Abstract
The total syntheses of the marine-derived lipopeptide natural product fellutamide B and deoxy-fellutamides B, C, and D are reported. These compounds were accessed through a novel solid-phase synthetic strategy using Weinreb amide-derived resin. As part of the synthesis, a new enantioselective route to (3R)-hydroxy lauric acid was developed utilizing a Brown allylation reaction followed by an oxidative cleavage-oxidation sequence as the key steps. The activity of these natural products, and natural product analogues was also assessed against Mycobacterium tuberculosis in vitro.
1. Introduction
Fellutamides A–D (Figure 1, 1–4) are a family of marine-derived lipopeptide natural products, characterized by a C-terminal aldehyde and a (3R)-β-hydroxy alkanoate tail (Figure 1). Fellutamides A (1), C (3) and D (4) contain the non-ribosomal amino acid β-l-threo-hydroxy-glutamine, while fellutamides A (1) and B (2) both possess a β-hydroxylated fatty chain amide derived from (3R)-hydroxy lauric acid (5).
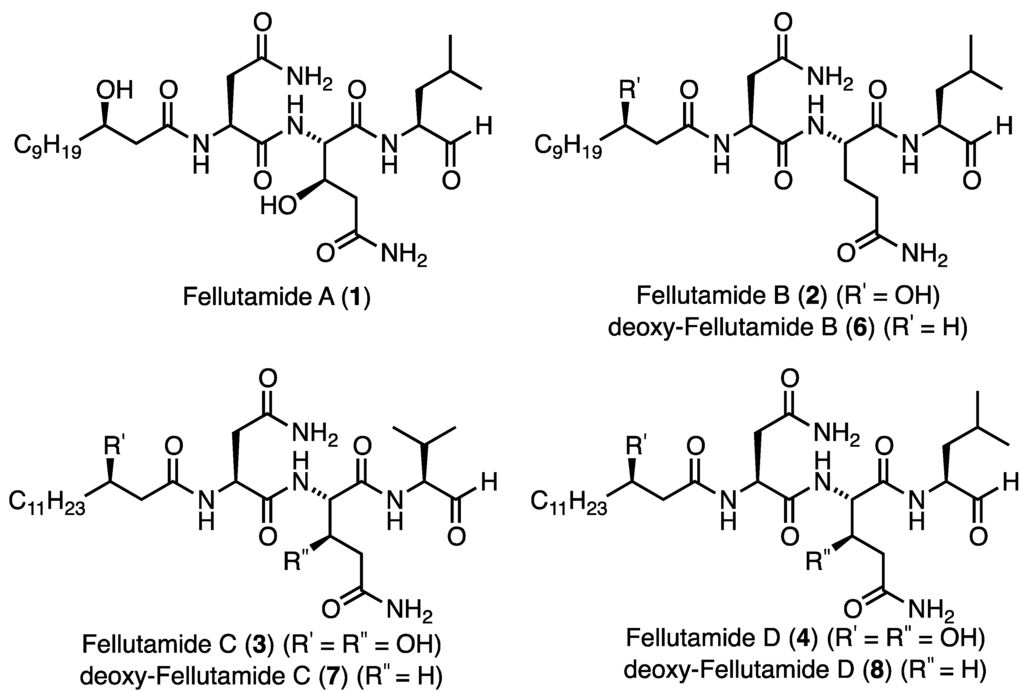
Figure 1.
Fellutamides A–D (1–4), and deoxy-fellutamides B (6), C (7), and D (8).
Fellutamides A (1) and B (2) were first isolated by Shigemori and co-workers in 1991 from the fungus Penicillium fellutanum, found in the gastrointestine of the Candystripe Cardinalfish Apogon endekataenia, located off the coast of Japan [1]. Both of these compounds were found to be cytotoxic against human epidermoid carcinoma KB, exhibiting half maximal inhibitory concentration (IC50) values of 0.5 μg/mL and 0.7 μg/mL for fellutamide A (1) and fellutamide B (2), respectively. From this family of natural products only the total synthesis of fellutamide B (2) has been achieved which was reported by Crews and co-workers in 2006 [2]. In 2011, Xu and co-workers reported the isolation of fellutamides C (3) and D (4) from an undescribed species of Metulocladosporiella, a fungus isolated from Equatorial Guinean soil [3]. It was reported that the cellular target of fellutamides C (3) and D (4) is the fungal proteasome, and these natural products were found to be active against a number of Candida fungal species with minimal inhibitory concentration (MIC) values between 2 and 32 μg/mL. Interestingly, fellutamide C (3) was consistently more potent than fellutamide D (4). In addition, screening against Bortezomib sensitive (PC-3) and insensitive (A549) human cancer cell lines revealed that these compounds were potent inhibitors of the PC-3 cell line with no inhibition of the A549 cell line.
Since their isolation, fellutamides A (1) and B (2) have been found to have a number of other interesting activities, across a range of biological systems [4]. In particular, fellutamide B (2) is a potent inhibitor of the Mycobacterium tuberculosis proteasome with an inhibition constant (KI) of 6.8 nM against the M. tuberculosis 20S core particle (CP). In fact, fellutamide B (2) is the most potent peptide inhibitor of the M. tuberculosis proteasome reported to date [5]. Additionally, fellutamide B (2) is also a potent inhibitor of the human 20S CP with a KI of 11.5 nM [5]. It has since been discovered that fellutamide B (2) binds to the M. tuberculosis 20S proteasome via a different mechanism to the human 20S proteasome [5]. As such, it can be envisaged that modifications to the structure may result in higher affinity and selectivity for the M. tuberculosis proteasome. Two potent irreversible oxathiazol-2-one inhibitors of the M. tuberculosis proteasome were recently elucidated from a high throughput screen of 20,000 compounds [6]. These compounds have been shown to cross the cell wall and inhibit non-replicating M. tuberculosis in vitro, which may therefore provide inspiration for the development of new TB drug leads which operate via novel modes of action.
Given the interesting structural features and biological activities of the fellutamide natural products, particularly as inhibitors of the M. tuberculosis proteasome, we sought to develop a general synthesis of fellutamide B (2) and the deoxy-fellutamide analogues 6–8 (Figure 2). Additionally, we were interested in the potential of these compounds as inhibitors of M. tuberculosis growth and in the future, as M. tuberculosis proteasome inhibitors. Access to these compounds may help to establish the necessity of the hydroxyl groups for binding to the proteasome and for activity against M. tuberculosis and lead to the development of selective M. tuberculosis proteasome inhibitors as potential anti-tubercular agents.
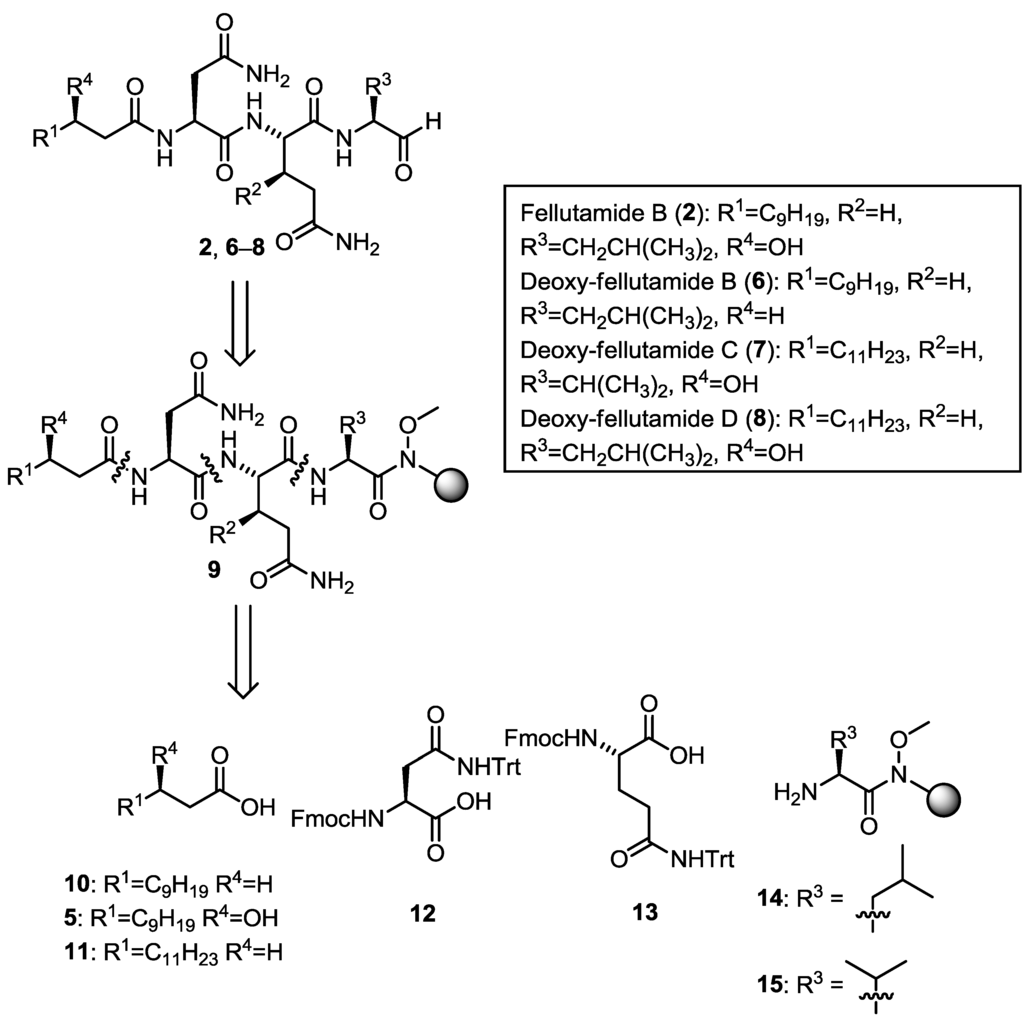
Figure 2.
Retrosynthetic analysis for Fellutamide B (2) and deoxy-analogues 6–8.
2. Results and Discussion
2.1. Synthetic Plan
We envisaged that fellutamide B (2) and deoxy-fellutamide analogues 6–8 could be synthesized by solid-phase peptide synthesis on Weinreb amide resin [7,8,9] which upon reduction would afford either the C-terminal leucinal, present in fellutamide B (2), and analogues (6 and 8) or a valinal moiety, found in deoxy-fellutamide C (7) (Figure 1). The C-terminal aldehyde is one of the most interesting structural features present in the fellutamide natural product family, as it is essential for the inhibitory activity of fellutamide B (2) against the M. tuberculosis proteasome [5]. Due to the inherent reactivity of this functional group, we envisioned that the aldehyde would ideally be introduced in the final step of the synthesis, without the need for subsequent purification. As such, we planned to introduce the aldehyde functionality from resin bound Weinreb amide precursors (general structure 9) which could be assembled following standard solid-phase peptide synthesis (SPPS) protocols, starting with the fragments 5, 10–15, which would provide access to fellutamide B (2) as well as deoxy-analogues 6–8 (Figure 2). The (3R)-hydroxy lauric acid (5) fragment (R1 = C9H19 and R4 = OH) required for the synthesis of fellutamide B (2) is not commercially available and, as such, required preparation.
Synthesis of (3R)-Hydroxy Lauric Acid (5)
Although there have been a number of syntheses of (3R)-hydroxy alkanoic acids reported in the literature to date, [10,11] we proposed a shorter route whereby the desired acid 5 could be accessed from the chiral homoallylic alcohol 16 via an oxidative cleavage-oxidation sequence (Scheme 1). Homoallylic alcohol 16 in turn could be prepared via an enantioselective allylation reaction as the key stereochemistry determining step from the commercially available aldehyde, decanal (17). To this end, decanal (17) was treated with the chiral boron reagent (−)-B-allyl-diisopinocampheylborane [(−)-B(Ipc)2(allyl)] [(−)-18] which was preformed from (–)-B-methoxy-diisopinocampheylborane and allyl magnesium bromide at −78 °C (See Supplementary Information). After removal of the resulting magnesium salts by filtration under an inert atmosphere, decanal (17) was added to the solution of reagent (−)-18 at −100 °C. Oxidation of this adduct by addition of basic hydrogen peroxide liberated the free homoallylic alcohol 16, which was isolated in excellent yield (83%, 85% ee, Scheme 1) [12]. The absolute configuration was confirmed by comparison with the literature value for this compound (+2.9, lit. +11.2) [13], and the enantiomeric excess was determined from the corresponding Mosher’s ester of 16 (See Supplementary Information) [14]. Furthermore, dihydroxylation of the olefin 16 employing catalytic osmium(VIII) tetroxide and N-methylmorpholine-N-oxide, and subsequent oxidative cleavage using sodium periodate provided the intermediate aldehyde, which was subjected to Pinnick oxidation conditions to afford the desired acid 5 in good yield (77%, Scheme 1). Finally, recrystallization of the corresponding dicyclohexylammonium salt of acid 5 from acetonitrile (3 times) following the procedure of Tai and co-workers [15] led to enantiomeric enrichment, providing the acid 5 in 58% overall yield, and >95% ee, as determined from the Mosher’s ester of the corresponding methyl ester of 5 (See Supplementary Information).
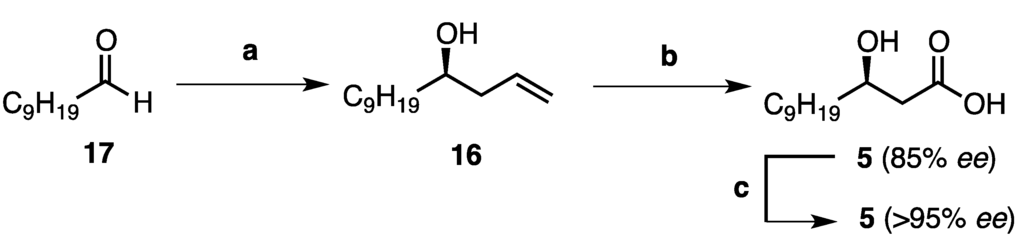
Scheme 1.
Synthesis of (3R)-hydroxy lauric acid (5). Reagents and conditions: (a) (i) (−)-B(Ipc)2(allyl) [(−)-18], Et2O, −100 °C; (ii) NaOH, H2O2, reflux to r.t., 83%, 85% ee; (b) (i) OsO4, NMO, (CH3)2CO, H2O, r.t.; (ii) NaIO4, THF, H2O, r.t.; (iii) NaClO2, NaH2PO4, t-BuOH, H2O, r.t. (77%, 85% ee, over three steps); (c) (i) HN(C6H11)2, MeCN, r.t. (recrystallised 3 times from MeCN); (ii) 1M HCl, 75%, >95% ee.
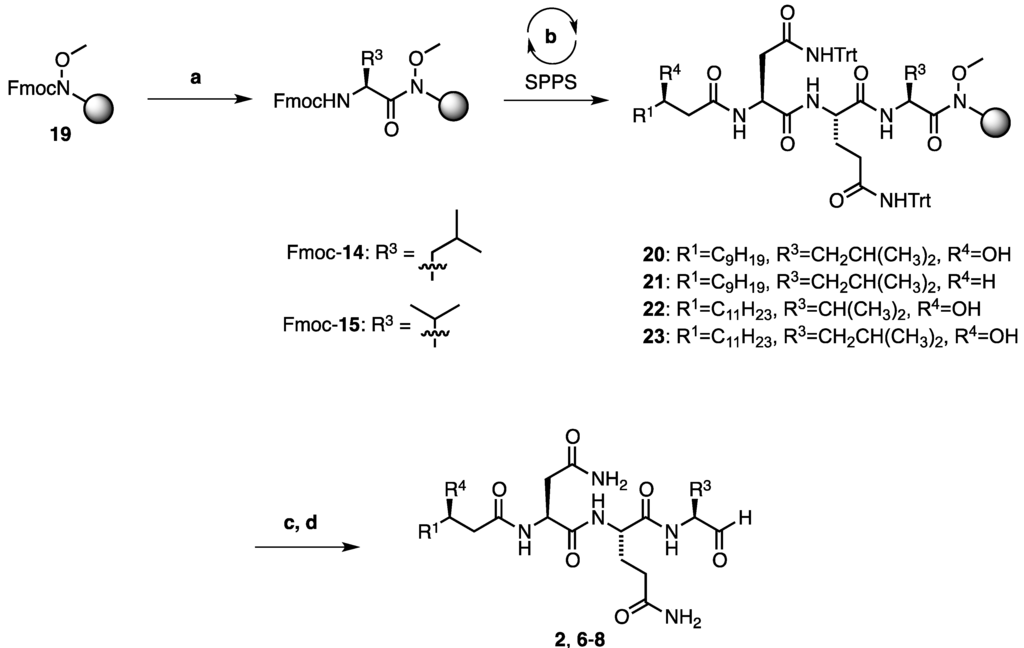
Scheme 2.
Fmoc-SPPS of resin bound intermediates 20–23. Reagents and conditions: (a) Deprotection: 10 vol% piperidine/DMF, 2 × 3 min Coupling: 5 eq Fmoc-Leu-OH or Fmoc-Val-OH, 5 equiv. HATU, 10 equiv. iPr2EtN, 2 × 6 h; (b) (One cycle) Deprotection: 10 vol% piperidine/DMF, 2 × 3 min; Coupling 1 [Fmoc-Gln(Trt)-OH and Fmoc-Asn(Trt)-OH]: 4 eq AA, 4 eq PyBOP, 8 eq NMM, 1–2 h; or Coupling 2 (for fatty acid 5, 10, or 11): 1.2 eq acid, 1.2 eq HATU, 2.4 eq iPr2EtN; and Capping: 10 vol% acetic anhydride/pyridine, 2 min; (c) TFA:TIS:H2O, (9:0.5:0.5, v/v/v), 1 h; (d) LiAlH4, THF, 0 °C, then KHSO4, NaKTartrate, 0 °C to r.t.
2.2. Total Synthesis of Fellutamide B (2) and Deoxy-Fellutamides B (6), C (7), and D (8)
With enantiopure (3R)-hydroxy lauric acid (5) in hand (Section 2.1, Scheme 1), the synthesis of fellutamide B (2) and the deoxy-fellutamide analogues 6–8 was next undertaken (Scheme 2). Beginning with Weinreb amide resin 19 and after Fmoc-deprotection, either Fmoc-protected leucine or Fmoc-protected valine was coupled onto the resin using 2-(1H-7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU) as the coupling reagent and N,N-diisopropylethylamine as the base. The couplings were carried out twice for extended times (6 h) to facilitate complete coupling to the secondary Weinreb amine. After deprotection of the resulting N-terminal Fmoc group of Fmoc-protected 14 or 15, resin-bound precursors 20–23 were synthesized following standard SPPS protocols (Scheme 2), whereby fatty acids 5, 10, or 11 were coupled in the final step. After deprotection of the trityl protecting groups of 20–23 under acidolytic conditions, the resulting resin-bound peptides were dried and swelled in anhydrous tetrahydrofuran before treatment with LiAlH4 solution to liberate the free aldehyde functionalities and afford the desired peptide products 2, 6–8 which were produced in good yield (44%–60%) over the six steps (Table 1).

Table 1.
Yields of fellutamide B (2) and deoxy-fellutamides B, C, and D (6–8) after cleavage from resin based on the original loading of the Weinreb amide resin (6 steps).
| Compound | R1 | R3 | R4 | Yield (%) |
|---|---|---|---|---|
| Fellutamide B (2) | C9H19 | CH2CH(CH3)2 | OH | 55 |
| Deoxyfellutamide B (6) | C9H19 | CH2CH(CH3)2 | H | 44 (24% after purification) |
| Deoxyfellutamide C (7) | C11H23 | CH(CH3)2 | OH | 53 |
| Deoxyfellutamide D (8) | C11H23 | CH2CH(CH3)2 | OH | 60 |
Gratifyingly, the 1H NMR data obtained for synthetic fellutamide B (2) matched that of the isolated material with the exception of one signal in the 13C NMR spectrum (See Supplementary Information) [1]. Furthermore, fellutamide B (2) and deoxy-fellutamide C (7) and D (8) were of sufficient purity for biological testing without the need for purification (up to 95% purity). However, deoxyfellutamide B (6) was purified by reverse-phase HPLC which provided the desired product with no observable epimerisation of the C-terminal aldehyde, despite the presence of TFA in the eluent.
2.3. Testing of Fellutamide B (2) and Deoxy-Fellutamides B (6), C (7), and D (8) against Mycobacterium tuberculosis
Fellutamide B (2) and the three deoxy-fellutamides 6–8 were tested against replicating virulent M. tuberculosis (H37Rv) in vitro. Unfortunately these compounds did not demonstrate any inhibitory activity at concentrations up to 1 mM. Given that the proteasome is essential for resistance to reactive nitrogen species, which are induced following T cell activation of infected macrophages [16], we next screened 2 and 6–8 in an in vitro model of dormant (non-replicating) M. tuberculosis [17]. This was achieved by treating M. tuberculosis cultures with the diethylenetriamine-nitric oxide (DETA-NO) adduct as a nitric oxide source during incubation with 2, 6, 7 or 8. Unfortunately these compounds also failed to inhibit the growth of the bacteria at concentrations up to 1 mM in the presence of sub-lethal concentrations of NO. Given that the oxathiazol-2-one class of potent M. tuberculosis inhibitors were able to inhibit the growth of M. tuberculosis under these conditions in vitro [6], this result may suggest that fellutamide B (2) and the deoxyfellutamides 6–8 are not capable of crossing the cell wall of M. tuberculosis and, as such, do not exhibit any measurable in vitro activity. Future work in our laboratories will involve assessing the activity of these synthetic fellutamides against the M. tuberculosis proteasome, the results of which will be reported in due course.
3. Experimental Section
3.1. General Experimental Methods
All reactions were conducted under an atmosphere of argon or nitrogen unless otherwise stated, or if water was present in the reaction. Commercially available chemicals were used as purchased or purified by standard methods where necessary according to Armarego and Chai [18]. The following temperature baths were used: ice/water/salt (0 °C), MeCN/dry ice (−42 °C), acetone/dry ice (−78 °C), diethyl ether/dry ice (−100 °C). Silicon oil baths were used for high temperature reactions. THF was distilled over sodium wire and benzophenone. CH2Cl2 was distilled over calcium hydride. Other anhydrous solvents were purchased from Sigma-Aldrich.
NMR spectra were recorded at 300 K, unless otherwise stated, using Bruker Avance DRX200, DRX300, DPX400 or DPX500 spectrometers at a frequency of 200.1, 300.2, 400.2 or 500.2 MHz respectively. 1H-NMR chemical shifts are reported in parts per million (ppm) and are referenced to solvent residual signals: CDCl3 (δ = 7.26 ppm), C6D6 (δ = 7.16 ppm) and d6-DMSO (δ = 2.50 ppm). 1H-NMR data is reported as chemical shift (δH), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet), coupling constant (J in Hz), relative integral and assignment where possible with the help of COSY and HMBC experiments where necessary. 13C-NMR chemical shifts are reported in parts per million and were referenced to the solvent residual signals: CDCl3 (δ = 77.16 ppm) and d6-DMSO (δ = 39.52 ppm).
Low resolution mass spectra (LRMS) were recorded on a Finnigan LCQ Deca ion trap mass spectrometer. High resolution mass spectra (HRMS) were recorded on a Bruker 7T Fourier Transform Ion Cyclotron Resonance (FTICR) mass spectrometer. Melting points were recorded using a Stanford Research Systems Optimelt Automated Melting Point System. Infrared (IR) absorption spectra were recorded on a Bruker Alpha Spectrometer with attenuated total reflection (ATR) capability and were processed with Opus 6.5 software. Optical rotations were measured at 23 °C at 589 nm (Na D line) using a Perkin Elmer Model 341 polarimeter or an Optical Activity PoLAAR 2001 polarimeter, concentrations (c) in g/100 mL.
Thin layer chromatography was carried out on Merck Kieselgel 60 F254 pre-coated aluminium sheets. Compounds were visualized by UV light at 254 nm or by staining with vanillin, or phosphomolybdic acid. Flash chromatography was carried out using 230–400 mesh Kieselgel 60 silica get with solvents as described. Ratios of solvent systems used for flash chromatography are expressed in v/v as specified. Preparative reverse phase HPLC was performed using a Waters 2535 Multisolvent Delivery System with a Waters 2489 UV/visible detector operating at 245 and 254 nm. A Waters SunFire™ series C18 column (150 × 19 mm, 5 μm particle size) was used with a flow rate of 7 mL min−1. Compounds were loaded in DMSO and eluted with 0.1% TFA in water (solvent A) and 0.1% TFA in MeCN (solvent B) using a linear gradient of 0%–100% B over 40 min. LC-MS was performed on a Shimadzu LCMS 2020 with LC-20AD Pump and a SPD-20A detector on a Waters SunFire™ series C18 column (150 × 2.1 mm, 5 μm particle size), at a flow rate of 0.2 mL min−1. All separations involved a mobile phase of 0.1% formic acid in water (solvent A) and 0.1% formic acid in MeCN (solvent B) using a linear gradient of 0%–100% B over 10 or 30 min.
3.2. (3R)-Hydroxy Lauric Acid (5)
3.2.1. (4R)-Tridec-1-en-4-ol (16)
To a solution of (−)-B(Ipc)2OMe (6.01 g, 19.2 mmol) in Et2O (70 mL) at −78 °C was added allyl magnesium bromide (19.2 mL, 19.2 mmol, 1 M in Et2O) dropwise over 20 min and left to stir at this temperature for 10 min. The reaction mixture was then warmed to room temperature over 2 h, and then cooled to −100 °C. Distilled decanal (17) (2.41 mL, 12.8 mmol) in Et2O (13 mL) was added dropwise by cannula and the reaction mixture was left to stir at −100 °C for 1.25 h and then warmed to room temperature over 1.25 h. The mixture was quenched at 0 °C with 30% v/v aqueous H2O2 (15 mL) and aqueous 3 M NaOH (20 mL) and heated to reflux for 1 h, and then stirred at room temperature for 16 h. The mixture was poured onto water (100 mL) and extracted with Et2O (3 × 100 mL). The combined organic layers were washed with saturated aqueous NaHCO3 (100 mL), dried (Na2SO4), filtered and concentrated in vacuo. The crude product was purified twice by flash chromatography (eluent: 5:95 v/v EtOAc/hexanes) and (eluent: 3:97 v/v→5:95 v/v EtOAc/hexanes) to yield homoallylic alcohol 16 as a colourless oil (2.10 g, 83%).
1H-NMR (CDCl3, 400 MHz) δ (ppm) 5.89–5.77 (m, 1H, OCCH2CHCH2), 5.17–5.13 (m, 1H, OCCH2CHCHH), 5.13–5.10 (m, 1H, CH2CHCHH), 3.68–3.60 (m, 1H, CHOH), 2.34–2.26 (m, 1H, OCHCHH), 2.19–2.09 (m, 1H, OCHCHH), 1.59 (br. s, 1H, OH), 1.50–1.19 (m, 16H, alkyl chain), 0.88 (t, J = 6.74 Hz, 3H, CH3); [α]D = +2.9 (c 0.99, CH2Cl2); These data are in agreement with those previously reported by Zhu et al. [13].
3.2.2. (3R)-3-Hydroxydodecanoic Acid (5)
To a solution of homoallylic alcohol 16 (2.10 g, 10.6 mmol) in acetone/water (8:1 v/v, 100 mL) was added OsO4 (0.13 mL, 11.0 μmol, 2.5 wt% in t-BuOH) and N-methylmorpholine-N-oxide (2.48 g, 21.2 mmol). The mixture was left to stir at room temperature for 16 h after which time OsO4 (0.1 mL, 8.00 μmol, 2.5 wt% in t-BuOH) was added and it was left to stir for another 16 h. The reaction mixture was poured onto water (150 mL) and extracted with EtOAc (3 × 100 mL). The combined organic layers were washed with water (70 mL) and saturated aqueous NaCl (70 mL), dried (MgSO4), filtered and concentrated in vacuo. The crude solid was used without further purification. To a solution of the crude product in THF/water (9:1.25 v/v, 103 mL) was added sodium periodate (3.40 g, 15.9 mmol). The mixutre was left to stir at room temperature for 2 h. The reaction mixture was then poured onto water (150 mL) and extracted with EtOAc (3 × 100 mL). The combined organic layers were washed with saturated aqueous NaHCO3 (70 mL), dried (MgSO4), filtered and concentrated in vacuo. The crude oil was used without further purification. To a solution of the crude product in t-BuOH/water (1:1 v/v, 100 mL) was added sodium chlorite (4.80 g, 53 mmol) and sodium dihydrogen phosphate (6.35 g, 53 mmol). The mixture was left to stir at room temperature for 16 h. The reaction mixture was then poured onto water (150 mL) which was acidified to pH = 1 with aqueous 1 M HCl. This was then extracted with EtOAc (3 × 100 mL). The combined organic layers were washed with saturated aqueous NaCl (70 mL), dried (MgSO4), filtered and concentrated in vacuo. The crude product with purified by flash chromatography (eluent: 1:99:0.1 v/v/v→3.5:96.5:0.1 v/v/v MeOH/CH2Cl2/AcOH) to yield acid 5 as a white solid (1.77 g, 77%).
The purified acid 5 (1.70 g, 7.89 mmol) was dissolved in MeCN (56 mL) and dicyclohexylamine (1.72 mL, 8.68 mmol) was added. The mixture was then recrystallized from MeCN three times. The salt was then dissolved in water (40 mL) and aqueous 1 M HCl was added until the solution reached pH = 1. This solution was extracted with Et2O (5 × 40 mL). The combined organic layers were dried (Na2SO4), filtered and concentrated in vacuo to yield acid 5 as a white solid (1.34 g, 75%).
1H-NMR (CDCl3, 400 MHz) δ (ppm) 4.08–3.99 (m, 1H, HOCH), 2.58 (dd, J = 3.1, 16.5 Hz, 1H, CHHCOOH), 2.48 (dd, J = 8.9, 17.0 Hz, 1H, CHHCOOH), 1.61–1.18 (m, 16H, alkyl chain), 0.88 (t, J = 6.5 Hz, 3H, CH3); [α]D = −21.0 (c 0.11, CH2Cl2); LRMS (−ESI) m/z 215 [M − H]–; mp. 62.3 °C. These data are in agreement with those previously reported by Guaragna et al. [11].
3.3. Mosher’s Ester Determination of Enantiomeric Excess
3.3.1. Methyl (3R)-3-Hydroxydodecanoate (Methyl Ester-5)
To a solution of acid 5 (50 mg, 0.23 mmol) in MeOH (1 mL) at 0 °C was added thionyl chloride (84 μL, 1.16 mmol) dropwise. The solution was stirred at room temperature for 16 h. The reaction mixture was concentrated in vacuo and the crude product was purified by flash chromatography (eluent: 20:80 v/v EtOAc/petroleum ether) to yield the methyl ester of 5 as a white solid (47 mg, 89%).
1H-NMR (CDCl3, 300 MHz) δ (ppm) 4.05–3.95 (m, 1H, CHOH), 3.71 (s, 3H, OCH3), 2.52 (dd, J = 3.3, 16.3, 1H, CHHCOOMe), 2.40 (dd, J = 8.9, 16.6 Hz, 1H, CHHCOOMe), 1.58–1.20 (m, 16H, alkyl chain), 0.87 (t, J = 6.4 Hz, 3H, CH3); [α]D = −9.6 (c 0.37 , CH2Cl2); HRMS (+ESI) Calc. for C13H26O3Na [M + Na]+: 253.1774, found: 253.1775; mp. 28.5 °C. These data are in agreement with those previously reported by Hasdemir and co-workers [19].
3.3.2. Tridec-1-en-4-ol (rac-16)
To a solution of distilled decanal (17) (1.20 mL, 6.40 mmol) in Et2O (11.5 mL) at −42 °C was added allyl magnesium bromide (7.68 mL, 7.68 mmol, 1 M in Et2O) dropwise. The mixture was left to stir at this temperature before being warmed to room temperature for 1 h. The reaction mixture was quenched at 0 °C with saturated aqueous NH4Cl (5 mL), poured onto saturated aqueous NH4Cl (40 mL) and extracted with Et2O (3 × 40 mL). The combined organic layers were washed with saturated aqueous NaHCO3 (30 mL) and water (30 mL), dried (Na2SO4), filtered and concentrated in vacuo to yield homoallylic alcohol rac-16 as a colourless oil (1.26 g, 99%).
1H-NMR (CDCl3, 300 MHz) δ (ppm) 5.92–5.75 (m, 1H, OCCH2CHCH2), 5.19–5.13 (m, 1H, OCCH2CHCHH), 5.13–5.09 (m, 1H, OCCH2CHCHH), 3.70–3.59 (m, 1H, CHOH), 2.37–2.25 (m, 1H, OCHCHH), 2.20–2.08 (m, 1H, HOCHCHH), 1.52–1.20 (m, 16H, alkyl chain), 0.88 (t, J = 6.36 Hz, 3H, CH3); LRMS (+APCI) m/z 181 [M − OH]+. These data are in agreement with those previously reported by Dommisse et al. [20].
3.3.3. 3-Hydroxydodecanoic Acid (rac-5)
To a solution of homoallylic alcohol rac-16 (217 mg, 1.10 mmol) in acetone/water (8:1 v/v, 15 mL) was added OsO4 (0.69 mL, 0.055 mmol, 2.5 wt% in t-BuOH) and N-methylmorpholine-N-oxide (257 mg, 2.19 mmol). The mixture was left to stir at room temperature for 16 h. The reaction mixture was poured onto water (15 mL) and extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with water (20 mL) and saturated aqueous NaCl (20 mL), dried (MgSO4), filtered and concentrated in vacuo. The crude solid was used without further purification. To a solution of the crude product in THF/water (9:1.25 v/v, 10.25 mL) was added sodium periodate (351 mg, 1.64 mmol). The mixutre was stirred at room temperature for 16 h. The reaction mixture was then poured onto water (15 mL) and extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with saturated aqueous NaCl (20 mL), dried (MgSO4), filtered and concentrated in vacuo. The crude oil was used without further purification. To a solution of the crude product in t-BuOH/water (1:1 v/v, 12 mL) was added sodium chlorite (992 mg, 11.0 mmol) and sodium dihydrogen phosphate (1.31 g, 11.0 mmol). The mixture was stirred at room temperature for 16 h. The reaction mixture was then poured onto water (15 mL) which was acidified to pH = 2 with aqueous 1 M HCl. This was then extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with saturated aqueous NaCl (1 × 20 mL), dried (MgSO4), filtered and concentrated in vacuo. The crude product with purified by flash chromatography (eluent: 3:97:0.1 v/v/v→5:95:0.1 v/v/v MeOH/CH2Cl2/AcOH) to yield acid rac-5 as a white solid (142 mg, 60%).
1H-NMR (CDCl3, 400 MHz) δ (ppm) 4.07–3.99 (m, 1H, HOCH), 2.57 (dd, J = 3.1, 16.6 Hz, 1H, CHHCOOH), 2.47 (dd, J = 9.0, 16.4 Hz, 1H, CHHCOOH), 1.61–1.19 (m, 16H, alkyl chain), 0.88 (t, J = 6.7 Hz, 3H, CH3); LRMS (−ESI) m/z 215 [M − H]−; mp. 69.0 °C. These data are in agreement with those previously reported by Skogh and Guaragna et al. [11,21].
3.3.4. Methyl 3-Hydroxydodecanoate (rac-Methyl Ester 5)
To a solution of acid rac-5 (50 mg, 0.231 mmol) in MeOH (1 mL) at 0 °C was added thionyl chloride (84 μL, 1.16 mmol) dropwise. The solution was stirred at room temperature for 16 h. The reaction mixture was concentrated in vacuo and the crude product was purified by flash chromatography (eluent: 20:80 v/v EtOAc/petroleum ether) to yield the methyl ester of rac-5 as a colourless oil (48 mg, 90%).
1H-NMR (CDCl3, 300 MHz) δ (ppm) 4.05–3.94 (m, 1H, CHOH), 3.71 (s, 3H, OCH3), 2.52 (dd, J = 3.3, 16.5, 1H, CHHCOOMe), 2.40 (dd, J = 8.9, 16.7 Hz, 1H, CHHCOOMe), 1.59–1.2 (m, 16H, alkyl chain), 0.87 (t, J = 6.4 Hz, 3H, CH3); HRMS (+ESI) Calc. for C13H26O3Na [M + Na]+: 253.1774, found: 253.1774. These data are in agreement with those previously reported by Hasdemir and co-workers [19].
3.3.5. General Procedure for Mosher’s Esterification
To a solution of alcohol methyl ester-5, rac-methyl ester-5, 16 or rac-16, (1 equiv., ~5–10 mg) in CH2Cl2 (0.025 M) was added MTPA (2 equiv.), DIC (3 equiv.) and DMAP (1.5 equiv.). The solution was stirred at room temperature for 16 h and analysed by TLC. If starting material was present then additional α-methoxy-α-trifluoromethylphenylacetic acid (MTPA) (1 equiv.), DIC (1 equiv.) and DMAP (1 equiv.) were added and the reaction was left to stir at room temperature until complete consumption of starting material, and more reagents were added if necessary. The reaction mixture was then poured onto water (5 mL) and extracted with CH2Cl2 (3 × 4 mL). The combined organic layers were washed with aqueous citric acid (5 mL, 10% wt/v) and saturated aqueous NaHCO3 (5 mL), dried (MgSO4), filtered and concentrated in vacuo. The crude product was then analysed by 1H-NMR and 19F-NMR to determine the ee (See Supplementary Information) [14].
3.4. Solid Phase Peptide Synthesis (0.1 mmol)
Loading: Weinreb amide resin (139 mg, 0.1 mmol) was swollen in DMF (3 mL) for 5 min. The resin was then shaken in a solution of piperdine/DMF (3 mL, 1:9 v/v) for 3 min and this procedure repeated. The resin was filtered and washed with DMF (5 × 3 mL), DCM (5 × 3 mL) and DMF (5 × 3 mL). A solution of Fmoc-Leu-OH (177 mg, 0.5 mmol) or Fmoc-Val-OH (167 mg, 0.5 mmol), HATU (190 mg, 0.5 mmol) and Hünig’s base (170 μL, 1 mmol) in DMF (1 mL) was added to the resin and shaken at room temperature for 6 h. The resin was filtered and washed with DMF (5 × 3 mL), CH2Cl2 (5 × 3 mL) and DMF (5 × 3 mL). The procedure was repeated.
Fmoc-deprotection: The resin was then shaken in a solution of piperdine/DMF (3 mL, 1:9 v/v) for 3 min and the procedure repeated. The resin was filtered and washed with DMF (5 × 3 mL), CH2Cl2 (5 × 3 mL) and DMF (5 × 3 mL).
Fmoc-Gln(Trt)-OH, Fmoc-Asn(Trt)-OH and lauric acid (10) coupling: A solution of acid (0.4 mmol), PyBOP (208 mg, 0.4 mmol) and NMM (88 μL, 0.8 mmol) in DMF (1 mL) was added to the resin and shaken at room temperature for 2 h. The resin was filtered and washed with DMF (5 × 3 mL), CH2Cl2 (5 × 3 mL) and DMF (5 × 3 mL).
(3R)-hydroxy lauric acid (5) and (3R)-hydroxy myristic acid (11) coupling: A solution of acid (0.12 mmol), HATU (45 mg, 0.12 mmol) and Hünig’s base (42 μL, 2.4 mmol) in DMF (1 mL) was added to the resin and shaken at room temperature for 16 h. The resin was filtered and washed with DMF (5 × 3 mL), CH2Cl2 (5 × 3 mL) and DMF (5 × 3 mL).
Capping: The resin was treated with a solution of pyridine/Ac2O (3 mL, 9:1 v/v) for 2 min. The resin was filtered and washed with DMF (5 × 3 mL), CH2Cl2 (5 × 3 mL) and DMF (5 × 3 mL).
Side-chain deprotection: The resin was treated with a solution of TFA:TIS:H2O (9:0.5:0.5, v/v/v) (3 mL) and shaken for 1 h. The resin was then washed with CH2Cl2 (20 × 3 mL) and then dried thoroughly under vacuum.
3.5. Resin Cleavage Conditions (0.1 mmol)
The resin bound peptide (0.1 mmol) was swollen in dry THF (7.5 mL) in a round bottom flask for 1 h. To this mixture at 0 °C was added a solution of LiAlH4 (0.5 mL, 1 mmol, 2 M in THF). The mixture was stirred at this temperature for 40 min, before diluting with THF (5 mL) and quenching with saturated aqueous KHSO4 solution (2.25 mL) and saturated aqueous NaKTartrate solution (1.5 mL). The solution was then warmed to room temperature over 40 min. The supernatant was decanted before CH2Cl2 (30 mL) was added to the resultant solid and filtered through Celite. The filtrate was combined with the supernatant liquid, and a white solid precipitated. Water (50 mL) was added and the solution was extracted with further CH2Cl2 (100 mL). The combined organic extracts were then dried (MgSO4), filtered and concentrated in vacuo to yield the desired peptide.
3.6. Synthesis of Peptides 2 and 6–8
3.6.1. Fellutamide B (2)
Weinreb amide resin (115 μmol) was loaded with Fmoc-Leu-OH, and then Fmoc-Gln(Trt)-OH, Fmoc-Asn(Trt)-OH and (3R)-hydroxy lauric acid (5) were coupled sequentially according to the SPPS protocol. Cleavage from the resin as detailed in Section 3.5, yielded fellutamide B (2) as a white solid (35 mg, 55%).
1H-NMR (d6-DMSO, 500 MHz) δ (ppm) 9.35 (s, 1H, H-1), 8.26 (d, J = 7.5 Hz, 1H, H-6), 8.15–8.08 (m, 2H, H-13 + H-19), 7.43 (s, 1H, H-18), 7.21 (s, 1H, H-12), 6.92 (s, 1H, H-18), 6.75 (s, 1H, H-12), 4.64 (d, J = 4.7 Hz, 1H, OH), 4.53–4.44 (m, 1H, H-15), 4.21–4.15 (m, 1H, H-8), 4.07–4.00 (m, 1H, H-2), 3.78 (br s, 1H, H-22), 2.55 (dd, J = 6.6, 15.3, 1H, H-16), 2.45 (dd, J = 7.0, 15.3 Hz, 1H, H-16), 2.27–2.15 (m, 2H, 2 × H-21), 2.15–2.03 (m, 2H, 2 × H-10), 2.01–1.86 (m, 1H, H-9), 1.81–1.70 (m, 1H, H-9), 1.67–1.58 (m, 1H, H-4), 1.56–1.45 (m, 2H, 2 × H-3), 1.42–1.17 (m, 16H, H-23-H-30), 0.93–0.75 (m, 9H, 3 × H-5+3 × H-5′ + 3 × H-31); 13C-NMR (d6-DMSO, 125 MHz) δ (ppm) 201.6, 173.8, 171.9, 171.8, 171.2, 171.0, 67.5, 56.6, 52.4, 49.9, 43.3, 37.0, 36.9, 31.4, 31.3, 29.1, 29.0, 28.7, 25.1, 23.9, 23.0, 22.1, 21.3, 13.9, (3 signals obscured); [α]D = −9.3 (c 0.17, MeOH); LRMS (+ESI) 578 m/z [M + Na]+; HRMS (+ESI) Calc. for C28H53N5O8Na [M + CH3OH + Na]+: 610.3786, found: 610.3785; IR νmax (ATR): 3338 (O–H), 1655 (C=O), 1090; mp. decomposition. These data are in agreement with those previously reported by Shigemori et al. and Schneekloth Jr. et al. [1,2].
3.6.2. Deoxy-fellutamide B (6)
Weinreb amide resin (200 μmol) was loaded with Fmoc-Leu-OH and then Fmoc-Gln(Trt)-OH, Fmoc-Asn(Trt)-OH and lauric acid (10) were coupled sequentially according to the SPPS protocol. Cleavage from the resin as detailed in Section 3.5, yielded the title compound 6 as a white solid (48 mg, 44% crude). A portion (11 mg) was purified by reverse phase HPLC to yield deoxy-fellutamide B (6) as a white solid (6 mg, 55%).
1H-NMR (d6-DMSO, 500 MHz) δ (ppm) 9.35 (s, 1H, H-1), 8.29 (d, J = 7.3 Hz, 1H, H-6), 8.10 (d, J = 7.6, 1H, H-13), 8.03 (d, J = 7.4, 1H, H-19), 7.40 (s, 1H, H-18), 7.20 (s, 1H, H-12), 6.91 (s, 1H, H-18), 6.74 (s, 1H, H-12), 4.49 (app. q, J = 7, 1H, H-15), 4.22–4.16 (m, 1H, H-8), 4.07–4.02 (m, 1H, H-2), 2.54 (dd, J = 6.5, 15.7 Hz, 1H, H-16), 2.41 (dd, J = 6.7, 15.7 Hz, 1H, H-16), 2.15–2.03 (m, 4H, 2 × H-10 + 2 × H-21), 2.00–1.91 (m, 1H, H-9), 1.82–1.71 (m, 1H, H-9), 1.68–1.59 (m, 1H, H-4), 1.53–1.42 (m, 4H, 2 × H-3 + 2 × H-22), 1.32–1.18 (m, 16H, H-23-H-30), 0.90–0.82 (m, 9H, 3 × H-5 + 3 × H5′ + 3 × H-31); 13C-NMR (d6-DMSO, 100 MHz) δ (ppm) 201.6, 173.7, 172.5, 171.8, 171.7, 171.2, 56.6, 52.4, 49.8, 37.1, 36.3, 35.1, 31.3, 29.0, 29.0, 28.9, 28.8, 28.7, 28.7, 27.4, 25.1, 23.9, 23.1, 22.1, 21.3, 13.9, (1 signal obscured); [α]D = −23.7 (c 0.14, MeOH); LRMS (+ESI) 562 m/z [M + Na]+; HRMS (+ESI) Calc. for C27H49N5O6Na [M + Na]+: 562.3575, found: 562.3564; IR νmax (ATR): 2925, 1742 (C=O), 1627 (C=O), 1547, 1365, 1216; mp. decomposition
3.6.3. Deoxy-fellutamide C (7)
Weinreb amide resin (100 μmol) was loaded with Fmoc-Val-OH and then Fmoc-Gln(Trt)-OH, Fmoc-Asn(Trt)-OH and (3R)-hydroxy myristic acid (11) were coupled sequentially according to the SPPS protocol. Cleavage from the resin as detailed in Section 3.5, yielded deoxy-fellutamide C (7) as a white solid (30 mg, 53%).
1H-NMR (d6-DMSO, 500 MHz) δ (ppm) 9.40 (d, J = 1.3 Hz, 1H, H-1), 8.14 (d, J = 7.6, 1H, H-6), 8.09 (d, J = 7.8 Hz, 1H, H-19), 8.07 (d, J = 7.8 Hz, 1H, H-13), 7.40 (s, 1H, H-18), 7.23 (s, 1H, H-12), 6.91 (s, 1H, H-18), 6.76 (s, 1H, H-12), 4.65 (d, J = 4.9 Hz, 1H, OH), 4.55–4.47 (m, 1H, H-15), 4.27–4.22 (m, 1H, H-8), 3.98 (ddd, J = 1.3, 5.7, 7.9 Hz, 1H, H-2), 3.81–3.73 (m, 1H, H-22), 2.55 (dd, J = 6.4, 15.0 Hz, 1H, H-16), 2.44 (dd, J = 7.0, 15.4 Hz, 1H, H-16), 2.24–2.05 (m, 5H, 2 × H-21 + 2 × H-10 + H-3), 2.02–1.90 (m, 1H, H-9), 1.82–1.72 (m, 1H, H-9), 1.39–1.16 (m, 20H, H-23-H-32), 0.90 (d, J = 6.9 Hz, 3H, 3 × H-4), 0.87–0.83 (m, 6H, 3 × H-5 + 33 × H-33) 13C-NMR (d6-DMSO, 125 MHz) δ (ppm) 201.4, 173.9, 171.9, 171.8, 171.2, 171.1, 67.5, 63.1, 52.4, 49.8, 43.4, 37.0, 36.9, 31.4, 31.3, 29.1, 29.1, 29.0, 29.0, 28.7, 25.1, 22.1, 19.0, 18.0, 13.9, (3 signals obscured); [α]D = −13.5 (c 0.12, MeOH); LRMS (+ESI) 592 m/z [M + Na]+; HRMS (+ESI) Calc. for C28H51N5O7Na [M + Na]+: 592.3681, found: 592.3678; IR νmax (ATR): 3332 (O–H), 1654 (C=O), 1083; mp. decomposition.
3.6.4. Deoxy-fellutamide D (8)
Weinreb amide resin (100 μmol) was loaded with Fmoc-Leu-OH and then Fmoc-Gln(Trt)-OH, Fmoc-Asn(Trt)-OH (48) and (3R)-hydroxy myristic acid (11) were coupled sequentially according to the SPPS protocol. Cleavage from the resin as detailed in Section 3.5, yielded deoxy-fellutamide D (8) as a white solid (35 mg, 60%).
1H-NMR (d6-DMSO, 500 MHz) δ (ppm) 9.35 (s, 1H, H-1), 8.26 (d, J = 7.6 Hz, 1H, H-6), 8.10 (app. dd, J = 1.7, 7.3 Hz, 2H, H-13 + H-19), 7.42 (s, 1H, H-18), 7.21 (s, 1H, H-12), 6.92 (s, 1H, H-18), 6.75 (s, 1H, H-12), 4.64 (d, J = 5.1 Hz, 1H, OH), 4.53–4.46 (m, 1H, H-15), 4.22–4.16 (m, 1H, H-8), 4.06–4.01 (m, 1H, H-2), 3.82–3.73 (m, 1H, H-22), 2.56 (dd, J = 6.7, 15.7 Hz, 1H, H-16), 2.45 (dd, J = 7.0, 15.3 Hz, 1H, H-16), 2.26–2.16 (m, 2H, 2 × H-21), 2.14–2.03 (m, 2H, 2 × H-10), 2.01–1.91 (m, 1H, H-9), 1.81–1.69 (m, 1H, H-9), 1.68–1.57 (m, 1H, H-4), 1.51–1.45 (m, 2H, 2 × H-3), 1.39–1.16 (m, 20H, H-23-H-32), 0.91–0.76 (m, 9H, 3 × H-5 + 3 × H-5′+3 × H-33) 13C-NMR (d6-DMSO, 125 MHz) δ (ppm) 201.6, 173.8, 171.9, 171.8, 171.2, 171.1, 67.5, 56.6, 52.4, 49.8, 43.4, 36.9, 36.3, 31.4, 31.3, 29.1, 29.1, 29.0, 28.7, 25.12, 23.9, 23.1, 22.1, 21.3, 13.9, (4 signals obscured); [α]D = −15.0 (c 0.13, MeOH); LRMS (+ESI) 606 m/z [M + Na]+; HRMS (+ESI) Calc. for C29H53N5O7Na [M + Na]+: 606.3837, found: 606.3839; IR νmax (ATR): 3356 (O–H), 1740 (C=O), 1648 (C=O), 1084; mp. decomposition.
3.7. Mycobacterium tuberculosis Inhibition Assays
M. tuberculosis H37Rv (ATCC 27294) was grown in Middlebrook 7H9 broth medium supplemented with ADC (Difco Laboratories, Detroit, MI, USA), 0.5% glycerol, and 0.05% Tween-80. Freshly seeded cultures were grown at 37 °C to mid-exponential phase (OD600 0.4–0.8) for use in resazurin reduction inhibition assay [22,23,24]. M. tuberculosis H37Rv, grown to mid-exponential phase, was diluted to OD600 0.002 in 7H9S media (Middlebrook 7H9 with ADC, 0.05% glycerol, 0.05% Tween-80, 1% tryptone) and ~2 × 104 CFU/mL added to the wells of 96-well microtiter plates with serial dilutions of the inhibitors, which were originally dissolved in 100% DMSO. Plates were incubated for 5 days at 37 °C in a humidified incubator prior to the addition of a 0.02% resazurin solution (30 μL) and 20% Tween-80 (12.5 μL) to each well. Sample fluorescence was measured after 48 h on a BMG Labtech Polarstar Omega instrument with an excitation wave-length of 530 nm and emission at 590 nm. Changes in fluorescence relative to positive control wells (H37Rv with no inhibitor) minus negative control wells (no H37Rv) were plotted for determination of MIC50 values.
As M. tuberculosis proteasome inhibitors were more active in the presence of non-lethal levels of NO [6], M. tuberculosis was cultured with the synthetic fellutamides in the presence of DETA-NO, which releases NO with T1/2 of ~20 h [17]. M. tuberculosis H37Ra (ATCC 25177) was grown to mid-exponential phase (OD600 0.4–0.8) in ADC-supplemented Middlebrook 7H9 medium, and then ~2 × 105 bacilli in 100 μL were incubated with serial dilutions of the inhibitors and 50 μM DETA-NO in 100 μL in 96-well microtiter plates. Plates were incubated for 5 days at 37 °C with the addition of DETA-NO at 24, 48, 72, 96 and 120 h to a final concentration of 50 μM. Absorbance was measured daily on BMG Labtech Polarstar Omega instrument at OD600 prior to the addition of the DETA-NO.
4. Conclusions
The work described here represents a significant step towards the synthesis of the fellutamide class of natural products and related analogues. The total synthesis of fellutamide B (2) and deoxy-fellutamides B (6), C (7) and D (8) was achieved. The synthesis employed a novel Weinreb amide solid-phase peptide synthesis approach whereby the sensitive C-terminal aldehyde could be generated in the final step of the synthesis. An improved enantioselective synthesis of (3R)-hydroxy lauric acid (5) was also described and was successfully incorporated into a resin bound peptide for the total synthesis of fellutamide B (2). Despite the fact that fellutamide B and the natural product analogues did not exhibit activity against M. tuberculosis in vitro, the reported activity of this natural product class against the M. tuberculosis proteasome warrants further investigation. Indeed, it is possible that synthetic analogues which are capable of penetrating the cell wall of M. tuberculosis to reach the proteasome may serve as lead compounds for the development of new chemotherapies against latent M. tuberculosis infections.
Acknowledgments
The authors would like to thank the National Health and Medical Research Council of Australia for funding.
Conflict of Interest
The authors declare no conflict of interest.
References
- Shigemori, H.; Wakuri, S.; Yazawa, K.; Nakamura, T.; Sasaki, T.; Kobayashi, J. Fellutamide-A and fellutamide-B, cytotoxic peptides from a marine fish-possessing fungus Penicillium fellutanum. Tetrahedron 1991, 47, 8529–8534. [Google Scholar] [CrossRef]
- Schneekloth, J.S., Jr.; Sanders, J.L.; Hines, J.; Crews, C.M. Neurotrophic peptide aldehydes: Solid phase synthesis of fellutamide B and a simplified analog. Bioorg. Med. Chem. Lett. 2006, 16, 3855–3858. [Google Scholar] [CrossRef]
- Xu, D.; Ondeyka, J.; Harris, G.H.; Zink, D.; Kahn, J.N.; Wang, H.; Bills, G.; Platas, G.; Wang, W.; Szewczak, A.A.; et al. Isolation, structure, and biological activities of fellutamides C and D from an undescribed Metulocladosporiella (chaetothyriales) using the genome-wide Candida albicans fitness test. J. Nat. Prod. 2011, 74, 1721–1730. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Tsuji, T.; Wakuri, S.; Yazawa, K.; Kondo, K.; Shigemori, H.; Kobayashi, J. Stimulation of nerve growth-factor synthesis and secretion by fellutamide-A in vitro. Biosci. Biotechnol. Biochem. 1993, 57, 195–199. [Google Scholar] [CrossRef]
- Lin, G.; Li, D.; Chidawanyika, T.; Nathan, C.; Li, H. Fellutamide B is a potent inhibitor of the Mycobacterium tuberculosis proteasome. Arch. Biochem. Biophys. 2010, 501, 214–220. [Google Scholar] [CrossRef]
- Lin, G.; Li, D.Y.; de Carvalho, L.P.S.; Deng, H.T.; Tao, H.; Vogt, G.; Wu, K.Y.; Schneider, J.; Chidawanyika, T.; Warren, J.D.; et al. Inhibitors selective for mycobacterial versus human proteasomes. Nature 2009, 461, 621–626. [Google Scholar] [CrossRef]
- Nahm, S.; Weinreb, S.M. N-Methoxy-N-methylamides as effective acylating agents. Tetrahedron Lett. 1981, 22, 3815–3818. [Google Scholar] [CrossRef]
- Fehrentz, J.A.; Paris, M.; Heitz, A.; Velek, J.; Liu, C.F.; Winternitz, F.; Martinez, J. Improved solid-phase synthesis of C-terminal peptide aldehydes. Tetrahedron Lett. 1995, 36, 7871–7874. [Google Scholar] [CrossRef]
- Dinh, T.Q.; Armstrong, R.W. Synthesis of ketones and aldehydes via reactions of Weinreb-type amides on solid support. Tetrahedron Lett. 1996, 37, 1161–1164. [Google Scholar] [CrossRef]
- Bauer, J.; Brandenburg, K.; Zahringer, U.; Rademann, J. Chemical synthesis of a glycolipid library by a solid-phase strategy allows elucidation of the structural specificity of immunostimulation by rhamnolipids. Chem. Eur. J. 2006, 12, 7116–7124. [Google Scholar] [CrossRef]
- Guaragna, A.; Nisco, M.D.; Pedatella, S.; Palumbo, G. Studies towards lipid A: A synthetic strategy for the enantioselective preparation of 3-hydroxy fatty acids. Tetrahedron Asymmetry 2006, 17, 2839–2841. [Google Scholar] [CrossRef]
- Brown, H.C.; Jadhav, P.K. Asymmetric carbon-carbon bond formation via β-allyldiisopinocampheylborane. Simple synthesis of secondary homoallylic alcohols with excellent enantiomeric purities. J. Am. Chem. Soc. 1983, 105, 2092–2093. [Google Scholar] [CrossRef]
- Zhu, S.F.; Qiao, X.C.; Zhang, Y.Z.; Wang, L.X.; Zhou, Q.L. Highly enantioselective palladium-catalyzed umpolung allylation of aldehydes. Chem. Sci. 2011, 2, 2428–2428. [Google Scholar] [CrossRef]
- Dale, J.A.; Dull, D.L.; Mosher, H.S. α-Methoxy-α-trifluoromethylphenylacetic acid, a versatile reagent for the determination of enantiomeric composition of alcohols and amines. J. Org. Chem. 1969, 34, 2543–2549. [Google Scholar] [CrossRef]
- Nakahata, M.; Imaida, M.; Ozaki, H.; Harada, T.; Tai, A. The preparation of optically pure 3-hydroxyalkanoic acid—The enantioface-differentiating hydrogenation of the C=O double-bond with modified raney-nickel. Bull. Chem. Soc. Jpn. 1982, 55, 2186–2189. [Google Scholar] [CrossRef]
- Darwin, K.H.; Ehrt, S.; Gutierrez-Ramos, J.C.; Weich, N.; Nathan, C.F. The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science 2003, 302, 1963–1966. [Google Scholar] [CrossRef]
- Bryk, R.; Gold, B.; Venugopal, A.; Singh, J.; Samy, R.; Pupek, K.; Cao, H.; Popescu, C.; Gurney, M.; Hotha, S.; et al. Selective killing of nonreplicating mycobacteria. Cell Host Microbe 2008, 3, 137–145. [Google Scholar] [CrossRef]
- Armarego, W.L.F.; Chai, C.L.L. Purification of Laboratory Chemicals, 5th ed; Elsevier: Waltham, MA, USA, 2003. [Google Scholar]
- Hasdemir, B.; Onar, H.C.; Yusufoglu, A. Asymmetric synthesis of long chain β-hydroxy fatty acid methyl esters as new elastase inhibitors. Tetrahedron Asymmetry 2012, 23, 1100–1105. [Google Scholar] [CrossRef]
- Dommisse, A.; Wirtz, J.; Koch, K.; Barthlott, W.; Kolter, T. Synthesis of (S)-nonacosan-10-ol, the major component of tubular plant wax crystals. Eur. J. Org. Chem. 2007, 21, 3508–3511. [Google Scholar]
- Skogh, M. The higher normal chain dl-β-hydroxy acids—Synthesis and investigation of the crystal behaviour of 17 homologous acids with 8 to 24 carbon atoms. Acta Chem. Scand. 1952, 6, 809–817. [Google Scholar] [CrossRef]
- Taneja, N.K.; Tyagi, J.S. Resazurin reduction assays for screening of anti-tubercular compounds against dormant and actively growing Mycobacterium tuberculosis, Mycobacterium bovis BCG and Mycobacterium smegmatis. J. Antimicrob. Chemother. 2007, 60, 288–293. [Google Scholar] [CrossRef]
- Manos-Turvey, A.; Cergol, K.M.; Salam, N.K.; Bulloch, E.M.M.; Chi, G.; Pang, A.; Britton, W.J.; West, N.P.; Baker, E.N.; Lott, J.S.; et al. Synthesis and evaluation of M. tuberculosis salicylate synthase (MbtI) inhibitors designed to probe plasticity in the active site. Org. Biomol. Chem. 2012, 10, 9223–9236. [Google Scholar] [CrossRef]
- West, N.P.; Cergol, K.M.; Xue, M.; Randall, E.J.; Britton, W.J.; Payne, R.J. Inhibitors of an essential mycobacterial cell wall lipase (Rv3802c) as tuberculosis drug leads. Chem. Commun. 2011, 47, 5166–5168. [Google Scholar] [CrossRef]
Supplementary Files
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).