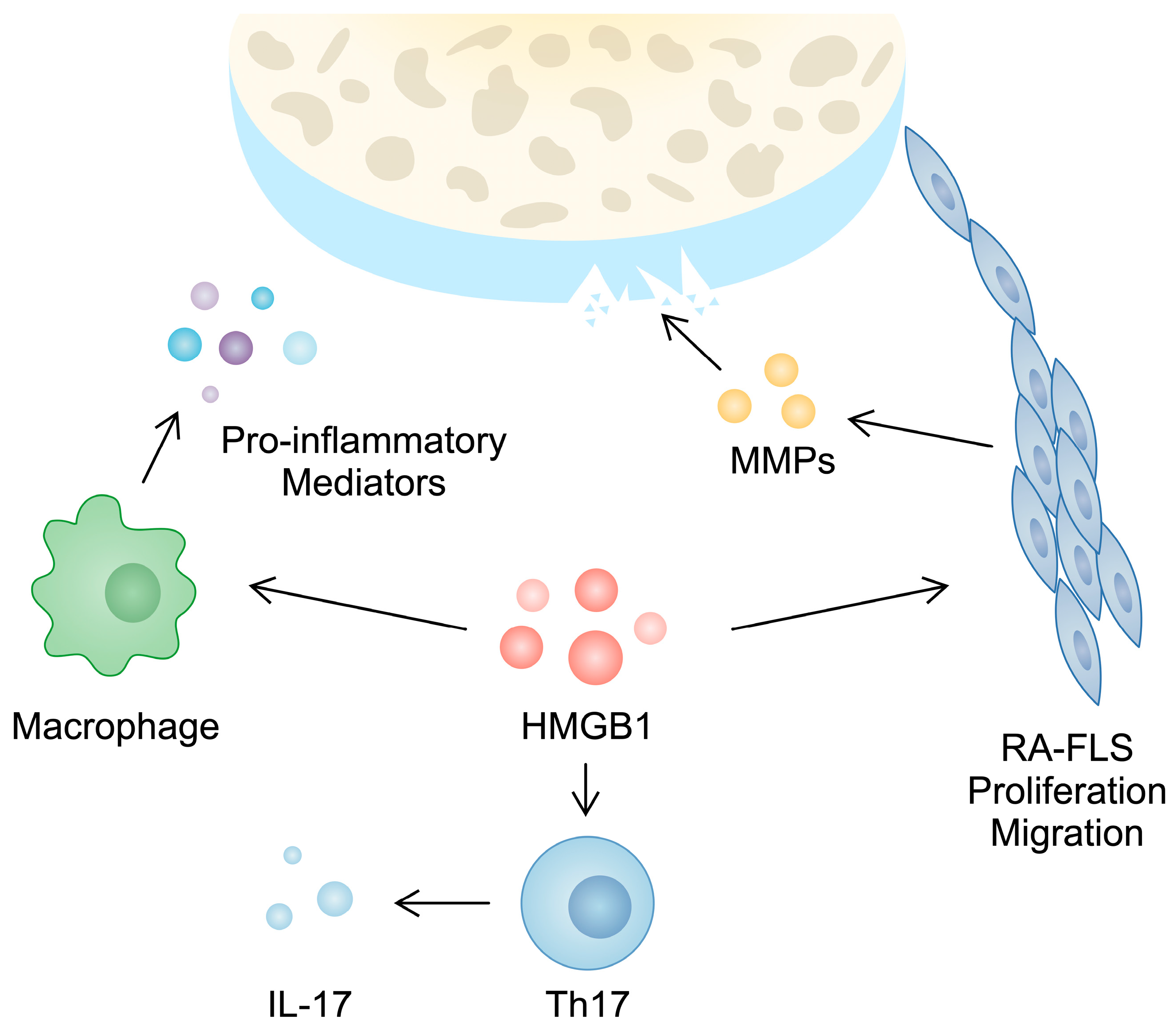
| HMGB1 | Increased | Elevated in untreated patients compared with patients receiving methotrexate | HMGB1 may promote RA-FLS proliferation, migration, invasiveness, and the expression of MMPs. HMGB1 synergizes with LPS to induce aggressive behavior in RA-FLSs. HMGB1 stimulates synovial angiogenesis. HMGB1 promotes the production of pro-inflammatory cytokines from macrophages. By forming a complex with CXCL12, HMGB1 enhances monocyte migration. HMGB1 promotes macrophage pyroptosis, a process that has been implicated in the pathogenesis of RA. HMGB1 enhances the Th17 cell population, which plays a role in RA pathogenesis. | [27,28,29,31,32,33,37,38,41,43] |
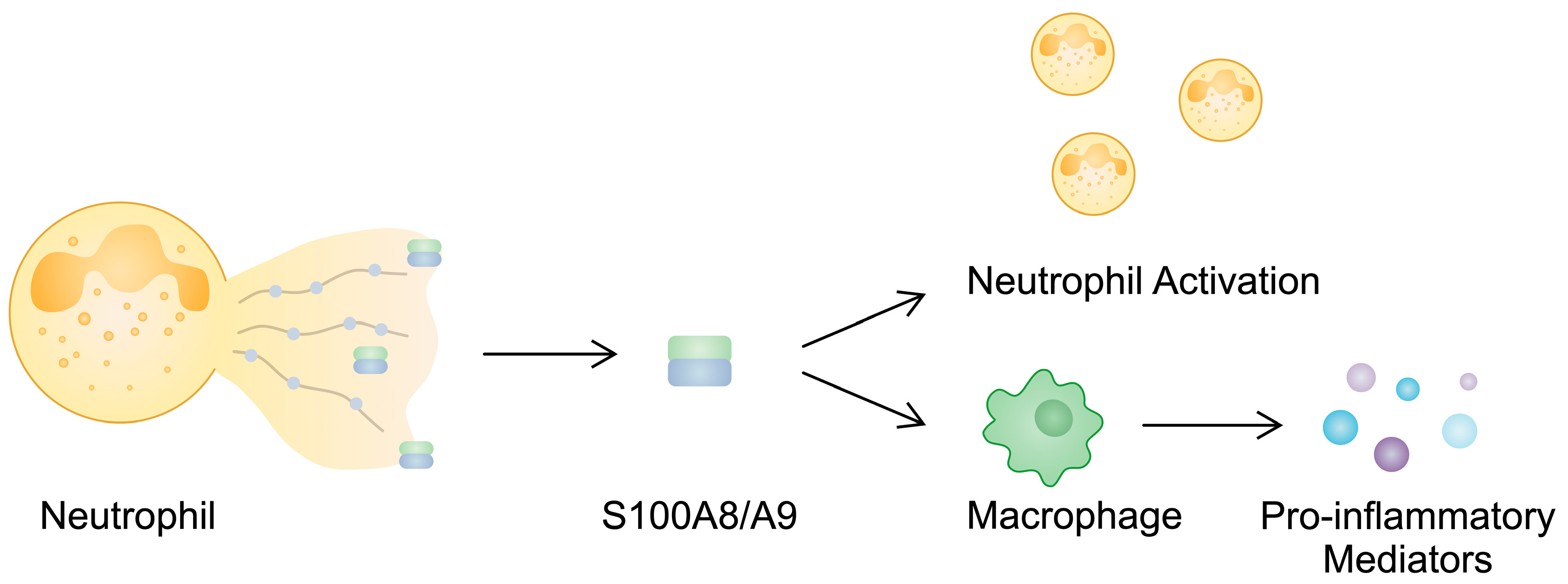
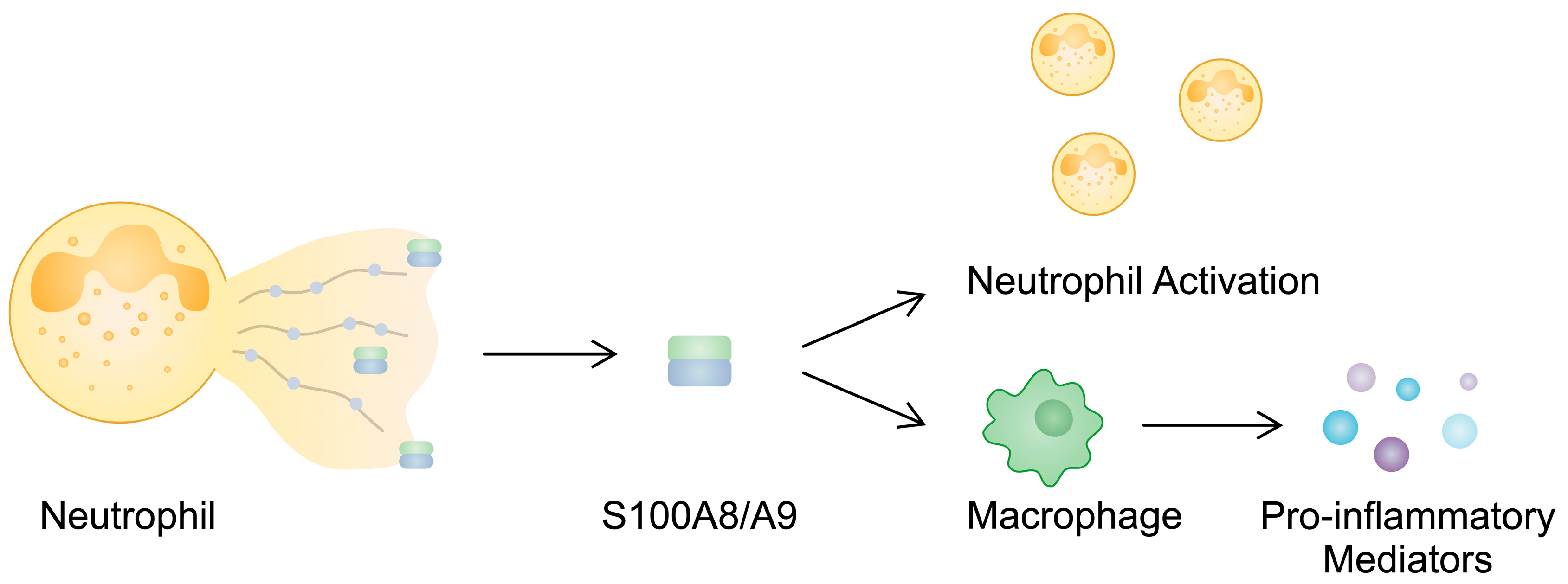
| S100A8/S100A9 | Blood: Increased | - | Calprotectin is released in NETs and contributes to neutrophil activation. S100A8/A9 stimulates the production of pro-inflammatory mediators in monocytes. The expression of S100A8/A9 in RA-FLSs increases after stimulation with IL-22, a cytokine involved in the pathogenesis of RA. | [57,58,59,72,75,78] |
| S100A11 | Synovial fluid: Increased (vs. OA) | - | S100A11 is released from neutrophils during NETosis, and this alarmin can contribute to the pro-inflammatory responses of other neutrophils. | [86,87] |
| S100A12 | Blood: Increased Synovial fluid: Increased (vs. OA) | - | S100A12 enhances the infiltration of neutrophiles. S100A12 has been found to enhance osteoclastogenesis. | [88,89,91] |
| IL-33 | Blood: Increased Synovial fluid: Increased (vs. OA) | IL-33 regulates inflammatory responses in RA-FLSs. | [94,95,100,101] |
| Alarmin | Blood/Synovial Fluid Level/PBMC Expression | Expression in Synovial Tissues/Chondrocytes | Potential Role in the Pathogenesis | References |
|---|---|---|---|---|
| HMGB1 | Increased | Increased | HMGB1 levels positively correlate with disease severity. Targeting HMGB1 was associated with beneficial effects in in vitro and in vivo studies. miR-410-3p targets and decreases the expression of HMGB1. Overexpression of miR-410-3p in OA mouse models is associated with reduced cartilage damage. Treatment of chondrocytes with IL-1β increases the expression of HMGB1. HMGB1 enhances the expression of β-catenin in the pre-chondral cell line. Wnt/β-catenin pathway is associated with the progression of OA. | [120,121,123,124,125,126,128,129] |
| S100A8/A9 | - | Increased | Serum levels of S100A8/A9 are positively correlated with the WOMAC score. Treatment of synovial tissue with S100A9 enhances the expression of pro-inflammatory cytokines. S100A9 enhances the production of cartilage-degrading enzymes. S100A8 and S100A9 are involved in the processes of osteophyte formation. S100A8/A9 can enhance signaling through the Wnt pathway. Using an S100A9/TLR-4 inhibitor is associated with suppressed OA. | [131,132,133,138,139,140,141,144] |
| IL-33 | - | Increased | Stimulation of articular chondrocytes with IL-33 is associated with elevated expression of pro-inflammatory mediators and cartilage-degrading enzymes. Chondrocyte-specific knockout of IL-33 and the use of ST2-neutralizing antibodies alleviates OA in mice. Chondrocytes treated with IL-33 show reduced expression of anti-inflammatory IL-37. | [148,149,150,151] |
| Alarmin | Serum Expression | Keratinocyte Expression | Potential Role in the Pathogenesis of Psoriasis | References |
|---|---|---|---|---|
| HMGB1 | Increased | Increased | HMGB1 promotes the excessive proliferation and expression of inflammatory cytokines by keratinocytes. HMGB1 promotes autophagy. HMGB1 released from keratinocytes shifts the polarization of macrophages toward the inflammatory M1 phenotype. | [161,162,167,169] |
| S100A2 | - | Increased | - | [177] |
| S100A7 | Increased | Increased | - | [161,174] |
| S100A8 | - | Increased | S100A8 decreases the expression of filaggrin and loricrin, impairing the skin barrier. S100A8 upregulates the expression of IL-6, IL-8, and MCP-1. Keratinocytes overexpressing S100A8 exhibit increased NF-κB activity. | [173,180,181] |
| S100A9 | - | Increased | S100A9 decreases the expression of filaggrin and loricrin, impairing the skin barrier. S100A9 upregulates the expression of IL-6, IL-8, and MCP-1. IL-22 is associated with the upregulation of S100A9 in psoriatic skin. Keratinocytes overexpressing S100A9 exhibit increased NF-κB activity. S100A9 has a positive correlation with Th17-related gene expression, including IL-1β, IL-6, IL-21, IL-22, IL-27, TNF, IL-12-beta, IL-23A, and IL-17A. | [173,179,180,181] |
| S100A12 | Increased | Increased | [161,174] | |
| S100A8/A9 | Increased | Increased | S100A8 and S100A9 increase the transcriptional level of IL-17A, mediating the development of autoreactive CD8+ T cells. S100A8 and A9 enhance the secretion of NF-κB pathway products from neutrophils and monocytes, contributing to plaque disease. In patients with psoriasis, S100A8/A9 potentiates the production of IL-8 induced by TNF-α. | [161,172,173,174,175,176,181] |
| Cathelicidin (LL-37) | Increased level of LL-37 autoantibodies | Increased | Cathelicidin is recognized as an autoantigen by circulating T cells. LL-37 activates dDCs via TLR7 and TLR9. Cathelicidin binds nucleic acids released from keratinocytes upon injury and forms a complex that activates LL-37-specific dDCs. LL-37 induces the proliferation of circulating CD3+ T cells, which produce IL-17. | [188,189,190,191,193,194,198,199] |
| HSP27 | - | Increased | HSP27 may be an autoantigen associated with immune response in streptococcal-induced psoriasis. An increase in HSP27 expression in response to stress may explain the role of a stressful environment in exacerbating psoriatic lesions. | [153,204] |
| HSP60 | - | Increased | High response to S. pyogenes HSP60 may suggest the involvement of that alarmin in the chronic form of psoriasis. An association between the response to S. pyogenes HSP60 and the chronic form of psoriasis has been observed. | [210] |
| HSP70 | - | Conflicting results | HSP70 acts via its receptor—CD91—expressed predominantly by activated antigen-presenting cells, observed in the process of the early development of psoriatic lesions. HSP70 downregulates mRNA of IL-4, IL-5, and IL-17F and upregulates that of IL-17A and IL-22. | [207,208,211] |
| HSP90 α | Higher levels of anti-Hsp90α antibodies | Increased | The immune reaction against microbial HSP may attack autologous HSP, leading to autoimmunity. HSP90 plays a key role in regulating Act 1, a molecule that is essential in IL-17-dependent signaling. | [200,201,202,203,206] |
| HSP65 | Elevated HSP65 antibody titers | - | HSP65 is an antigen of Mycobacteria species; it supports the mechanism of molecular mimicry and underlying infection-induced psoriasis. | [200] |
| Beta-defensins | Increased | Increased | Human beta-defensin 2 acts as a ligand for CCR6, thereby inducing Th17. | [221,222,223,226] |
| TSLP | Increased | Increased | TSLP stimulates the hyperproliferation of epidermal cells and their expression of VEGF-α. TSLP is responsible for the maturation of antigen-presenting cells. TSLP promotes IL-23 production via CD40L. | [216,227,231,232,233,236] |
| IL-33 | Increased | Increased | IL-33 stimulates keratinocytes into producing CXCL1 and CXCL8, which are responsible for recruiting neutrophils, and CCL20, which recruits IL-17-producing T cells to the epidermis. IL-33 lets DC mature, which enables the induction of differentiation into Th17 cells, producing IL-17A. IL-33 activates the release of IL-1, IL-6, and IL-13 by mast cells. This alarmin plays a role in inducing pruritus by directly stimulating nerves in psoriasis. IL-33 might trigger Th2 cells to produce IL-4, IL-13, and IL-31. | [20,240,241,243,244,248,249] |
| IL-1α | Lowered | Conflicting results | IL-1 can induce inflammation by regulating several genes in the skin. Propeptide of IL-1, via CCL-20, causes the development of inflammatory skin lesions and attracts neutrophils to infiltrate the tissue. The propeptide of IL-1 recruits neutrophils and monocytes as an initial step of inflammation IL-1 cytokines can stimulate the production of IL-6, TNF, and IL-33 by mast cells. IL-1 alpha acts synergistically with IL-17A, IL-22, OSM, and TNF alpha in inhibiting the differentiation of keratinocytes. The alarmin stimulates the release of IL-36 from macrophages. IL-1 activates dendritic cells, which release IL-12 and IL-23, leading subsequently to the differentiation of Th1 and Th17 cells. | [18,252,253,254,255,256,260,261] |
| Drug | Mechanism of Action | Impact on Alarmins | References |
|---|---|---|---|
| Etanercept | TNF-α inhibitor | Decreased expression of HMGB1 in the skin. Decreased serum levels of S100A7, S100A12, and S100A8/A9. | [170] [174,176] |
| Ustekinumab | IL-12/23 inhibitor | Decreased serum levels of S100A7, S100A12, and S100A8/A9. Downregulation of HSP90 in the skin.Increased level of lesional IL-1α in the skin. | [176] [214] [259] |
| Secukinumab | IL-17a inhibitor | Decreased expression of S10S0A7A, S100A12, and S100A9 in the skin. Decreased serum levels of S100A7, S100A12, and S100A8/A9. Higher serum levels of hBD2. Decreased expression of hBD2 in the skin. Increased level of lesional IL-1α in the skin. | [172,176,183] [183,227,228,259] |
| Risankizumab | IL-23 inhibitor | Decreased expression of S100A7 in the skin. Decreased expression of hBD2 in the skin. | [184] |
| Guselkumab | IL-23 inhibitor | Decreased expression of hBD2 in the skin. | [183,227] |
| Tacrolimus | Calcineurin inhibitor | Inhibition of TNF-α/IL-17A-induced S100A9 expression in the skin. | [185] |
| Deucravacitinib | TYK2 inhibitor | Decreased expression of S100A9, S100A8, and β-defensin 4A/B. | [186] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiełbowski, K.; Stańska, W.; Bakinowska, E.; Rusiński, M.; Pawlik, A. The Role of Alarmins in the Pathogenesis of Rheumatoid Arthritis, Osteoarthritis, and Psoriasis. Curr. Issues Mol. Biol. 2024, 46, 3640-3675. https://doi.org/10.3390/cimb46040228
Kiełbowski K, Stańska W, Bakinowska E, Rusiński M, Pawlik A. The Role of Alarmins in the Pathogenesis of Rheumatoid Arthritis, Osteoarthritis, and Psoriasis. Current Issues in Molecular Biology. 2024; 46(4):3640-3675. https://doi.org/10.3390/cimb46040228
Chicago/Turabian StyleKiełbowski, Kajetan, Wiktoria Stańska, Estera Bakinowska, Marcin Rusiński, and Andrzej Pawlik. 2024. "The Role of Alarmins in the Pathogenesis of Rheumatoid Arthritis, Osteoarthritis, and Psoriasis" Current Issues in Molecular Biology 46, no. 4: 3640-3675. https://doi.org/10.3390/cimb46040228