Bioprospecting from Marine Sediments of New Brunswick, Canada: Exploring the Relationship between Total Bacterial Diversity and Actinobacteria Diversity

Abstract
:1. Introduction
2. Results and Discussion
2.1. Culture-Independent Analysis of Bacterial Diversity in New Brunswick Marine Sediments

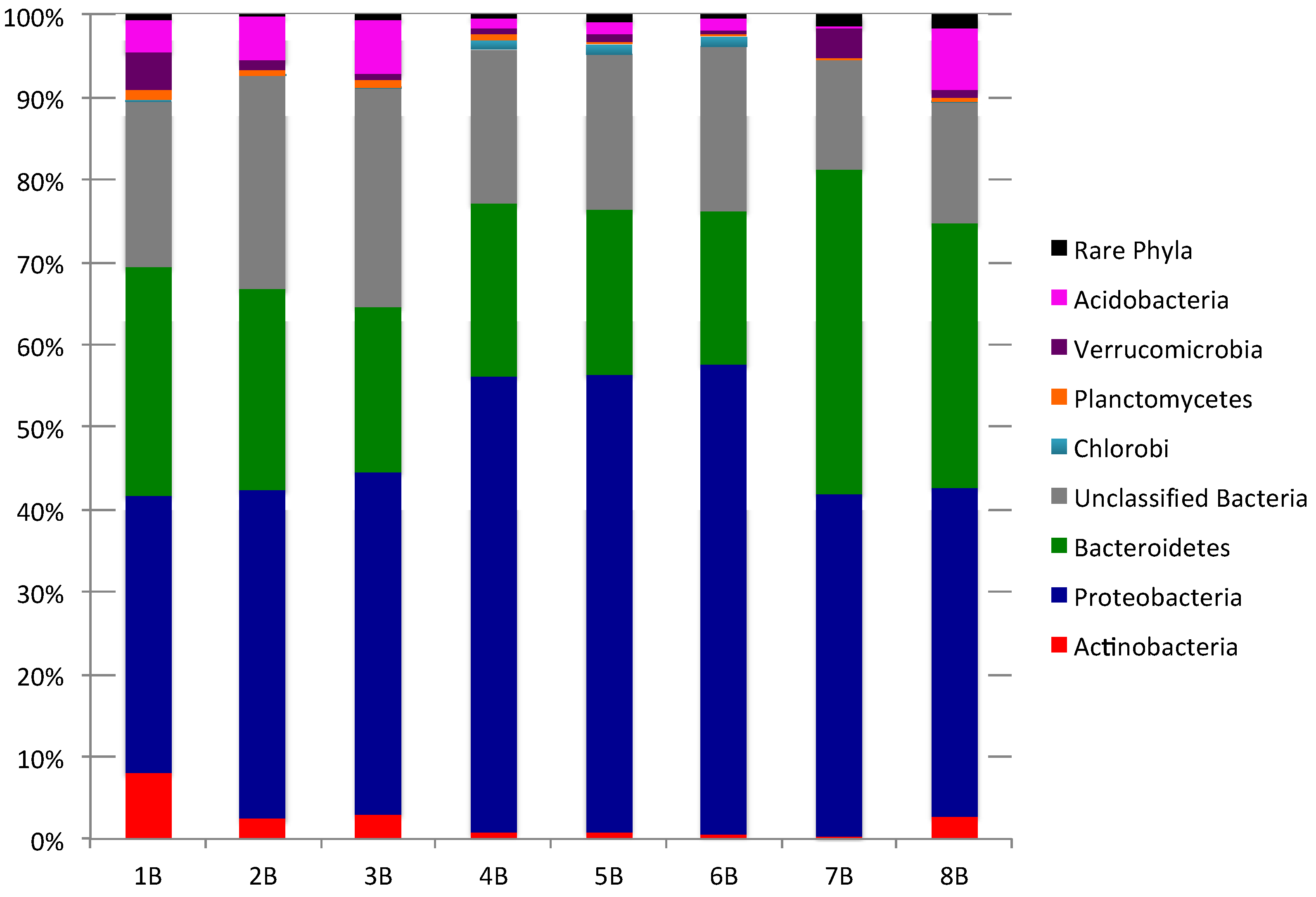{kind=link}
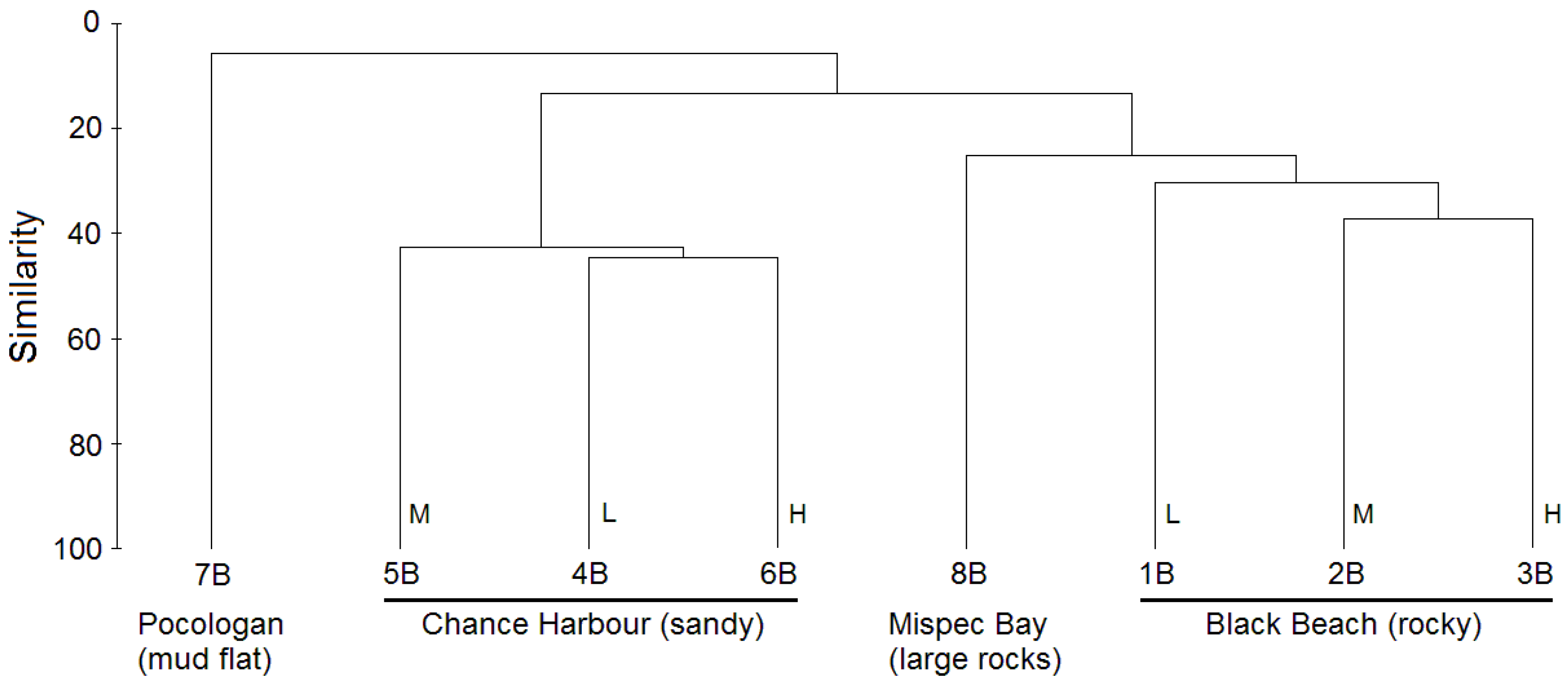{kind=link}
{kind=link}
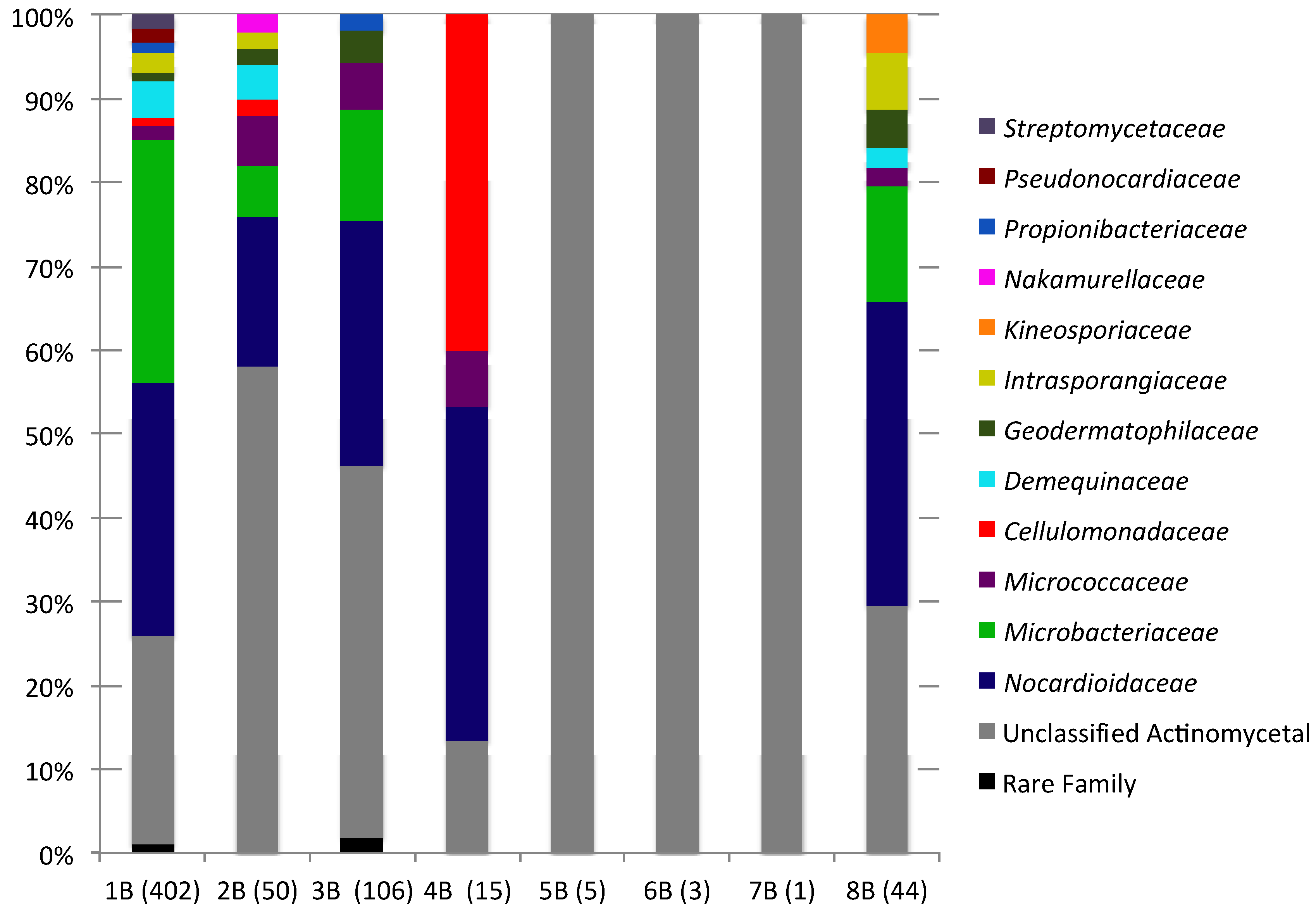{kind=link}
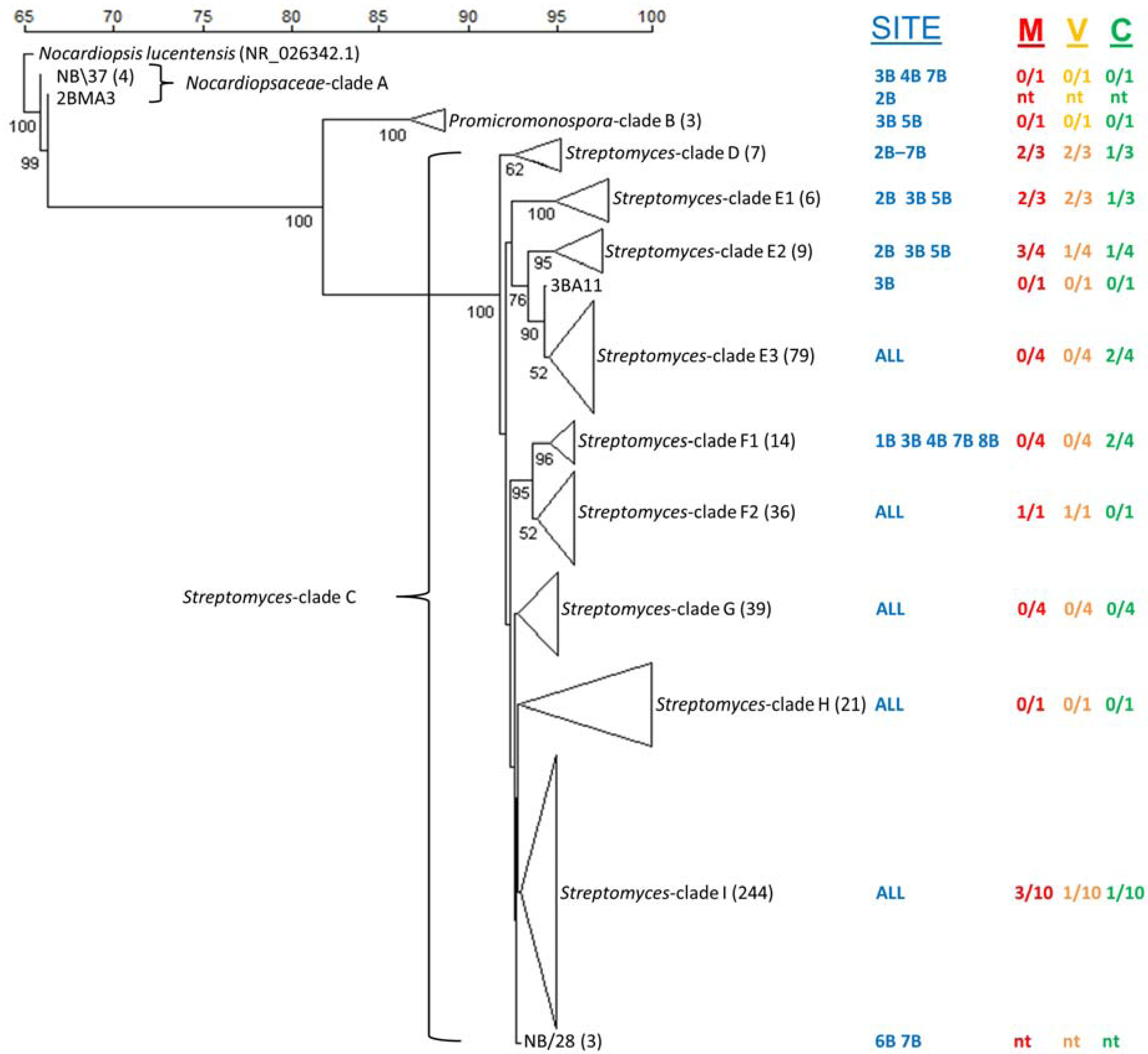{kind=link}
| Sample | Sample size | Average sequence length (bp) | Richness | Chao1 | C (%) a | Shannon diversity index (H’) | Shannon equitability index (E) |
|---|---|---|---|---|---|---|---|
| 1B | 6791 | 244 | 2102 | 3812 | 83 | 6.74 | 0.88 |
| 3500 | - | 1412 | 3123 | 74 | 6.55 | 0.90 | |
| 2B | 4634 | 246 | 1555 | 3247 | 79 | 6.39 | 0.87 |
| 3500 | - | 1301 | 2990 | 76 | 6.31 | 0.88 | |
| 3B | 5957 | 246 | 2040 | 4419 | 79 | 6.69 | 0.88 |
| 3500 | - | 1456 | 3459 | 73 | 6.52 | 0.90 | |
| 4B | 7856 | 247 | 1841 | 3806 | 86 | 5.73 | 0.76 |
| 3500 | - | 1098 | 2750 | 79 | 5.54 | 0.80 | |
| 5B | 4383 | 247 | 1276 | 2674 | 82 | 5.72 | 0.80 |
| 3500 | - | 1108 | 2435 | 80 | 5.66 | 0.81 | |
| 6B | 5306 | 247 | 1264 | 2743 | 86 | 5.51 | 0.77 |
| 3500 | - | 976 | 2255 | 82 | 5.41 | 0.76 | |
| 7B | 3672 | 246 | 1232 | 2294 | 81 | 6.38 | 0.90 |
| 3500 | - | 1199 | 2268 | 80 | 6.36 | 0.90 | |
| 8B | 6283 | 244 | 1659 | 2888 | 86 | 6.38 | 0.86 |
| 3500 | - | 1195 | 2403 | 80 | 6.24 | 0.88 |


| Sample | Concentration in parts per million (ppm) | Particle size (mm) as a percentage of total sample weight | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S2− | Zn2+ | Na+ | Fe2/3+ | Cu2+ | B | Mg2+ | Ca2+ | K2O | P2O2 | pH | OM (%) | <0.6 | 0.6–1.0 | 1.0–2.0 | >2 | |
| 1B | 57 | 3.3 | 1088 | 233 | 1.7 | 2.3 | 270 | 248 | 194 | 51 | 7.2 | 1.6 | 22.2 | 57.5 | 18.9 | 1.4 |
| 2B | 77 | 3 | 1164 | 236 | 1.7 | 4.1 | 330 | 314 | 213 | 45 | 7.9 | 1.3 | 12.4 | 51.8 | 32 | 3.9 |
| 3B | 53 | 2.9 | 1003 | 220 | 1.7 | 3.3 | 283 | 259 | 198 | 39 | 7.7 | 1.4 | 0.5 | 23 | 72.7 | 3.9 |
| 4B | 81 | 1 | 1095 | 132 | 0.1 | 1.6 | 192 | 141 | 111 | 42 | 7.8 | 0.4 | 4.4 | 34.1 | 61.2 | 0.4 |
| 5B | 97 | 1 | 1264 | 170 | 0.1 | 1.8 | 214 | 304 | 122 | 50 | 7.9 | 0.3 | 2.8 | 20.4 | 75.6 | 1.3 |
| 6B | 107 | 1.1 | 1293 | 147 | 0.2 | 1.9 | 229 | 163 | 125 | 46 | 7.7 | 0.3 | 0.7 | 17.1 | 82.3 | 0 |
| 7B | 128 | 2.4 | 2895 | 575 | 0.3 | 6 | 508 | 485 | 387 | 187 | 7.9 | 1.5 | 14.6 | 5.5 | 79.9 | 0 |
| 8B | 49 | 1.6 | 740 | 137 | 0.3 | 1.4 | 169 | 194 | 118 | 27 | 7.9 | 3.4 | 0.1 | 4.4 | 54 | 41 |

2.2. Culture-Independent Analysis of Actinomycete Diversity

2.3. Culture-Dependent Actinomycete Diversity

3. Experimental Section
3.1. Sample Collection and Processing
3.2. Sediment Analysis
3.3. Isolation of Sediment Metagenomic DNA
3.4. 16S rRNA Amplicon Pyrosequencing and Sequence Analysis
3.5. Cluster Analysis of (a) Culture-Independent Bacterial Communities (b) Comparison of Sediment Chemical Composition
3.6. Analysis of Similarities (ANOSIM) of (a) Culture-Independent Bacterial Communities (b) Culturable Actinomycete Diversity by Location
3.7. Canonical-Correspondence Analysis (CCA) Testing the Influence of Sediment Parameters on (a) Culture-Independent Bacterial Communities (b) Culturable Actinomycete Diversity by Location
3.8. Actinomycete Culturable Diversity
3.9. Identification of Bacterial Isolates
3.10. Phylogenetic Analysis of Cultured Actinomycetes
3.11. Fermentation and Extraction
3.12. Antimicrobial Testing
3.13. Chemical Analysis of Fermentation Extracts
4. Conclusions
Supplementary Files
Acknowledgments
Conflicts of Interest
References
- Bérdy, J. Bioactive microbial metabolites. J. Antibiot. (Tokyo) 2005, 58, 1–26. [Google Scholar] [CrossRef]
- Kieser, T.; Bibb, M.J.; Buttner, M.J.; Chater, K.F.; Hopwood, D.A. Practical Streptomyces Genetics, 2nd ed.; John Innes Foundation: Norwich, UK, 2000. [Google Scholar]
- Fischbach, M.A.; Walsh, C.T. Antibiotics for emerging pathogens. Science 2009, 325, 1089–1093. [Google Scholar] [CrossRef]
- Koehn, F.E.; Carter, G.T. Rediscovering natural products as a source of new drugs. Discov. Med. 2005, 5, 159–164. [Google Scholar]
- Bredholt, H.; Fjaervik, E.; Johnsen, G.; Zotchev, S.B. Actinomycetes from sediments in the Trondheim Fjord, Norway: Diversity and biological activity. Mar. Drugs 2008, 6, 12–24. [Google Scholar] [CrossRef]
- Pathom-Aree, W.; Stach, J.E.M.; Ward, A.C.; Horikoshi, K.; Bull, A.T.; Goodfellow, M. Diversity of actinomycetes isolated from challenger deep sediment (10,898 M) from the Mariana Trench. Extremophiles 2006, 10, 181–189. [Google Scholar]
- Jensen, P.R.; Williams, P.G.; Dong-Chan, O.; Zeigler, L.; Fenical, W. Species-Specific secondary metabolite production in marine actinomycetes of the genus Salinispora. Appl. Environ. Microbiol. 2007, 73, 1146–1152. [Google Scholar] [CrossRef]
- Maldonado, L.A.; Fragoso-Yáñez, D.; Pérez-García, A.; Rosellón-Druker, J.; Quintana, E.T. Actinobacterial diversity from marine sediments collected in Mexico. Antonie Van Leeuwenhoek 2009, 95, 111–120. [Google Scholar] [CrossRef]
- Ravenschlag, K.; Sahm, K.; Pernthaler, J.; Amann, R. High bacterial diversity in permanently cold marine sediments. Appl. Environ. Microbiol. 1999, 65, 3982–3989. [Google Scholar]
- Feling, R.H.; Buchan, G.O.; Mincer, T.J.; Kauffman, C.A.; Jensen, P.R.; Fenical, W. Salinosporamide A: A highly cytotoxic proteasome inhibitor from a novel microbial source, a marine bacterium of the new genus Salinospora. Angew. Chem. 2003, 42, 355–357. [Google Scholar] [CrossRef]
- Kwon, H.C.; Kauffman, C.A.; Jensen, P.R.; Fenical, W. Marinomycins A–D, antitumor-antibiotics of a new structure class from a marine actinomycete of the recently discovered genus “Marinispora”. J. Am. Chem. Soc. 2006, 128, 1622–1632. [Google Scholar] [CrossRef]
- Olano, C.; Méndez, C.; Salas, J.A. Antitumor compounds from marine actinomycetes. Mar. Drugs 2009, 7, 210–248. [Google Scholar] [CrossRef]
- Hughes, C.C.; Prieto-Davo, A.; Jensen, P.R.; Fenical, W. The marinopyrroles, antibiotics of an unprecedented structure class from a marine Streptomyces sp. Org. Lett. 2008, 10, 629–631. [Google Scholar] [CrossRef]
- Fenical, W. Chemical studies of marine bacteria: Developing a new resource. Chem. Rev. 1993, 93, 1673–1683. [Google Scholar] [CrossRef]
- Liu, X.; Ashforth, E.; Ren, B.; Song, F.; Dai, H.; Liu, M.; Wang, J.; Xie, Q.; Zhang, L. Bioprospecting microbial natural product libraries from the marine environment for drug discovery. J. Antibiot. (Tokyo) 2010, 63, 415–422. [Google Scholar] [CrossRef]
- Freel, K.C.; Edlund, A.; Jensen, P.R. Microdiversity and evidence for high dispersal rates in the marine actinomycete ‘Salinispora pacifica’. Environ. Microbiol. 2012, 14, 480–493. [Google Scholar] [CrossRef]
- Jensen, P.R.; Dwight, R.; Fenical, W. Distribution of actinomycetes in near-shore tropical marine sediments. Appl. Environ. Microbiol. 1991, 57, 1102–1108. [Google Scholar]
- Jorgensen, S.L.; Hannisdal, B.; Lanzén, A.; Baumberger, T.; Flesland, K.; Fonseca, R.; Ovreås, L.; Steen, I.H.; Thorseth, I.H.; Pedersen, R.B.; et al. Correlating microbial community profiles with geochemical data in highly stratified sediments from the arctic mid-ocean ridge. Proc. Natl. Acad. Sci. USA 2012, 109, E2846–E2855. [Google Scholar]
- Fu, P.; Wang, S.; Hong, K.; Li, X.; Liu, P.; Wang, Y.; Zhu, W. Cytotoxic bipyridines from the marine-derived actinomycete Actinoalloteichus cyanogriseus WH1-2216-6. J. Nat. Prod. 2011, 74, 1751–1756. [Google Scholar] [CrossRef]
- Jensen, P.R.; Fenical, W. The relative abundance and seawater requirements of gram-positive bacteria in near-shore tropical marine samples. Microb. Ecol. 1995, 29, 249–257. [Google Scholar] [CrossRef]
- Ward, A.C. Diversity and biogeography of marine actinobacteria. Curr. Opin. Microbiol. 2006, 9, 279–286. [Google Scholar] [CrossRef]
- Jensen, P.R.; Gontang, E.; Mafnas, C.; Mincer, T.J.; Fenical, W. Culturable marine actinomycete diversity from tropical pacific ocean sediments. Environ. Microbiol. 2005, 7, 1039–1048. [Google Scholar] [CrossRef]
- Lim, J.; Woodward, J.; Tulaczyk, S.; Christoffersen, P.; Cummings, S.P. Analysis of the microbial community and geochemistry of a sediment core from Great Slave Lake, Canada. Antonie Van Leeuwenhoek 2011, 99, 423–430. [Google Scholar] [CrossRef]
- Forschner, S.R.; Sheffer, R.; Rowley, D.C.; Smith, D.C. Microbial diversity in cenozoic sediments recovered from the Lomonosov Ridge in the central arctic basin. Environ. Microbiol. 2009, 11, 630–639. [Google Scholar] [CrossRef]
- Turley, C. Bacteria in the cold deep-sea benthic boundary layer and sediment-water interface of the NE Atlantic. FEMS Microbiol. Ecol. 2000, 33, 89–99. [Google Scholar]
- Burzynski, M. A Guide to Fundy National Park; Douglas & McIntyre: Madeira Park, BC, USA, 1985. [Google Scholar]
- Das, S.; Lyla, P.S.; Khan, S.A. Marine microbial diversity and ecology: Importance and future perspectives. Curr. Sci. 2006, 90, 1325–1335. [Google Scholar]
- Wang, Y.; Sheng, H.-F.; He, Y.; Wu, J.-Y.; Jiang, Y.-X.; Tam, N.F.-Y.; Zhou, H.-W. Comparison of the levels of bacterial diversity in freshwater, intertidal wetland, and marine sediments by using millions of illumina tags. Appl. Environ. Microbiol. 2012, 78, 8264–8271. [Google Scholar] [CrossRef]
- Babalola, O.O.; Kirby, B.M.; Le Roes-Hill, M.; Cook, A.E.; Cary, S.C.; Burton, S.G.; Cowan, D.A. Phylogenetic analysis of actinobacterial populations associated with Antarctic Dry Valley mineral soils. Environ. Microbiol. 2009, 11, 566–576. [Google Scholar] [CrossRef]
- Tringe, S.G.; Rubin, E.M.; Tringe, S.G.; Rubin, E.M. Metagenomics: DNA sequencing of environmental samples. Nat. Rev. Gen. 2005, 6, 805–814. [Google Scholar] [CrossRef]
- Sogin, M.L.; Morrison, H.G.; Huber, J.A.; Mark Welch, D.; Huse, S.M.; Neal, P.R.; Arrieta, J.M.; Herndl, G.J. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc. Natl. Acad. Sci. USA 2006, 103, 12115–12120. [Google Scholar]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar]
- Bull, A.T. Search and discovery strategies for biotechnology: The paradigm shift. Microbiol. Mol. Biol. Rev. 2000, 64, 573–606. [Google Scholar] [CrossRef]
- Chao, A.; Lee, S. Estimating the number of classes via sample coverage. J. Am. Stat. Assoc. 1992, 87, 210–217. [Google Scholar] [CrossRef]
- Chao, A. Nonparametric estimation of the number of classes in a population. Scand. J. Stat. 1984, 11, 265–270. [Google Scholar]
- Schloss, P.D.; Handelsman, J. Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl. Environ. Microbiol. 2005, 71, 1501–1506. [Google Scholar] [CrossRef]
- Kemp, P.F.; Aller, J.Y. Bacterial diversity in aquatic and other environments: What 16S rDNA libraries can tell us. FEMS Microbiol. Ecol. 2004, 47, 161–177. [Google Scholar] [CrossRef]
- Nacke, H.; Thürmer, A.; Wollherr, A.; Will, C.; Hodac, L.; Herold, N.; Schöning, I.; Schrumpf, M.; Daniel, R. Pyrosequencing-based assessment of bacterial community structure along different management types in German forest and grassland soils. PLoS One 2011, 6, 1–12. [Google Scholar]
- Fierer, N.; Jackson, R.B. The diversity and biogeography of soil bacterial communities. Proc. Natl. Acad. Sci. USA 2006, 103, 626–631. [Google Scholar] [CrossRef]
- Bushaw-Newton, K.; Ewers, E.; Velinsky, D.; Ashley, J.; MacAvoy, S. Bacterial community profiles from sediments of the Anacostia River using metabolic and molecular analyses. Environ. Sci. Pollut. Res. 2012, 19, 1271–1279. [Google Scholar] [CrossRef]
- Duncan, K.; University of Prince Edward Island, Charlottetown, PE, Canada. Unpublished work. 2013.
- Sørensen, K.B.; Glazer, B.; Hannides, A.; Gaidos, E. Spatial structure of the microbial community in sandy carbonate sediment. Mar. Ecol. Prog. Ser. 2007, 346, 61–74. [Google Scholar] [CrossRef]
- Musat, N.; Werner, U.; Knittel, K.; Kolb, S.; Dodenhof, T.; van Beusekom, J.E.; de Beer, D.; Dubilier, N.; Amann, R. Microbial community structure of sandy intertidal sediments in the North Sea, Sylt-Rømø Basin, Wadden Sea. Syst. Appl. Microbiol. 2006, 29, 333–348. [Google Scholar] [CrossRef]
- Gobet, A.; Böer, S.I.; Huse, S.M.; van Beusekom, J.E.; Quince, C.; Sogin, M.L.; Boetius, A.; Ramette, A. Diversity and dynamics of rare and of resident bacterial populations in coastal sands. ISME J. 2012, 6, 542–553. [Google Scholar] [CrossRef]
- Hollister, E.B.; Engledow, A.S.; Hammett, A.J.; Provin, T.L.; Wilkinson, H.H.; Gentry, T.J. Shifts in microbial community structure along an ecological gradient of hypersaline soils and sediments. ISME J. 2010, 4, 829–838. [Google Scholar] [CrossRef]
- Certini, G.; Campbell, C.D.; Edwards, A.C. Rock fragments in soil support a different microbial community from the fine earth. Soil Biol. Biochem. 2004, 36, 1119–1128. [Google Scholar] [CrossRef]
- Kirchman, D.L.; Cottrell, M.T.; Lovejoy, C. The structure of bacterial communities in the western Arctic Ocean as revealed by pyrosequencing of 16S rRNA genes. Environ. Microbiol. 2010, 12, 1132–1143. [Google Scholar] [CrossRef]
- Giovannoni, S.J.; Stingl, U. Molecular diversity and ecology of microbial plankton. Nature 2005, 437, 343–348. [Google Scholar] [CrossRef]
- Ghiglione, J.; Galand, P.E.; Pommier, T.; Pedrós-Alió, C.; Maas, E.W.; Bakker, K.; Bertilson, S.; Kirchmanj, D.L.; Lovejoy, C.; Yager, P.L.; et al. Pole-to-pole biogeography of surface and deep marine bacterial communities. Proc. Natl. Acad. Sci. USA 2012, 109, 17633–17638. [Google Scholar]
- Mincer, T.J.; Fenical, W.; Jensen, P.R. Culture-dependent and culture-independent diversity within the obligate marine actinomycete genus Salinispora. Appl. Environ. Microbiol. 2005, 71, 7019–7028. [Google Scholar] [CrossRef]
- Bull, A.T. Marine actinobacteria: Perspectives, challenges, future directions. Antonie Van Leeuwenhoek 2005, 87, 65–79. [Google Scholar] [CrossRef]
- Eccleston, G.P.; Brooks, P.R.; Kurtböke, D.I. The occurrence of bioactive Micromonosporae in aquatic habitats of the sunshine coast in Australia. Mar. Drugs 2008, 6, 243–261. [Google Scholar] [CrossRef]
- Maldonado, L.A. Characterisation of micromonosporae from aquatic environments using molecular taxonomic methods. Antonie van Leeuwenhoek 2008, 94, 289–298. [Google Scholar] [CrossRef]
- Schwientek, P.; Szczepanowski, R.; Rückert, C.; Stoye, J.; Pühler, A. Sequencing of high G+C microbial genomes using the ultrafast pyrosequencing technology. J. Biotechnol. 2011, 155, 68–77. [Google Scholar] [CrossRef]
- Jensen, P.R. An assessment of actinobacterial diversity in the marine environment. Antonie Van Leeuwenhoek 2008, 94, 51–62. [Google Scholar] [CrossRef]
- Newman, D.J.; Hill, R.T. New drugs from marine microbes: The tide is turning. J. Ind. Microbiol. Biotechnol. 2006, 33, 539–544. [Google Scholar] [CrossRef]
- Guo, Y.; Zheng, W.; Rong, X.; Huang, Y. A multilocus phylogeny of the Streptomyces griseus 16S rRNA gene clade: Use of multilocus sequence analysis for streptomycete systematics. Int. J. Syst. Evol. Microbiol. 2008, 58, 149–159. [Google Scholar] [CrossRef]
- Wawrik, B.; Kutliev, D.; Abdivasievna, U.A.; Kukor, J.J.; Zylstra, G.J.; Kerkhof, L. Biogeography of actinomycete communities and type II polyketide synthase genes in soils collected in New Jersey and Central Asia. Appl. Environ. Microbiol. 2007, 73, 2982–2989. [Google Scholar] [CrossRef]
- Gontang, E.A.; Fenical, W.; Jensen, P.R. Phylogenetic diversity of gram-positive bacteria cultured from marine sediments. Appl. Environ. Microbiol. 2007, 73, 3272–3282. [Google Scholar] [CrossRef]
- Takizawa, M.; Colwell, R.R.; Hill, R.T. Isolation and diversity of actinomycetes in the Chesapeake Bay. Appl. Environ. Microbiol. 1993, 59, 997–1002. [Google Scholar]
- Weyland, H. Actinomycetes of the Bottom Sediments of Various Sea; France, B., Ed.; Actes de colloques: Brest, France, 1986; p. 79. [Google Scholar]
- Omura, S.; Iwai, Y.; Hirano, A.; Nakagawa, A.; Awaya, J.; Tsuchiya, H.; Takahashi, Y.; Asuma, R. A new alkaloid AM-2282 of Streptomyces origin taxonomy, fermentation, isolation and preliminary characterization. J. Antibiot. 1977, 30, 275–282. [Google Scholar] [CrossRef]
- Schleissner, C.; Pérez, M.; Losada, A.; Rodríguez, P.; Crespo, C.; Zúñiga, P.; Fernández, R.; Reyes, F.; de la Calle, F. Antitumor actinopyranones produced by Streptomyces albus POR-04-15-053 isolated from a marine sediment. J. Nat. Prod. 2011, 74, 1590–1596. [Google Scholar] [CrossRef]
- Jensen, P.R. Linking species concepts to natural product discovery in the post-genomic era. J. Ind. Microbiol. Biotechnol. 2010, 37, 219–224. [Google Scholar] [CrossRef]
- Watve, M.G.; Tickoo, R.; Jog, M.M.; Bhole, B.D. How many antibiotics are produced by the genus Streptomyces? Arch. Microbiol. 2001, 176, 386–390. [Google Scholar] [CrossRef]
- Magarvey, N.A; Keller, J.M.; Bernan, V.; Dworkin, M.; Sherman, D.H. Isolation and characterization of novel marine-derived actinomycete taxa rich in bioactive metabolites. Appl. Environ. Microbiol. 2004, 70, 7520–7529. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the last 25 years. J. Nat. Prod. 2007, 70, 461–477. [Google Scholar] [CrossRef]
- Hou, Y.; Braun, D.R.; Michel, C.R.; Klassen, J.L.; Adnani, N.; Wyche, T.P.; Bugni, T.S. Microbial strain prioritization using metabolomics tools for the discovery of natural products. Anal. Chem. 2012, 84, 4277–4283. [Google Scholar]
- Penn, K.; Jenkins, C.; Nett, M.; Udwary, D.W.; Gontang, E.A.; McGlinchey, R.P.; Foster, B.; Lapidus, A.; Podell, S.; Allen, E.E.; et al. Genomic islands link secondary metabolism to functional adaptation in marine actinobacteria. Int. J. Syst. Evol. Microbiol. 2009, 3, 1193–1203. [Google Scholar]
- Donadio, S.; Monciardini, P.; Sosio, M. Polyketide synthases and nonribosomal peptide synthetases: The emerging view from bacterial genomics. Nat. Prod. Rep. 2007, 24, 1073–1109. [Google Scholar] [CrossRef]
- Burzynski, M.; Marceau, A. Fundy Bay of the Giant Tides; Fundy Guild Inc.: Alma, NB, Canada, 1984. [Google Scholar]
- Tiessen, H.; Bettany, J.R.; Stewart, J.W.B. An improved method for the determination of carbon in soils and soil extracts by dry combustion. Commun. Soil Sci. Plant Anal. 1981, 12, 211–218. [Google Scholar] [CrossRef]
- Mehlich, A. Mehlich 3 soil test extractant: A modification of Mehlich 2 extractant. Commun. Soil Sci. Plant Anal. 1984, 15, 1409–1416. [Google Scholar]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual, 2nd ed.; Cold Springs Harbour Laboratory Press: Cold Springs Harbour, NY, USA, 1989. [Google Scholar]
- Dowd, S.E.; Callaway, T.R.; Wolcott, R.D.; Sun, Y.; McKeehan, T.; Hagevoort, R.G.; Edrington, T.S. Evaluation of the bacterial diversity in the feces of cattle using 16S rDNA bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP). BMC Microbiol. 2008, 8, 1–8. [Google Scholar] [CrossRef]
- Research and Testing Laboratory Homepage. Available online: http://www.researchandtesting.com (accessed on 5 September 2013).
- Acosta-Martínez, V.; Dowd, S.; Sun, Y.; Allen, V. Tag-encoded pyrosequencing analysis of bacterial diversity in a single soil type as affected by management and land use. Soil Biol. Biochem. 2008, 40, 2762–2770. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- Schloss, P.D.; Gevers, D.; Westcott, S.L. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS One 2011, 6, e2731. [Google Scholar]
- Schloss. Mothur Wiki SOP. Available online: http://Www.Mothur.Org/Wiki/454_SOP (accessed on 1 October 2013).
- Mothur. Available online: http://www.mothur.org/wiki/Silva_reference (accessed on 1 October 2013).
- Clarke, K.R.; Gorley, R.N. PRIMER (Plymouth Routines in Multivariate Ecological Research) V6: User Manual/Tutorial; PRIMER-E Ltd.: Plymouth, UK, 2006. [Google Scholar]
- Warwick, R.M.; Clarke, K.R. Changes in Marine Communities: An Approach to Statistical Analysis and Interpretation, 2nd ed.; PRIMER-E Ltd.: Plymouth, UK, 2001. [Google Scholar]
- Mincer, T.J. Widespread and persistent populations of a major new marine actinomycete taxon in ocean sediments. Appl. Environ. Microbiol. 2002, 68, 5005–5011. [Google Scholar] [CrossRef]
- Maldonado, L.A. Diversity of cultivable actinobacteria in geographically widespread marine sediments. Antonie Van Leeuwenhoek 2005, 87, 11–18. [Google Scholar] [CrossRef]
- Atlas, R.M.; Parks, L.C. Handbook of Microbiological Media; CRC Press: Boca Raton, FL, USA, 1993; p. 1079. [Google Scholar]
- Shirling, E.B.; Gottlieb, D. Methods for characterization of Streptomyces species. Int. J. Syst. Evol. Microbiol. 1966, 16, 313–349. [Google Scholar]
- Hopwood, D.A. Genetic Manipulation of Streptomyces: A Laboratory Manual; The John Innes Foundation: Norwich, UK, 1986. [Google Scholar]
- Weisburg, W.G.; Barns, S.M.; Pelletier, D.A.; Lane, D.J. 16S ribosomal DNA amplification for phylogenetic study. J. Bacteriol. 1991, 173, 697–703. [Google Scholar]
- Kaplan, C.W.; Astaire, J.C.; Sanders, M.E.; Reddy, B.S.; Kitts, C.L. 16S ribosomal DNA terminal restriction fragment pattern analysis of bacterial communities in feces of rats fed Lactobacillus acidophilus NCFM. Appl. Environ. Microbiol. 2001, 67, 1935–1939. [Google Scholar] [CrossRef]
- Basic Local Alignment Search tool. Available online: http://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 1 June 2013).
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar]
- ClustalW2. Available online: http://www.ebi.ac.uk/tools/clustalw2 (accessed on 15 June 2013).
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Duncan, K.; Haltli, B.; Gill, K.A.; Kerr, R.G. Bioprospecting from Marine Sediments of New Brunswick, Canada: Exploring the Relationship between Total Bacterial Diversity and Actinobacteria Diversity. Mar. Drugs 2014, 12, 899-925. https://doi.org/10.3390/md12020899
Duncan K, Haltli B, Gill KA, Kerr RG. Bioprospecting from Marine Sediments of New Brunswick, Canada: Exploring the Relationship between Total Bacterial Diversity and Actinobacteria Diversity. Marine Drugs. 2014; 12(2):899-925. https://doi.org/10.3390/md12020899
Chicago/Turabian StyleDuncan, Katherine, Bradley Haltli, Krista A. Gill, and Russell G. Kerr. 2014. "Bioprospecting from Marine Sediments of New Brunswick, Canada: Exploring the Relationship between Total Bacterial Diversity and Actinobacteria Diversity" Marine Drugs 12, no. 2: 899-925. https://doi.org/10.3390/md12020899