Preparative Separation and Purification of Trichothecene Mycotoxins from the Marine Fungus Fusarium sp. LS68 by High-Speed Countercurrent Chromatography in Stepwise Elution Mode


 ,
,  and
and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. HPLC Analysis of the Crude Extract
2.2. Optimization of Suitable HSCCC Solvent System
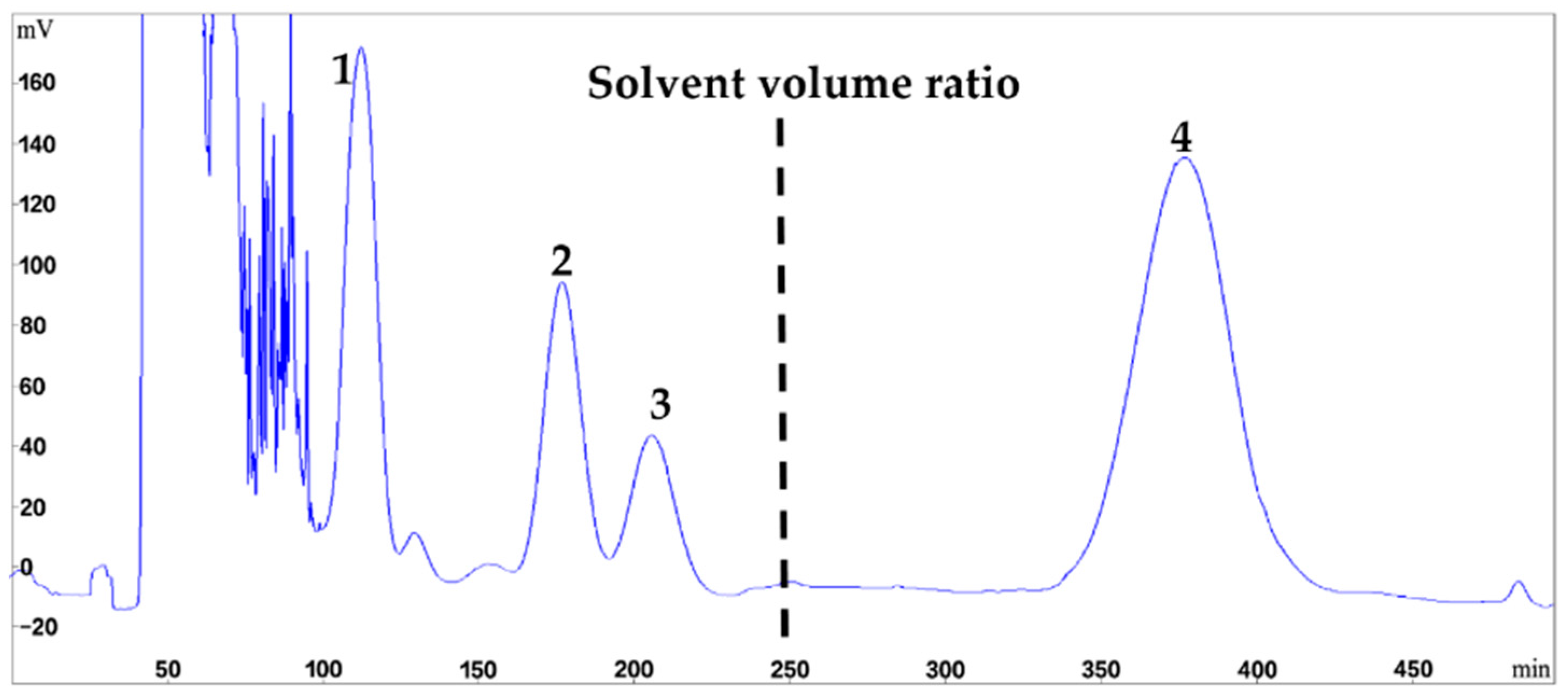
2.3. Stepwise HSCCC Separation
2.4. Structural Identification
3. Materials and Methods
3.1. Reagents and Materials
3.2. Fungal Material
3.3. Apparatus
3.4. Culturing and Extraction
3.5. Determination of Partition Coefficients (KD)
3.6. HSCCC Separation
3.7. HPLC Analysis and Identification of the Peaks
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cast, I. Mycotoxins: Risks in Plant, Animal, And Human Systems; Task Force Report No. 139; Council for Agriculture Science and Technology: Ames, IA, USA, 2003. [Google Scholar]
- Berthiller, F.; Crews, C.; Dall’Asta, C.; Saeger, S.D.; Haesaert, G.; Karlovsky, P.; Oswald, I.P.; Seefelder, W.; Speijers, G.; Stroka, J. Masked mycotoxins: A review. Mol. Nutr. Food Res. 2013, 57, 165–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMullen, M.; Jones, R.; Gallenberg, D. Scab of wheat and barley: A re-emerging disease of devastating impact. Plant Dis. 1997, 81, 1340–1348. [Google Scholar] [CrossRef]
- Goswami, R.S.; Kistler, H.C. Heading for disaster: Fusarium graminearum on cereal crops. Mol. Plant Pathol. 2004, 5, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Starkey, D.E.; Ward, T.J.; Aoki, T.; Gale, L.R.; Kistler, H.C.; Geiser, D.M.; Suga, H.; Tóth, B.; Varga, J.; O’Donnell, K. Global molecular surveillance reveals novel fusarium head blight species and trichothecene toxin diversity. Fungal Genet. Biol. 2007, 44, 1191–1204. [Google Scholar] [CrossRef] [PubMed]
- Kazan, K.; Gardiner, D.M.; Manners, J.M. On the trail of a cereal killer: Recent advances in Fusarium graminearum pathogenomics and host resistance. Mol. Plant Pathol. 2012, 13, 399–413. [Google Scholar] [CrossRef] [PubMed]
- Grove, J.F. Macrocyclic trichothecenes. Nat. Prod. Rep. 1993, 10, 429–448. [Google Scholar] [CrossRef]
- Grove, J.F. The trichothecenes and their biosynthesis. Prog. Chem. Org. Nat. Prod. 2007, 38, 63–130. [Google Scholar] [CrossRef]
- Wilkins, K.; Nielsen, K.F.; Din, S.U. Patterns of volatile metabolites and nonvolatile trichothecenes produced by isolates of Stachybotrys, Fusarium, Trichoderma, Trichothecium and Memnoniella. Environ. Sci. Pollut. Res. 2003, 10, 162–166. [Google Scholar] [CrossRef]
- Trapp, S.; Hohn, T.; McCormick, S.; Jarvis, B. Characterization of the gene cluster for biosynthesis of macrocyclic trichothecenes in Myrothecium roridum. Mol. Gen. Genet. 1998, 257, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, B.B.; Mazzola, E.P. Macrocyclic and other novel trichothecenes: Their structure, synthesis, and biological significance. Acc. Chem. Res. 1982, 15, 388–395. [Google Scholar] [CrossRef]
- Desjardins, A.E.; Mccormick, S.P.; Appell, M. Structure-activity relationships of trichothecene toxins in an Arabidopsis thaliana leaf assay. J. Agric. Food Chem. 2007, 55, 6487–6492. [Google Scholar] [CrossRef] [PubMed]
- Alexander, N.J.; McCormick, S.P.; Ziegenhorn, S.L. Phytotoxicity of selected trichothecenes using Chlamydomonas reinhardtii as a model system. Nat. Toxins 1999, 7, 265–269. [Google Scholar] [CrossRef]
- Eudes, F.; Comeau, A.; Rioux, S.; Collin, J. Phytotoxicité de huit mycotoxines associées à la fusariose de l’épi chez le blé. Can. J. Plant Pathol. 2000, 22, 286–292. [Google Scholar] [CrossRef]
- Veprikova, Z.; Zachariasova, M.; Dzuman, Z.; Zachariasova, A.; Fenclova, M.; Slavikova, P.; Vaclavikova, M.; Mastovska, K.; Hengst, D.; Hajslova, J. Mycotoxins in plant-based dietary supplements: Hidden health risk for consumers. J. Agric. Food Chem. 2015, 63, 6633–6643. [Google Scholar] [CrossRef] [PubMed]
- Cutler, H.G.; Jarvis, B.B. Preliminary observations on the effects of macrocyclic trichothecenes on plant growth. Environ. Exp. Bot. 1985, 25, 115–128. [Google Scholar] [CrossRef]
- Ryu, S.M.; Lee, H.M.; Song, E.G.; Seo, Y.H.; Lee, J.; Guo, Y.Q.; Kim, B.S.; Kim, J.J.; Jin, S.H.; Ryu, K.H. Antiviral activities of trichothecenes isolated from Trichoderma albolutescens against pepper mottle virus. J. Agric. Food Chem. 2017, 65, 4273–4279. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhao, Y.; Gu, D.; Ayupbek, A.; Huang, Y.; Dou, J.; Ito, Y.; Zhang, T.; Aisa, H.A. Separation of the minor flavonols from flos gossypii by high-speed countercurrent chromatography. J. Liq. Chromatogr. Relat. Technol. 2010, 33, 1502–1515. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, I.A.; Fisher, D. Role of counter-current chromatography in the modernisation of chinese herbal medicines. J. Chromatogr. A 2009, 1216, 740–753. [Google Scholar] [CrossRef] [PubMed]
- Sticher, O. Natural product isolation. Nat. Prod. Rep. 2008, 25, 517–554. [Google Scholar] [CrossRef] [PubMed]
- Oka, F.; Oka, H.; Ito, Y. Systematic search for suitable two-phase solvent systems for high-speed counter-current chromatography. J. Chromatogr. A 1991, 538, 99–108. [Google Scholar] [CrossRef]
- Ito, Y. Golden rules and pitfalls in selecting optimum conditions for high-speed counter-current chromatography. J. Chromatogr. A 2005, 1065, 145–168. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Wang, H.; Yan, X.; Zhu, P.; Chen, J.; Yang, R. Preparative isolation and purification of macrolactin antibiotics from marine bacterium Bacillus amyloliquefaciens using high-speed counter-current chromatography in stepwise elution mode. J. Chromatogr. A 2013, 1272, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.Y.; Ignatova, S.; Hewitson, P.; Di, D.L. An overview of recent progress in elution mode of counter current chromatography. Trac-Trend Anal. Chem. 2016, 77, 214–225. [Google Scholar] [CrossRef]
- Ridge, C.D.; Mazzola, E.P.; Colesb, M.P.; Hinkley, S.F.R. Isolation and characterization of roridin E. Magn. Reson. Chem. 2016, 55, 337–340. [Google Scholar] [CrossRef] [PubMed]
- Isaka, M.; Punya, J.; Lertwerawat, Y.; Tanticharoen, M.; Thebtaranonth, Y. Antimalarial activity of macrocyclic trichothecenes isolated from the fungus Myrothecium verrucaria. J. Nat. Prod. 1999, 62, 329–331. [Google Scholar] [CrossRef] [PubMed]
- Namikoshi, M.; Kobayashi, H.; Yoshimoto, T.; Meguro, S.; Akano, K. Isolation and characterization of bioactive metabolites from marine-derived filamentous fungi collected from tropical and sub-tropical coral reefs. Chem. Pharm. Bull. 2000, 48, 1452–1457. [Google Scholar] [CrossRef] [PubMed]
- Temba, B.A.; Sultanbawa, Y.; Kriticos, D.J.; Fox, G.P.; Harvey, J.J.; Fletcher, M.T. Tools for defusing a major global food and feed safety risk: Nonbiological postharvest procedures to decontaminate mycotoxins in foods and feeds. J. Agric. Food Chem. 2016, 64, 8959–8972. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent System (v/v) Hexanes–EtOAc–CH3OH–H2O | KD | |||
|---|---|---|---|---|
| Compound 1 | Compound 2 | Compound 3 | Compound 4 | |
| 1:1:1:1 | 2.08 | 2.61 | 2.33 | 10.58 |
| 5.5:4.5:5:5 | 0.93 | 2.25 | 2.14 | 4.73 |
| 6:4:5:5 | 0.61 | 1.83 | 1.26 | 3.32 |
| 6.5:3.5:5:5 | 0.37 | 1.43 | 1.12 | 2.58 |
| 7:3:5:5 | 0.45 | 1.25 | 0.80 | 3.21 |
| 7.5:2.5:5:5 | 0.36 | 1.10 | 0.68 | 2.73 |
| 8:2:5:5 | 0.19 | 0.58 | 0.54 | 1.87 |
| 8.5:1.5:5:5 | 0.11 | 0.37 | 0.32 | 1.12 |
| 9:1:5:5 | 0.10 | 0.30 | 0.25 | 0.83 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Zhou, X.; Naman, C.B.; Lu, Y.; Ding, L.; He, S. Preparative Separation and Purification of Trichothecene Mycotoxins from the Marine Fungus Fusarium sp. LS68 by High-Speed Countercurrent Chromatography in Stepwise Elution Mode. Mar. Drugs 2018, 16, 73. https://doi.org/10.3390/md16020073
Liu Y, Zhou X, Naman CB, Lu Y, Ding L, He S. Preparative Separation and Purification of Trichothecene Mycotoxins from the Marine Fungus Fusarium sp. LS68 by High-Speed Countercurrent Chromatography in Stepwise Elution Mode. Marine Drugs. 2018; 16(2):73. https://doi.org/10.3390/md16020073
Chicago/Turabian StyleLiu, Yong, Xuezhen Zhou, C. Benjamin Naman, Yanbin Lu, Lijian Ding, and Shan He. 2018. "Preparative Separation and Purification of Trichothecene Mycotoxins from the Marine Fungus Fusarium sp. LS68 by High-Speed Countercurrent Chromatography in Stepwise Elution Mode" Marine Drugs 16, no. 2: 73. https://doi.org/10.3390/md16020073
APA StyleLiu, Y., Zhou, X., Naman, C. B., Lu, Y., Ding, L., & He, S. (2018). Preparative Separation and Purification of Trichothecene Mycotoxins from the Marine Fungus Fusarium sp. LS68 by High-Speed Countercurrent Chromatography in Stepwise Elution Mode. Marine Drugs, 16(2), 73. https://doi.org/10.3390/md16020073