Seven New and Two Known Lipopeptides as well as Five Known Polyketides: The Activated Production of Silent Metabolites in a Marine-Derived Fungus by Chemical Mutagenesis Strategy Using Diethyl Sulphate

Abstract
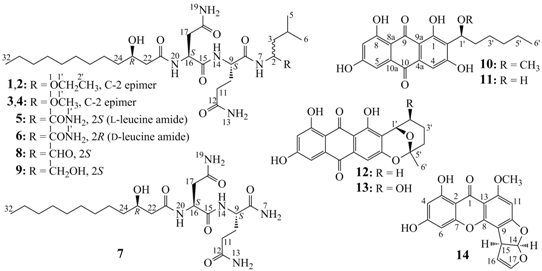
:
1. Introduction
2. Results and Discussion
2.1. Fermentation, Isolation of 1–14, and Identification of Known Compounds 8–14
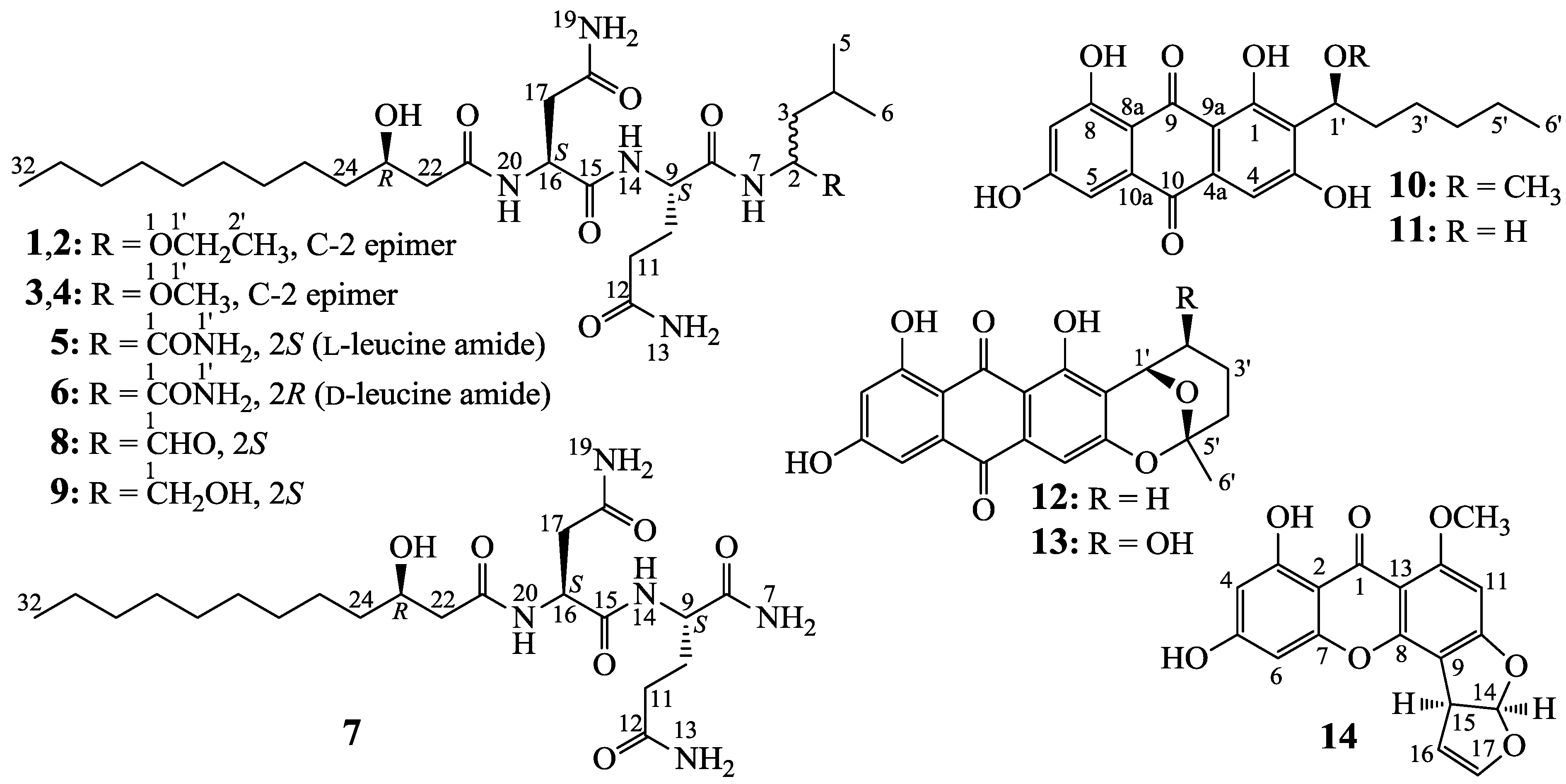
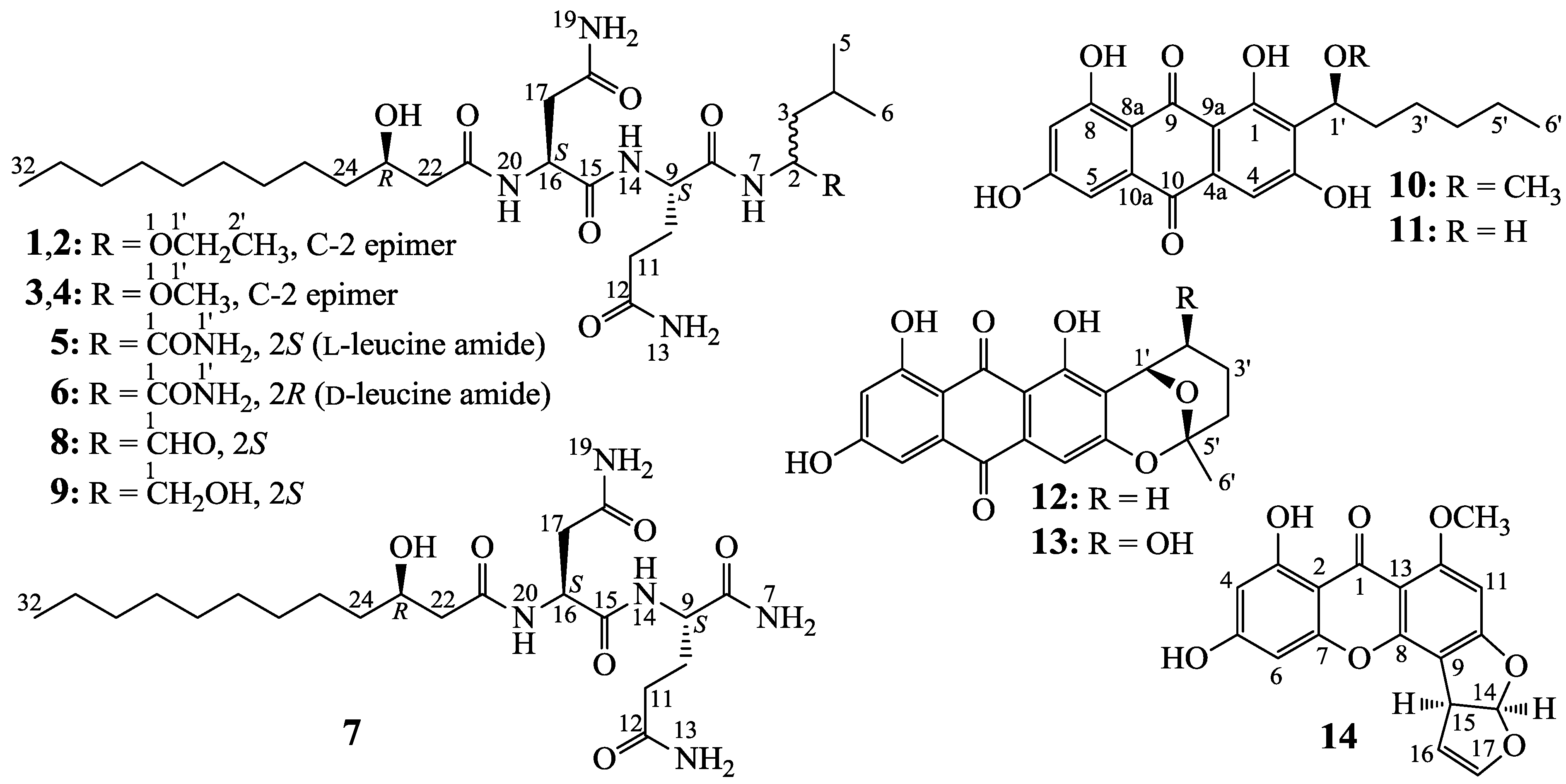
2.2. Structure Determination of New Compounds 1–7
 −18.0 (c 0.1, MeOH), and penicimutalide B (2), −17.0 (c 0.1, MeOH), were obtained as amorphous powders from MeOH, and their molecular compositions were assigned to be C28H53N5O7 by HRESIMS (measured 594.3834 [M + Na]+ for both 1 and 2, calcd for C28H53N5O7Na [M + Na]+ 594.3843). Penicimutalides C (3), −23.7 (c 0.3, MeOH), and D (4), −24.8 (c 0.3, MeOH), amorphous powders (MeOH), were assigned the same molecular formula C27H51N5O7 (HRESIMS: measured 580.3695 [M + Na]+ for 3 and 580.3684 [M + Na]+ for 4; calcd for C27H51N5O7Na [M + Na]+ 580.3686). Penicimutalides E (5), −15.0 (c 0.04, MeOH), and F (6), −18.8 (c 0.04, MeOH), amorphous powders (MeOH), had also the same molecular composition C27H50N6O7 (HRESIMS: measured 571.3819 [M + H]+ for 5 and 571.3805 [M + H]+ for 6, calcd for C27H51N6O7 [M + H]+ 571.3819). Penicimutalide G (7), an amorphous powder (MeOH), −7.5 (c 0.1, MeOH), was assigned the molecular formula C21H39N5O6 by HRESIMS (measured 458.2970 [M + H]+, calcd for C21H40N5O6 [M + H]+ 458.2979).
−18.0 (c 0.1, MeOH), and penicimutalide B (2), −17.0 (c 0.1, MeOH), were obtained as amorphous powders from MeOH, and their molecular compositions were assigned to be C28H53N5O7 by HRESIMS (measured 594.3834 [M + Na]+ for both 1 and 2, calcd for C28H53N5O7Na [M + Na]+ 594.3843). Penicimutalides C (3), −23.7 (c 0.3, MeOH), and D (4), −24.8 (c 0.3, MeOH), amorphous powders (MeOH), were assigned the same molecular formula C27H51N5O7 (HRESIMS: measured 580.3695 [M + Na]+ for 3 and 580.3684 [M + Na]+ for 4; calcd for C27H51N5O7Na [M + Na]+ 580.3686). Penicimutalides E (5), −15.0 (c 0.04, MeOH), and F (6), −18.8 (c 0.04, MeOH), amorphous powders (MeOH), had also the same molecular composition C27H50N6O7 (HRESIMS: measured 571.3819 [M + H]+ for 5 and 571.3805 [M + H]+ for 6, calcd for C27H51N6O7 [M + H]+ 571.3819). Penicimutalide G (7), an amorphous powder (MeOH), −7.5 (c 0.1, MeOH), was assigned the molecular formula C21H39N5O6 by HRESIMS (measured 458.2970 [M + H]+, calcd for C21H40N5O6 [M + H]+ 458.2979).
{kind=link}
{kind=link}
{kind=link}
| Proton | 1 b | 2 b | 3 c | 4 c | 5 b | 6 b | 7 c | 8 c | 9 c |
|---|---|---|---|---|---|---|---|---|---|
| 1 | — | — | — | — | — | — | — | 9.35 s | 3.30 ddd (10.6, 5.6, 4.8) |
| — | — | — | — | — | — | — | — | 3.23 ddd (10.6, 6.0, 4.8) | |
| 2 | 5.07 dt (9.2, 6.9) | 5.06 dt (9.1, 6.9) | 4.97 dt (9.1, 6.9) | 4.96 dt (9.4, 7.2) | 4.07–4.02 m | 4.15–4.07 m | — | 4.08–4.00 m | 3.84–3.72 (overlapped) |
| 3 | 1.48 dt (13.8, 6.9) | 1.45 dt (13.8, 6.9) | 1.49 dt (13.8, 6.9) | 1.49–1.38 (2H) m | 1.59–1.50 m | 1.59–1.49 m | — | 1.56–1.44 (2H) m | 1.40–1.25 (overlapped) |
| 1.40–1.33 m | 1.40 dt (13.8, 6.9) | 1.42–1.30 m | — | 1.48–1.42 m | 1.49–1.40 m | — | — | — | |
| 4 | 1.65–1.57 m | 1.60–1.52 m | 1.65–1.54 m | 1.61–1.49 m | 1.59–1.50 m | 1.59–1.49 m | — | 1.68–1.58 m | 1.63–1.52 m |
| 5 | 0.83 d (6.8) | 0.81 d (6.6) | 0.83 d (6.7) | 0.81 d (6.6) | 0.80 d (6.6) | 0.79 d (6.2) | — | 0.84 d (6.4) | 0.82 d (6.4) |
| 6 | 0.86 d (6.7) | 0.84 d (6.8) | 0.85 d (6.7) | 0.84 d (6.6) | 0.87 d (6.6) | 0.84 d (5.6) | — | 0.89 d (6.8) | 0.86 d (6.4) |
| 7 | 8.02 d (9.2) | 8.04 d (9.1) | 8.03 d (9.1) | 8.04 d (9.4) | 7.86 d (8.4) | 7.87 d (8.4) | 7.34 br s | 8.27 d (7.2) | 7.43 d (7.6) |
| 7.08 br s | |||||||||
| 9 | 4.13 td (7.8, 4.6) | 4.13–4.05 m | 4.18–4.08 m | 4.13–4.05 m | 4.13–4.09 m | 4.15–4.07 m | 4.06 td (8.0, 4.6) | 4.21–4.16 m | 4.15–4.05 (overlapped) |
| 10 | 1.99–1.89 m | 1.97–1.90 m | 2.02–1.88 m | 2.00–1.89 m | 1.95–1.88 m | 2.00–1.92 m | 1.95 dtd (13.2, 7.5, 4.6) | 2.02–1.90 m | 1.97–1.85 m |
| 1.78–1.68 m | 1.78–1.70 m | 1.80–1.68 m | 1.81–1.69 m | 1.78–1.72 m | 1.77–1.67 m | 1.70 ddt (13.2, 8.0, 7.5) | 1.82–1.68 m | 1.78–1.65 m | |
| 11 | 2.12–2.04 m | 2.13–2.03 m | 2.14–2.03 m | 2.13–2.05 m | 2.21–2.16 m | 2.08–2.00 m | 2.06 t (7.5) | 2.14–2.04 m | 2.11–2.02 m |
| 13 | 7.21 br s | 7.20 br s | 7.25 br s | 7.24 br s | 7.22 br s | 7.18 br s | 7.21 br s | 7.21 br s | 7.20 br s |
| 6.75 br s | 6.74 br s | 6.79 br s | 6.78 br s | 6.76 br s | 6.71 br s | 6.75 br s | 6.76 br s | 6.74 br s | |
| 14 | 7.94 d (7.8) | 8.06 d (7.6) | 8.05 d (8.0) | 8.12 d (7.6) | 8.10 d (7.2) | 8.09 d (8.4) | 7.98 d (8.0) | 8.10 d (7.6) | 8.00 d (8.0) |
| 16 | 4.49 q (7.0) | 4.50 q (7.0) | 4.49 q (6.9) | 4.50 q (7.0) | 4.48 q (7.2) | 4.45 q (6.9) | 4.48 q (7.0) | 4.50 q (7.2) | 4.48 br q (6.9) |
| 17 | 2.54 dd (15.6, 7.0) | 2.55 dd (15.6, 7.0) | 2.54 dd (15.5, 6.9) | 2.55 dd (15.6, 7.0) | 2.58–2.42 m | 2.54 dd (15.6, 6.9) | 2.57 dd (15.5, 7.0) | 2.55 dd (15.5, 7.2) | 2.55 dd (15.6, 6.9) |
| 2.43 dd (15.6, 7.0) | 2.43 dd (15.6, 7.0) | 2.43 dd (15.5, 6.9) | 2.42 dd (15.6, 7.0) | 2.58–2.42 m | 2.43 dd (15.6, 6.9) | 2.43 dd (15.5, 7.0) | 2.44 dd (15.5, 7.2) | 2.43 dd (15.6, 6.9) | |
| 19 | 7.37 br s | 7.40 br s | 7.41 br s | 7.43 br s | 7.46 br s | 7.41 br s | 7.42 br s | 7.42 br s | 7.42 br s |
| 6.90 br s | 6.94 br s | 6.93 br s | 7.24 br s | 6.95 br s | 6.91 br s | 6.96 br s | 6.93 br s | 6.94 br s | |
| 20 | 8.08 d (7.0) | 8.05 d (7.0) | 8.12 d (6.9) | 8.08 d (7.0) | 8.26 d (7.2) | 8.15 d (7.2) | 8.09 d (7.0) | 8.09 d (7.2) | 8.10 d (6.9) |
| 22 | 2.26–2.16 m | 2.25–2.16 m | 2.27-2.16 m | 2.25–2.16 m | 2.24–2.16 m | 2.25–2.15 m | 2.25–2.14 m | 2.24–2.16 m | 2.25–2.17 m |
| 23 | 3.82–3.75 m | 3.80–3.75 m | 3.84–3.73 m | 3.82–3.72 m | 3.80–3.74 m | 3.81–3.74 m | 3.82–3.73 m | 3.82–3.72 m | 3.84–3.72 (overlapped) |
| 24 | 1.40–1.33 m | 1.39–1.30 m | 1.42–1.30 m | 1.38–1.30 m | 1.40–1.18 m | 1.40–1.18 m | 1.40–1.20 m | 1.40–1.20 m | 1.40–1.20 m |
| 25 | 1.40–1.33 m | 1.39–1.30 m | 1.42–1.30 m | 1.38–1.30 m | 1.40–1.18 m | 1.40–1.18 m | 1.40–1.20 m | 1.40–1.20 m | 1.40–1.20 m |
| 1.30–1.18 m | 1.30–1.18 m | 1.30–1.18 m | 1.30–1.18 m | 1.40–1.18 m | 1.40–1.18 m | 1.40–1.20 m | 1.40–1.20 m | 1.40–1.20 m | |
| 26 | 1.30–1.18 m | 1.30–1.18 m | 1.30–1.18 m | 1.30–1.18 m | 1.40–1.18 m | 1.40–1.18 m | 1.40–1.20 m | 1.40–1.20 m | 1.40–1.20 m |
| 27 | 1.30–1.18 m | 1.30–1.18 m | 1.30–1.18 m | 1.30–1.18 m | 1.40–1.18 m | 1.40–1.18 m | 1.40–1.20 m | 1.40–1.20 m | 1.40–1.20 m |
| 28 | 1.30–1.18 m | 1.30–1.18 m | 1.30–1.18 m | 1.30–1.18 m | 1.40–1.18 m | 1.40–1.18 m | 1.40–1.20 m | 1.40–1.20 m | 1.40–1.20 m |
| 29 | 1.30–1.18 m | 1.30–1.18 m | 1.30–1.18 m | 1.30–1.18 m | 1.40–1.18 m | 1.40–1.18 m | 1.40–1.20 m | 1.40–1.20 m | 1.40–1.20 m |
| 30 | 1.30–1.18 m | 1.30–1.18 m | 1.30–1.18 m | 1.30–1.18 m | 1.40–1.18 m | 1.40–1.18 m | 1.40–1.20 m | 1.40–1.20 m | 1.40–1.20 m |
| 31 | 1.30–1.18 m | 1.30–1.18 m | 1.30–1.18 m | 1.30–1.18 m | 1.40–1.18 m | 1.40–1.18 m | 1.40–1.20 m | 1.40–1.20 m | 1.40–1.20 m |
| 32 | 0.85 t (7.0) | 0.86 t (7.2) | 0.85 t (7.2) | 0.86 t (6.8) | 0.85 t (6.9) | 0.85 t (6.9) | 0.86 t (6.8) | 0.85 t (6.8) | 0.86 t (6.8) |
| 1' | 3.44 dq (14.1, 7.0) | 3.46 dq (14.1, 7.0) | 3.11 (3H) s | 3.12 (3H) s | 6.97 br s | 7.17 br s | — | — | — |
| 3.27 dq (14.1, 7.0) | 3.28 dq (14.1, 7.0) | — | — | 6.95 br s | 6.97 br s | — | — | — | |
| 2' | 1.05 t (7.0) | 1.05 t (7.0) | — | — | — | — | — | — | — |
| 1–O H | — | — | — | — | — | — | — | — | 4.54 br t (4.8) |
| 23–O H | 4.64 d (4.8) | 4.62 d (4.8) | 4.68 d (4.8) | 4.65 d (5.1) | 4.61 d (5.4) | 4.66 br s | 4.65 d (5.1) | 4.63 d (5.2) | 4.64 br d (4.8) |
| Carbon | 1 b | 2 b | 3 c | 4 c | 5 b | 6 b | 7 c | 8 c | 9 c |
|---|---|---|---|---|---|---|---|---|---|
| 1 | — | — | — | — | 174.2 s | 174.3 s | — | 201.6 s | 63.7 t |
| 2 | 77.2 d | 77.3 d | 78.9 d | 78.9 d | 49.9 d | 48.7 d | — | 56.6 d | 48.8 d |
| 3 | 43.3 t | 43.1 t | 43.0 t | 42.8 t | 40.0 t | 40.2 t | — | 36.3 t | 40.2 t |
| 4 | 24.0 d | 23.9 d | 24.1 d | 24.0 d | 24.2 d | 24.1 d | — | 23.9 d | 24.1 d |
| 5 | 22.3 q | 22.2 q | 22.4 q | 22.2 q | 21.2 q | 21.8 q | — | 21.3 q | 22.1 q |
| 6 | 22.4 q | 22.5 q | 22.5 q | 22.7 q | 23.0 q | 23.4 q | — | 23.1 q | 23.4 q |
| 8 | 171.5 s | 171.6 s | 171.7 s | 171.8 s | 170.9 s | 170.5 s | 173.3 s | 171.9 s | 170.5 s |
| 9 | 52.6 d | 52.8 d | 52.8 d | 53.9 d | 53.2 d | 52.6 d | 52.3 d | 52.4 d | 52.7 d |
| 10 | 27.6 t | 27.5 t | 27.6 t | 27.5 t | 27.1 t | 27.7 t | 27.5 t | 27.5 t | 27.8 t |
| 11 | 31.4 t | 31.4 t | 31.5 t | 31.5 t | 31.4 t | 31.5 t | 31.5 t | 31.4 t | 31.5 t |
| 12 | 173.7 s | 173.7 s | 173.9 s | 173.8 s | 173.8 s | 173.9 s | 173.8 s | 173.8 s | 173.9 s |
| 15 | 171.0 s | 171.1 s | 171.3 s | 171.2 s | 170.9 s | 170.9 s | 170.8 s | 171.1 s | 170.9 s |
| 16 | 49.9 d | 49.7 d | 50.0 d | 49.8 d | 49.9 d | 49.8 d | 49.8 d | 49.8 d | 49.9 d |
| 17 | 36.9 t | 37.0 t | 36.9 t | 36.9 t | 36.9 t | 36.8 t | 36.9 t | 37.0 t | 37.0 t |
| 18 | 171.6 s | 171.7 s | 171.8 s | 171.9 s | 171.8 s | 171.8 s | 171.9 s | 171.8 s | 171.8 s |
| 21 | 171.2 s | 171.1 s | 171.2 s | 171.1 s | 171.2 s | 171.2 s | 171.1 s | 171.2 s | 171.2 s |
| 22 | 43.4 t | 43.4 t | 43.5 t | 43.5 t | 43.4 t | 43.4 t | 43.5 t | 43.5 t | 43.5 t |
| 23 | 67.4 d | 67.4 d | 67.5 d | 67.5 d | 67.4 d | 67.4 d | 67.5 d | 67.5 d | 67.4 d |
| 24 | 36.8 t | 36.8 t | 37.0 t | 37.1 t | 37.0 t | 36.9 t | 36.9 t | 36.9 t | 36.9 t |
| 25 | 25.1 t | 25.1 t | 25.2 t | 25.2 t | 25.0 t | 25.1 t | 25.1 t | 25.1 t | 25.1 t |
| 26 | 29.1t | 29.1t | 29.2 t | 29.2 t | 29.1 t | 29.1 t | 28.7 t | 29.1 t | 29.1 t |
| 27 | 29.1t | 29.1t | 29.1 t | 29.2 t | 29.1 t | 29.0 t | 29.0 t | 29.1 t | 29.1 t |
| 28 | 29.0 t | 29.0 t | 29.1 t | 29.1 t | 28.9 t | 28.9 t | 29.1 t | 29.0 t | 29.0 t |
| 29 | 28.7 t | 28.7 t | 28.8 t | 28.8 t | 28.7 t | 28.7 t | 29.1 t | 28.7 t | 28.7 t |
| 30 | 31.3 t | 31.3 t | 31.4 t | 31.4 t | 31.2 t | 31.3 t | 31.3 t | 31.3 t | 31.3 t |
| 31 | 22.0 t | 22.0 t | 22.2 t | 22.2 t | 22.0 t | 22.1 t | 22.1 t | 22.1 t | 21.8 t |
| 32 | 13.9 q | 13.9 q | 14.0 q | 14.0 q | 13.9 q | 13.9 q | 14.0 q | 14.0 q | 14.0 q |
| 1′ | 61.8 t | 61.8 t | 54.3 q | 54.4 q | — | — | — | — | — |
| 2′ | 15.0 q | 15.1 q | — | — | — | — | — | — | — |
−16.0 (c 0.05, CHCl3); lit. −17.8 (c 1.2, CHCl3) [24]) and established the 23R configuration in 1–7. Therefore, absolute configurations have been defined for all chiral carbons in 1–7 except for the C-2 carbons in 1–4. Thus, 1/2 and 3/4 were each pairs of epimers at C-2.2.3. HPLC-PDAD-UV/HPLC-ESI-MS Analyses of G59 and AD-2-1 Extracts for Detecting 1–14

2.4. Inhibitory Effects of 1–14 on Several Human Cancer Cell lines
| Compound | K562 | HL-60 | HeLa | BGC-823 | MCF-7 |
|---|---|---|---|---|---|
| 1 | 37.6% | 14.1% | 12.8% | 19.9% | 33.0% |
| 2 | 34.4% | 28.0% | 27.6% | 28.1% | 34.5% |
| 3 | 40.7% | 19.8% | 19.1% | 13.5% | 27.0% |
| 4 | 40.0% | 29.6% | 29.0% | 27.9% | 38.0% |
| 5 | 17.0% | 42.6% | 42.0% | 43.9% | 12.5% |
| 6 | 10.1% | 38.8% | 37.9% | 49.0% | 21.8% |
| 7 | 16.1% | 12.0% | 11.5% | 10.3% | 16.5% |
| 9 | 48.3% | 33.4% | 33.0% | 33.3% | 35.5% |
3. Experimental Section
3.1. General Experimental
3.2. MTT Assay
3.3. Fermentation and EtOAc Extract Preparation
3.3.1. Initial Fungal Strain and its Mutant the 1–14 Producing Strain
3.3.2. Preparation of Spore Suspensions
3.3.3. Fermentation and Extraction
3.4. Isolation of Compounds 1–14
3.5. Physicochemical and Spectral Data for Compounds 1–14
−18.0 (c 0.1, MeOH). Positive ESI-MS m/z: 594 [M + Na]+; negative ESI-MS m/z: 570 [M − H]−, 606 [M + Cl]−, 616 [M + HCOO]−, 660 [M + HC2O4]−. Positive HR-ESI-MS m/z: measured 594.3834 [M + Na]+, calcd for C28H53N5O7Na [M + Na]+ 594.3843, measured 610.3626 [M + K]+, calcd for C28H53N5O7Na [M + K]+ 610.3582. IR νmax cm−1 (KBr): 3422, 3283, 2958, 2922, 2852, 1663, 1626, 1552, 1536, 1516, 1443, 1384, 1311, 1261, 1095, 1018, 928, 879. 1H and 13C NMR data: see Table 1 and Table 2 and see also Table S1 in the Supplementary Information. −17.0 (c 0.1, MeOH). Positive ESI-MS m/z: 594 [M + Na]+; negative ESI-MS m/z: 570 [M − H]−, 606 [M + Cl]−, 616 [M + HCOO]−, 660 [M + HC2O4]−. Positive HR-ESI-MS m/z: measured 594.3847 [M + Na]+, calcd for C28H53N5O7Na [M + Na]+ 594.3843, measured 610.3612 [M + K]+, calcd for C28H53N5O7K [M + K]+ 610.3582. IR νmax cm−1 (Diamond ATR crystal): 3283, 3215, 2934, 2862, 1654, 1540, 1419, 1320, 1290, 1255, 1147, 1122, 1063, 883, 865. 1H and 13C NMR data: see Table 1 and Table 2 and see also Table S2 in the Supplementary Information. −23.7 (c 0.3, MeOH). Positive ESI-MS m/z: 558 [M + H]+, 580 [M + Na]+; negative ESI-MS m/z: 592 [M + Cl]−. Positive HR-ESI-MS m/z: measured 580.3695 [M + Na]+, calcd for C27H51N5O7Na [M + Na]+ 580.3686. IR νmax cm−1 (Diamond ATR crystal): 3268, 2934, 2862, 1650, 1536, 1418, 1325, 1282, 1256, 1196, 1145, 1114, 1062, 979, 949. 1H and 13C NMR data: see Table 1 and Table 2 and see also Table S3 in the Supplementary Information. −24.8 (c 0.3, MeOH). Positive ESI-MS m/z: 558 [M + H]+, 580 [M + Na]+, 596 [M + K]+; negative ESI-MS m/z: 556 [M − H]−,592 [M + Cl]−. Positive HR-ESI-MS m/z: measured 580.3684 [M + Na]+, calcd for C27H51N5O7Na [M + Na]+ 580.3686, measured 596.3426 [M + K]+, calcd for C28H53N5O7K [M + K]+ 596.3426. IR νmax cm−1 (Diamond ATR crystal): 3274, 2930, 2859, 1650, 1632, 1537, 1417, 1325, 1281, 1255, 1197, 1139, 1112, 1056, 946, 865. 1H and 13C NMR data: see Table 1 and Table 2 and see also Table S4 in the Supplementary Information. −15.0 (c 0.04, MeOH). Positive ESI-MS m/z: 571 [M + H]+; negative ESI-MS m/z: 569 [M − H]−. Positive HR-ESI-MS m/z: measured 571.3819 [M + H]+, calcd for C27H51N6O7 [M + H]+ 571.3819; measured 593.3634 [M + Na]+, calcd for C27H50N6O7Na [M + Na]+ 593.3639. IR νmax cm−1 (Diamond ATR crystal): 3273, 2931, 2859, 1642, 1546, 1537, 1418, 1324, 1283, 1156, 1134, 1074, 864, 783. 1H and 13C NMR data: see Table 1 and Table 2 and see also Table S5 in the Supplementary Information. −18.8 (c 0.04, MeOH). Positive ESI-MS m/z: 571 [M + H]+; negative ESI-MS m/z: 569 [M − H]−. Positive HR-ESI-MS m/z: measured 571.3805 [M + H]+, calcd for C27H51N6O7 [M + H]+ 571.3819. IR νmax cm−1 (Diamond ATR crystal): 3273, 2933, 2860, 1653, 1628, 1540, 1418, 1177, 1132, 1024. 1H and 13C NMR data: see Table 1 and Table 2 and see also Table S6 in the Supplementary Information. −7.5 (c 0.1, MeOH). Positive ESI-MS m/z: 458 [M + H]+, 480 [M + Na]+; negative ESI-MS m/z: 456 [M − H]−, 492 [M + Cl]−. Positive HR-ESI-MS m/z: measured 458.2970 [M + H]+, calcd for C21H40N5O6 [M + H]+ 458.2979; measured 480.2798 [M + Na]+, calcd for C21H39N5O6Na [M + Na]+ 480.2798; measured 496.2526 [M + K]+, calcd for C27H5N5O7K [M + K]+ 496.2537. IR νmax cm−1 (Diamond ATR crystal): 3274, 3205, 2929, 2857, 1651, 1632, 1540, 1414, 1320, 1281, 1254, 1200, 1133, 1095, 1070, 1042, 958, 866. 1H and 13C NMR data: see Table 1 and Table 2 and see also Table S7 in the Supplementary Information. −28.0 (c 0.5, MeOH). Positive ESI-MS m/z: 556 [M + H]+; negative ESI-MSm/z: 554 [M − H]−. 1H and 13C NMR data: see Table 1 and Table 2. −33.0 (c 0.2, MeOH). Positive ESI-MS m/z: 558 [M + H]+, 580 [M + Na]+; negative ESI-MS m/z: 592 [M + Cl]−. Positive HR-ESI-MS m/z: measured 558.3862 [M + H]+, calcd for C27H52N5O7 [M + H]+ 558.3867; measured 580.3692 [M + Na]+, calcd for C27H51N5O7Na [M + Na]+ 580.3686; measured 596.3413 [M + K]+, calcd for C27H51N5O7K [M + K]+ 596.3426. 1H and 13C NMR data: see Table 1 and Table 2. +1.8 (c 0.4, MeOH), ESI-MS m/z: 387 [M + H]+. 1H NMR (acetone-d6, 400MHz) δ: 12.78 (1H, s, 1-OH), 12.18 (1H, s, 8-OH), 10.10 (1H, br s, 6-OH), 9.69 (1H, br s, 3-OH) 7.23 (1H, d, J = 2.4 Hz, H-5), 7.13 (1H, s, H-4), 6.64 (1H, d, J = 2.4 Hz, H-7), 4.97 (1H, dd, J = 7.9, 5.1 Hz, H-1′), 3.49 (3H, s, 1′-OCH3), 1.95–1.75 (2H, m, H2-3′), 1.58–1.38 (2H, m, H2-2′), 1.38–1.24 (4H, m, H2-4′, H2-5′), 0.89 (3H, t, J = 6.8 Hz, H3-6′). −130.0 (c 0.52, MeOH). ESI-MS m/z: 373 [M + H]+. 1H NMR (acetone-d6, 400MHz) δ: 12.63 (1H, s, 1-OH), 12.08 (1H, s, 8-OH), 9.00 (2H, br s, 1-OH, 3-OH), 7.09 (1H, d, J = 2.4 Hz, H-5), 7.00 (1H, s, H-4), 6.50 (1H, d, J = 2.4 Hz, H-7), 5.29 (1H, dd, J = 7.8, 4.5 Hz, H-1′), 1.80–1.63 (2H, m, H2-3′), 1.50–1.30 (2H, m, H2-2′), 1.30–1.10 (4H, m, H2-4′, H2-5′), 0.76 (3H, t, J = 6.8 Hz, H3-6′). 13C NMR (acetone-d6, 100 MHz) δ: 190.7 (C-9), 181.8 (C-10), 166.0 (C-8), 165.9 (C-1), 165.0 (C-6),161.7 (C-3), 136.4 (C-4a), 134.5 (C-10a), 122.0 (C-2), 110.3 (C-5), 110.2 (C-9a), 109.4 (C-8a), 108.9 (C-7), 108.8 (C-4), 69.9 (C-1′), 36.9 (C-2′), 32.3 (C-3′), 25.6 (C-4′), 23.2 (C-5′), 14.3 (C-6′). −1.7 (c 0.3, CHCl3). 1H NMR (acetone-d6, 400MHz) δ: 12.51 (1H, s, 1-OH), 12.19 (1H, s, 8-OH), 10.09 (1H, s, 6-OH), 7.22 (1H, d, J = 2.5 Hz, H-5), 7.09 (1H, s, H-4), 6.63 (1H, d, J = 2.5 Hz, H-7), 5.29 (1H, d, J = 2.8 Hz, H-1′), 2.15–1.25 (6H, m, H2-2′,3′,4′), 1.55 (3H, s, H3-6′). 13C NMR (acetone-d6, 100MHz) δ: 190.7 (C-9), 181.6 (C-10), 166.0 (C-8), 165.8 (C-1), 161.5 (C-6), 159.8 (C-3), 136.3 (C-4a), 134.4 (C-10a), 117.1 (C-2), 110.2 (C-5), 109.5 (C-9a), 109.4 (C-8a), 108.9 (C-7), 108.4 (C-4), 102.2 (C-5′), 67.4 (C-1′), 36.3 (C-2′), 28.0 (C-4′), 27.9 (C-6′), 16.5 (C-3′). +0.3 (c 0.2, CHCl3). ESI-MS m/z: 385 [M + H]+. 1H NMR (acetone-d6, 400MHz) δ: 12.58 (1H, s, 1-OH), 12.22 (1H, s, 8-OH), 10.17 (1H, s, 6-OH), 7.26 (1H, d, J = 2.5 Hz, H-5), 7.14 (1H, s, H-4), 6.67 (1H, d, J = 2.5 Hz, H-7), 5.17 (1H, br s, H-1′), 3.95 (1H, br s, H-2′), 2.30–1.30 (4H, m, H2-3′, H2-4′), 1.58 (3H, s, H3-6′). −384 (c 0.5, CHCl3), ESI-MS m/z: 325 [M + H]+, 347 [M + Na]+. 1H NMR (400 MHz, CDCl3) δ: 13.23(1H, s, 3-OH), 7.47 (1H, dd, J = 8.4, 8.0 Hz, H-5), 6.80 (1H, d, J = 7.0 Hz, H-14), 6.79 (1H, dd, J = 8.0, 1.0 Hz, H-6), 6.72 (1H, dd, J = 8.4, 1.0 Hz, H-4), 6.50 (1H, t, J = 2.5 Hz, H-17), 6.40 (1H, s, H-11), 5.43 (1H, t, J = 2.5 Hz, H-16), 4.76 (1H, dt, J = 7.0, 2.5 Hz, H-15), 3.98 (3H, s, H3-18). 13C NMR (100 MHz, CDCl3) δ: 181.2 (C-1), 164.4 (C-10), 163.1 (C-12), 162.1 (C-3), 154.8 (C-7), 153.8 (C-8), 145.2 (C-17), 135.6 (C-5), 113.2 (C-14), 111.1 (C-4), 108.7 (C-2), 106.4 (C-6), 105.8 (C-9), 105.7 (C-13), 102.4 (C-16), 90.4 (C-11), 56.7 (C-18), 47.9 (C-15).3.6. Marfey Analysis for 1–9
3.6.1. Hydrolysis and Derivatization with FDAA
3.6.2. Oxidization of Leucinal in 8 to Leucine, then Hydrolysis and Derivatization with FDAA
3.6.3. HPLC Analysis of FDAA Derivatives of 1–9 to Determine Absolute Configurations
| FDAA derivative | tR (min) | FDAA derivative | tR (min) |
|---|---|---|---|
| l-Aspartic acid | 28.60 | d-Aspartic acid | 30.55 |
| l-Glutamic acid | 30.85 | d-Glutamic acid | 33.38 |
| l-leucinol | 51.14 | d-leucinol | 59.53 |
| l-leucine | 53.11 | d-leucine | 59.85 |
3.7. Hydrolysis of 1–9 for Determination of the 23R Absolute Configuration in 1–9
−16 (c 0.05, CHCl3). Positive ESI-MS m/z: 239 [M + Na]+; negative ESI-MS m/z: 215 [M − H]−. 1H NMR (400 MHz, CDCl3) δ: 4.02 (1H, m), 2.57 (2H, dd, J = 16.6, 3.0 Hz), 2.47 (1H, dd, J = 16.6, 9 Hz), 1.40–1.58 (2H, m), 1.20–1.36 (14H, m), 0.87 (3H, t, J = 6.6 Hz). 13C NMR (CDCl3) δ: 176.8, 68.0, 40.8, 36.5, 31.9, 29.7, 29.5, 29.4, 29.3, 25.4, 22.7, 14.1. The and 1H and 13C NMR data are identical with those reported for 3R-hydroxydodecanoic acid in the literature [24].3.8. HPLC-PDAD-UV Analysis for Detecting 1–14 in the G59 and AD-2-1 Extracts
3.9. HPLC-ESI-MS Analysis for Detecting 10–14 in the G59 and AD-2-1 Extracts
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Brakhage, A.A.; Schroeckh, V. Fungal secondary metabolites—Strategies to activate silent gene clusters. Fungal Genet. Biol. 2011, 48, 15–22. [Google Scholar]
- Cichewicz, R.H.; Henrikson, J.C.; Wang, X.; Branscum, K.M. Strategies for accessing microbial secondary metabolites from silent biosynthetic pathways. In Manual of Industrial Microbiology and Biotechnology, 3rd ed.; Baltz, R.H., Davies, J.E., Demain, A.L., Bull, A.T., Junker, B., Katz, L., Lynd, L.R., Masurekar, P.C., Reeves, D., Zhao, H., Eds.; ASM Press: Washington, DC, USA, 2010; pp. 78–95. [Google Scholar]
- Ochi, K.; Hosaka, T. New strategies for drug discovery: Activation of silent or weakly expressed microbial gene clusters. Appl. Microbiol. Biotechnol. 2013, 97, 87–98. [Google Scholar]
- Hertweck, C. Hidden biosynthetic treasures brought to light. Nat. Chem. Biol. 2009, 5, 450–452. [Google Scholar] [CrossRef]
- Bode, H.B.; Beth, B.; Höfs, R.; Zeek, A. Big effects from small changes: Possible ways to explore nature’s chemical diversity. ChemBioChem 2002, 3, 619–627. [Google Scholar]
- Cichewicz, R.H. Epigenome manipulation as a pathway to new natural product scaffolds and their congeners. Nat. Prod. Rep. 2010, 27, 11–22. [Google Scholar] [CrossRef]
- Takahashi, J.A.; Teles, A.P.C.; Bracarense, A.A.P.; Gomes, D.C. Classical and epigenetic approaches to metabolite diversification in filamentous fungi. Phytochem. Rev. 2013. [Google Scholar] [CrossRef]
- Ochi, K. From microbial differentiation to ribosome engineering. Biosci. Biothenol. Biochem. 2007, 71, 1373–1386. [Google Scholar] [CrossRef]
- Hosaka, T.; Ohnishi-Kameyama, M.; Muramatsu, H.; Murakami, K.; Tsurumi, Y.; Kodani, S.; Yoshida, M.; Fujie, A.; Ochi, K. Antibacterial discovery in actinomycetes strains with mutations in RNA polymerase or ribosomal protein S12. Nat. Biotechnol. 2009, 27, 462–464. [Google Scholar] [CrossRef]
- Chai, Y.J.; Cui, C.B.; Li, C.W.; Wu, C.J.; Tian, C.K.; Hua, W. Activation of the dormant secondary metabolite production by introducing gentamicin-resistance in a marine-derived Penicillium purpurogenum G59. Mar. Drugs 2012, 10, 559–582. [Google Scholar]
- Wu, C.J.; Cui, C.B.; Tian, C.K.; Li, C.W. Antitumor metabolites produced by two Penicillium purpurogenum G59 mutants. J. Int. Pharm. Res. 2010, 37, 122–126. [Google Scholar]
- Fang, S.M.; Wu, C.J.; Li, C.W.; Cui, C.B. A practical strategy to discover new antitumor compounds by activating silent metabolite production in fungi by diethyl sulphate mutagenesis. Mar. Drugs 2014, 12, 1788–1814. [Google Scholar]
- Fang, S.M.; Cui, C.B.; Li, C.W.; Wu, C.J.; Zhang, Z.J.; Li, L.; Huang, X.J.; Ye, W.C. Purpurogemutantin and purpurogemutantidin, new drimenyl cyclohexenone derivatives produced by a mutant obtained by diethyl sulfate mutagenesis of a marine-derived Penicillium purpurogenum G59. Mar. Drugs 2012, 10, 1266–1287. [Google Scholar]
- Tian, C.K.; Cui, C.B.; Han, X.X. Isolation of fungal strains in unusual environment and screening for their antitumor activity. J. Int. Pharm. Res. 2008, 35, 401–405. [Google Scholar]
- Shigemori, H.; Wakuri, S.; Yazawa, K.; Nakamura, T.; Sasaki, T.; Kobayashi, J. Fellutamides A and B, cytotoxic peptides from a marine fish-possessing fungus Penicillium fellutanum. Tetrahedron 1991, 47, 8529–8534. [Google Scholar]
- Lee, Y.M.; Dang, H.T.; Hon, J.; Lee, C.O.; Bae, K.S.; Kim, D.K.; Jung, J.H. A cytotoxic lipopeptide from the sponge-derived fungus Aspergillus versicolor. Bull. Korean Chem. Soc. 2010, 31, 205–208. [Google Scholar]
- Shao, C.; Wang, C.; Wei, M.; Li, S.; She, Z.; Gu, Y.; Lin, Y. Structural and spectral assignments of six anthraquinone derivatives from the mangrove fungus (ZSUH-36). Magn. Reson. Chem. 2008, 46, 886–889. [Google Scholar]
- Ren, H.; Gu, Q.-Q.; Cui, C.-B. Anthraquinone derivatives produced by marine-derived Penicillium flavidorsum SHK1227 and their antitumor activities. Chin. J. Med. Chem. 2007, 77, 148–154. [Google Scholar]
- O’Malley, G.J.; Murphy, R.A., Jr.; Cava, M.P. Aflatoxin precursors: Total synthesis of (±)-averufin and (±)-nidurufin. J. Org. Chem. 1985, 50, 5533–5537. [Google Scholar]
- Muphy, R.A., Jr.; Cava, M.P. Stereochemistry of nidurufin: Synthesis of 6,8-dideoxynidurufin and 6,8-dideoxyepinidurufin. J. Am. Chem. Soc. 1984, 106, 7630–7632. [Google Scholar]
- Pachler, K.G.R.; Steyn, P.S.; Vleggaar, R.; Wessels, P.L.; Scott, D.B. Carbon-13 nuclear magnetic resonance assignments and biosynthesis of aflatoxin B1 and sterigmatocystin. J. Chem. Soc. Perkin Trans. 1 1976, 11, 1182–1189. [Google Scholar]
- Marfey, P. Determination of d-amino acids. II. Use of a bifunctional reagent, 1,5-difluoro-2,4-dinitrobenzene. Carlsberg Res. Commun. 1984, 49, 591–596. [Google Scholar]
- Xu, D.; Ondeyka, J.; Harris, G.H.; Zink, D.; Kahn, J.N.; Wang, H.; Bills, G.; Platas, G.; Wang, W.; Szewczak, A.A.; et al. Isolation, structure, and biological activities of fellutamides C and D from an undescribed Metulocladosporiella (Chaetothyriales) using the genome-wide Candida albicans fitness test. J. Nat. Prod. 2011, 74, 1721–1730. [Google Scholar]
- Guaragna, A.; Nisco, D.M.; Pedatella, S.; Palumbo, G. Studies towards lipid A: A synthetic strategy for the enantioselective preparation of 3-hydroxy fatty acids. Tetrahedron: Asymmetry 2006, 17, 2839–2841. [Google Scholar]
- Rateb, M.E.; Ebel, R. Secondary metabolites of fungi from marine habitats. Nat. Prod. Rep. 2011, 28, 290–344. [Google Scholar] [CrossRef]
- Saleem, M.; Ali, M.S.; Hussain, S.; Jabbar, A.; Ashraf, M.; Lee, Y.S. Marine natural products of fungal origin. Nat. Prod. Rep. 2007, 24, 1142–1152. [Google Scholar]
- Yamakuchi, K; Tsuji, T.; Wakuri, S.; Yazawa, K.; Kondo, K.; Shigemori, H.; Kobayashi, J. Stimulation of nerve growth factor synthesis and secretion by fellutamide A in vitro. Biosci. Biotechnol. Biochem. 1993, 57, 195–199. [Google Scholar]
- Schneekloth, J.S., Jr.; Sanders, J.L.; Hines, J.; Crews, C.M. Neurotrophic peptide aldehydes: Solid phase synthesis of fellutamide B and a simplified analog. Bioorg. Med. Chem. Lett. 2006, 16, 3855–3858. [Google Scholar] [CrossRef]
- Hines, J.; Groll, M.; Fahnestock, M.; Crews, C.M. Proteasome inhibition by fellutamide B induces nerve growth factor synthesis. Chem. Biol. 2008, 15, 501–512. [Google Scholar]
- Moore, B.S.; Eustáquio, A.S.; McGlinchey, R.P. Advances in and applications of proteasome inhibitors. Curr. Opin. Chem. Biol. 2008, 12, 434–440. [Google Scholar] [CrossRef]
- Lin, G.; Li, D.; Chidawanyika, T.; Nathan, C.; Li, H. Fellutamide B is a potent inhibitor of the Mycobacterium tuberculosis proteasome. Arch. Biochem. Biophys. 2010, 501, 214–220. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wu, C.-J.; Li, C.-W.; Cui, C.-B. Seven New and Two Known Lipopeptides as well as Five Known Polyketides: The Activated Production of Silent Metabolites in a Marine-Derived Fungus by Chemical Mutagenesis Strategy Using Diethyl Sulphate. Mar. Drugs 2014, 12, 1815-1838. https://doi.org/10.3390/md12041815
Wu C-J, Li C-W, Cui C-B. Seven New and Two Known Lipopeptides as well as Five Known Polyketides: The Activated Production of Silent Metabolites in a Marine-Derived Fungus by Chemical Mutagenesis Strategy Using Diethyl Sulphate. Marine Drugs. 2014; 12(4):1815-1838. https://doi.org/10.3390/md12041815
Chicago/Turabian StyleWu, Chang-Jing, Chang-Wei Li, and Cheng-Bin Cui. 2014. "Seven New and Two Known Lipopeptides as well as Five Known Polyketides: The Activated Production of Silent Metabolites in a Marine-Derived Fungus by Chemical Mutagenesis Strategy Using Diethyl Sulphate" Marine Drugs 12, no. 4: 1815-1838. https://doi.org/10.3390/md12041815
APA StyleWu, C.-J., Li, C.-W., & Cui, C.-B. (2014). Seven New and Two Known Lipopeptides as well as Five Known Polyketides: The Activated Production of Silent Metabolites in a Marine-Derived Fungus by Chemical Mutagenesis Strategy Using Diethyl Sulphate. Marine Drugs, 12(4), 1815-1838. https://doi.org/10.3390/md12041815