Marine-Sourced Anti-Cancer and Cancer Pain Control Agents in Clinical and Late Preclinical Development †

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Treatment of Pain Associated with Cancer
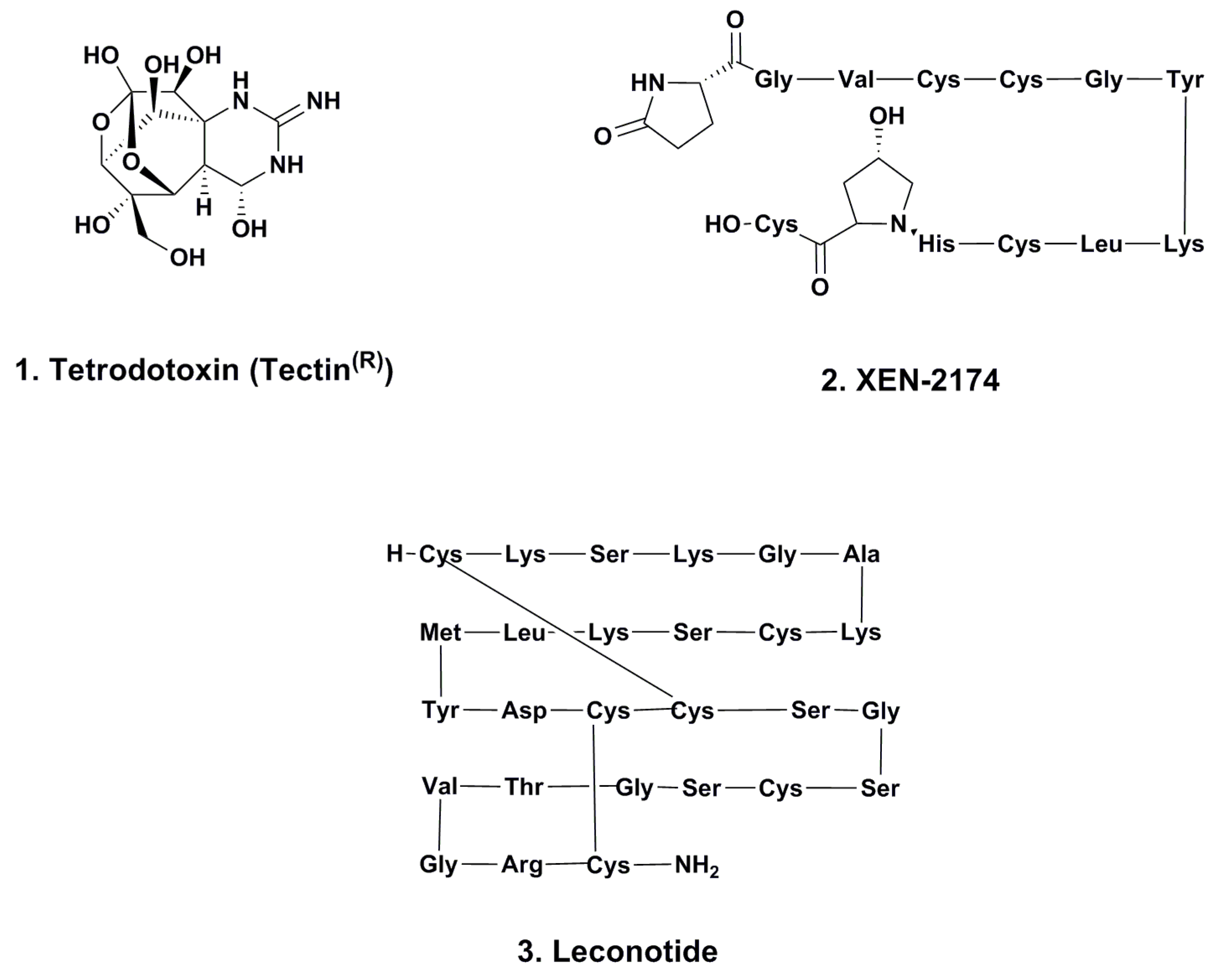
2.1. Tetrodotoxin (Tectin ®, Phase III; Figure 1, 1)
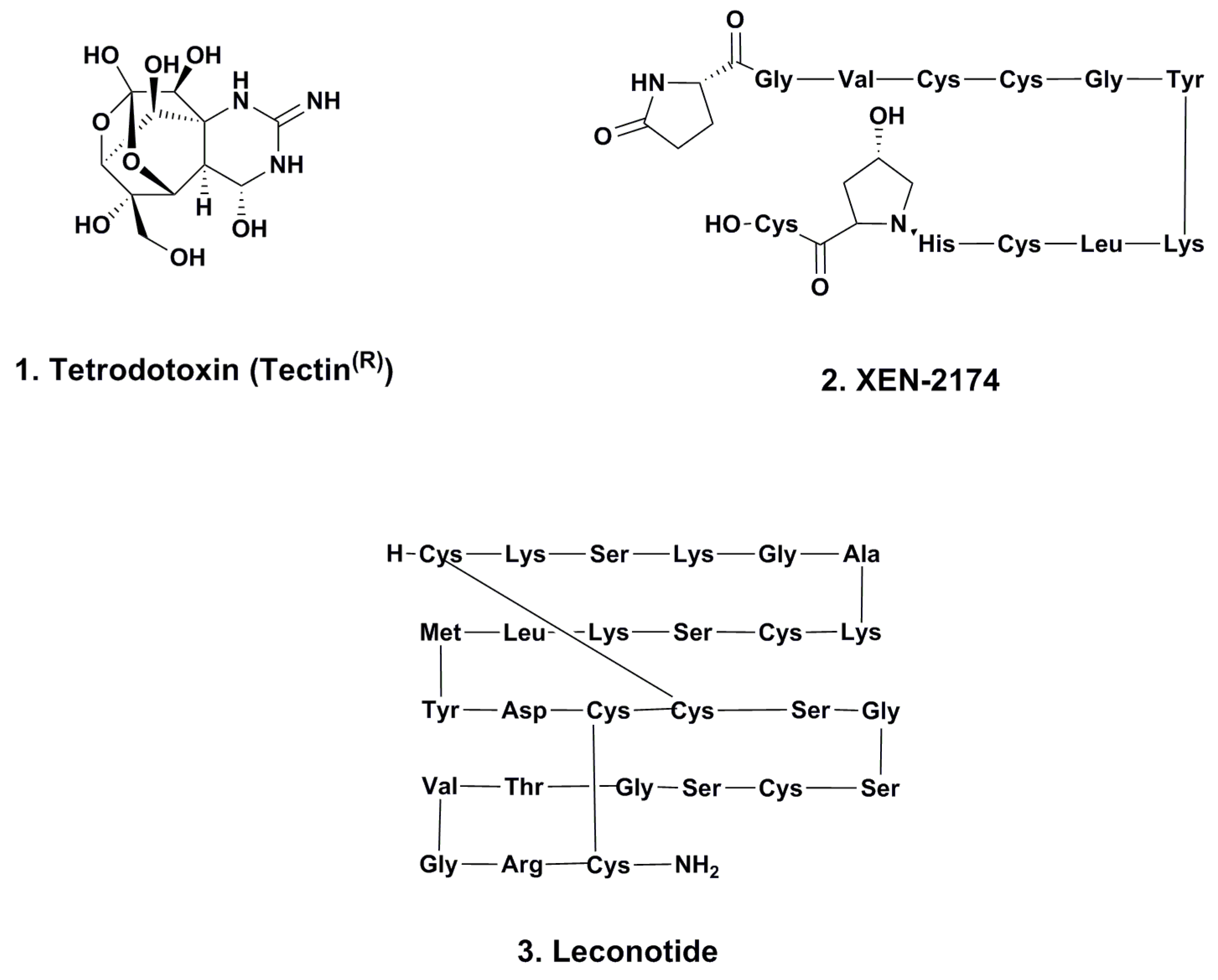
2.2. XEN-2174 (Phase II; Figure 1, 2)
2.3. Leconotide (AM-336, ω-Conotoxin CVID; Phase I; Figure 1, 3)
2.4. Immunological Use of Keyhole Limpet Hemocyanin (KLH; Phase I–III)
3. Approved Marine-Derived Antitumor Agents Still in Clinical Trials (and Close Chemical Relatives)
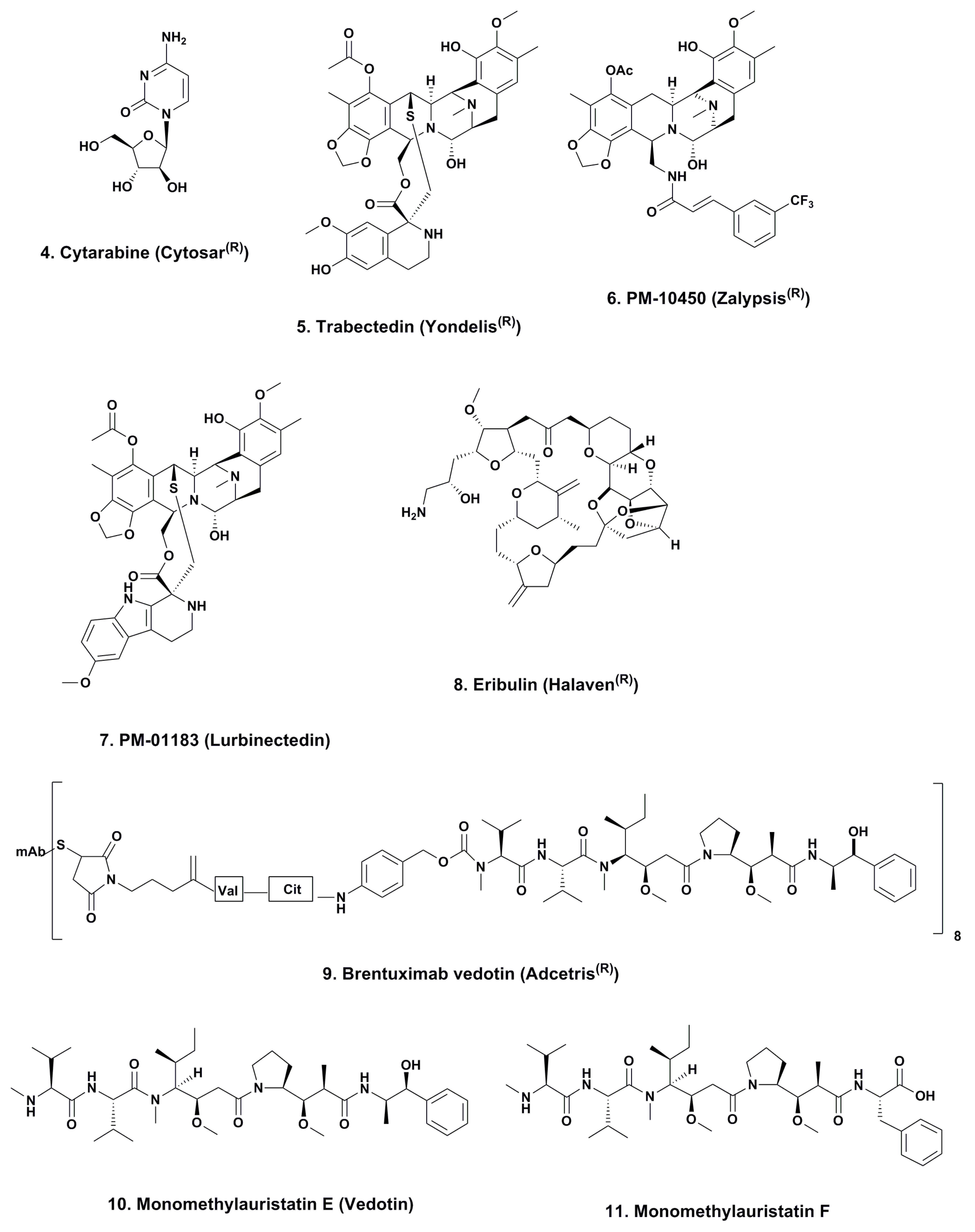
3.1. Cytarabine (Phases I to IV; Figure 2, 4)
3.2. ET743 (Trabectedin; Yondelis®; Phases I to III; Figure 2, 5)
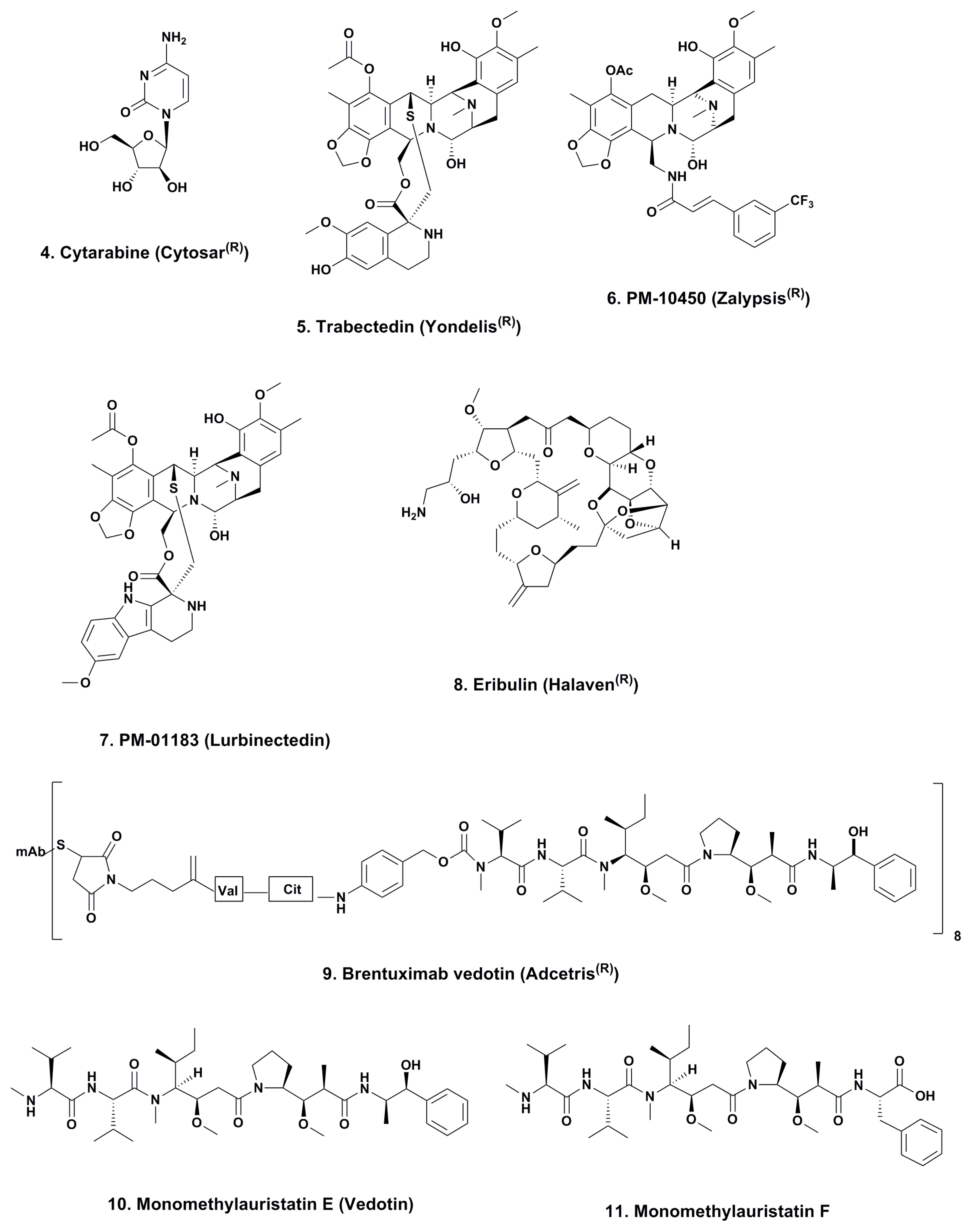
3.2.1. PM-10450 (Zalypsis®; Phases I–II; Figure 2, 6)
3.2.2. Lurbinectedin (PM-01183; Phases I–II; Figure 2, 7)
3.3. Eribulin (Halaven®; Phases I–IV; Figure 2, 8)
3.4. Brentuximab Vedotin (Adcetris®; Phases 0 to IV; Figure 2, 9)
3.4.1. Glembatumumab Vedotin (Phase II)
3.4.2. ABT-414 (Phase I–II)
3.4.3. PSMA-ADC (Phase II)
3.4.4. DCDT-2980S (Phase II)
3.4.5. DCDS-4501A (Phase II)
3.4.6. Enfortumab vedotin (Phase I)
3.4.7. Vorsetuzumab Mafdotin (SGN-75; Phase I)
3.4.8. SGN-19A (SGN-CD19A; Phase I)
3.4.9. BAY 79-4620 (3ee9/MMAE; Phase I)
3.4.10. AGS-16C3F (AGS-16M8F; Phase I)
3.4.11. DMUC-5754A (RG-7458; Phase I)
3.4.12. DNIB-0600A (RG-7599; Phase I)
3.4.13. A1-mcMMAF (PF-06263507; Phase I)
3.4.14. DMOT-4039A (Phase I)
3.4.15. RG-7600 (Phase I)
3.4.16. DEDN-6526A (RG-7636; Phase I)
3.4.17. DSTP-3086S (RG-7450; thio-antiSTEAP1-MC-vc-PAB-MMAE; Phase I)
3.4.18. MLN-0264 (Phase I)
3.4.19. RG-7598 (Phase I)
3.4.20. SGN-LIV1A (Phase I)
3.4.21. AGS-15E (AGS-15ME; Phase I)
3.5. Preclinical Auristatin-Linked ADCs
3.5.1. CDX-014 (CR-014-vcMMAE)
3.5.2. HuMax-CD74
3.5.3. HuMab-TF-011-vcMMAE (HuMax-TF-ADC; TF-011-MMAE IND Filed)
4. Other Marine-Derived Compounds in Clinical Trials against Cancer (Phases I–III)
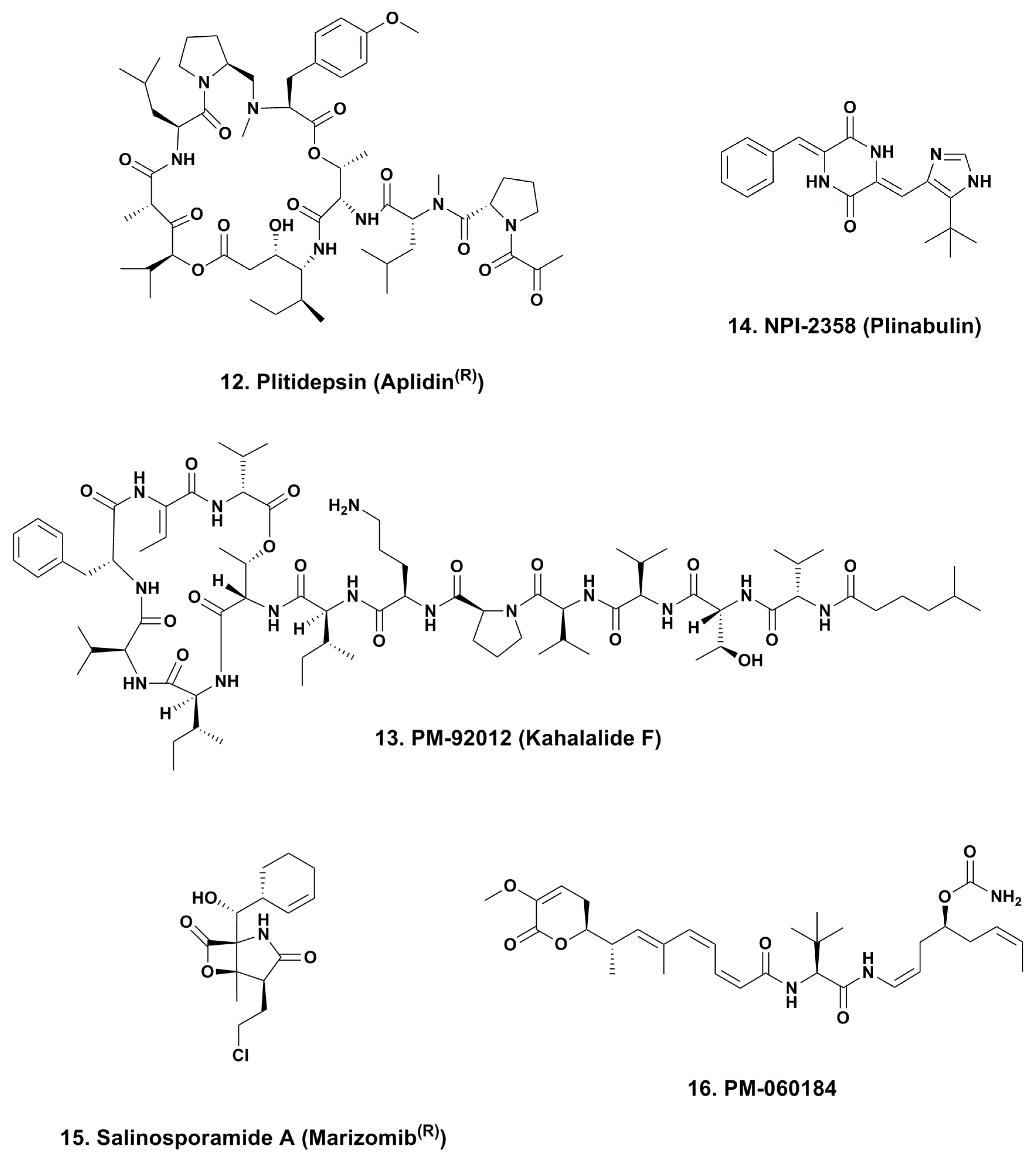
4.1. Aplidine (Ptilidepsin, Aplidin®; Phase II–III; Figure 3, 12)
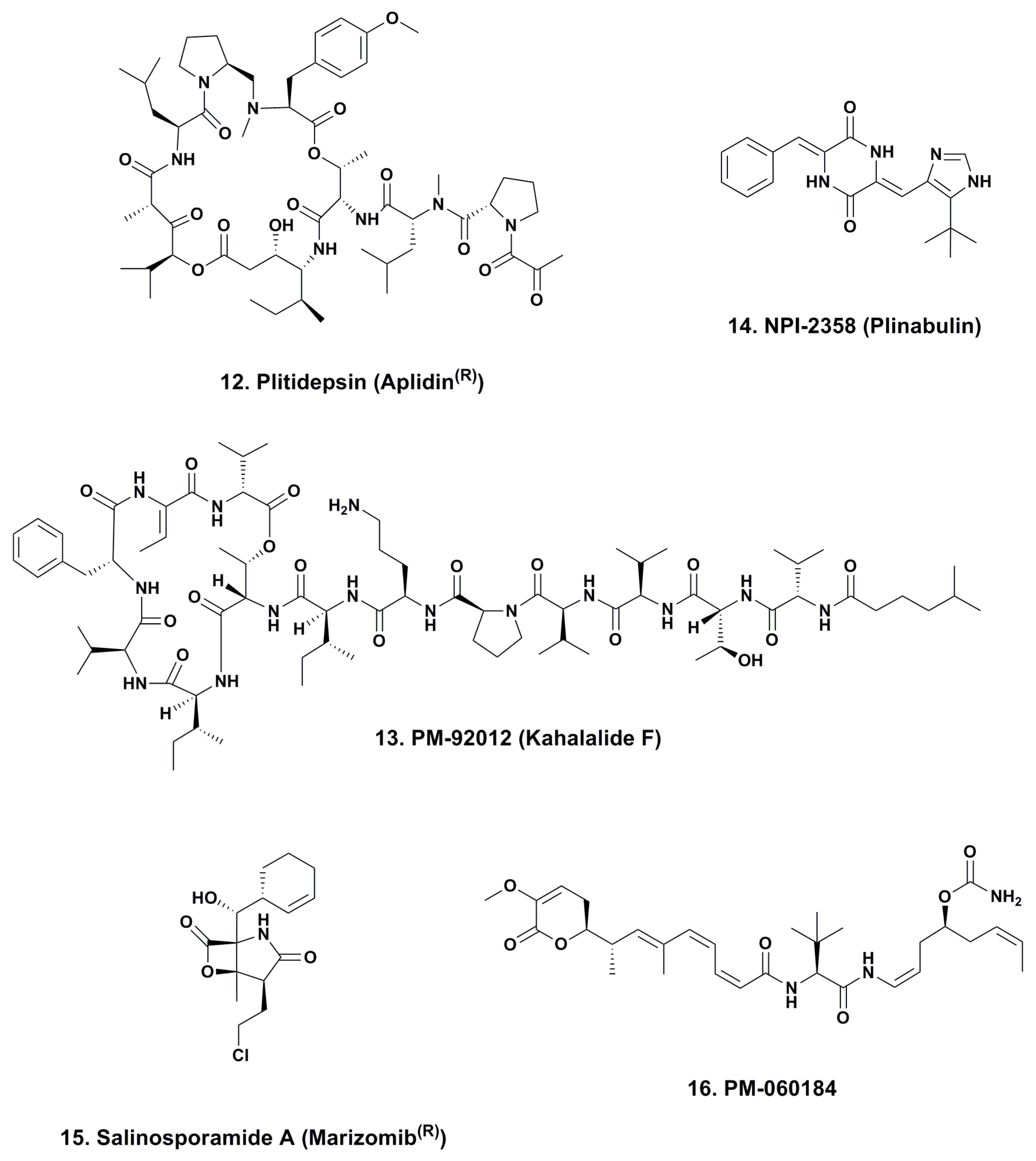
4.2. Kahalalide F (PM-92102, Phase II; Figure 3, 13)
4.3. Plinabulin (NPI-2358, Phase II; Figure 3, 14)
4.4. Marizomib® (Salinosporamide A; NPI-0052; Phase I, Figure 3, 15)
4.5. PM-060184 (Phase I; Figure 3, 16)
5. Conclusions
Conflicts of Interest
References
- Turabi, A.; Plunkett, A.R. The application of genomic and molecular data in the treatment of chronic cancer pain. J. Surg. Oncol. 2012, 105, 494–501. [Google Scholar] [CrossRef]
- Nieto, F.R.; Cobos, E.J.; Tejada, M.Á.; Sánchez-Fernández, C.; González-Cano, R.; Cendán, C.M. Tetrodotoxin (TTX) as a therapeutic agent for pain. Mar. Drugs 2012, 10, 281–305. [Google Scholar] [CrossRef]
- Moczydlowski, E.G. The molecular mystique of tetrodotoxin. Toxicon 2013, 63, 165–183. [Google Scholar] [CrossRef]
- Chau, R.; Kalaitzis, J.A.; Neilan, B.A. On the origins and biosynthesis of tetrodotoxin. Aquat. Toxicol. 2011, 104, 61–72. [Google Scholar] [CrossRef]
- Nishikawa, T.; Isobe, M. Synthesis of tetrodotoxin, a classic but still fascinating natural product. Chem. Rec. 2013, 13, 286–302. [Google Scholar] [CrossRef]
- Brust, A.; Palant, E.; Croker, D.E.; Colless, B.; Drinkwater, R.; Patterson, B.; Schroeder, C.I.; Wilson, D.; Nielsen, C.K.; Smith, M.T.; et al. χ-Conopeptide pharmacophore development: Toward a novel class of norepinephrine transporter inhibitor (Xen2174) for pain. J. Med. Chem. 2009, 52, 6991–7002. [Google Scholar] [CrossRef]
- Sharpe, I.A.; Palant, E.; Schroeder, C.I.; Kaye, D.M.; Adams, D.J.; Alewood, P.F.; Lewis, R.J. Inhibition of the norepinephrine transporter by the venom peptide chi-MrIA. Site of Action, Na+ dependence, and structure-activity relationship. J. Biol. Chem. 2003, 278, 40317–40322. [Google Scholar]
- Jayamanne, A.; Jeong, H.J.; Schroeder, C.J.; Lewis, R.J.; Christie, M.J.; Vaughan, C.W. Spinal actions of omega-conotoxins, CVID, MVIIA and related peptides in a rat neuropathic pain model. Br. J. Pharmacol. 2013, 170, 245–254. [Google Scholar] [CrossRef]
- Daly, N.L.; Craik, D.J. Conopeptides as novel options for pain management. Drugs Future 2011, 36, 25–32. [Google Scholar]
- Jurincic, C.D.; Engelmann, U.; Gasch, J.; Klippel, K.F. Immunotherapy in bladder cancer with keyhole limpet hemocyanin: A randomized study. J. Urol. 1988, 139, 723–726. [Google Scholar]
- Miles, D.; Roche, H.; Martin, M.; Perren, T.J.; Cameron, D.A.; Glaspy, J.; Dodwell, D.; Parker, J.; Mayordomo, J.; Tres, A.; et al. Phase III multicenter clinical trial of the Sialyl-TN (STn)-Keyhole limpet hemocyanin (KLH) vaccine for metastatic breast cancer. Oncologist 2011, 16, 1092–1100. [Google Scholar] [CrossRef]
- Lammers, R.J.; Witjes, W.P.; Janzing-Pastors, M.H.; Caris, C.T.; Witjes, J.A. Intracutaneous and intravesical immunotherapy with keyhole limpet hemocyanin compared with intravesical mitomycin in patients with non-muscle-invasive bladder cancer: Results from a prospective randomized phase III trial. J. Clin. Oncol. 2012, 30, 2273–2279. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Available online: http://clinicaltrials.gov/ (accessed on 15 October 2013).
- Newman, D.J.; Cragg, G.M.; Snader, K.M. The influence of natural products upon drug discovery. Nat. Prod. Rep. 2000, 17, 215–234. [Google Scholar] [CrossRef]
- Suckling, C.J. Chemical approaches to the discovery of new drugs. Sci. Prog. 1991, 75, 323–359. [Google Scholar]
- Bergmann, W.; Feeney, R.J. Isolation of a new thymine pentoside from sponges. J. Am. Chem. Soc. 1950, 72, 2809–2810. [Google Scholar]
- Bergmann, W.; Feeney, R.J. Marine products. XXXII. The nucleosides of sponges. I. J. Org. Chem. 1951, 16, 981–987. [Google Scholar]
- Bergmann, W.; Burke, D.C. Marine products. XXXIX. The nucleosides of sponges. III. Spongothymidine and spongouridine. J. Org. Chem. 1955, 20, 1501–1507. [Google Scholar] [CrossRef]
- Löwenberg, B. Sense and nonsense of high-dose cytarabine for acute myeloid leukemia. Blood 2013, 121, 26–28. [Google Scholar] [CrossRef]
- Sigel, M.M.; Wellham, L.L.; Lichter, W.; Dudeck, L.E.; Gargus, J.L.; Lucas, L.H. Food-Drugs from the Sea: Proceedings 1969; Marine Technology Society: Washington, DC, USA, 1970.
- Holt, T.G. The Isolation and Structural Characterization of the Ecteinascidins. Ph.D Thesis, Department of Chemistry, University of Illinois at Urbana-Champaign, Champaign, IL, USA, 1986. [Google Scholar]
- Rinehart, K.; Holt, T.G.; Fregeau, N.L.; Stroh, J.G.; Kiefer, P.A.; Sun, F.; Li, L.H.; Martin, D.G. Ecteinascidins 729, 743, 745, 759A, 759B and 770: Potent antitumor agents from the Caribbean tunicate Ecteinascidia turbinata. J. Org. Chem. 1990, 55, 4512–4515. [Google Scholar] [CrossRef]
- Wright, A.E.; Forleo, D.A.; Gunawardana, G.P.; Gunasekera, S.P.; Koehn, F.E.; McConnell, O.J. Antitumor tetrahydroisoquinoline alkaloids from the colonial ascidian Ecteinascidia turbinata. J. Org. Chem. 1990, 55, 4508–4512. [Google Scholar]
- Cuevas, C.; Francesch, A.; Reports, N.P. Development of Yondelis® (trabectedin, ET-743). A semisynthetic process solves the supply problem. Nat. Prod. Rep. 2009, 26, 322–337. [Google Scholar] [CrossRef]
- Martínez, S.; Pérez, L.; Galmarini, C.M.; Aracil, M.; Tercero, J.C.; Gago, F.; Albella, B.; Bueren, J.A. Inhibitory effects of marine-derived DNA-binding anti-tumour tetrahydroisoquinolines on the Fanconi anaemia pathway. Br. J. Pharmacol. 2013, 170, 871–882. [Google Scholar] [CrossRef]
- Romano, M.; Frapolli, R.; Zangarini, M.; Bello, E.; Porcu, L.; Galmarini, C.M.; Garcia-Fernandez, L.F.; Cuevas, C.; Allavena, P.; Erba, E.; D’Incalci, M. Comparison of in vitro and in vivo biological effects of trabectedin, lurbinectedin (PM01183) and Zalypsis® (PM00104). Int. J. Cancer 2013, 133, 2024–2033. [Google Scholar] [CrossRef]
- Yap, T.A.; Cortes-Funes, H.; Shaw, H.; Rodriguez, R.; Olmo, S.D.; Lal, R.; Fong, P.C.; Tan, D.S.; Harris, D.; Capdevila, J.; et al. First-in-man phase I trial of two schedules of the novel synthetic tetrahydroisoquinoline alkaloid PM00104 (Zalypsis) in patients with advanced solid tumours. Br. J. Cancer 2012, 106, 1379–1385. [Google Scholar] [CrossRef]
- Giddings, L.-A.; Newman, D.J. Microbial natural products: Molecular blueprints for antitumor drugs. J. Ind. Microbiol. Biotechnol. 2013, 40, 1181–1210. [Google Scholar] [CrossRef]
- Fontana, A.; Cavaliere, P.; Wahidullah, S.; Naik, C.G.; Cimino, G. A new antitumor isoquinoline alkaloid from teh marine numdibranch Jorunna funebris. Tetrahedron 2000, 56, 7305–7308. [Google Scholar] [CrossRef]
- Perez, M.; Fernandez, C.; Chicharro, J.L.; Zarzuelo, M.; de la Calle, F.; Cuevas, C.; Francesch, A.; Gallego, P.; Manzanares, I. Hemisynthetic Methods and New Compounds. WO 20000069862, 23 November 2000. [Google Scholar]
- Leal, J.F.M.; Garcia-Hernandez, V.; Moneo, V.; Domingo, A.; Bueren-Calabuig, J.A.; Negri, A.; Gago, F.; Gulillen-Navarro, M.J.; Aviles, P.; Cuevas, C.; et al. Molecular pharmacology and antitumor activity of Zalypsis® in several human cell lines. Biochem. Pharmacol. 2009, 78, 162–170. [Google Scholar] [CrossRef]
- Massard, C.; Margetts, J.; Amellal, N.; Drew, Y.; Bahleda, R.; Stevens, P.; Armand, J.P.; Calvert, H.; Soria, J.C.; Coronado, C.; et al. Phase I study of PM00104 (Zalypsis®) administered as a 1-hour weekly infusion resting every fourth week in patients with advanced solid tumors. Invest. New Drugs 2013, 31, 623–630. [Google Scholar] [CrossRef]
- Capdevila, J.; Clive, S.; Casado, E.; Michie, C.; Piera, A.; Sicart, E.; Carreras, M.J.; Coronado, C.; Kahatt, C.; Soto Matos-Pita, A.; et al. A phase I pharmacokinetic study of PM00104 (Zalypsis) administered as a 24-h intravenous infusion every 3 weeks in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2013, 71, 1247–1254. [Google Scholar] [CrossRef]
- Martin, L.P.; Krasner, C.; Rutledge, T.; Luque Ibanes, M.; Fernandez-Garcıa, E.M.; Kahatt, C.; Siguero Gomez, M.; McMeekin, S. Phase II study of weekly PM00104 (ZALYPSIS®) in patients with pretreated advanced/metastatic endometrial or cervical cancer. Med. Oncol. 2013, 30, 627–631. [Google Scholar] [CrossRef]
- Leal, J.F.; Martínez-Díez, M.; García-Hernández, V.; Moneo, V.; Domingo, A.; Bueren-Calabuig, J.A.; Negri, A.; Gago, F.; Guillén-Navarro, M.J.; Avilés, P.; et al. PM01183, a new DNA minor groove covalent binder with potent in vitro and in vivo anti-tumour activity. Br. J. Pharmacol. 2010, 161, 1099–1110. [Google Scholar] [CrossRef]
- Soares, D.G.; Machado, M.S.; Rocca, C.J.; Poindessous, V.; Ouaret, D.; Sarasin, A.; Galmarini, C.M.; Henriques, J.A.; Escargueil, A.E.; Larsen, A.K. Trabectedin and its C subunit modified analogue PM01183 attenuate nucleotide excision repair and show activity toward platinum-resistant cells. Mol. Cancer Ther. 2011, 10, 1481–1489. [Google Scholar] [CrossRef]
- Yu, M.J.; Kishi, Y.; Littlefield, B.A. Discovery of E7389, a Fully Synthetic Macrocyclic Ketone Analog of Halichondrin B. In Anticancer Agents from Natural Products, 2nd ed.; Cragg, G.M., Kingston, D.G.I., Newman, D.J., Eds.; Taylor and Francis: Boca Raton, FL, USA, 2012; pp. 317–345. [Google Scholar]
- Yu, M.J.; Zheng, W.; Seletsky, B.M. From micrograms to grams: Scale-up synthesis of eribulin mesylate. Nat. Prod. Rep. 2013, 30, 1158–1164. [Google Scholar] [CrossRef]
- Austad, B.C.; Calkins, T.L.; Chase, C.E.; Fang, F.G.; Horstmann, T.E.; Hua, Y.; Lewis, B.M.; Niu, X.; Noland, T.A.; Orr, J.D.; et al. Commercial manufacture of Halaven®: Chemoselective transformations en route to structurally complex macrocyclic ketones. Synlett 2013, 24, 333–337. [Google Scholar] [CrossRef]
- Udwary, D.W.; Zeigler, L.; Asolkar, R.N.; Singan, V.; Lapidus, A.; Fenical, W.; Jensen, P.R.; Moore, B.S. Genome sequencing reveals complex secondary metabolome in the marine actinomycete Salinispora tropica. Proc. Natl. Acad. Sci. USA 2007, 104, 10376–10381. [Google Scholar] [CrossRef]
- Doronina, S.O.; Mendelsohn, B.A.; Bovee, T.D.; Cerveny, C.G.; Alley, S.C.; Meyer, D.L.; Oflazoglu, E.; Toki, B.E.; Sanderson, R.J.; Zabinski, R.F.; et al. Enhanced activity of monomethylauristatin F through monoclonal antibody delivery: Effects of linker technology on efficacy and toxicity. Bioconjug. Chem. 2006, 17, 114–124. [Google Scholar] [CrossRef]
- Copeland, A.; Younes, A. Brentuximab vedotin; Anti-CD30 antibody-drug conjugate oncolytic. Drugs Future 2010, 35, 797–801. [Google Scholar] [CrossRef]
- Ansell, S.M. Brentuximab vedotin: Delivering an antimitotic drug to activated lymphoma cells. Expert Opin. Investig. Drugs. 2011, 20, 99–105. [Google Scholar] [CrossRef]
- Haddley, K. Brentuximab vedotin: Its role in the treatment of anaplastic large cell and Hodgkin’s lymphoma. Drugs Today 2012, 48, 259–270. [Google Scholar]
- Newland, A.M.; Li, J.X.; Wasco, L.E.; Aziz, M.T.; Lowe, D.K. Brentuximab vedotin: A CD30-directed antibody-cytotoxic drug conjugate. Pharmacother 2013, 33, 93–104. [Google Scholar] [CrossRef]
- DeSchuytner, B.; Kuvalanka, K.; Hibner, B.; Bolen, J. Takeda’s oncology discovery strategy. Jpn. J. Clin. Oncol. 2013, 43, 357–361. [Google Scholar] [CrossRef]
- Tse, K.F.; Jeffers, M.; Pollack, V.A.; McCabe, D.A.; Shadish, M.L.; Khramtsov, N.V.; Hackett, C.S.; Shenoy, S.G.; Kuang, B.; Boldog, F.L.; et al. CR011, a fully human monoclonal antibody-auristatin E conjugate, for the treatment of melanoma. Clin. Cancer Res. 2006, 12, 1373–1382. [Google Scholar] [CrossRef]
- Pollack, V.A.; Alvarez, E.; Tse, K.F.; Torgov, M.Y.; Xie, S.; Shenoy, S.G.; MacDougall, J.R.; Arrol, S.; Zhong, H.; Gerwien, R.W.; et al. Treatment parameters modulating regression of human melanoma xenografts by an antibody-drug conjugate (CR011-vcMMAE) targeting GPNMB. Cancer Chemother. Pharmacol. 2007, 60, 423–435. [Google Scholar] [CrossRef]
- Rose, A.A.; Grosset, A.A.; Dong, Z.; Russo, C.; Macdonald, P.A.; Bertos, N.R.; St-Pierre, Y.; Simantov, R.; Hallett, M.; Park, M.; et al. Glycoprotein nonmetastatic B is an independent prognostic indicator of recurrence and a novel therapeutic target in breast cancer. Clin. Cancer Res. 2010, 16, 2147–2156. [Google Scholar] [CrossRef]
- Zhou, L.T.; Liu, F.Y.; Li, Y.; Peng, Y.M.; Liu, Y.H.; Li, J. Gpnmb/osteoactivin, an attractive target in cancer immunotherapy. Neoplasma 2012, 59, 1–5. [Google Scholar] [CrossRef]
- Yardley, D.A.; Weaver, R.; Melisko, M.E.; Saleh, M.N.; Arena, F.P.; Forero, A.; Cigler, T.; Stopeck, A.; Citron, D.; Oliff, I.; et al. A Randomized Phase 2 Study of the Antibody-Drug Conjugate CDX-011 in Advanced GPNMB-Overexpressing Breast Cancer: The EMERGE Study. In Proceedings of the CTRC-AACR San Antonio Breast Cancer Symposium, San Antonio, TX, USA, 4–8 December 2012.
- Phillips, A.C.; Boghaert, E.R.; Vaidya, K.S.; Ansell, P.J.; Shalinsky, D.R.; Zhang, Y.; Voorbach, M.J.; Mudd, S.; Holen, K.D.; Humerickhouse, R.A.; et al. ABT-414: An Anti-EGFR Antibody-Drug Conjugate as a Potential Therapeutic for the Treatment of Patients with Squamous Cell Tumors. In Proceedings of the 25th EORTC-NCI-AACR Symposium on Molecular Targets Cancer Therapeutics; Boston, MA, USA: 19–23 October 2013.
- Ma, D.; Hopf, C.E.; Malewicz, A.D.; Donovan, G.P.; Senter, P.D.; Goeckeler, W.F.; Maddon, P.J.; Olson, W.C. Potent antitumor activity of an auristatin-conjugated, fully human monoclonal antibody to prostate-specific membrane antigen. Clin. Cancer Res. 2006, 12, 2591–2596. [Google Scholar] [CrossRef]
- Wang, X.; Ma, D.; Olson, W.C.; Heston, W.D. In vitro and in vivo responses of advanced prostate tumors to PSMA ADC, an auristatin-conjugated antibody to prostate-specific membrane antigen. Mol. Cancer Ther. 2011, 10, 1728–1739. [Google Scholar] [CrossRef]
- Li, D.; Poon, K.A.; Yu, S.F.; Dere, R.; Go, M.; Lau, J.; Zheng, B.; Elkins, K.; Danilenko, D.; Kozak, K.R.; et al. DCDT2980S, an anti-CD22-monomethyl auristatin E antibody-drug conjugate, is a potential treatment for non-Hodgkin lymphoma. Mol. Cancer Ther. 2013, 12, 1255–1265. [Google Scholar]
- Yanagita, Y.; Takenaka, T. Astellas’ drug discovery strategy: Focus on oncology. Jpn. J. Clin. Oncol. 2012, 42, 241–246. [Google Scholar] [CrossRef]
- Satpayev, D.; Torgov, M.; Yang, P.; Morrison, K.; Shostak, Y.; Raitano, A.; Liu, W.; Lortie, D.; An, Z.; Capo, L.; et al. Development of AGS-22M6E, a Novel Antibody Drug Conjugate (ADC) Targeting Nectin-4 for the Treatment of Solid Tumors. In Proceedings of the 102nd Annual Meeting American Association Cancer Research (AACR), Orlando, FL, 2–6 April 2011. Abstract 2832.
- Iyer, U.; Kadambi, V.J. Antibody drug conjugates—Trojan horses in the war on cancer. J. Pharmacol. Toxicol. Methods 2011, 64, 207–212. [Google Scholar] [CrossRef]
- Albertson, T.M.; Sandalic, L.; Law, C.-L.; Broglio, K.; Berry, S. Phase 1,Open Label, Dose-Escalation Studies of SGN-CD19A in Patients with Relapsed or Refractory B-Lineage Acute Leukemia and Non-Hodgkin Lymphoma. In Proceedings of the 104th Annual Meeting American Association Cancer Research (AACR), Washington, DC, USA, 6–10 April 2013. Abstract 2412.
- Francisco, J.A.; Cerveny, C.G.; Meyer, D.L.; Mixan, B.J.; Klussman, K.; Chace, D.F.; Rejniak, S.X.; Gordon, K.A.; DeBlanc, R.; Toki, B.E.; et al. cAC10-vcMMAE, an anti-CD30-monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood 2003, 102, 1458–1465. [Google Scholar] [CrossRef]
- Gudas, J.M.; Torgov, M.; An, Z.; Jia, X.C.; Morrison, K.J.; Morrison, R.K.; Kanner, S.B.; Raitano, A.B.; Jakobovits, A. AGS-16M8F: A Novel Antibody Drug Conjugate (ADC) for Treating Renal and Liver Cancers. In Proceedings of the Genitourinary Cancers Symposium, San Francisco, CA, USA, 5–7 March 2010. Abstract 328.
- Liu, J.; Moore, K.; Birrer, M.; Berlin, S.; Matulonis, U.; Infante, J.; Xi, J.; Kahn, R.; Wang, Y.; Wood, K.; et al. Targeting MUC16 with the Antibody-Drug Conjugate (ADC) DMUC5754A in Patients with Platinum-Resistant Ovarian Cancer: A Phase I Study of Safety and Pharmacokinetics. In Proceedings of the 104th Annual Meeting American Association Cancer Research (AACR), Washington, DC, USA, 6–10 April 2013. Abstract LB-290.
- Gordon, M.S.; Gerber, D.E.; Infante, J.R.; Xu, J.; Shames, D.S.; Choi, Y.; Kahn, R.S. A Phase I Study of the Safety and Pharmacokinetics of DNIB0600A, an Anti-NaPi2b Antibody-Drug-Conjugate (ADC),in Patients (pts) with Non-Small Cell Lung Cancer (NSCLC) and Platinum-Resistant Ovarian Cancer (OC). In proceedings of the 2013 ASCO Annual Meeting, Chicago, IL, USA, 31 May–4 June 2013. Abstract 2507.
- Sapra, P.; Damelin, M.; Dijoseph, J.; Marquette, K.; Geles, K.G.; Golas, J.; Dougher, M.; Narayanan, B.; Giannakou, A.; Khandke, K.; et al. Long-term tumor regression induced by an antibody-drug conjugate that targets 5T4, an oncofetal antigen expressed on tumor-initiating cells. Mol. Cancer Ther. 2013, 12, 38–47. [Google Scholar] [CrossRef]
- Product Development Portfolio. Available online: http://www.roche.com/research_and_development/who_we_are_how_we_work/pipeline.htm (accessed on 12 November 2013).
- Bhakta, S.; Junutula, J.R. Cysteine Engineered Antibodies and Conjugates. US 2011/0301334 A1, 7 June 2011. [Google Scholar]
- Boswell, C.A.; Mundo, E.E.; Zhang, C.; Bumbaca, D.; Valle, N.R.; Kozak, K.R.; Fourie, A.; Chuh, J.; Koppada, N.; Saad, O.; et al. Impact of drug conjugation on pharmacokinetics and tissue distribution of anti-STEAP1 antibody-drug conjugates in rats. Bioconjug. Chem. 2011, 22, 1994–2004. [Google Scholar]
- Lin, K.; Tibbitts, J. Pharmacokinetic considerations for antibody drug conjugates. Pharm. Res. 2012, 29, 2354–2366. [Google Scholar] [CrossRef]
- Danila, D.C.; Szmulewitz, R.Z.; Higano, C.S.; Gilbert, H.; Kahn, R.S.; Wood, K.; Agarwal, P.; Lin, K.; Kabbarah, O.; Fine, B.M.; et al. A Phase I Study of the Safety and Pharmacokinetics of DSTP3086S, an Anti-STEAP1 Antibody-Drug Conjugate (ADC),in Patients (pts) with Metastatic Castration-Resistant Prostate Cancer (CRPC). In proceedings of the 2013 ASCO Annual Meeting, Chicago, IL, USA, 31 May–4 June 2013. Abstract 5020.
- Veiby, P.; Zhang, J.; Yang, J.; McDonald, A.; Fasanmade, A.; Wyant, T.; Almhanna, K.; Kalebic, T. The Investigational Drug MLN0264 First-in-Human, First in Class ADC Targeting GCC: Phase I Dose-Escalation Study and Supportive Scientific Rationale. In Proceedings of the 24th EORTC-NCI-AACR Symposium Molecular Targets Cancer Therapeutics, Dublin, Ireland, 6–9 November 2012. Abstract 329.
- Zhang, J.; Gallery, M.; Wyant, T.; Stringer, B.; Manfredi, M.; DAnaee, H.; Veiby, P. MLN0264, an Investigational, First-in-Class Antibody-Drug Conjugate (ADC) Targeting Guanylyl Cyclase C (GCC),Demonstrates Antitumor Activity Alone and in Combination with Gemcitabine in Human Pancreatic Cancer Xenograft Models Expressing GCC. In Proceedings of the 25th EORTC-NCI-AACR Symposium Molecular Targets Cancer Therapeutics, Boston, MA, USA, 19–23 October 2013. Abstract B194.
- Sussman, D.; Smith, L.M.; Anderson, M.E.; Duniho, S.; Hunter, J.H.; Kostner, H.; Miyamoto, J.B.; Nesterova, A.; Westendorf, L.; van Epps, H.A.; et al. SGN-LIV1A: A Development Stage Antibody Drug-Conjugate Targeting LIV-1 for the Treatment of Metastatic Breast Cancer. In Proceedings of the 104th Annual Meeting American Association Cancer Research (AACR), Washington, DC, USA, 6–10 April 2013. Abstract 3962.
- Yang, P.; Coleman, J.; Li, Y.; Zhang, Y.; Junge, C.; Morrison, K.; Donate, F.; Stover, D.; Morrison, K. SLITRK6, the Target of a Novel Antibody Drug Conjugate AGS15E, is Expressed in Bladder and Other Cancers. In Proceedings of the 104th Annual Meeting American Association Cancer Research (AACR), Washington, DC, USA, 6–10 April 2013. Abstract 1274.
- Verploegen, S.; Overdijk, M.; van Dijkhuizen, R.; Bleeker, W.K.; van Berkel, P.; Parren, P.; Lisby, S. Human Antibodies and Antibody-Drug Conjugates Against CD74. WO 2012/104344A1, 9 August 2012. [Google Scholar]
- Breij, E.C.W.; Satijn, D.; Verploegen, S.; de Goeij, B.E.; Schuurhuis, D.H.; Bleeker, W.K.; Houtkamp, M.; Parren, P.W. Use of an Antibody-Drug Conjugate Targeting Tissue Factor to Induce Complete Tumor Regression in Xenograft Models with Heterogeneous Target Expression. In Proceedings of the 2013 ASCO Annual Meeting, hicago, IL, USA, 31 May–4 June 2013.
- Rinehart, K.L., Jr.; Lithgow-Bertelloni, A.M. Novel Antiviral and Cytotoxic Agent. WO 9104985 A1, 19 April 1991. [Google Scholar]
- Rinehart, K.L., Jr.; Katauskas, A.J. Semi-synthetic studies toward didemnin analogues. WO 98/17275, 24 October 1997. [Google Scholar]
- Rinehart, K.L., Jr.; Lithgow-Bertelloni, A.M. Dehydrodidemnin B. US5834586, 10 November 1998. [Google Scholar]
- Jou, G.; González, I.; Albericio, F.; Lloyd-Williams, P.; Giralt, E. Total synthesis of dehydrodidemnin B. Use of uronium and phosphonium salt coupling reagents in peptide synthesis in solution. J. Org. Chem. 1997, 62, 354–366. [Google Scholar] [CrossRef]
- Giralt, E.; Albericio, F.; Lloyd-Williams, P.; González-Valcarcel, I.; Jou, G.; Gómez, A.; Manzanares, I. Procedimiento de Preparación de Didemnina A. ES 2102322, 16 July 1997. (in Spanish). [Google Scholar]
- Lee, J.; Currano, J.N.; Carroll, P.J.; Joullie, M.M. Didemnins, tamandarins and related natural products. Nat. Prod. Rep. 2012, 29, 404–424. [Google Scholar] [CrossRef]
- Cuevas, C.; Francesch, A.; Galmarini, C.M.; Aviles, P.; Munt, S. Ecteinascidin-743 (Yondelis®). Aplidin® and Irvalec®. In Anticancer Agents from Natural Products, 2nd ed.; Cragg, G.M., Kingston, D.G.I., Newman, D.J., Eds.; Taylor and Francis: Boca Raton, FL, USA, 2012; pp. 291–316. [Google Scholar]
- Geoerger, B.; Estlin, E.J.; Aerts, I.; Kearns, P.; Gibson, B.; Corradini, N.; Doz, F.; Lardelli, P.; Miguel, B.D.; Soto, A.; et al. A phase I and pharmacokinetic study of plitidepsin in children with advanced solid tumours: An innovative therapies for children with cancer (ITCC) study. Eur. J. Cancer 2012, 48, 289–296. [Google Scholar] [CrossRef]
- Xu, Y.; Kersten, R.D.; Nam, S.-J.; Lu, L.; Al-Suwailem, A.M.; Zheng, H.; Fenical, W.; Dorrestein, P.C.; Moore, B.S.; Qian, P.-Y. Bacterial biosynthesis and maturation of the didemnin anti-cancer agents. J. Am. Chem. Soc. 2012, 134, 8625–8632. [Google Scholar] [CrossRef]
- Ashour, M.; Edrada, R.; Ebel, R.; Wray, V.; Wätjen, W.; Padmakumar, K.; Müller, W.E.; Lin, W.H.; Proksch, P. Kahalalide derivatives from the Indian sacoglossan mollusk Elysia grandifolia. J. Nat. Prod. 2006, 69, 1547–1553. [Google Scholar] [CrossRef]
- Von Schwarzenberg, K.; Vollmar, A.M. Targeting apoptosis pathways by natural compounds in cancer: Marine compounds as lead structures and chemical tools for cancer therapy. Cancer Lett. 2013, 332, 295–303. [Google Scholar] [CrossRef]
- Shilabin, A.G.; Hamann, M.T. In vitro and in vivo evaluation of select kahalalide F analogs with antitumor and antifungal activities. Bioorg. Med. Chem. 2011, 19, 6628–6632. [Google Scholar] [CrossRef]
- Hayashi, Y.; Yamazaki-Nakamura, Y.; Yakushiji, F. Medicinal chemistry and chemical biology of diketopiperazine-type antimicrotubule and vascular-disrupting agents. Chem. Pharm. Bull. 2013, 61, 889–901. [Google Scholar] [CrossRef]
- Fenical, W.; Scripps Institution of Oceanography, La Jolla, CA, USA. Personal Communication, 2013.
- Nett, M.; Gulder, T.A.; Kale, A.J.; Hughes, C.C.; Moore, B.S. Function-oriented biosynthesis of beta-lactone proteasome inhibitors in Salinispora tropica. J. Med. Chem. 2009, 52, 6163–6167. [Google Scholar] [CrossRef]
- Fenical, W.; Jensen, P.R.; Palladino, M.A.; Lam, K.S.; Lloyd, G.K.; Potts, B.C. Discovery and development of the anticancer agent salinosporamide A (NPI-0052). Bioorg. Med. Chem. 2009, 17, 2175–2180. [Google Scholar] [CrossRef]
- Nguyen, H.; Ma, G.; Gladysheva, T.; Fremgen, T.; Romo, D. Bioinspired total synthesis and human proteasome inhibitory activity of (−)-salinosporamide A, (−)-homosalinosporamide A, and derivatives obtained via organonucleophile promoted bis-cyclizations. J. Org. Chem. 2011, 76, 2–12. [Google Scholar] [CrossRef]
- Lechner, A.; Eustáquio, A.S.; Gulder, T.A.; Hafner, M.; Moore, B.S. Selective overproduction of the proteasome inhibitor salinosporamide A via precursor pathway regulation. Chem. Biol. 2011, 18, 1527–1536. [Google Scholar] [CrossRef]
- Eustáquio, A.S.; Nam, S.J.; Penn, K.; Lechner, A.; Wilson, M.C.; Fenical, W.; Jensen, P.R.; Moore, B.S. The discovery of salinosporamide K from the marine bacterium “Salinispora pacifica” by genome mining gives insight into pathway evolution. Chembiochem 2011, 12, 61–64. [Google Scholar] [CrossRef]
- Martin, M.J.; Coello, L.; Fernandez, R.; Reyes, F.; Rodriguez, A.; Murcia, C.; Garranzo, M.; Mateo, C.; Sanchez-Sancho, F.; Bueno, S.; et al. Isolation and first total synthesis of PM050489 and PM060184, two new marine anticancer compounds. J. Am. Chem. Soc. 2013, 135, 10164–10171. [Google Scholar] [CrossRef]
- Pera, B.; Barasoain, I.; Canales, A.; Matesanz, R.; Rodrı́guez-Salarichs, J.; Garcı́a-Fernández, L.F.; Moneo, V.; Jiménez-Barbero, J.; Galmarini, C.M.; Cuevas, C.; et al. New interfacial microtubule inhibitors of marine origin with potent antitumor activity and a distinct mechanism. ACS Chem. Biol. 2013, 8, 2084–2094. [Google Scholar] [CrossRef]
- Bhatnagar, I.; Kim, S.K. Marine antitumor drugs: Status, shortfalls and strategies. Mar. Drugs 2010, 8, 2702–2720. [Google Scholar] [CrossRef]
- Petit, K.; Biard, J.-F. Marine natural products and related compounds as anticancer agents: An overview of their clinical status. Anticaner Agents Med. Chem. 2013, 13, 603–631. [Google Scholar] [CrossRef]
- Flahive, E.; Srirangam, J. The dolastatins: Novel Antitumor Agents from Dolabella auricularia. In Anticancer Agents from Natural Products, 2nd ed.; Cragg, G.M., Kingston, D.G.I., Newman, D.J., Eds.; Taylor and Francis: Boca Raton, FL, USA, 2012; pp. 263–289. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Newman, D.J.; Cragg, G.M. Marine-Sourced Anti-Cancer and Cancer Pain Control Agents in Clinical and Late Preclinical Development. Mar. Drugs 2014, 12, 255-278. https://doi.org/10.3390/md12010255
Newman DJ, Cragg GM. Marine-Sourced Anti-Cancer and Cancer Pain Control Agents in Clinical and Late Preclinical Development. Marine Drugs. 2014; 12(1):255-278. https://doi.org/10.3390/md12010255
Chicago/Turabian StyleNewman, David J., and Gordon M. Cragg. 2014. "Marine-Sourced Anti-Cancer and Cancer Pain Control Agents in Clinical and Late Preclinical Development" Marine Drugs 12, no. 1: 255-278. https://doi.org/10.3390/md12010255