Synthesis of PPAR-γ Activators Inspired by the Marine Natural Product, Paecilocin A

Abstract
:1. Introduction

2. Results and Discussion
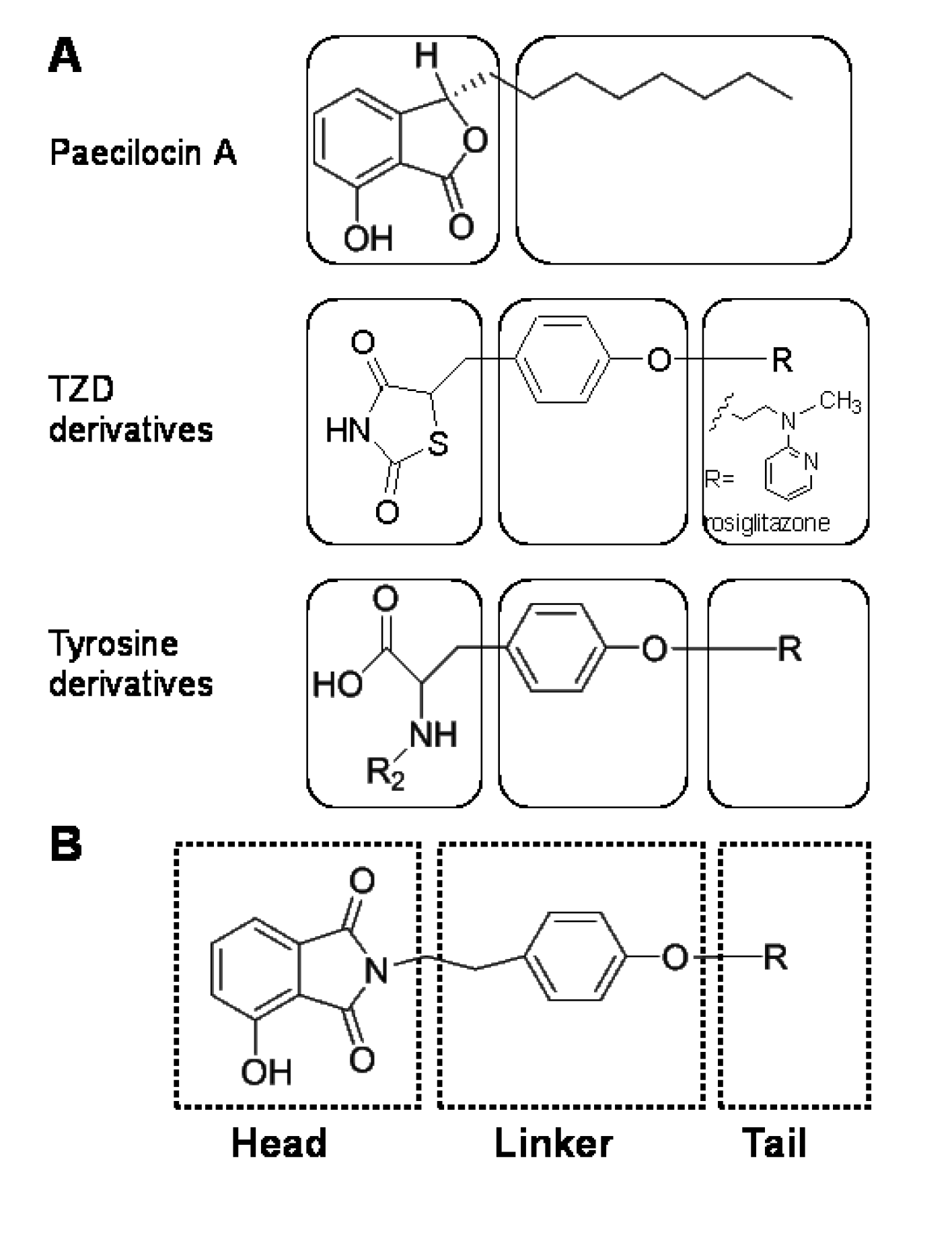
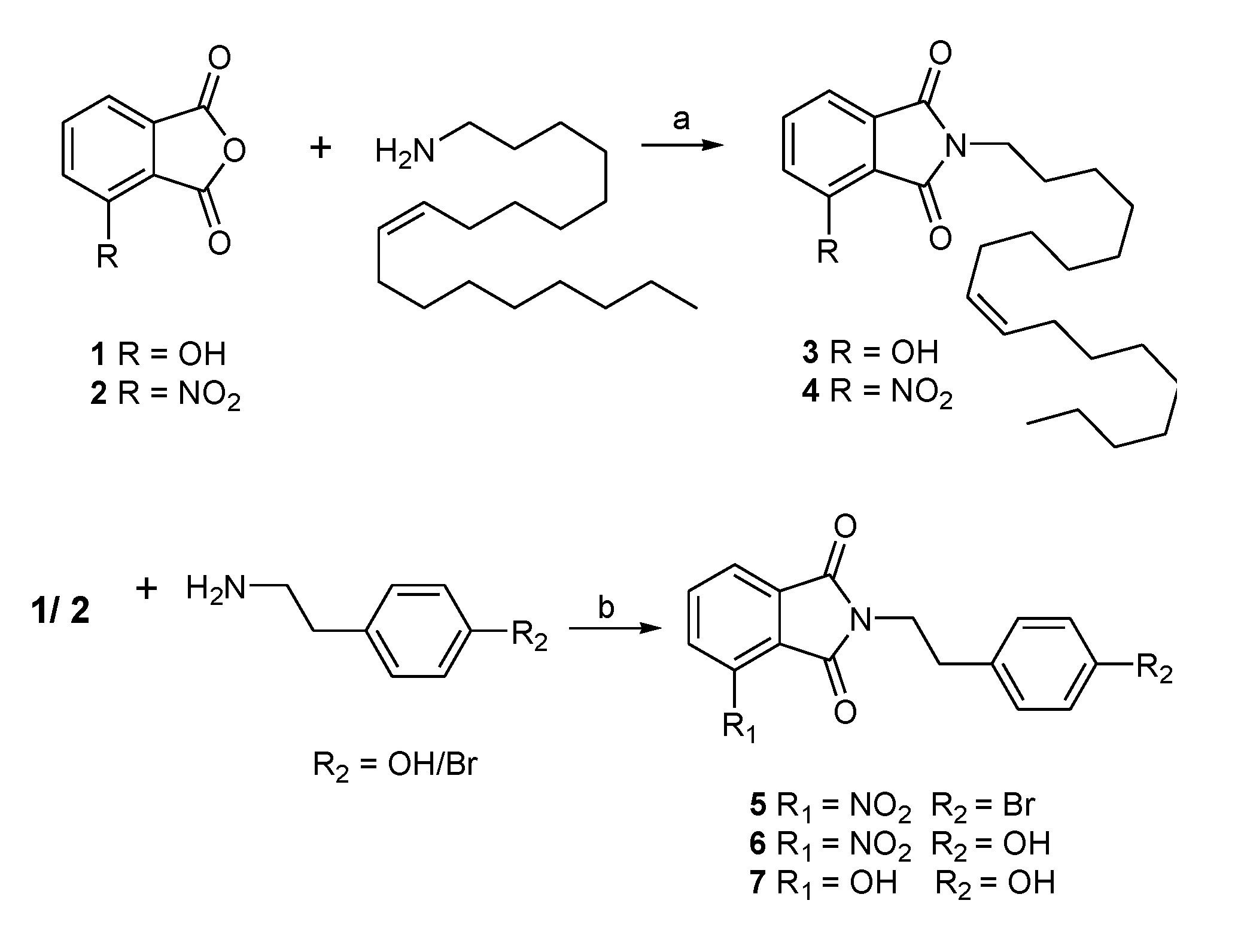
2.1. Chemistry


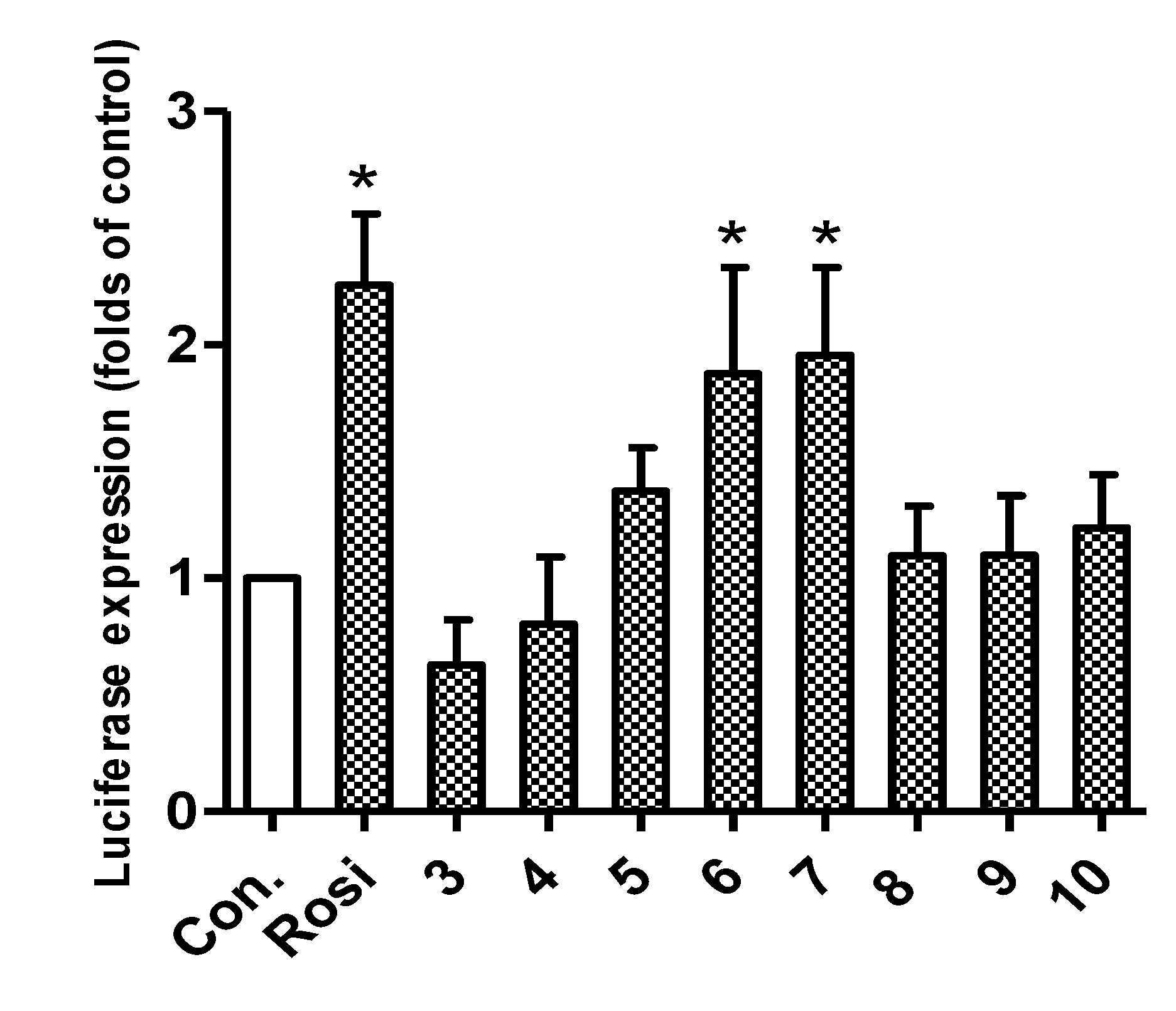
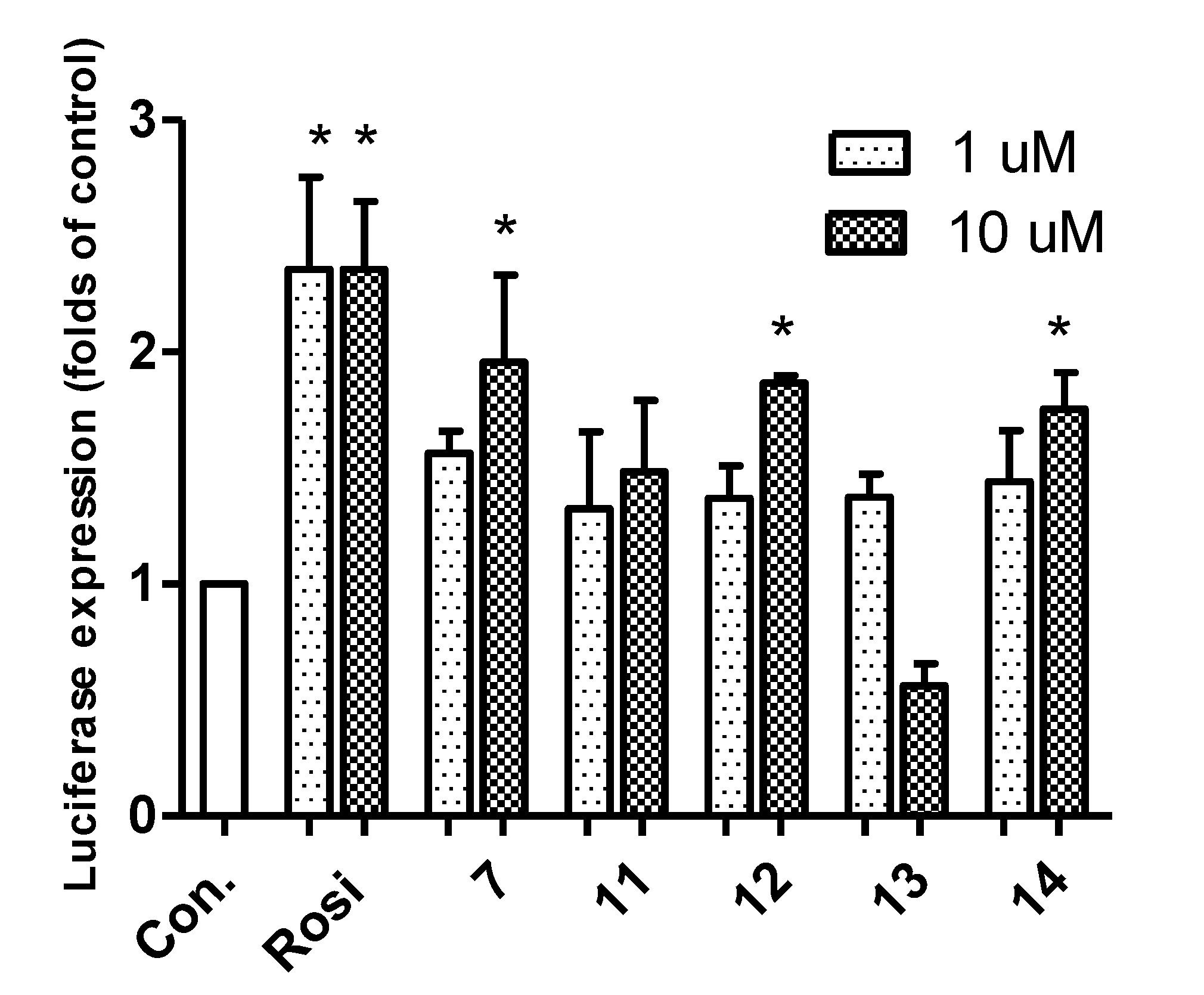
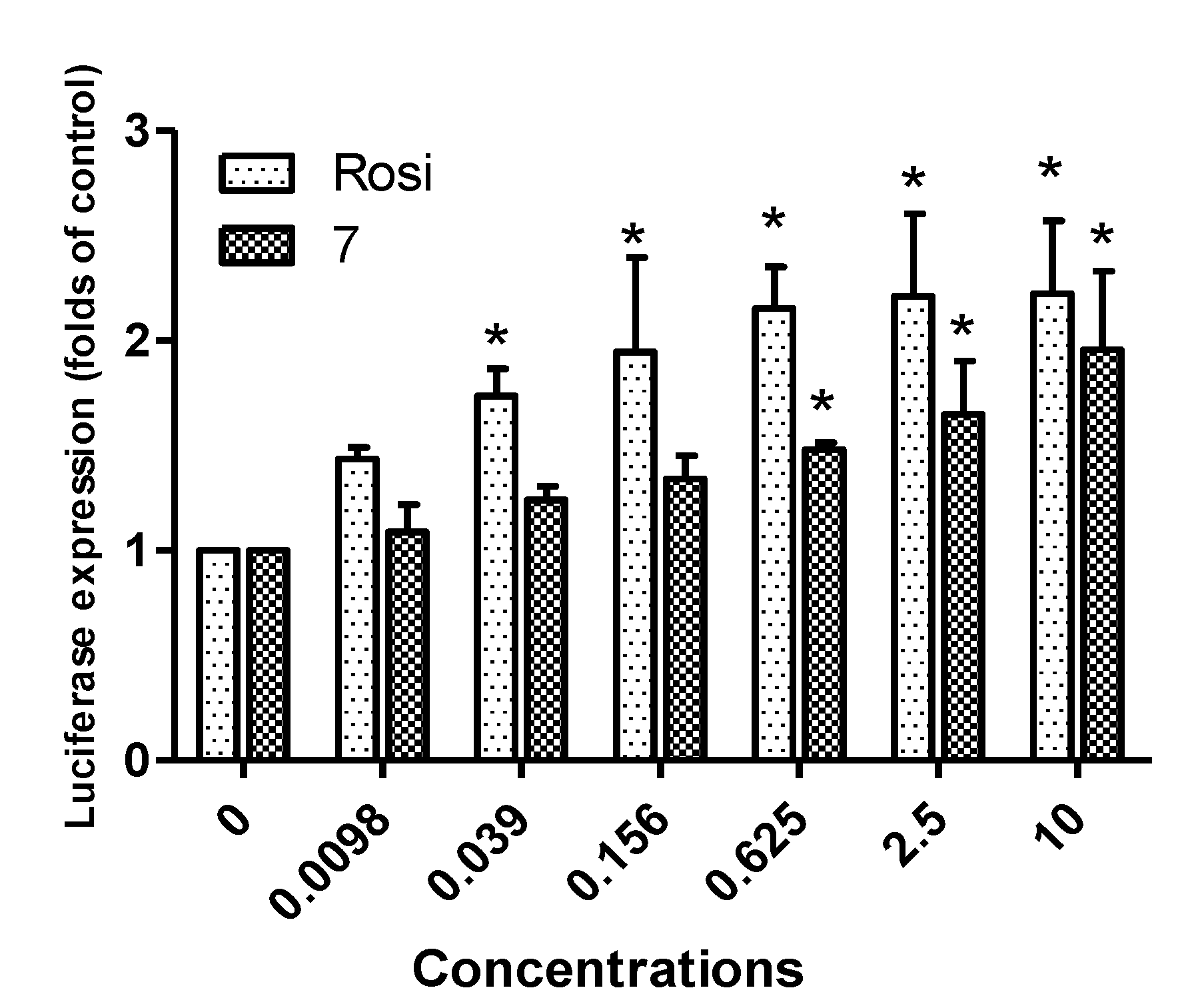
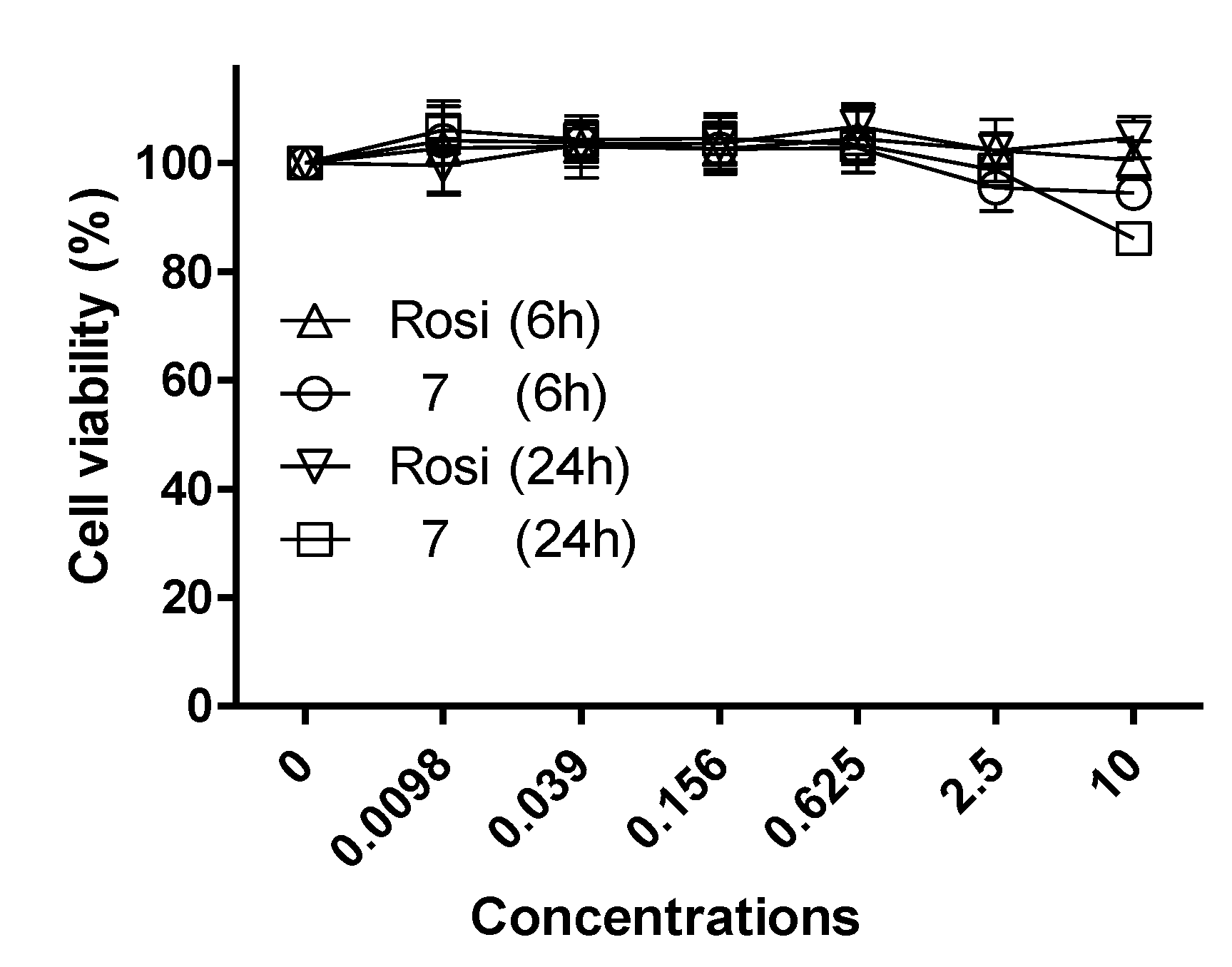
2.2. Biological Activity





3. Experimental Section
3.1. Chemistry
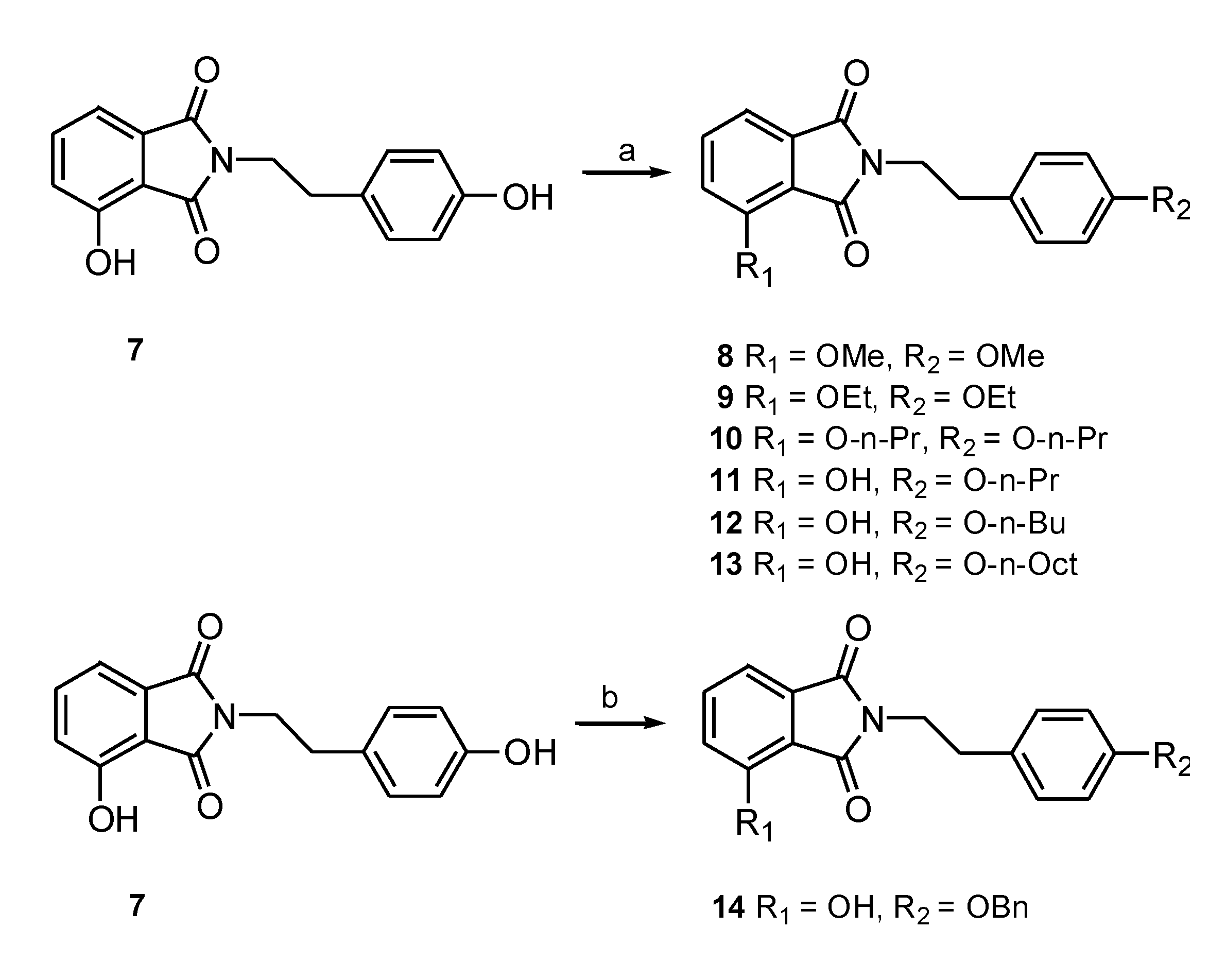
3.1.1. Preparation of N-Substituted Phthalimides 3–7
3.1.2. General Procedure for the Synthesis of N-substituted Phthalimides 8–13
3.1.3. General Procedure for the Synthesis of 3-Hydroxy-N-(p-benzyl-phenethyl) Phthalimide (14)
3.2. Luciferase Assay
3.3. Cell Proliferation Assay
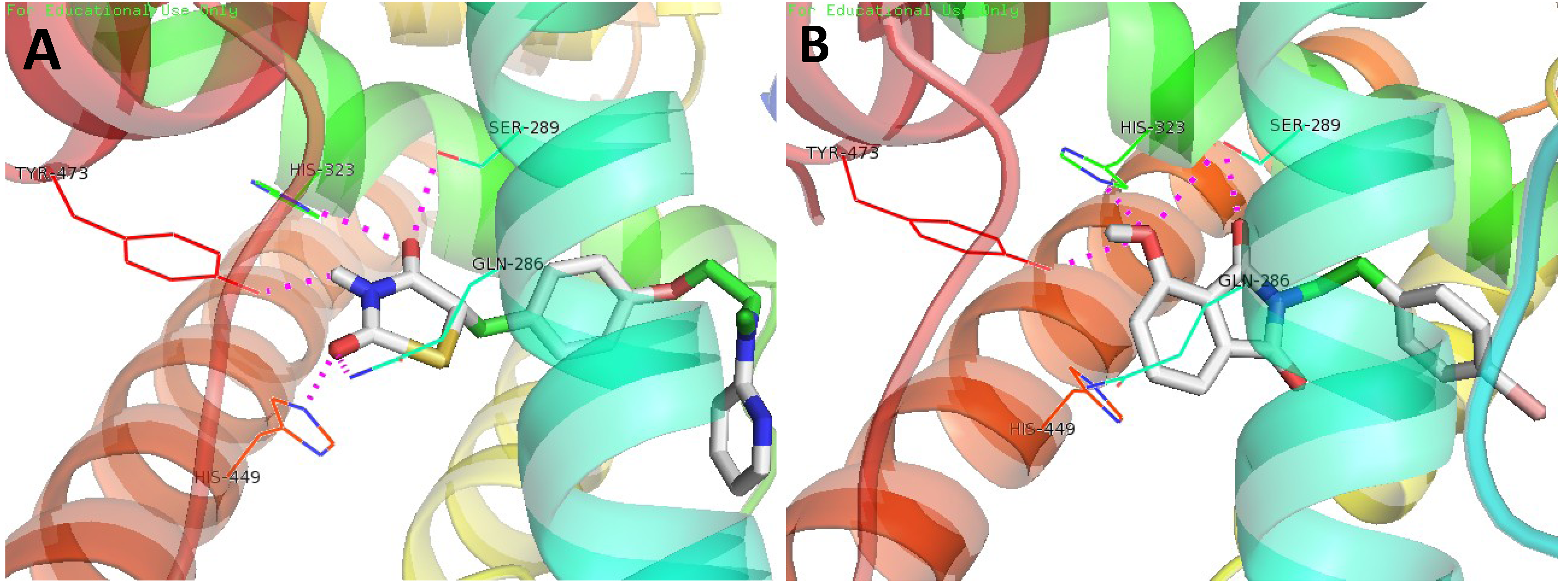
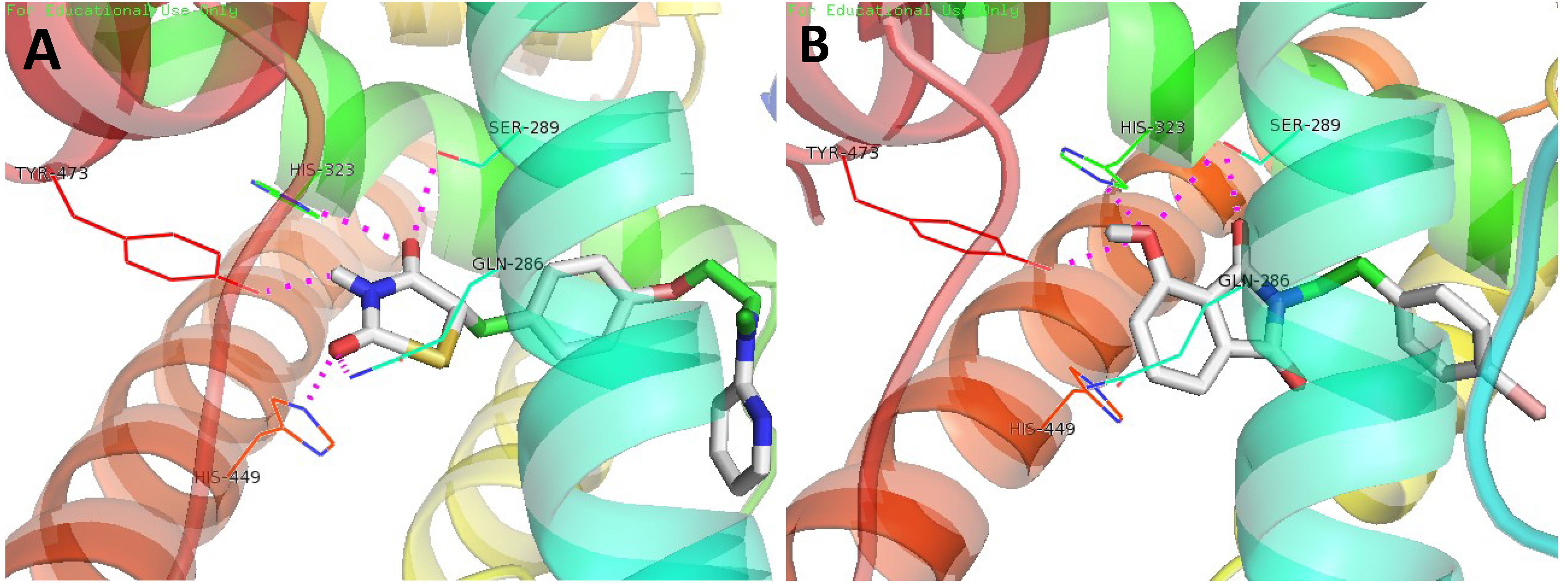
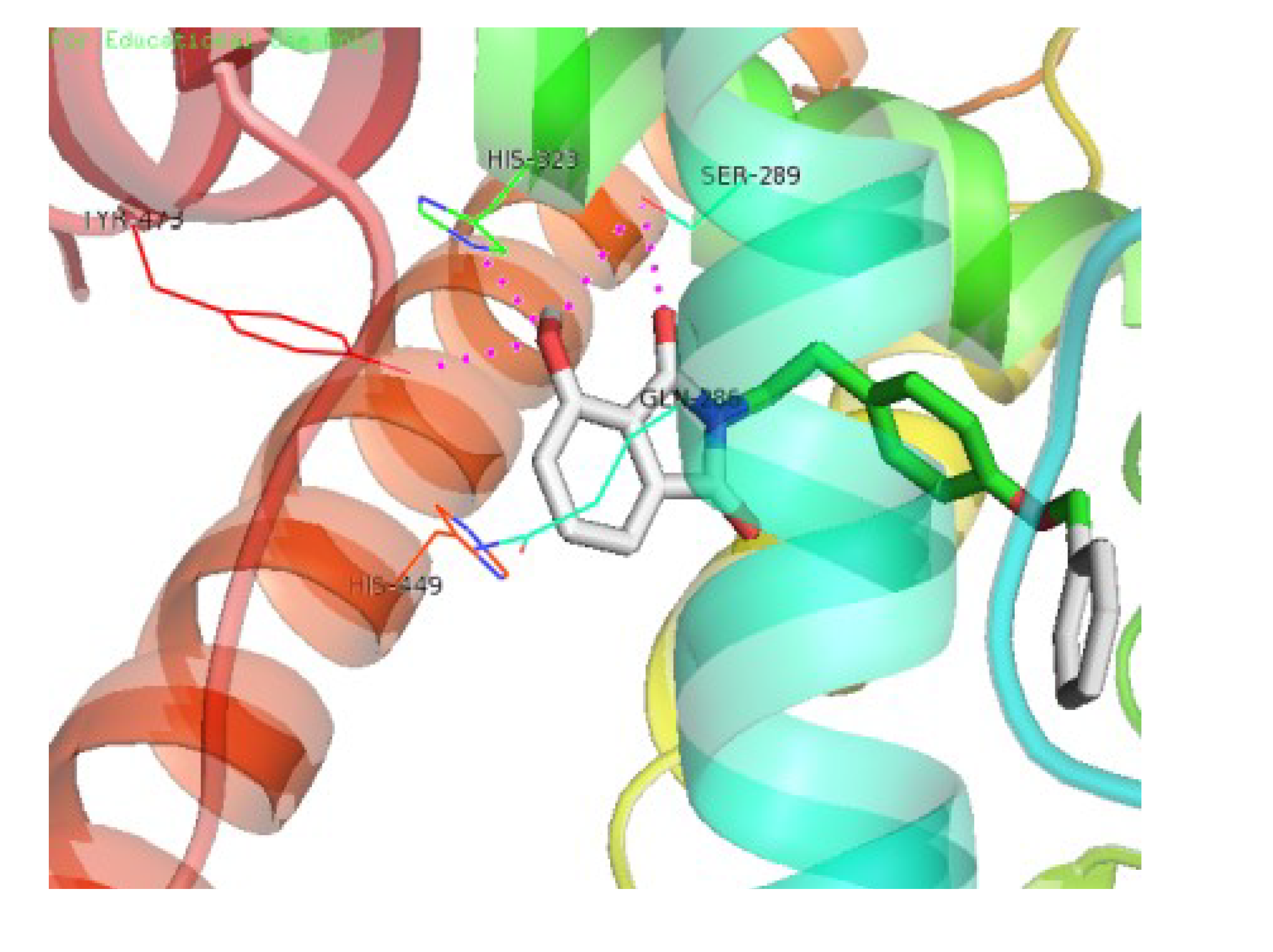
3.4. Molecular Docking Study
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Evans, R.M.; Barish, G.D.; Wang, Y.X. PPARs and the complex journey to obesity. Nat. Med. 2004, 10, 355–361. [Google Scholar] [CrossRef]
- Berger, J.; Moller, D.E. The mechanisms of action of PPARs. Annu. Rev. Med. 2002, 53, 409–435. [Google Scholar] [CrossRef]
- Mangelsdorf, D.J.; Thummel, C.; Beato, M.; Herrlich, P.; Schötz, G.; Umesono, K.; Blumberg, B.; Kastner, P.; Mark, M.; Chambon, P. The nuclear receptor superfamily: The second decade. Cell 1995, 83, 835–839. [Google Scholar] [CrossRef]
- Nuclear Receptors Nomenclature Committee. A unified nomenclature system for the nuclear receptor superfamily. Cell 1999, 97, 161–163. [Google Scholar] [CrossRef]
- Spiegelman, B.M. PPAR-γ: Adipogenic regulator and thiazolidinedione receptor. Diabetes 1998, 47, 507–514. [Google Scholar] [CrossRef]
- Mayerson, A.B.; Hundal, R.S.; Dufour, S.; Lebon, V.; Befroy, D.; Cline, G.W.; Enocksson, S.; Inzucchi, S.E.; Shulman, G.I.; Petersen, K.F. The effects of rosiglitazone on insulin sensitivity, lipolysis, and hepatic and skeletal muscle triglyceride content in patients with type 2 diabetes. Diabetes 2002, 51, 797–802. [Google Scholar] [CrossRef]
- Henke, B.R.; Blanchard, S.G.; Brackeen, M.F.; Brown, K.K.; Cobb, J.E.; Collins, J.L.; Harrington, W.W., Jr.; Hashim, M.A.; Hull-Ryde, E.A.; Kaldor, I. N-(2-Benzoylphenyl)-l-tyrosine PPARgamma agonists. 1. Discovery of a novel series of potent antihyperglycemic and antihyperlipidemic agents. J. Med. Chem. 1998, 41, 5020–5036. [Google Scholar] [CrossRef]
- Collins, J.L.; Blanchard, S.G.; Boswell, G.E.; Charifson, P.S.; Cobb, J.E.; Henke, B.R.; Hull-Ryde, E.A.; Kazmierski, W.M.; Lake, D.H.; Leesnitzer, L.M. N-(2-Benzoylphenyl)-l-tyrosine PPARγ agonists. 2. Structure-activity relationship and optimization of the phenyl alkyl ether moiety. J. Med. Chem. 1998, 41, 5037–5054. [Google Scholar] [CrossRef]
- Cobb, J.E.; Blanchard, S.G.; Boswell, E.G.; Brown, K.K.; Charifson, P.S.; Cooper, J.P.; Collins, J.L.; Dezube, M.; Henke, B.R.; Hull-Ryde, E.A. N-(2-Benzoylphenyl)-l-tyrosine PPARγ agonists. 3. Structure-activity relationship and optimization of the N-aryl substituent. J. Med. Chem. 1998, 41, 5055–5069. [Google Scholar] [CrossRef]
- Krey, G.; Braissant, O.; L’Horset, F.; Kalkhoven, E.; Perroud, M.; Parker, M.G.; Wahli, W. Fatty acids, eicosanoids, and hypolipidemic agents identified as ligands of peroxisome proliferator-activated receptors by coactivator-dependent receptor ligand assay. Mol. Endocrinol. 1997, 11, 779–791. [Google Scholar] [CrossRef]
- Forman, B.M.; Tontonoz, P.; Chen, J.; Brun, R.P.; Spiegelman, B.M.; Evans, R.M. 15-Deoxy-δ 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell 1995, 83, 803–812. [Google Scholar] [CrossRef]
- Nolte, R.T.; Wisely, G.B.; Westin, S.; Cobb, J.E.; Lambert, M.H.; Kurokawa, R.; Rosenfeld, M.G.; Willson, T.M.; Glass, C.K.; Milburn, M.V. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature 1998, 395, 137–143. [Google Scholar] [CrossRef]
- Xiao, B.; Yin, J.; Park, M.H.; Liu, J.; Li, J.L.; Kim, E.L.; Hong, J.K.; Chung, H.Y.; Jung, J.H. Design and synthesis of marine fungal phthalide derivatives as PPAR-γ agonists. Bioorg. Med. Chem. 2012, 20, 4954–4961. [Google Scholar] [CrossRef]
- Willson, T.M.; Brown, P.J.; Sternbach, D.D.; Henke, B.R. The PPARs: From orphan receptors to drug discovery. J. Med. Chem. 2000, 43, 527–550. [Google Scholar] [CrossRef]
- Kuhn, B.; Hilpert, H.; Benz, J.; Binggeli, A.; Grether, U.; Humm, R.; Marki, H.P.; Meyer, M.; Moh, P. Structure-based design of indole propionic acids as novel PPARα/γ co-agonists. Bioorg. Med. Chem. Lett. 2006, 16, 4016–4020. [Google Scholar] [CrossRef]
- Liu, J.; Li, F.M.; Kim, E.L.; Li, J.L.; Hong, J.K.; Bae, K.S.; Chung, H.Y.; Kim, H.S.; Jung, J.H. Antibacterial polyketides from the Jellyfish-dirived fungus Pacecilomyces variotii. J. Nat. Prod. 2011, 74, 1826–1829. [Google Scholar] [CrossRef]
- Pessoa, C.; Ferreira, P.M.P.; Lotufo, L.V.; Moraes, M.O.; Cavalcanti, S.M.T.; Coêlho, L.C.D.; Hernandes, M.Z.; Leite, A.C.L.; DeSimone, C.A.; Costa, V.M.A. Discovery of phthalimides as immunomodulatory and antitumor drug prototypes. Chem. Med. Chem. 2010, 5, 523–528. [Google Scholar] [CrossRef]
- Motoshima, K.; Ishikawa, M.; Hashimoto, Y.; Sugita, K. Peroxisome proliferator-activated receptor agonists with phenethylphenylphthalimide skeleton derived from thalidomide-related liver X receptor antagonists: Relationship between absolute configuration and subtype selectivity. Bioorg. Med. Chem. 2011, 19, 3156–3172. [Google Scholar] [CrossRef]
- Pluempanupat, W.; Adisakwattana, S.; Yibchok-Anun, S.; Chavasiri, W. Synthesis of N-phenylphthalimide derivatives as alpha-glucosidase inhibitors. Arch. Pharm. Res. 2007, 30, 1501–1506. [Google Scholar]
- Santini, C.; Berger, G.D.; Han, W.; Mosley, R.; MacNaul, K.; Berger, J.; Boebber, T.; Wu, M.; Moller, D.E.; Tolman, R.L. Phenylacetic acid derivatives as hPPAR agonists. Bioorg. Med. Chem. 2003, 13, 1277–1280. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar]
Appendix
- center_x = 49.666
- center_y = −29.084
- center_z = 15.217
- size_x = 48
- size_y = 56
- size_z = 44
- exhaustiveness = 100
- mode = 9

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | AT a | AA b | NP c | AI d |
|---|---|---|---|---|
| 3 | −7.5 | −6.756 | 4 | 3.50 |
| 4 | −7.4 | −6.733 | 4 | 2.35 |
| 5 | −7.2 | −6.944 | 0 | 0.00 |
| 6 | −8.3 | −7.311 | 3 | 2.33 |
| 7 | −8.4 | −7.356 | 4 | 1.75 |
| 8 | −7.6 | −6.944 | 4 | 0.75 |
| 9 | −7.8 | −7.467 | 4 | 1.00 |
| 10 | −6.9 | −6.656 | 3 | 0.67 |
| 11 | −8.6 | −7.189 | 4 | 1.00 |
| 12 | −9.3 | −8.378 | 4 | 1.11 |
| 13 | −8.9 | −8.400 | 4 | 1.67 |
| 14 | −9.4 | −8.133 | 3 | 1.67 |
| Rosi | −8.2 | −7.033 | 4 | 1.50 |


© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Xiao, B.; Su, M.; Kim, E.L.; Hong, J.; Chung, H.Y.; Kim, H.S.; Yin, J.; Jung, J.H. Synthesis of PPAR-γ Activators Inspired by the Marine Natural Product, Paecilocin A. Mar. Drugs 2014, 12, 926-939. https://doi.org/10.3390/md12020926
Xiao B, Su M, Kim EL, Hong J, Chung HY, Kim HS, Yin J, Jung JH. Synthesis of PPAR-γ Activators Inspired by the Marine Natural Product, Paecilocin A. Marine Drugs. 2014; 12(2):926-939. https://doi.org/10.3390/md12020926
Chicago/Turabian StyleXiao, Bin, Mingzhi Su, Eun La Kim, Jongki Hong, Hae Young Chung, Hyung Sik Kim, Jun Yin, and Jee H. Jung. 2014. "Synthesis of PPAR-γ Activators Inspired by the Marine Natural Product, Paecilocin A" Marine Drugs 12, no. 2: 926-939. https://doi.org/10.3390/md12020926