
Specific Learning Disorders: Variation Analysis of 15 Candidate Genes in 9 Multiplex Families

 ,
,  , , ,
, , ,  and
and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. SLD Diagnosis of the Subject
2.2. Genomic DNA Preparation and Mutational Analysis
2.3. Bioinformatic Analysis
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brimo, K.; Dinkler, L.; Gillberg, C.; Lichtenstein, P.; Lundström, S.; Åsberg Johnels, J. The co-occurrence of neurodevelopmental problems in dyslexia. Dyslexia 2021, 27, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Grigorenko, E.L.; Compton, D.L.; Fuchs, L.S.; Wagner, R.K.; Willcutt, E.G.; Fletcher, J.M. Understanding, educating, and supporting children with specific learning disabilities: 50 years of science and practice. Am. Psychol. 2020, 75, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Shaywitz, S.E.; Shaywitz, B.A. Dyslexia (specific reading disability). Biol. Psychiatry 2005, 57, 1301–1309. [Google Scholar] [CrossRef]
- Smart, D.; Youssef, G.J.; Sanson, A.; Prior, M.; Toumbourou, J.W.; Olsson, C.A. Consequences of childhood reading difficulties and behaviour problems for educational achievement and employment in early adulthood. Br. J. Educ. Psychol. 2017, 87, 288–308. [Google Scholar] [CrossRef]
- Ziegler, J.C.; Goswami, U. Reading acquisition, developmental dyslexia, and skilled reading across languages: A psycholinguistic grain size theory. Psychol. Bull. 2005, 131, 3–29. [Google Scholar] [CrossRef] [PubMed]
- APA—American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Publishing: Arlington, VA, USA, 2013. [Google Scholar]
- Leonardi, M.M.; Di Blasi, F.D.; Savelli, E.; Buono, S. Reading and spelling disorders in a school-based population screening in Sicily (Italy). Dyslexia 2021, 27, 452–467. [Google Scholar] [CrossRef] [PubMed]
- ISS-Istituto Superiore di Sanità. Consensus Conference. Disturbi Specifici dell’apprendimento. Sistema Nazionale per le Linee Guida. 2022. Available online: https://www.iss.it/documents/20126/8331678/LG-389-AIP_DSA (accessed on 14 April 2023).
- Erbeli, F.; Hart, S.A.; Taylor, J. Longitudinal associations among reading-related skills and reading comprehension: A twin study. Child. Dev. 2018, 89, e480–e493. [Google Scholar] [CrossRef]
- Gayan, J.; Olson, R.K. Genetic and environmental influences on ortho- graphic and phonological skills in children with reading disabilities. Dev. Neuropsychol. 2001, 20, 483–507. [Google Scholar] [CrossRef]
- Harlaar, N.; Spinath, F.M.; Dale, P.S.; Plomin, R. Genetic influences on early word recognition abilities and disabilities: A study of 7-year-old twins. J. Child. Psychol. Psychiatry 2005, 46, 373–384. [Google Scholar] [CrossRef]
- Friend, A.; DeFries, J.C.; Wadsworth, S.J.; Olson, R.K. Genetic and environmental influences on word recognition and spelling deficits as a function of age. Behav. Genet. 2007, 37, 477–486. [Google Scholar] [CrossRef]
- Wray, N.R.; Lee, S.H.; Mehta, D.; Vinkhuyzen, A.A.E.; Dudbridge, F.; Middeldorp, C. Research review: Polygenic methods and their application to psychiatric traits. J. Child. Psychol. Psychiatry 2014, 55, 1068–1087. [Google Scholar] [CrossRef]
- Hannula-Jouppi, K.; Kaminen-Ahola, N.; Taipale, M.; Eklund, R.; Nopola-Hemmi, J.; Kääriäinen, H.; Kere, J. The axon guidance receptor gene ROBO1 is a candidate gene for develop- mental dyslexia. PLoS Genet. 2005, 1, e50. [Google Scholar] [CrossRef]
- Elbert, A.; Lovett, M.W.; Cate-Carter, T.; Pitch, A.; Kerr, E.N.; Barr, C.L. Genetic variation in the KIAA0319 5′ region as a possible contribu- tor to dyslexia. Behav. Genet. 2011, 41, 77–89. [Google Scholar] [CrossRef]
- Kidd, T.; Brose, K.; Mitchell, K.J.; Fetter, R.D.; Tessier-Lavigne, M.; Goodman, C.S.; Tear, G. Roundabout controls axon cross- ing of the CNS midline and defines a novel subfamily of evolution- arily conserved guidance receptors. Cell 1998, 92, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Poon, M.W.; Tsang, W.H.; Chan, S.O.; Li, H.M.; Ng, H.K.; Waye, M.M.Y. Dys- lexia-associated kiaa0319-like protein interacts with axon guidance receptor nogo receptor 1. Cell. Mol. Neurobiol. 2011, 31, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Burbridge, T.; Wang, Y.; Volz, A.; Peschansky, V.; Lisann, L.; Galaburda, A.; Turco, J.L.; Rosen, G. Postnatal analysis of the effect of embryonic knockdown and overexpression of candidate dyslexia susceptibility gene homolog Dcdc2 in the rat. Neuroscience 2008, 152, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Tran, C.; Wigg, K.G.; Zhang, K.; Cate-Carter, T.D.; Kerr, E.; Field, L.L.; Kaplan, B.J.; Lovett, M.W.; Barr, C.L. Association of the ROBO1 gene with reading disabilities in a family-based analysis. Genes. Brain Behav. 2014, 13, 430–438. [Google Scholar] [CrossRef]
- Platko, J.V.; Wood, F.B.; Pelser, I.; Meyer, M.; Gericke, G.S.; O’Rourke, J.; Birns, J.; Purcell, S.; Pauls, D.L. Association of reading disability on chromosome 6p22 in the Afrikaner population. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2008, 147B, 1278–1287. [Google Scholar] [CrossRef]
- Francks, C.; Paracchini, S.; Smith, S.D.; Richardson, A.J.; Scerri, T.S.; Cardon, L.R.; Marlow, A.J.; MacPhie, I.L.; Walter, J.; Pennington, B.F.; et al. A 77-kilobase region of chromosome 6p22.2 is associated with dyslexia in families from the United Kingdom and from the United States. Am. J. Hum. Genet. 2004, 75, 1046–1058. [Google Scholar] [CrossRef]
- Smith, S.D.; Kimberling, W.J.; Pennington, B.F.; Lubs, H.A. Specific reading disability: Identification of an inherited form through linkage analysis. Science 1983, 219, 1345–1347. [Google Scholar] [CrossRef]
- Chapman, N.H.; Igo, R.P.; Thomson, J.B.; Matsushita, M.; Brkanac, Z.; Holzman, T.; Berninger, V.W.; Wijsman, E.M.; Raskind, W.H. Linkage analyses of four regions previously implicated in dyslexia: Confirmation of a locus on chromosome 15q. Am. J. Med Genet. 2004, 131B, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Grigorenko, E.L.; Wood, F.B.; Meyer, M.S.; Hart, L.A.; Speed, W.C.; Shuster, A.; Pauls, D. Susceptibility loci for dis- tinct components of developmental dyslexia on chromosomes 6 and 15. Am. J. Hum. Genet. 1997, 60, 27–39. [Google Scholar] [PubMed]
- Erbeli, F.; Rice, M.; Paracchini, S. Insights into Dyslexia Genetics Research from the Last Two Decades. Brain Sci. 2022, 12, 27. [Google Scholar] [CrossRef]
- Diaz, R.; Kronenberg, N.M.; Martinelli, A.; Liehm, P.; Riches, A.C.; Gather, M.C.; Paracchini, S. KIAA0319 influences cilia length, cell migration and mechanical cell–substrate interaction. Sci. Rep. 2022, 12, 722. [Google Scholar] [CrossRef]
- Paniagua, S.; Cakir, B.; Hu, Y.; Kiral, F.R.; Tanaka, Y.; Xiang, Y.; Patterson, B.; Gruen, J.R.; Park, I.-H. Dyslexia associated gene KIAA0319 regulates cell cycle during human neuroepithelial cell development. Front. Cell. Dev. Biol. 2022, 10, 967147. [Google Scholar] [CrossRef] [PubMed]
- Tarkar, A.; Loges, N.T.; Slagle, C.E.; Francis, R.; Dougherty, G.W.; Tamayo, J.V.; Shook, B.; Cantino, M.; Schwartz, D.; Jahnke, C.; et al. DYX1C1 is required for axonemal dynein assembly and ciliary motility. Nat. Genet. 2013, 45, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Schueler, M.; Braun, D.A.; Chandrasekar, G.; Gee, H.Y.; Klasson, T.D.; Halbritter, J.; Bieder, A.; Porath, J.D.; Airik, R.; Zhou, W.; et al. DCDC2 mutations cause a renal-hepatic ciliopathy by disrupting Wnt signaling. Am. J. Hum. Genet. 2015, 96, 81–92. [Google Scholar] [CrossRef]
- Doust, C.; Fontanillas, P.; Eising, E.; Gordon, S.D.; Wang, Z.; Molz, B.; Pourcain, B.S.; Francks, C.; Marioni, R.E.; Zhao, J.; et al. Discovery of 42 genome-wide significant loci associated with dyslexia. Nat. Genet. 2022, 54, 1621–1629. [Google Scholar] [CrossRef]
- Gialluisi, A.; Andlauer, T.F.M.; Mirza-Schreiber, N.; Moll, K.; Becker, J.; Hoffmann, P.; Ludwig, K.U.; Czamara, D.; Pourcain, B.S.; Honbolygó, F.; et al. Genome-wide association study reveals new insights into the heritability and genetic correlates of developmental dyslexia. Mol. Psychiatry 2021, 26, 3004–3017. [Google Scholar] [CrossRef]
- Lyytinen, H.; Erskine, J.; Hämäläinen, J.; Torppa, M.; Ronimus, M. Dyslexia—Early Identification and Prevention: Highlights from the Jyväskylä Longitudinal Study of Dyslexia. Curr. Dev. Disord. Rep. 2015, 2, 330–338. [Google Scholar] [CrossRef]
- Kovas, Y.; Plomin, R. Learning Abilities and Disabilities: Generalist Genes, Specialist Environments. Curr. Dir. Psychol. Sci. 2007, 16, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, J.; Abbondanza, F.; Paracchini, S. Genome-wide association study and polygenic risk score analysis for hearing measures in children. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2021, 186, 318–328. [Google Scholar] [CrossRef]
- Brandler, W.M.; Paracchini, S. The genetic relationship between handedness and neurodevelopmental disorders. Trends Mol. Med. 2014, 20, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Kere, J. The molecular genetics and neurobiology of developmental dyslexia as model of a complex phenotype. Biochem. Biophys. Res. Commun. 2014, 452, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Yang, Q.; Cheng, C.; Wang, Z. Cumulative genetic score of KIAA0319 affects reading ability in Chinese children: Moderation by parental education and mediation by rapid automatized naming. Behav. Brain Funct. 2023, 19, 10. [Google Scholar] [CrossRef] [PubMed]
- Benchek, P.; Igo, R.P.; Voss-Hoynes, H.; Wren, Y.; Miller, G.; Truitt, B.; Zhang, W.; Osterman, M.; Freebairn, L.; Tag, J.; et al. Association between genes regulating neural pathways for quantitative traits of speech and language disorders. NPJ Genom. Med. 2021, 6, 64. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekar, G.; Vesterlund, L.; Hultenby, K.; Tapia-Páez, I.; Kere, J. The zebrafish orthologue of the dyslexia candidate gene DYX1C1 is essential for cilia growth and function. PLoS ONE 2013, 8, e63123. [Google Scholar] [CrossRef]
- Anthoni, H.; Sucheston, L.E.; Lewis, B.A.; Tapia-Páez, I.; Fan, X.; Zucchelli, M.; Taipale, M.; Stein, C.M.; Hokkanen, M.-E.; Castrén, E.; et al. The aromatase gene CYP19A1: Several genetic and functional lines of evidence supporting a role in reading, speech and language. Behav. Genet. 2012, 42, 509–527. [Google Scholar] [CrossRef]
- Matsson, H.; Huss, M.; Persson, H.; Einarsdottir, E.; Tiraboschi, E.; Nopola-Hemmi, J.; Schumacher, J.; Neuhoff, N.; Warnke, A.; Lyytinen, H.; et al. Polymorphisms in DCDC2 and S100B associate with developmental dyslexia. J. Hum. Genet. 2015, 60, 399–401. [Google Scholar] [CrossRef]
- Luciano, M.; Gow, A.J.; Pattie, A.; Bates, T.C.; Deary, I.J. The Influence of Dyslexia Candidate Genes on Reading Skill in Old Age. Behav. Genet. 2018, 48, 351–360. [Google Scholar] [CrossRef]
- Matsson, H.; Tammimies, K.; Zucchelli, M.; Anthoni, H.; Onkamo, P.; Nopola-Hemmi, J.; Lyytinen, H.; Leppanen, P.H.T.; Neuhoff, N.; Warnke, A.; et al. SNP variations in the 7q33 region containing DGKI are associated with dyslexia in the Finnish and German populations. Behav. Genet. 2011, 41, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Kong, R.; Shao, S.; Wang, J.; Zhang, X.; Guo, S.; Zou, L.; Zhong, R.; Lou, J.; Zhou, J.; Zhang, J.; et al. Genetic variant in DIP2A gene is associated with developmental dyslexia in Chinese population. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2016, 171B, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Scerri, T.S.; Paracchini, S.; Morris, A.; MacPhie, I.L.; Talcott, J.; Stein, J.; Smith, S.D.; Pennington, B.F.; Olson, R.K.; DeFries, J.C.; et al. Identification of candidate genes for dyslexia susceptibility on chromosome 18. PLoS ONE 2010, 5, e13712. [Google Scholar] [CrossRef]
- Eicher, J.D.; Gruen, J.R. Language impairment and dyslexia genes influence language skills in children with autism spectrum disorders. Autism Res. 2015, 8, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Mueller, B.; Ahnert, P.; Burkhardt, J.; Brauer, J.; Czepezauer, I.; Quente, E.; Boltze, J.; Wilcke, A.; Kirsten, H. Genetic risk variants for dyslexia on chromosome 18 in a German cohort. Genes. Brain Behav. 2014, 13, 350–356. [Google Scholar] [CrossRef]
- Yanpallewar, S.; Wang, T.; Koh, D.C.I.; Quarta, E.; Fulgenzi, G.; Tessarollo, L. Nedd4-2 haploinsufficiency causes hyperactivity and increased sensitivity to inflammatory stimuli. Sci. Rep. 2016, 6, 32957. [Google Scholar] [CrossRef]
- WHO—World Health Organization. International Statistical Classification of Diseases and Related Health Problems: 10th Revision (ICD-10); WHO: Geneva, Switzerland, 1992.
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Aguilera, M.A.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef]
- Taipale, M.; Kaminen, N.; Nopola-Hemmi, J.; Haltia, T.; Myllyluoma, B.; Lyytinen, H.; Muller, K.; Kaaranen, M.; Lindsberg, P.J.; Hannula-Jouppi, K.; et al. A candidate gene for developmental dyslexia encodes a nuclear tetratricopeptid.de repeat domain protein dynamically regulated in brain. Proc. Natl. Acad. Sci. USA 2003, 100, 11553–11558. [Google Scholar] [CrossRef]
- Ulfarsson, M.O.; Walters, G.B.; Gustafsson, O.; Steinberg, S.; Silva, A.; Doyle, O.M.; Brammer, M.; Gudbjartsson, D.F.; Arnarsdottir, S.; Jonsdottir, G.A.; et al. 15q11.2 CNV affects cognitive, structural and functional correlates of dyslexia and dyscalculia. Transl. Psychiatry 2017, 7, e1109. [Google Scholar] [CrossRef]
- Martinelli, A.; Rice, M.L.; Talcott, J.B.; Diaz, R.; Smith, S.; Raza, M.H.; Snowling, M.J.; Hulme, C.; Stein, J.; E Hayiou-Thomas, M.; et al. A rare missense variant in the ATP2C2 gene is associated with language impairment and related measures. Hum. Mol. Genet. 2021, 30, 1160–1171. [Google Scholar] [CrossRef]
- Grimm, T.; Garshasbi, M.; Puettmann, L.; Chen, W.; Ullmann, R.; Müller-Myhsok, B.; Klopocki, E.; Herbst, L.; Haug, J.; Jensen, L.R.; et al. A novel locus and candidate gene for familial developmental dyslexia on chromosome 4q. Z. Für Kinder-Und Jugendpsychiatrie Und Psychother. 2020, 48, 478–489. [Google Scholar] [CrossRef] [PubMed]
- Galesi, O.; Di Blasi, F.D.; Grillo, L.; Elia, F.; Giambirtone, M.C.; Figura, M.G.; Rizzo, B.; Buono, S.; Romano, C. Dyslexia and Attention Deficit Hyperactivity Disorder Associated to a De Novo 1p34.3 microdeletion. Genes 2022, 13, 1926. [Google Scholar] [CrossRef] [PubMed]
- Gerber, P.J. The Impact of Learning Disabilities on Adulthood. J. Learn. Disabil. 2011, 45, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Visser, M.; Kayser, M.; Palstra, R.J. HERC2 rs12913832 modulates human pigmentation by attenuating chromatin-loop for-mation between a long-range enhancer and the OCA2 promoter. Genome Res. 2012, 22, 446–455. [Google Scholar] [CrossRef]
- Brancato, D.; Coniglio, E.; Bruno, F.; Agostini, V.; Saccone, S.; Federico, C. Forensic DNA Phenotyping: Genes and Genetic Variants for Eye Color Prediction. Genes 2023, 14, 1604. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Gene | Gene Product Function | Refs. (a) | Position (b) (chr Band) | Genomic Size (b) (Kb) | RefSeq. Transcript (c) |
|---|---|---|---|---|---|
| CCPG1 | Related to the cell cycle regulation and cell division processes. | [39] | 15q21.3 | 53.121 | NR_037923.1 |
| CYP19A1 | Product is an enzyme involved in the steroid hormone conversion. | [40,41,42] | 15q21.2 | 130.540 | NM_000103.4 |
| DCDC2 | Involved in the formation of neuronal circuits, and neuronal migration. | [9,26] | 6p22.3 | 186.305 | NM_016356.5 |
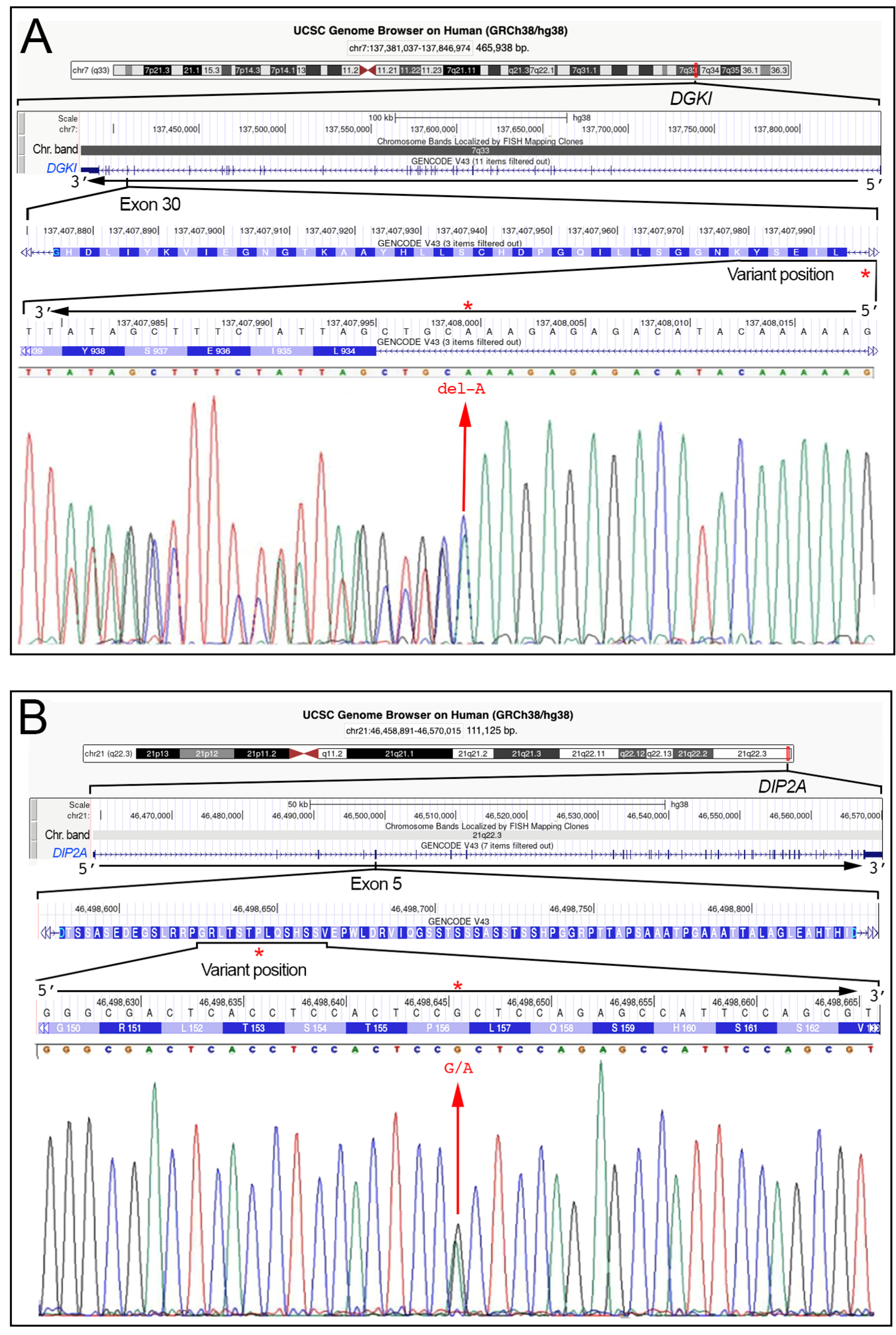
| DGKI | Involved in cellular signaling pathway by diacylglycerol phosphorylation, such as cell proliferation and differentiation, synaptic plasticity, neuronal signaling. | [43] | 7q33 | 465.938 | NM_001321708.2 |
| DIP2A | Involved in various cell processes, such as proliferation, differentiation processes; Implicated in neurogenesis and neuronal differentiation. | [41,44] | 21q22.3 | 111.125 | NM_015151.4 |
| DYM | Involved in various cellular processes related to the cellular homeostasis. | [45] | 18q21.1 | 419.329 | NM_001353214.3 |
| GCFC2 | Gene regulation and maintaining genome stability. | [41,46] | 2p12 | 48.211 | NM_003203.5 |
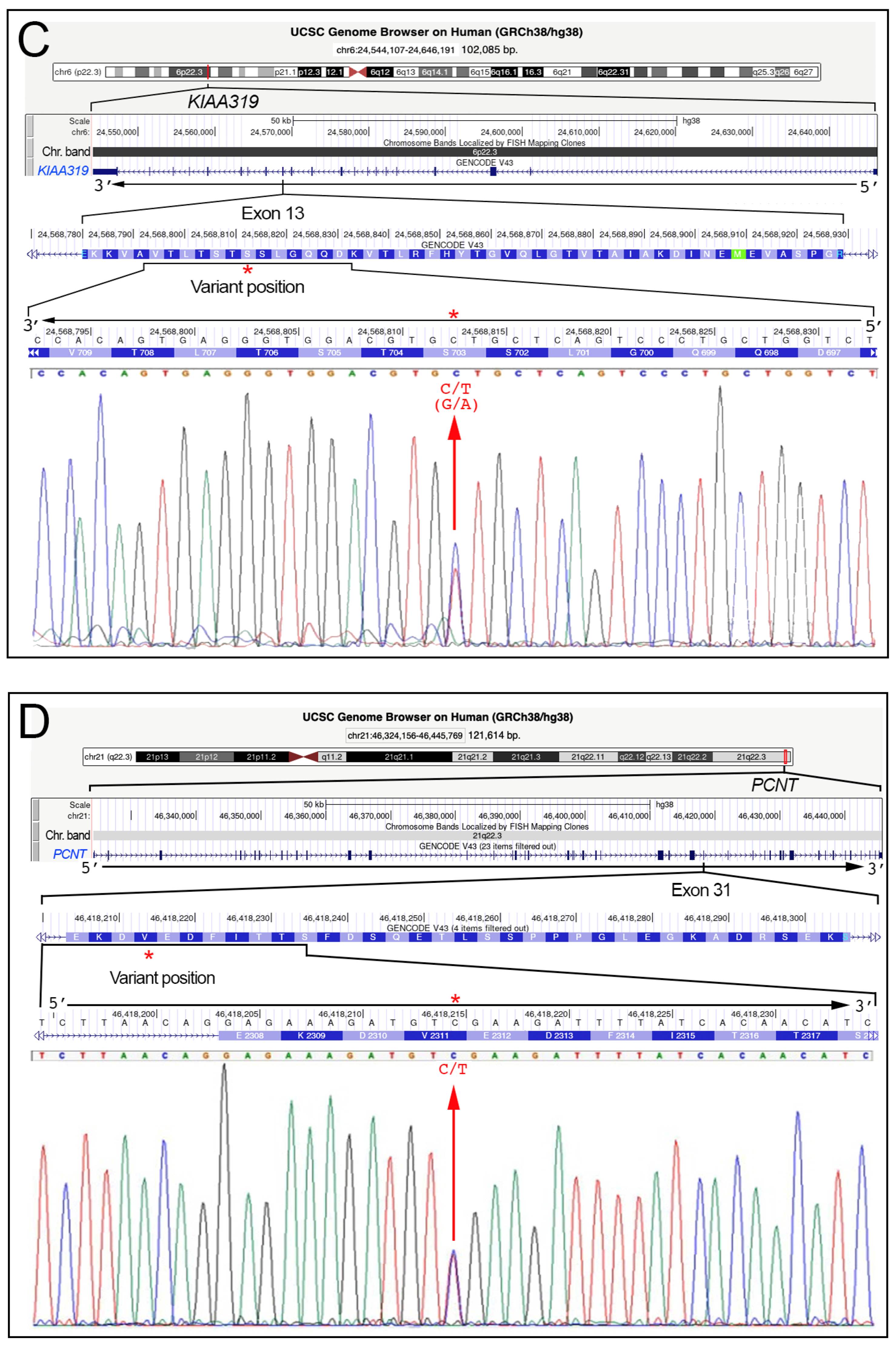
| KIAA0319 | Plays a role in brain development related to neuronal migration and neural connectivity. | [9,26,41] | 6p22.3 | 102.085 | NM_014809.4 |
| MC5R | Transmembrane protein involved in various cell processes, such as skin pigmentation, immunomodulation, thermoregulation. | [45] | 18p11.21 | 3.175 | NM_005913.3 |
| MRPL19 | Mitochondrial protein involved in mitochondrial function and cellular metabolism | [41] | 2p12 | 23.951 | NM_014763.4 |
| NEDD4L | Protein involved in ubiquitination of various proteins, regulating protein levels and functions within cells. | [45,47,48] | 18q21.31 | 357.314 | NM_001144967.3 |
| PCNT | A component of the centrosome and involved in various processes, such as cell division and organization of the microtubule network. | [41] | 21q22.3 | 121.614 | NM_006031.6 |
| PRMT2 | Methyltransferase involved in various processes, such as gene expression, RNA processing, cell signaling. | [41] | 21q22.3 | 29.451 | NM_206962.4 |
| ROBO1 | Cell surface receptor involved in axon guidance during neural development. | [9,26,41] | 3p12.3 | 1170.760 | NM_002941.4 |
| S100B | Calcium-binding protein involved in various cell processes, such as neurological function, immune response, cell cycle regulation. | [41] | 21q22.3 | 6.479 | NM_006272.3 |
| Gene | Ref. seq. | DNA Variant | Protein Variant | SNP-ID (a) | VarSome (ACMG) (b) | GnomAD Exomes (c) | gnomAD Genomes (c) | TSI 1000G (d) | Clinical Variant |
|---|---|---|---|---|---|---|---|---|---|
| DGKI | NM_004717.3 | c.2824-4del | == | rs1184296555 | L.B. | f = 0.0 | f = 0.0 | f = nf | N.D. |
| DIP2A | NM_015151.4 | c.468G>A | p.(Pro156=) | rs367616491 | L.B. | f = 0.00028 | f = 0.00042 | f = nf | N.D. |
| KIAA0319 | NM_014809.4 | c.2108G>A | p.(Ser703Asn) | rs138160539 | L.B. | f = 0.00134 | f = 0.00121 | f = 0.005 | L.B. |
| PCNT | NM_006031.6 | c.6933C>T | p.(Val2311=) | rs148444313 | Benign | f = 0.00372 | f = 0.00360 | f = 0.0 | C.i.p. |
| Family | Code | Sex and Parents | Age | Phenotype | DGKI c.2824-4del | DIP2A c.468G>A p.(Pro156=) | KIAA0319 c.2108G>A p.(Ser703Asn) | PCNT c.6933C>T p.(Val2311=) |
|---|---|---|---|---|---|---|---|---|
| F1 | 02008 | Female | 21 | D, DS | Heterozygous | Heterozygous | ||
| 04835 | Female | 25 | D, DY | |||||
| 02008M | Mother | 62 | LD | Heterozygous | ||||
| 02008P | Father | 62 | // | Heterozygous | ||||
| F2 | 04735 | Female | 22 | D, DS, DY | Heterozygous | |||
| 04735F | Male | 19 | D, DS, DY | Heterozygous | ||||
| 04735F1 | Male | 15 | D, DS, DY | |||||
| 04735M | Mother | 48 | // | |||||
| 04735P | Father | 53 | LD | Heterozygous | ||||
| F3 | 04802 | Male | 26 | D, ADHD | Heterozygous | |||
| 04883 | Male | 25 | D, DS, DY | Heterozygous | ||||
| F4 | 04833 | Male | 17 | D, DS, DY | ||||
| 04833F | Male | 17 | D, DS, DY | |||||
| 04833S | Female | 21 | D, DS, DY | |||||
| 04833S1 | Female | 16 | D, DS, DY | Heterozygous | ||||
| 04833M | Mother | 43 | // | Heterozygous | ||||
| 04833P | Father | 47 | LD | |||||
| F5 | 04966F | Male | 16 | D, DS, DY, ADHD | ||||
| 04966 | Female | 22 | D, DS | Heterozygous | ||||
| 04966M | Mother | 50 | // | |||||
| 04966P | Father | 52 | LD | Heterozygous | ||||
| F6 | 05034 | Male | 16 | D, DS | ||||
| 05034F | Male | 16 | D, DS | |||||
| 05034M | Mother | 40 | // | |||||
| 05034P | Father | 53 | // | |||||
| F7 | 05170 | Male | 22 | D, DS, DY | Heterozygous | |||
| 05170S | Female | 25 | D, DS, DY | |||||
| 05170M | Mother | 51 | LD | Heterozygous | ||||
| 05170P | Father | 55 | LD | |||||
| F8 | 05461 | Male | 26 | D, DS, DY | Heterozygous | |||
| 05461S | Female | 21 | D, DS, DY | Heterozygous | ||||
| 05461M | Mother | 52 | LD | Heterozygous | ||||
| 05461P | Father | 55 | // | Heterozygous | ||||
| F9 | 05640 | male | 18 | D, DS | Heterozygous | |||
| 05640S | female | 21 | D, DS | Heterozygous | ||||
| 05640M | Mother | 44 | LD | Heterozygous | ||||
| 05640P | Father | 50 | LD |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calì, F.; Di Blasi, F.D.; Avola, E.; Vinci, M.; Musumeci, A.; Gloria, A.; Greco, D.; Raciti, D.R.; Zagami, A.; Rizzo, B.; et al. Specific Learning Disorders: Variation Analysis of 15 Candidate Genes in 9 Multiplex Families. Medicina 2023, 59, 1503. https://doi.org/10.3390/medicina59081503
Calì F, Di Blasi FD, Avola E, Vinci M, Musumeci A, Gloria A, Greco D, Raciti DR, Zagami A, Rizzo B, et al. Specific Learning Disorders: Variation Analysis of 15 Candidate Genes in 9 Multiplex Families. Medicina. 2023; 59(8):1503. https://doi.org/10.3390/medicina59081503
Chicago/Turabian StyleCalì, Francesco, Francesco Domenico Di Blasi, Emanuela Avola, Mirella Vinci, Antonino Musumeci, Angelo Gloria, Donatella Greco, Daniela Rita Raciti, Alessandro Zagami, Biagio Rizzo, and et al. 2023. "Specific Learning Disorders: Variation Analysis of 15 Candidate Genes in 9 Multiplex Families" Medicina 59, no. 8: 1503. https://doi.org/10.3390/medicina59081503