
Unmasking a Silent Threat: Improving Pulmonary Hypertension Screening Methods for Interstitial Lung Disease Patients

Abstract
:1. Introduction
2. PH-ILD Pathophysiology
3. PH-ILD Disease Spectrum
4. PH-ILD Phenotypes
5. Challenges in Clinical Diagnosis of PH-ILD
6. PH-ILD Diagnostic Strategy
6.1. Pulmonary Function Testing
6.2. Cardiopulmonary Exercise Testing
6.3. 6 min Walking Test
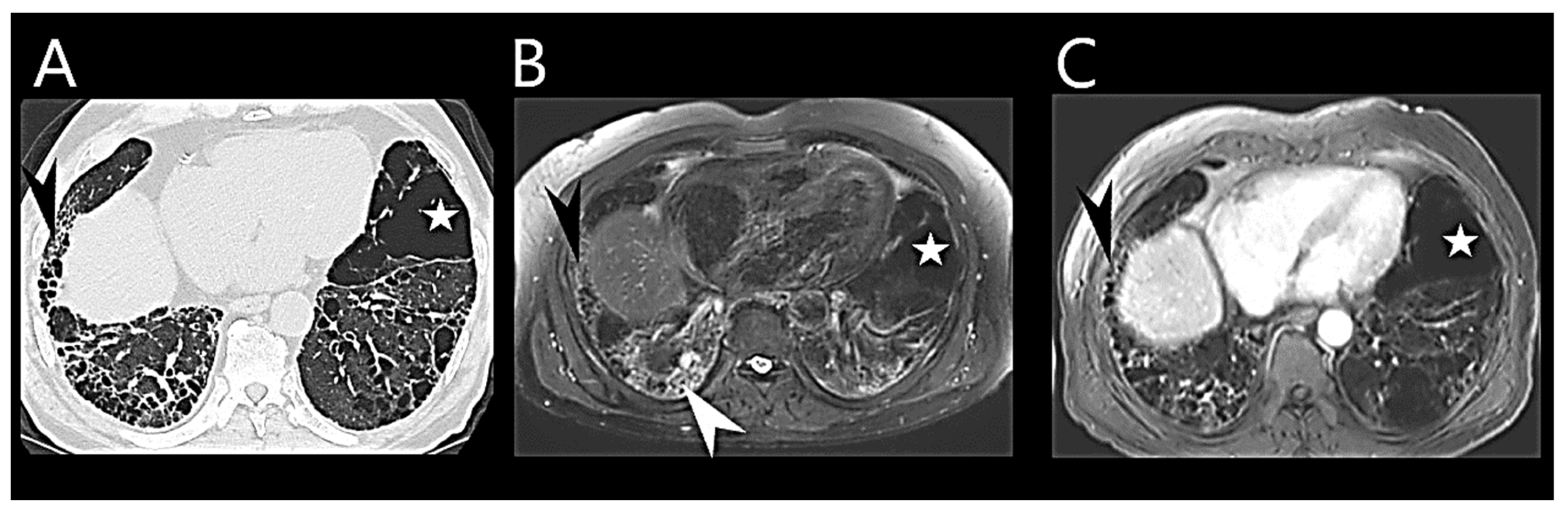
6.4. Chest Computed Tomography
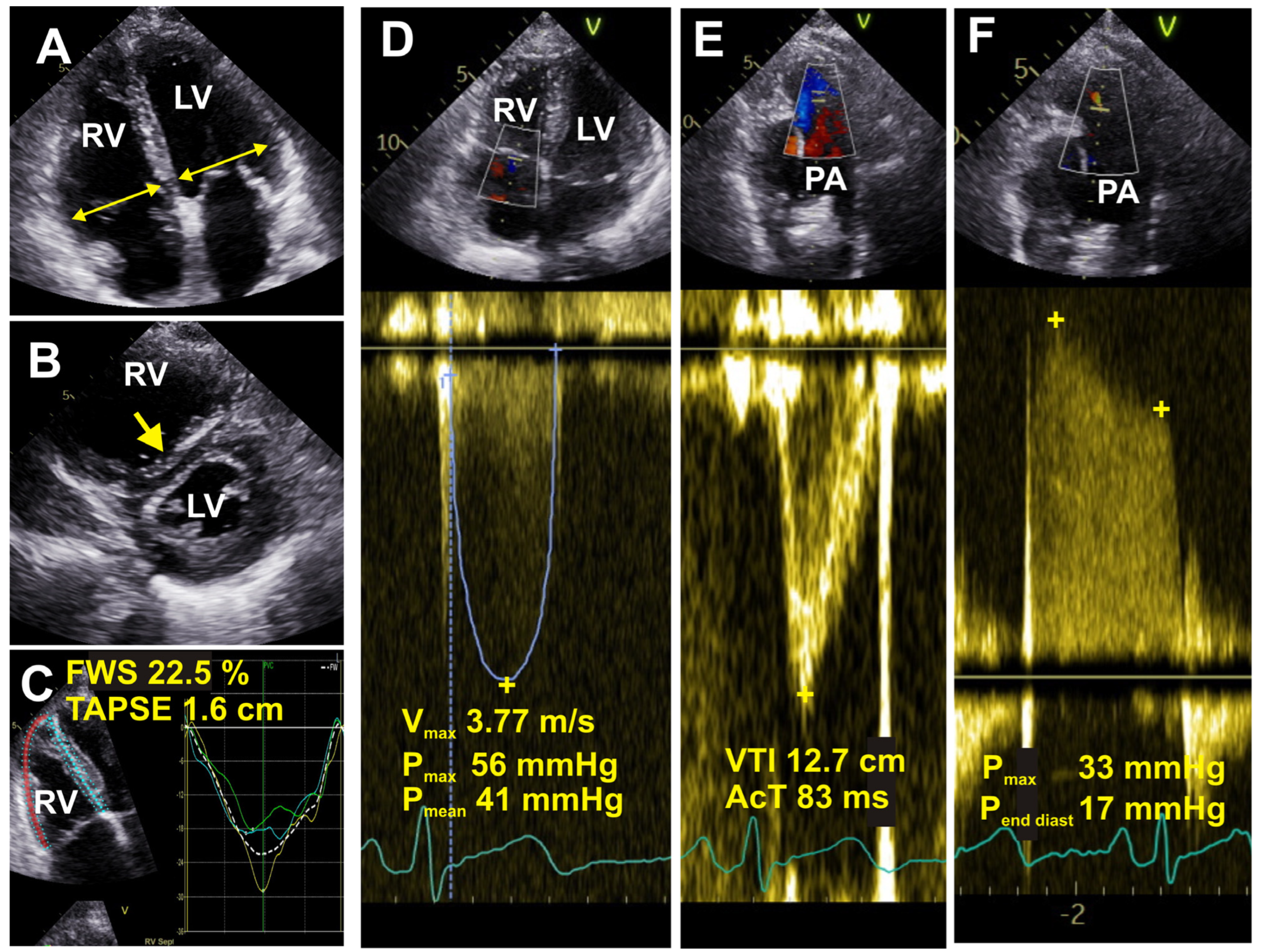
6.5. Echocardiography
6.6. Cardiac Magnetic Resonance Imaging
6.7. Biomarkers
7. Artificial Intelligence Applications
8. Management and Treatment
9. Prognosis and Outcomes
10. Conclusions
11. Key Points
- -
- PH-ILD encompasses a range of conditions, each with unique differences in clinical presentations and characteristics.
- -
- Increased suspicion of PH is warranted when the progression of dyspnoea does not align with a decline in pulmonary function parameters.
- -
- Diagnosing PH-ILD is challenging due to symptom overlap with ILDs without PH, emphasizing the need for non-invasive tests like echocardiography, cardiac MRI, and specific biomarkers. For now, RHC remains the gold standard for confirming PH-ILD diagnosis.
- -
- RHC is typically conducted primarily for ILD patients who meet the criteria for lung transplantation, are involved in clinical trials, or are being evaluated for specialized treatment.
- -
- There are currently limited specific treatments for PH-ILD, but recent trials with inhaled treprostinil show promise.
- -
- Managing PH-ILD involves supportive care, long-term oxygen therapy, and, in some cases, lung transplantation, with a focus on improving patient quality of life and prognosis.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 6MWT | 6-Minute Walk Test |
| ABG | Arterial Blood Gas |
| AI | Artificial Intelligence |
| AT | Anaerobic Threshold |
| BNP | Brain Natriuretic Peptide |
| CPET | Cardiopulmonary Exercise Testing |
| CT | Computed Tomography |
| CXCL13 | CXC-Chemokine Ligand 13 |
| COPD | Chronic Obstructive Pulmonary Disease |
| CPFE | Combined Pulmonary Fibrosis and Emphysema |
| CTDs | Connective Tissue Diseases |
| DLCO | Diffusing Capacity for Carbon Monoxide |
| FEV1 | Forced Expiratory Volume in 1 Second |
| FVC | Forced Vital Capacity |
| GDF-15 | Growth Differentiation Factor 15 |
| ILDs | Interstitial Lung Diseases |
| IIPs | Idiopathic Interstitial Pneumonias |
| iNO | Inhaled Nitric Oxide |
| IPAH | Idiopathic Pulmonary Arterial Hypertension |
| IPF | Idiopathic Pulmonary Fibrosis |
| LAM | Lymphangioleiomyomatosis |
| LHD | Left Heart Disease |
| LTOT | Long-Term Oxygen Therapy |
| LV | Left Ventricle |
| ML | Machine Learning |
| NSIP | Nonspecific Interstitial Pneumonitis |
| PA | Pulmonary Artery |
| PAH | Pulmonary Arterial Hypertension |
| PAWP | Pulmonary Artery Wedge Pressure |
| PAP | Pulmonary Arterial Pressure |
| PATE | Pulmonary Artery Thromboembolism |
| PH | Pulmonary Hypertension |
| PH-ILD | Pulmonary hypertension associated with Interstitial Lung Disease |
| PLCH | Pulmonary Langerhans Cell Histiocytosis |
| PVR | Pulmonary Vascular Resistance |
| RDW | Red Cell Distribution Width |
| RHC | Right-sided Heart Catheterization |
| RV | Right Ventricle |
| SSc | Systemic Sclerosis |
| TAPSE | Tricuspid Annular Plane Systolic Excursion |
| TRV | Tricuspid Regurgitation Velocity |
| VE/VCO2 | Ventilation/Carbon Dioxide |
| VO2 | Oxygen Uptake |
| WHO | World Health Organization |
References
- Humbert, M.; Kovacs, G.; Hoeper, M.M.; Badagliacca, R.; Berger, R.M.F.; Brida, M.; Carlsen, J.; Coats, A.J.S.; Escribano-Subias, P.; Ferrari, P.; et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Respir. J. 2023, 61, 2200879. [Google Scholar] [CrossRef] [PubMed]
- Teramachi, R.; Taniguchi, H.; Kondoh, Y.; Ando, M.; Kimura, T.; Kataoka, K.; Suzuki, A.; Furukawa, T.; Sakamoto, K.; Hasegawa, Y. Progression of mean pulmonary arterial pressure in idiopathic pulmonary fibrosis with mild to moderate restriction. Respirology 2017, 22, 986–990. [Google Scholar] [CrossRef] [PubMed]
- Olsson, K.M.; Hoeper, M.M.; Pausch, C.; Grünig, E.; Huscher, D.; Pittrow, D.; Rosenkranz, S.; Gall, H. Pulmonary vascular resistance predicts mortality in patients with pulmonary hypertension associated with interstitial lung disease: Results from the COMPERA registry. Eur. Respir. J. 2021, 58, 2101483. [Google Scholar] [CrossRef] [PubMed]
- Shorr, A.F.; Wainright, J.L.; Cors, C.S.; Lettieri, C.J.; Nathan, S.D. Pulmonary hypertension in patients with pulmonary fibrosis awaiting lung transplant. Eur. Respir. J. 2007, 30, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Andersen, C.U.; Mellemkjær, S.; Hilberg, O.; Nielsen-Kudsk, J.E.; Simonsen, U.; Bendstrup, E. Pulmonary hypertension in interstitial lung disease: Prevalence, prognosis and 6 min walk test. Respir. Med. 2012, 106, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Pulito-Cueto, V.; Genre, F.; López-Mejías, R.; Mora-Cuesta, V.M.; Iturbe-Fernández, D.; Portilla, V.; Sebastián Mora-Gil, M.; Ocejo-Vinyals, J.G.; Gualillo, O.; Blanco, R.; et al. Endothelin-1 as a Biomarker of Idiopathic Pulmonary Fibrosis and Interstitial Lung Disease Associated with Autoimmune Diseases. Int. J. Mol. Sci. 2023, 24, 1275. [Google Scholar] [CrossRef] [PubMed]
- Yanagihara, T.; Tsubouchi, K.; Zhou, Q.; Chong, M.; Otsubo, K.; Isshiki, T.; Schupp, J.C.; Sato, S.; Scallan, C.; Upagupta, C.; et al. Vascular–Parenchymal Cross-Talk Promotes Lung Fibrosis through BMPR2 Signaling. Am. J. Respir. Crit. Care Med. 2023, 207, 1498–1514. [Google Scholar] [CrossRef] [PubMed]
- Samarelli, A.V.; Tonelli, R.; Marchioni, A.; Bruzzi, G.; Gozzi, F.; Andrisani, D.; Castaniere, I.; Manicardi, L.; Moretti, A.; Tabbì, L.; et al. Fibrotic Idiopathic Interstitial Lung Disease: The Molecular and Cellular Key Players. Int. J. Mol. Sci. 2021, 22, 8952. [Google Scholar] [CrossRef]
- Miądlikowska, E.; Rzepka-Wrona, P.; Miłkowska-Dymanowska, J.; Białas, A.J.; Piotrowski, W.J. Review: Serum Biomarkers of Lung Fibrosis in Interstitial Pneumonia with Autoimmune Features—What Do We Already Know? J. Clin. Med. 2021, 11, 79. [Google Scholar] [CrossRef]
- Borek, I.; Birnhuber, A.; Voelkel, N.F.; Marsh, L.M.; Kwapiszewska, G. The vascular perspective on acute and chronic lung disease. J. Clin. Investig. 2023, 133, e170502. [Google Scholar] [CrossRef]
- Darwiche, T.; Collum, S.D.; Bi, W.; Reynolds, J.O.; Wilson, C.; Wareing, N.; Hernandez, A.M.; Mertens, T.C.J.; Zhou, Z.; Pandit, L.M.; et al. Alterations in cardiovascular function in an experimental model of lung fibrosis and pulmonary hypertension. Exp. Physiol. 2019, 104, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Dotan, Y.; Stewart, J.; Gangemi, A.; Wang, H.; Aneja, A.; Chakraborty, B.; Dass, C.; Zhao, H.; Marchetti, N.; D’Alonzo, G.; et al. Pulmonary vasculopathy in explanted lungs from patients with interstitial lung disease undergoing lung transplantation. BMJ Open Respir. Res. 2020, 7, e000532. [Google Scholar] [CrossRef] [PubMed]
- Colombat, M.; Mal, H.; Groussard, O.; Capron, F.; Thabut, G.; Jebrak, G.; Brugière, O.; Dauriat, G.; Castier, Y.; Lesèche, G.; et al. Pulmonary vascular lesions in end-stage idiopathic pulmonary fibrosis: Histopathologic study on lung explant specimens and correlations with pulmonary hemodynamics. Hum. Pathol. 2007, 38, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Kylhammar, D.; Rådegran, G. The principal pathways involved in the in vivo modulation of hypoxic pulmonary vasoconstriction, pulmonary arterial remodelling and pulmonary hypertension. Acta Physiol. 2017, 219, 728–756. [Google Scholar] [CrossRef] [PubMed]
- Maimon, N.; Salz, L.; Shershevsky, Y.; Matveychuk, A.; Guber, A.; Shitrit, D. Sarcoidosis-associated pulmonary hypertension in patients with near-normal lung function. Int. J. Tuberc. Lung Dis. 2013, 17, 406–411. [Google Scholar] [CrossRef] [PubMed]
- Duong, H.T.; Bonham, C.A. Sarcoidosis-associated Pulmonary Hypertension: Pathophysiology, Diagnosis, and Treatment. Clin. Pulm. Med. 2018, 25, 52–60. [Google Scholar] [CrossRef]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef]
- Collum, S.D.; Amione-Guerra, J.; Cruz-Solbes, A.S.; DiFrancesco, A.; Hernandez, A.M.; Hanmandlu, A.; Youker, K.; Guha, A.; Karmouty-Quintana, H. Pulmonary Hypertension Associated with Idiopathic Pulmonary Fibrosis: Current and Future Perspectives. Can. Respir. J. 2017, 2017, 1430350. [Google Scholar] [CrossRef]
- Klinger, J.R. Group III Pulmonary Hypertension. Cardiol. Clin. 2016, 34, 413–433. [Google Scholar] [CrossRef]
- Karampitsakos, T.; Tzouvelekis, A.; Chrysikos, S.; Bouros, D.; Tsangaris, I.; Fares, W.H. Pulmonary hypertension in patients with interstitial lung disease. Pulm. Pharmacol. Ther. 2018, 50, 38–46. [Google Scholar] [CrossRef]
- Hachulla, E.; Gressin, V.; Guillevin, L.; Carpentier, P.; Diot, E.; Sibilia, J.; Kahan, A.; Cabane, J.; Francès, C.; Launay, D.; et al. Early detection of pulmonary arterial hypertension in systemic sclerosis: A French nationwide prospective multicenter study. Arthritis Rheum. 2005, 52, 3792–3800. [Google Scholar] [CrossRef] [PubMed]
- Vonk, M.C.; Broers, B.; Heijdra, Y.F.; Ton, E.; Snijder, R.; van Dijk, A.P.J.; van Laar, J.M.; Bootsma, H.; van Hal, P.T.W.; van den Hoogen, F.H.J.; et al. Systemic sclerosis and its pulmonary complications in The Netherlands: An epidemiological study. Ann. Rheum. Dis. 2009, 68, 961–965. [Google Scholar] [CrossRef] [PubMed]
- Phung, S.; Strange, G.; Chung, L.P.; Leong, J.; Dalton, B.; Roddy, J.; Deague, J.; Playford, D.; Musk, M.; Gabbay, E. Prevalence of pulmonary arterial hypertension in an Australian scleroderma population: Screening allows for earlier diagnosis. Intern. Med. J. 2009, 39, 682–691. [Google Scholar] [CrossRef] [PubMed]
- Avouac, J.; Airò, P.; Meune, C.; Beretta, L.; Dieude, P.; Caramaschi, P.; Tiev, K.; Cappelli, S.; Diot, E.; Vacca, A.; et al. Prevalence of pulmonary hypertension in systemic sclerosis in European Caucasians and metaanalysis of 5 studies. J. Rheumatol. 2010, 37, 2290–2298. [Google Scholar] [CrossRef] [PubMed]
- Morrisroe, K.; Stevens, W.; Sahhar, J.; Rabusa, C.; Nikpour, M.; Proudman, S.; Australian Scleroderma Interest Group (ASIG). Epidemiology and disease characteristics of systemic sclerosis-related pulmonary arterial hypertension: Results from a real-life screening programme. Arthritis Res. Ther. 2017, 19, 42. [Google Scholar] [CrossRef] [PubMed]
- Coghlan, J.G.; Denton, C.P.; Grünig, E.; Bonderman, D.; Distler, O.; Khanna, D.; Müller-Ladner, U.; Pope, J.E.; Vonk, M.C.; Doelberg, M.; et al. Evidence-based detection of pulmonary arterial hypertension in systemic sclerosis: The DETECT study. Ann. Rheum. Dis. 2014, 73, 1340–1349. [Google Scholar] [CrossRef]
- Chauvelot, L.; Gamondes, D.; Berthiller, J.; Nieves, A.; Renard, S.; Catella-Chatron, J.; Ahmad, K.; Bertoletti, L.; Camara, B.; Gomez, E.; et al. Hemodynamic Response to Treatment and Outcomes in Pulmonary Hypertension Associated With Interstitial Lung Disease Versus Pulmonary Arterial Hypertension in Systemic Sclerosis: Data From a Study Identifying Prognostic Factors in Pulmonary Hypertension Associated With Interstitial Lung Disease. Arthritis Rheumatol. 2021, 73, 295–304. [Google Scholar]
- Piccari, L.; Allwood, B.; Antoniou, K.; Chung, J.H.; Hassoun, P.M.; Nikkho, S.M.; Saggar, R.; Shlobin, O.A.; Vitulo, P.; Nathan, S.D.; et al. Pathogenesis, clinical features, and phenotypes of pulmonary hypertension associated with interstitial lung disease: A consensus statement from the Pulmonary Vascular Research Institute’s Innovative Drug Development Initiative—Group 3 Pulmonary Hypertension. Pulm. Circ. 2023, 13, e12213. [Google Scholar]
- Hage, R.; Gautschi, F.; Steinack, C.; Schuurmans, M.M. Combined Pulmonary Fibrosis and Emphysema (CPFE) Clinical Features and Management. Int. J. Chronic Obstr. Pulm. Dis. 2021, 16, 167–177. [Google Scholar] [CrossRef]
- Ryerson, C.J.; Hartman, T.; Elicker, B.M.; Ley, B.; Lee, J.S.; Abbritti, M.; Jones, K.D.; King, T.E.; Ryu, J.; Collard, H.R. Clinical Features and Outcomes in Combined Pulmonary Fibrosis and Emphysema in Idiopathic Pulmonary Fibrosis. Chest 2013, 144, 234–240. [Google Scholar] [CrossRef]
- Tomioka, H.; Mamesaya, N.; Yamashita, S.; Kida, Y.; Kaneko, M.; Sakai, H. Combined pulmonary fibrosis and emphysema: Effect of pulmonary rehabilitation in comparison with chronic obstructive pulmonary disease. BMJ Open Respir. Res. 2016, 3, e000099. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Tsujino, I.; Tanino, M.; Ohira, H.; Nishimura, M. Broad and heterogeneous vasculopathy in pulmonary fibrosis and emphysema with pulmonary hypertension. Respirol. Case Rep. 2013, 1, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Cottin, V.; Le Pavec, J.; Prevot, G.; Mal, H.; Humbert, M.; Simonneau, G.; Cordier, J.-F. Pulmonary hypertension in patients with combined pulmonary fibrosis and emphysema syndrome. Eur. Respir. J. 2010, 35, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, R.P.; Ribeiro, R.; Melo, L.; Grima, B.; Oliveira, S.; Alves, J.D. Connective tissue disease-associated interstitial lung disease. Pulmonology 2022, 28, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Saggar, R.; Giri, P.C.; Deng, C.; Johnson, D.; McCloy, M.K.; Liang, L.; Shaikh, F.; Hong, J.; Channick, R.N.; Shapiro, S.S.; et al. Significance of autoimmune disease in severe pulmonary hypertension complicating extensive pulmonary fibrosis: A prospective cohort study. Pulm. Circ. 2021, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Cottin, V.; Harari, S.; Humbert, M.; Mal, H.; Dorfmüller, P.; Jaïs, X.; Reynaud-Gaubert, M.; Prevot, G.; Lazor, R.; Taillé, C.; et al. Pulmonary hypertension in lymphangioleiomyomatosis: Characteristics in 20 patients. Eur. Respir. J. 2012, 40, 630–640. [Google Scholar] [CrossRef] [PubMed]
- Freitas, C.S.G.; Baldi, B.G.; Jardim, C.; Araujo, M.S.; Sobral, J.B.; Heiden, G.I.; Kairalla, R.A.; Souza, R.; Carvalho, C.R.R. Pulmonary hypertension in lymphangioleiomyomatosis: Prevalence, severity and the role of carbon monoxide diffusion capacity as a screening method. Orphanet J. Rare Dis. 2017, 12, 74. [Google Scholar] [CrossRef]
- Wu, X.; Xu, W.; Wang, J.; Tian, X.; Tian, Z.; Xu, K. Clinical characteristics in lymphangioleiomyomatosis-related pulmonary hypertension: An observation on 50 patients. Front. Med. 2019, 13, 259–266. [Google Scholar] [CrossRef]
- Bhattacharyya, P.; Saha, D.; Bhattacherjee, P.; Das, S.; Bhattacharyya, P.; Dey, R. Tuberculosis associated pulmonary hypertension: The revelation of a clinical observation. Lung India 2016, 33, 135. [Google Scholar] [CrossRef]
- Park, S.Y.; Lee, S.M.; Shin, J.W.; Choi, B.W.; Kim, H.; Lee, J.S.; Lee, S.D.; Park, S.S.; Moon, H.S.; Park, Y.B. Epidemiology of chronic thromboembolic pulmonary hypertension in Korea: Results from the Korean registry. Korean J. Intern. Med. 2016, 31, 305–312. [Google Scholar] [CrossRef]
- Walsh, K.F.; Lui, J.K. Post-tuberculosis pulmonary hypertension: A case of global disparity in health care. Lancet Glob. Health 2022, 10, e476. [Google Scholar] [CrossRef] [PubMed]
- DuBrock, H.M.; Nathan, S.D.; Reeve, B.B.; Kolaitis, N.A.; Mathai, S.C.; Classi, P.M.; Nelsen, A.C.; Olayinka-Amao, B.; Norcross, L.N.; Martin, S.A. Pulmonary hypertension due to interstitial lung disease or chronic obstructive pulmonary disease: A patient experience study of symptoms and their impact on quality of life. Pulm. Circ. 2021, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Nikkho, S.M.; Richter, M.J.; Shen, E.; Abman, S.H.; Antoniou, K.; Chung, J.; Fernandes, P.; Hassoun, P.; Lazarus, H.M.; Olschewski, H.; et al. Clinical significance of pulmonary hypertension in interstitial lung disease: A consensus statement from the Pulmonary Vascular Research Institute’s innovative drug development initiative—Group 3 pulmonary hypertension. Pulm. Circ. 2022, 12, e12127. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; Lee, S.H.; Voswinckel, R.; Palazzini, M.; Jais, X.; Marinelli, A.; Barst, R.J.; Ghofrani, H.A.; Jing, Z.-C.; Opitz, C.; et al. Complications of right heart catheterization procedures in patients with pulmonary hypertension in experienced centers. J. Am. Coll. Cardiol. 2006, 48, 2546–2552. [Google Scholar] [CrossRef] [PubMed]
- Gläser, S.; Obst, A.; Koch, B.; Henkel, B.; Grieger, A.; Felix, S.B.; Halank, M.; Bruch, L.; Bollmann, T.; Warnke, C.; et al. Pulmonary Hypertension in Patients with Idiopathic Pulmonary Fibrosis—The Predictive Value of Exercise Capacity and Gas Exchange Efficiency. PLoS ONE 2013, 8, e65643. [Google Scholar] [CrossRef] [PubMed]
- Waxman, A.B.; Elia, D.; Adir, Y.; Humbert, M.; Harari, S. Recent advances in the management of pulmonary hypertension with interstitial lung disease. Eur. Respir. Rev. 2022, 31, 210220. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, T.; Kondoh, Y.; Taniguchi, H.; Yagi, M.; Matsuda, T.; Kimura, T.; Kataoka, K.; Johkoh, T.; Ando, M.; Hashimoto, N.; et al. A scoring system to predict the elevation of mean pulmonary arterial pressure in idiopathic pulmonary fibrosis. Eur. Respir. J. 2018, 51, 1701311. [Google Scholar] [CrossRef]
- Zisman, D.A.; Ross, D.J.; Belperio, J.A.; Saggar, R.; Lynch, J.P.; Ardehali, A.; Karlamangla, A.S. Prediction of pulmonary hypertension in idiopathic pulmonary fibrosis. Respir. Med. 2007, 101, 2153–2159. [Google Scholar] [CrossRef]
- Antoniou, K.M.; Margaritopoulos, G.A.; Goh, N.S.; Karagiannis, K.; Desai, S.R.; Nicholson, A.G.; Siafakas, N.M.; Coghlan, J.G.; Denton, C.P.; Hansell, D.M.; et al. Combined Pulmonary Fibrosis and Emphysema in Scleroderma-Related Lung Disease Has a Major Confounding Effect on Lung Physiology and Screening for Pulmonary Hypertension. Arthritis Rheumatol. 2016, 68, 1004–1012. [Google Scholar] [CrossRef]
- Gille, T.; Laveneziana, P. Cardiopulmonary exercise testing in interstitial lung diseases and the value of ventilatory efficiency. Eur. Respir. Rev. 2021, 30, 200355. [Google Scholar] [CrossRef]
- Boutou, A.K.; Pitsiou, G.G.; Trigonis, I.; Papakosta, D.; Kontou, P.K.; Chavouzis, N.; Nakou, C.; Argyropoulou, P.; Wasserman, K.; Stanopoulos, I. Exercise capacity in idiopathic pulmonary fibrosis: The effect of pulmonary hypertension: Pulmonary hypertension in lung fibrosis. Respirology 2011, 16, 451–458. [Google Scholar] [CrossRef] [PubMed]
- van der Plas, M.N.; van Kan, C.; Blumenthal, J.; Jansen, H.M.; Wells, A.U.; Bresser, P. Pulmonary vascular limitation to exercise and survival in idiopathic pulmonary fibrosis: Pulmonary vascular impairment in IPF. Respirology 2014, 19, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Ewert, R.; Ittermann, T.; Habedank, D.; Held, M.; Lange, T.J.; Halank, M.; Winkler, J.; Gläser, S.; Olschewski, H.; Kovacs, G. Prognostic value of cardiopulmonary exercise testing in patients with systemic sclerosis. BMC Pulm. Med. 2019, 19, 230. [Google Scholar] [CrossRef] [PubMed]
- Swigris, J.J.; Olson, A.L.; Shlobin, O.A.; Ahmad, S.; Brown, K.K.; Nathan, S.D. Heart rate recovery after six-minute walk test predicts pulmonary hypertension in patients with idiopathic pulmonary fibrosis: HRR and PH in IPF. Respirology 2011, 16, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Alhamad, E.H.; Cal, J.G.; Alrajhi, N.N.; Alharbi, W.M. Predictors of Mortality in Patients with Interstitial Lung Disease-Associated Pulmonary Hypertension. J. Clin. Med. 2020, 9, 3828. [Google Scholar] [CrossRef] [PubMed]
- Yoo, D.K.; Zompatori, M.; Barrile, A.; Rossi, G.; D’Amato, D.; Sergiacomi, G.; Rogliani, P.; Mura, M. Associated Pulmonary Hypertension Is an Independent Contributor to Exercise Intolerance in Chronic Fibrosing Interstitial Pneumonias. Respiration 2018, 96, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Goerne, H.; Batra, K.; Rajiah, P. Imaging of pulmonary hypertension: An update. Cardiovasc. Diagn. Ther. 2018, 8, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Foley, R.W.; Kaneria, N.; Ross, R.V.M.; Suntharalingam, J.; Hudson, B.J.; Rodrigues, J.C.; Robinson, G. Computed tomography appearances of the lung parenchyma in pulmonary hypertension. Br. J. Radiol. 2021, 94, 20200830. [Google Scholar] [CrossRef]
- Chen, R.; Liao, H.; Deng, Z.; He, Z.; Zheng, Z.; Lu, J.; Jiang, M.; Wu, X.; Guo, W.; Huang, Z.; et al. Efficacy of computed tomography in diagnosing pulmonary hypertension: A systematic review and meta-analysis. Front. Cardiovasc. Med. 2022, 9, 966257. [Google Scholar] [CrossRef]
- Tan, R.T.; Kuzo, R.; Goodman, L.R.; Siegel, R.; Haasler, G.R.; Presberg, K.W. Utility of CT Scan Evaluation for Predicting Pulmonary Hypertension in Patients With Parenchymal Lung Disease. Chest 1998, 113, 1250–1256. [Google Scholar] [CrossRef]
- Swift, A.J.; Dwivedi, K.; Johns, C.; Garg, P.; Chin, M.; Currie, B.J.; Rothman, A.M.; Capener, D.; Shahin, Y.; Elliot, C.A.; et al. Diagnostic accuracy of CT pulmonary angiography in suspected pulmonary hypertension. Eur. Radiol. 2020, 30, 4918–4929. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Wan, C.; Tian, P.; Wu, Y.; Li, X.; Yang, T.; An, J.; Wang, T.; Chen, L.; Wen, F. CT-Base Pulmonary Artery Measurementin the Detection of Pulmonary Hypertension: A Meta-Analysis and Systematic Review. Medicine 2014, 93, e256. [Google Scholar] [CrossRef] [PubMed]
- Zisman, D.A.; Karlamangla, A.S.; Ross, D.J.; Keane, M.P.; Belperio, J.A.; Saggar, R.; Lynch, J.P.; Ardehali, A.; Goldin, J. High-Resolution Chest CT Findings Do Not Predict the Presence of Pulmonary Hypertension in Advanced Idiopathic Pulmonary Fibrosis. Chest 2007, 132, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, S.; Matsuoka, S.; Yamashiro, T.; Fujikawa, A.; Yagihashi, K.; Kurihara, Y.; Nakajima, Y. Pulmonary arterial enlargement in patients with acute exacerbation of interstitial pneumonia. Clin. Imaging 2014, 38, 454–457. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Burns, A.T.; Yap, K.; Prior, D.L. The role of imaging in pulmonary hypertension. Cardiovasc. Diagn. Ther. 2021, 11, 859–880. [Google Scholar] [CrossRef] [PubMed]
- Scelsi, C.L.; Bates, W.B.; Melenevsky, Y.V.; Sharma, G.K.; Thomson, N.B.; Keshavamurthy, J.H. Egg-and-Banana Sign: A Novel Diagnostic CT Marker for Pulmonary Hypertension. Am. J. Roentgenol. 2018, 210, 1235–1239. [Google Scholar] [CrossRef]
- Frazier, A.A.; Burke, A.P. The Imaging of Pulmonary Hypertension. Semin. Ultrasound CT MRI 2012, 33, 535–551. [Google Scholar] [CrossRef]
- Kiely, D.G.; Levin, D.L.; Hassoun, P.M.; Ivy, D.; Jone, P.; Bwika, J.; Kawut, S.M.; Lordan, J.; Lungu, A.; Mazurek, J.A.; et al. Statement on imaging and pulmonary hypertension from the Pulmonary Vascular Research Institute (PVRI). Pulm. Circ. 2019, 9, 1–32. [Google Scholar] [CrossRef]
- Augustine, D.X.; Coates-Bradshaw, L.D.; Willis, J.; Harkness, A.; Ring, L.; Grapsa, J.; Coghlan, G.; Kaye, N.; Oxborough, D.; Robinson, S.; et al. Echocardiographic assessment of pulmonary hypertension: A guideline protocol from the British Society of Echocardiography. Echo Res. Pract. 2018, 5, G11–G24. [Google Scholar] [CrossRef]
- Nowak, J.; Hudzik, B.; Jastrzȩbski, D.; Niedziela, J.T.; Rozentryt, P.; Wojarski, J.; Ochman, M.; Karolak, W.; Żegleń, S.; Gierlotka, M.; et al. Pulmonary hypertension in advanced lung diseases: Echocardiography as an important part of patient evaluation for lung transplantation. Clin. Respir. J. 2018, 12, 930–938. [Google Scholar] [CrossRef]
- Mandoli, G.E.; De Carli, G.; Pastore, M.C.; Cameli, P.; Contorni, F.; D’Alessandro, M.; Bargagli, E.; Mondillo, S.; Cameli, M. Right cardiac involvement in lung diseases: A multimodality approach from diagnosis to prognostication. J. Intern. Med. 2021, 289, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Arcasoy, S.M.; Christie, J.D.; Ferrari, V.A.; Sutton, M.S.J.; Zisman, D.A.; Blumenthal, N.P.; Pochettino, A.; Kotloff, R.M. Echocardiographic Assessment of Pulmonary Hypertension in Patients with Advanced Lung Disease. Am. J. Respir. Crit. Care Med. 2003, 167, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Cowie, B.; Kluger, R.; Rex, S.; Missant, C. The utility of transoesophageal echocardiography for estimating right ventricular systolic pressure. Anaesthesia 2015, 70, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.-R.; Yan, P.-J.; Liu, S.-D.; Hu, Y.; Yang, K.-H.; Song, B.; Lei, J.-Q. Diagnostic accuracy of transthoracic echocardiography for pulmonary hypertension: A systematic review and meta-analysis. BMJ Open 2019, 9, e033084. [Google Scholar] [CrossRef] [PubMed]
- Saba, T.S.; Foster, J.; Cockburn, M.; Cowan, M.; Peacock, A.J. Ventricular mass index using magnetic resonance imaging accurately estimates pulmonary artery pressure. Eur. Respir. J. 2002, 20, 1519–1524. [Google Scholar] [CrossRef] [PubMed]
- Dellegrottaglie, S.; Sanz, J.; Poon, M.; Viles-Gonzalez, J.F.; Sulica, R.; Goyenechea, M.; Macaluso, F.; Fuster, V.; Rajagopalan, S. Pulmonary Hypertension: Accuracy of Detection with Left Ventricular Septal-to–Free Wall Curvature Ratio Measured at Cardiac MR. Radiology 2007, 243, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Shehata, M.L.; Harouni, A.A.; Skrok, J.; Basha, T.A.; Boyce, D.; Lechtzin, N.; Mathai, S.C.; Girgis, R.; Osman, N.F.; Lima, J.A.C.; et al. Regional and Global Biventricular Function in Pulmonary Arterial Hypertension: A Cardiac MR Imaging Study. Radiology 2013, 266, 114–122. [Google Scholar] [CrossRef]
- Freed, B.H.; Collins, J.D.; François, C.J.; Barker, A.J.; Cuttica, M.J.; Chesler, N.C.; Markl, M.; Shah, S.J. MR and CT Imaging for the Evaluation of Pulmonary Hypertension. JACC Cardiovasc. Imaging 2016, 9, 715–732. [Google Scholar] [CrossRef]
- Swift, A.; Capener, D.; Alex, R.; Charlie, E.; Robin, C.; Jim, W.; David, K. Prognostic value of MRI in pulmonary arterial hypertension: Early and late predictors. 43 Pulmonary Circulation and Pulmonary Vascular Disease. Eur. Respir. Soc. 2015, 46, OA3523. [Google Scholar] [CrossRef]
- Swift, A.J.; Capener, D.; Johns, C.; Hamilton, N.; Rothman, A.; Elliot, C.; Condliffe, R.; Charalampopoulos, A.; Rajaram, S.; Lawrie, A.; et al. Magnetic Resonance Imaging in the Prognostic Evaluation of Patients with Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2017, 196, 228–239. [Google Scholar] [CrossRef]
- Lewis, R.A.; Johns, C.S.; Cogliano, M.; Capener, D.; Tubman, E.; Elliot, C.A.; Charalampopoulos, A.; Sabroe, I.; Thompson, A.A.R.; Billings, C.G.; et al. Identification of Cardiac Magnetic Resonance Imaging Thresholds for Risk Stratification in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2020, 201, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Swift, A.J.; Rajaram, S.; Hurdman, J.; Hill, C.; Davies, C.; Sproson, T.W.; Morton, A.C.; Capener, D.; Elliot, C.; Condliffe, R.; et al. Noninvasive Estimation of PA Pressure, Flow, and Resistance With CMR Imaging. JACC Cardiovasc. Imaging 2013, 6, 1036–1047. [Google Scholar] [CrossRef] [PubMed]
- Lungu, A.; Wild, J.M.; Capener, D.; Kiely, D.G.; Swift, A.J.; Hose, D.R. MRI model-based non-invasive differential diagnosis in pulmonary hypertension. J. Biomech. 2014, 47, 2941–2947. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, A.J.; Solanki, R.; Johns, C.S.; Kiely, D.; Wild, J.; Swift, A.J. MRI Prediction of Precapillary Pulmonary Hypertension according to the Sixth World Symposium on Pulmonary Hypertension. Radiology 2020, 294, 482. [Google Scholar] [CrossRef]
- Alkhanfar, D.; Dwivedi, K.; Alandejani, F.; Shahin, Y.; Alabed, S.; Johns, C.; Garg, P.; Thompson, A.A.R.; Rothman, A.M.K.; Hameed, A.; et al. Non-invasive detection of severe PH in lung disease using magnetic resonance imaging. Front. Cardiovasc. Med. 2023, 10, 1016994. [Google Scholar] [CrossRef] [PubMed]
- Alenezi, F.; Covington, T.A.; Mukherjee, M.; Mathai, S.C.; Yu, P.B.; Rajagopal, S. Novel Approaches to Imaging the Pulmonary Vasculature and Right Heart. Circ. Res. 2022, 130, 1445–1465. [Google Scholar] [CrossRef] [PubMed]
- Santos-Gomes, J.; Gandra, I.; Adão, R.; Perros, F.; Brás-Silva, C. An Overview of Circulating Pulmonary Arterial Hypertension Biomarkers. Front. Cardiovasc. Med. 2022, 9, 924873. [Google Scholar] [CrossRef]
- Andersen, C.U.; Mellemkjær, S.; Nielsen-Kudsk, J.E.; Bendstrup, E.; Simonsen, U.; Hilberg, O. Diagnostic and prognostic role of biomarkers for pulmonary hypertension in interstitial lung disease. Respir. Med. 2012, 106, 1749–1755. [Google Scholar] [CrossRef]
- Vuga, L.J.; Tedrow, J.R.; Pandit, K.V.; Tan, J.; Kass, D.J.; Xue, J.; Chandra, D.; Leader, J.K.; Gibson, K.F.; Kaminski, N.; et al. C-X-C Motif Chemokine 13 (CXCL13) Is a Prognostic Biomarker of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2014, 189, 966–974. [Google Scholar] [CrossRef]
- Tzouvelekis, A.; Herazo-Maya, J.D.; Ryu, C.; Chu, J.-H.; Zhang, Y.; Gibson, K.F.; Adonteng-Boateng, P.K.; Li, Q.; Pan, H.; Cherry, B.; et al. S100A12 as a marker of worse cardiac output and mortality in pulmonary hypertension: S100A12 in pulmonary hypertension. Respirology 2018, 23, 771–779. [Google Scholar] [CrossRef]
- Parikh, R.; Konstantinidis, I.; O’Sullivan, D.M.; Farber, H.W. Pulmonary hypertension in patients with interstitial lung disease: A tool for early detection. Pulm. Circ. 2022, 12. [Google Scholar] [CrossRef] [PubMed]
- Ruocco, G.; Cekorja, B.; Rottoli, P.; Refini, R.M.; Pellegrini, M.; Di Tommaso, C.; Del Castillo, G.; Franci, B.; Nuti, R.; Palazzuoli, A. Role of BNP and echo measurement for pulmonary hypertension recognition in patients with interstitial lung disease: An algorithm application model. Respir. Med. 2015, 109, 406–415. [Google Scholar] [CrossRef] [PubMed]
- Kogan, E.; Didden, E.-M.; Lee, E.; Nnewihe, A.; Stamatiadis, D.; Mataraso, S.; Quinn, D.; Rosenberg, D.; Chehoud, C.; Bridges, C. A machine learning approach to identifying patients with pulmonary hypertension using real-world electronic health records. Int. J. Cardiol. 2023, 374, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.; Kim, K.-H.; Medina-Inojosa, J.; Jeon, K.-H.; Park, J.; Oh, B.-H. Artificial intelligence for early prediction of pulmonary hypertension using electrocardiography. J. Heart Lung Transplant. 2020, 39, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Kusunose, K.; Hirata, Y.; Tsuji, T.; Kotoku, J.; Sata, M. Deep learning to predict elevated pulmonary artery pressure in patients with suspected pulmonary hypertension using standard chest X ray. Sci. Rep. 2020, 10, 19311. [Google Scholar] [CrossRef]
- Sprecher, V.P.; Didden, E.; Swerdel, J.N.; Muller, A. Evaluation of code-based algorithms to identify pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension patients in large administrative databases. Pulm. Circ. 2020, 10, 2045894020961713. [Google Scholar] [CrossRef]
- Cordeiro, R.; Nunes, A.; Smith, O.; Renzoni, E.A. Oxygen in interstitial lung diseases. Breathe 2023, 19, 220271. [Google Scholar] [CrossRef]
- Dowman, L.; Hill, C.J.; May, A.; Holland, A.E. Pulmonary rehabilitation for interstitial lung disease. Cochrane Database Syst. Rev. 2021, 2, CD006322. [Google Scholar] [CrossRef]
- Kapnadak, S.G.; Raghu, G. Lung transplantation for interstitial lung disease. Eur. Respir. Rev. 2021, 30, 210017. [Google Scholar] [CrossRef]
- Zhao, N.; Chen, J.; Zhang, M.; Zhou, L.; Liu, L.; Yuan, J.; Pang, X.; Hu, D.; Ren, X.; Jin, Z. PAH-specific therapy for pulmonary hypertension and interstitial lung disease: A systemic review and meta-analysis. Front. Cardiovasc. Med. 2022, 9, 992879. [Google Scholar] [CrossRef]
- King, C.S.; Shlobin, O.A. The Trouble With Group 3 Pulmonary Hypertension in Interstitial Lung Disease. Chest 2020, 158, 1651–1664. [Google Scholar] [CrossRef] [PubMed]
- Idiopathic Pulmonary Fibrosis Clinical Research Network; Zisman, D.A.; Schwarz, M.; Anstrom, K.J.; Collard, H.R.; Flaherty, K.R.; Hunninghake, G.W. A controlled trial of sildenafil in advanced idiopathic pulmonary fibrosis. N. Engl. J. Med. 2010, 363, 620–628. [Google Scholar] [PubMed]
- Raghu, G. Treatment of Idiopathic Pulmonary Fibrosis With Ambrisentan: A Parallel, Randomized Trial. Ann. Intern. Med. 2013, 158, 641. [Google Scholar] [CrossRef] [PubMed]
- Nathan, S.; Behr, J.; Collard, H.R.; Cottin, V.; Hoeper, M.M.; Martinez, F.; Corte, T.; Keogh, A.; Leuchte, H.; Mogulkoc, N.; et al. RISE-IIP: Riociguat for the treatment of pulmonary hypertension associated with idiopathic interstitial pneumonia. Eur. Respir. Soc. 2017, 50 (Suppl. 61), OA1985. [Google Scholar] [CrossRef]
- Waxman, A.; Restrepo-Jaramillo, R.; Thenappan, T.; Ravichandran, A.; Engel, P.; Bajwa, A.; Allen, R.; Feldman, J.; Argula, R.; Smith, P.; et al. Inhaled Treprostinil in Pulmonary Hypertension Due to Interstitial Lung Disease. N. Engl. J. Med. 2021, 384, 325–334. [Google Scholar] [CrossRef]
- Kimura, M.; Taniguchi, H.; Kondoh, Y.; Kimura, T.; Kataoka, K.; Nishiyama, O.; Aso, H.; Sakamoto, K.; Hasegawa, Y. Pulmonary Hypertension as a Prognostic Indicator at the Initial Evaluation in Idiopathic Pulmonary Fibrosis. Respiration 2013, 85, 456–463. [Google Scholar] [CrossRef]
- Hayes, D.; Black, S.M.; Tobias, J.D.; Kirkby, S.; Mansour, H.M.; Whitson, B.A. Influence of Pulmonary Hypertension on Patients With Idiopathic Pulmonary Fibrosis Awaiting Lung Transplantation. Ann. Thorac. Surg. 2016, 101, 246–252. [Google Scholar] [CrossRef]
- Suzuki, A.; Taniguchi, H.; Watanabe, N.; Kondoh, Y.; Kimura, T.; Kataoka, K.; Matsuda, T.; Yokoyama, T.; Sakamoto, K.; Nishiyama, O.; et al. Significance of pulmonary arterial pressure as a prognostic indicator in lung-dominant connective tissue disease. PLoS ONE 2014, 9, e108339. [Google Scholar] [CrossRef]
- Kessler, R.; Faller, M.; Weitzenblum, E.; Chaouat, A.; Aykut, A.; Ducoloné, A.; Ehrhart, M.; Oswald-Mammosser, M. “Natural history” of pulmonary hypertension in a series of 131 patients with chronic obstructive lung disease. Am. J. Respir. Crit. Care Med. 2001, 164, 219–224. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Imaging Modality | PH Suggesting Features |
|---|---|
| Chest CT |
|
| Echocardiography |
|
| Cardiac MRI |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Averjanovaitė, V.; Gumbienė, L.; Zeleckienė, I.; Šileikienė, V. Unmasking a Silent Threat: Improving Pulmonary Hypertension Screening Methods for Interstitial Lung Disease Patients. Medicina 2024, 60, 58. https://doi.org/10.3390/medicina60010058
Averjanovaitė V, Gumbienė L, Zeleckienė I, Šileikienė V. Unmasking a Silent Threat: Improving Pulmonary Hypertension Screening Methods for Interstitial Lung Disease Patients. Medicina. 2024; 60(1):58. https://doi.org/10.3390/medicina60010058
Chicago/Turabian StyleAverjanovaitė, Vaida, Lina Gumbienė, Ingrida Zeleckienė, and Virginija Šileikienė. 2024. "Unmasking a Silent Threat: Improving Pulmonary Hypertension Screening Methods for Interstitial Lung Disease Patients" Medicina 60, no. 1: 58. https://doi.org/10.3390/medicina60010058