Predicting Blood–Brain Barrier Permeability of Marine-Derived Kinase Inhibitors Using Ensemble Classifiers Reveals Potential Hits for Neurodegenerative Disorders


Abstract
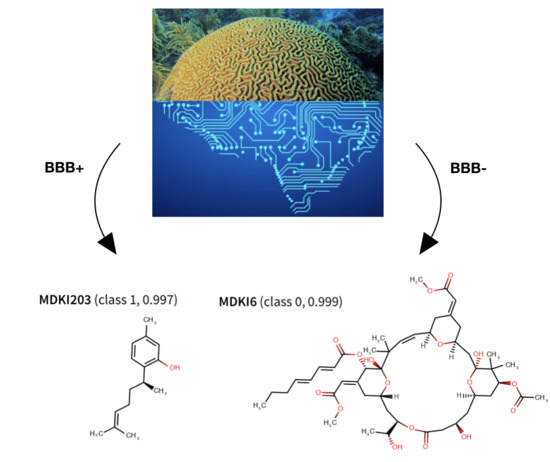
:
1. Introduction
2. Results and Discussion
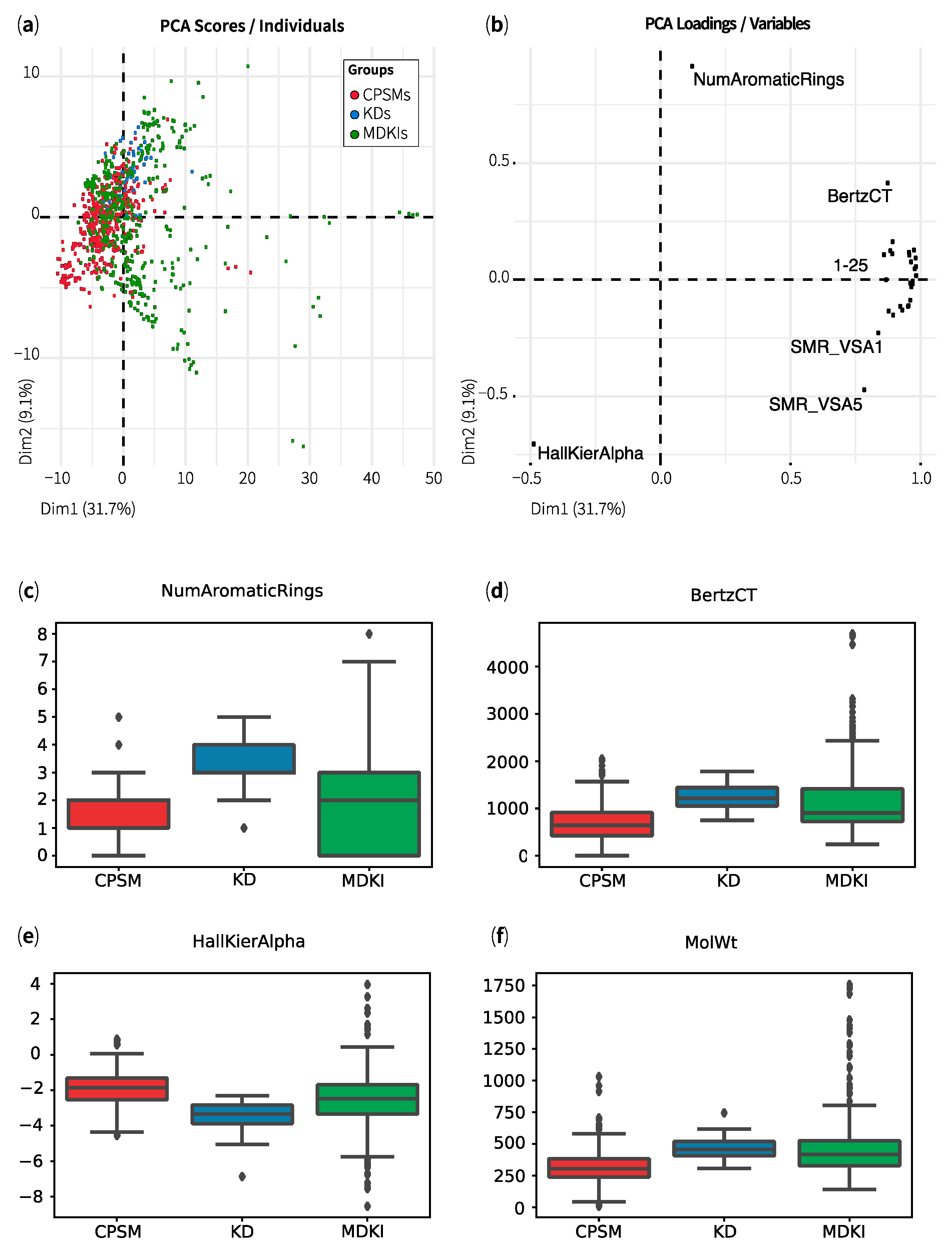
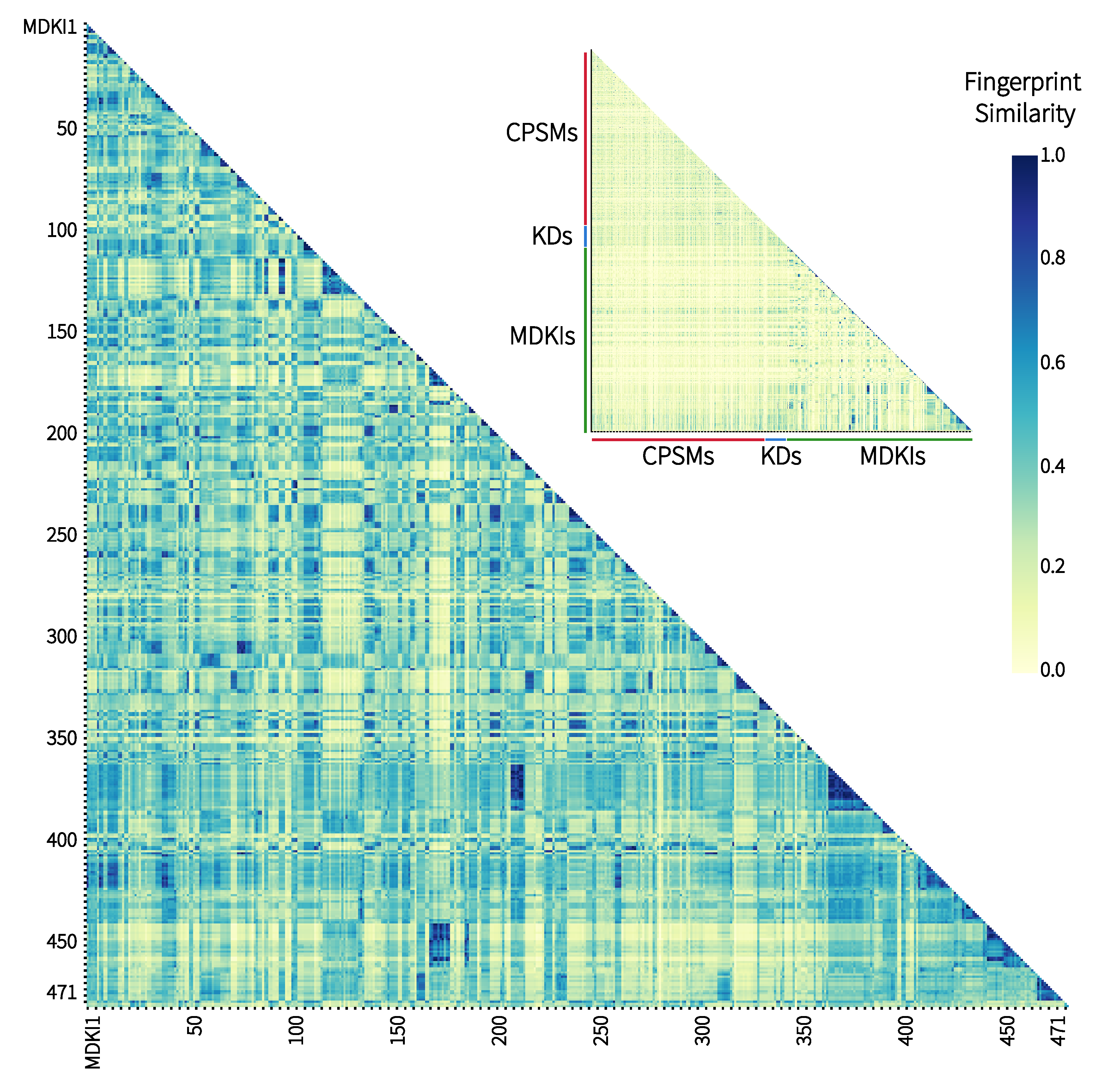
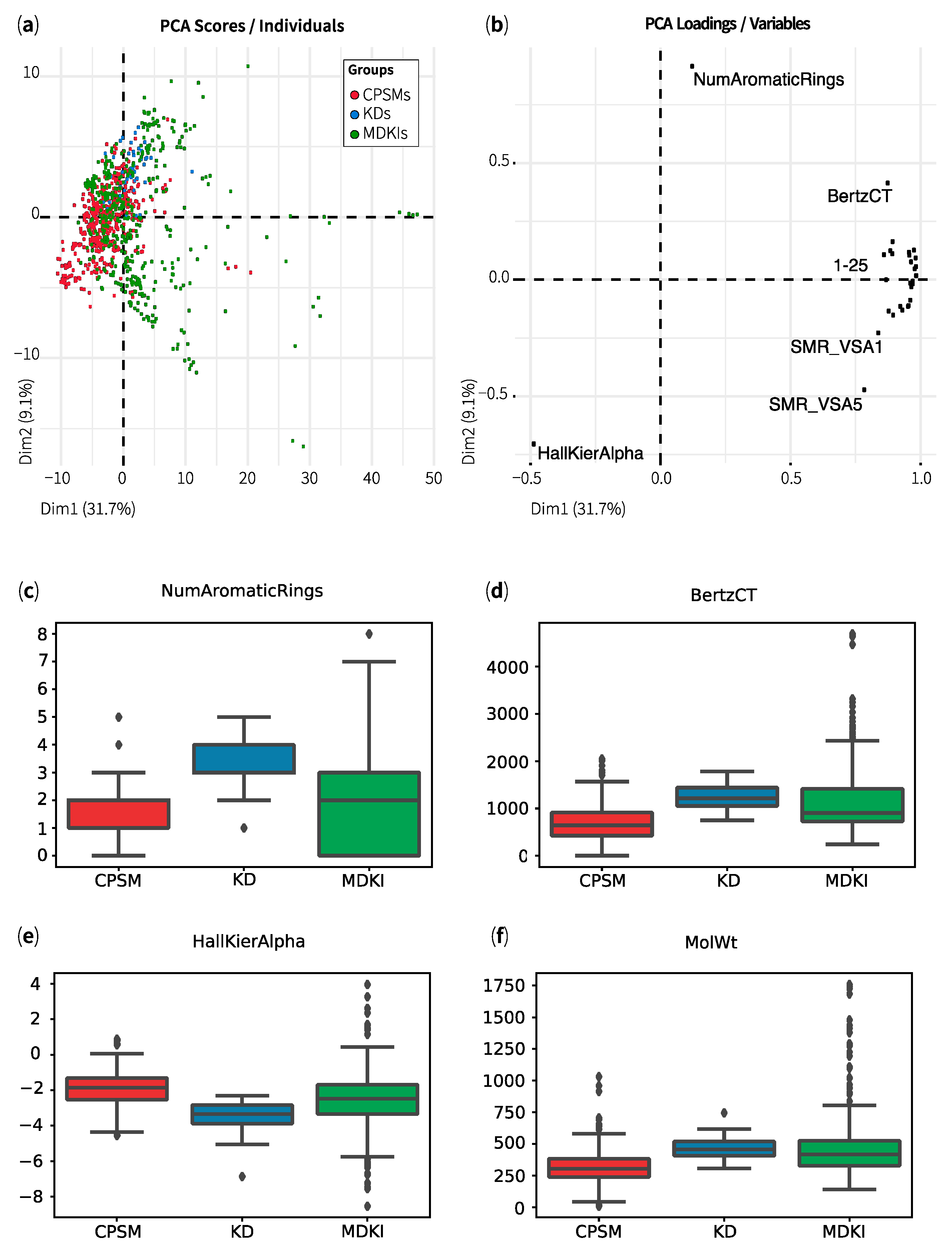
2.1. Exploratory Data Analysis: Similarities, Differences and Multicollinearity between Compounds
2.2. Comparing Classification and Regression QSPR Models
2.3. Optimising Top Performing QSPR Binary Classifiers
2.4. Identifying the Applicability Domain of our QSPR Models
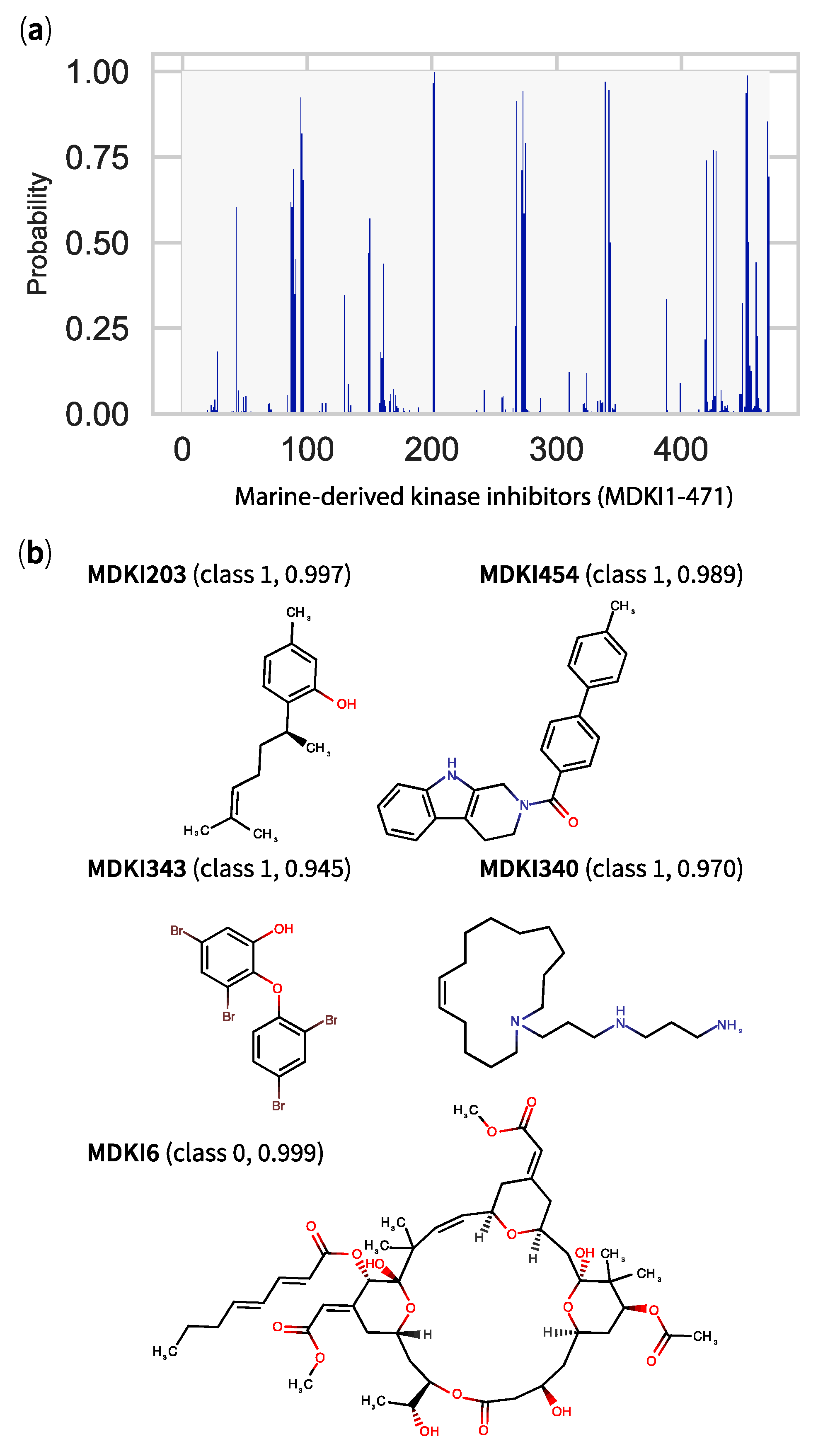
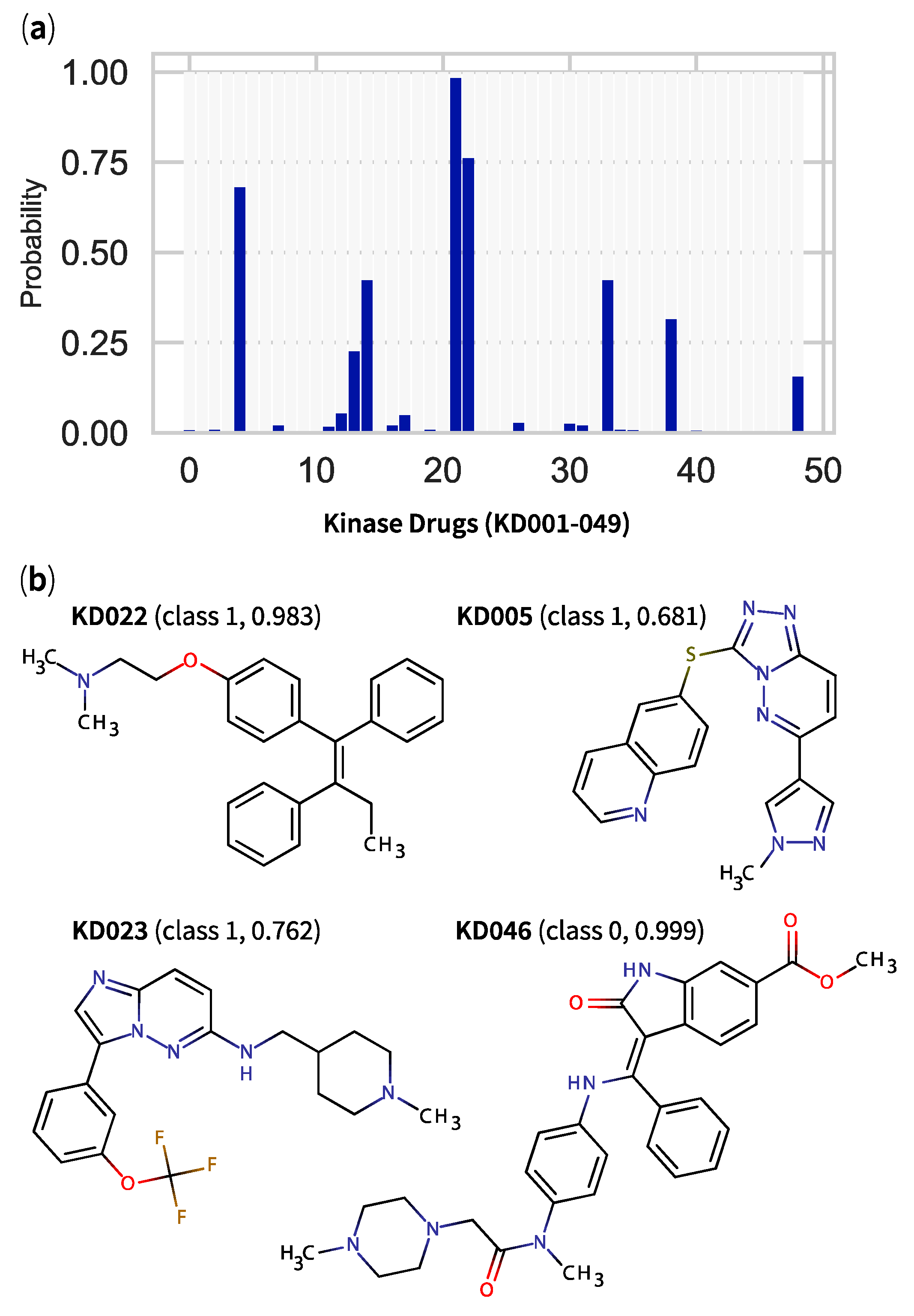
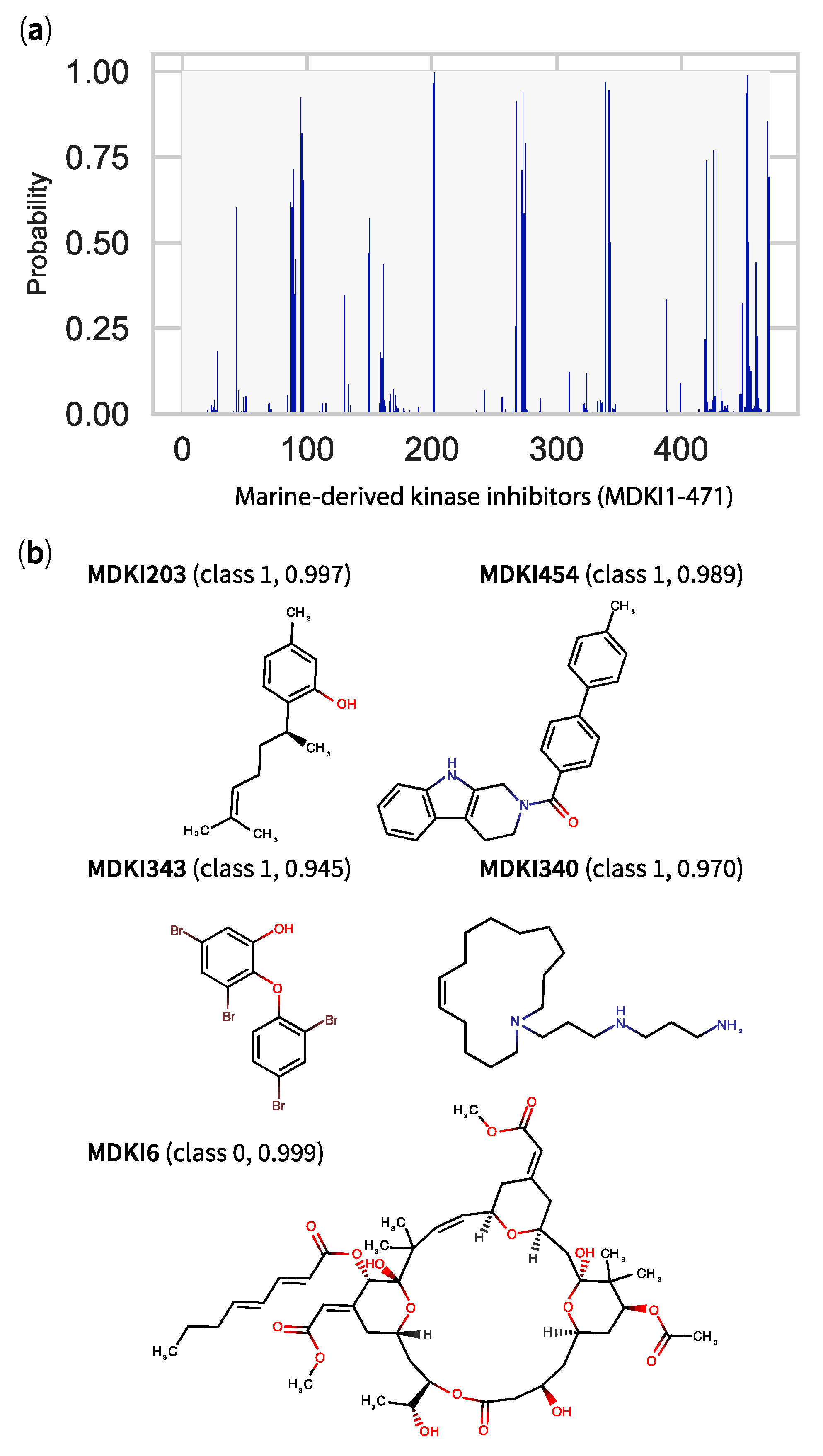
2.5. QSPR Model Results Identified Hits among KDs and MDKIs
3. Materials and Methods
3.1. Data Collection
3.2. Data Preparation
3.3. Model Building
3.4. Variable Selection and Hyperparameter Tuning
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
References
- Pettit, G.R.; Herald, C.L.; Doubek, D.L.; Herald, D.L.; Arnold, E.; Clardy, J. Isolation and Structure of Bryostatin 1. J. Am. Chem. Soc. 1982, 104, 6846–6848. [Google Scholar] [CrossRef]
- Hennings, H.; Blumberg, P.M.; Pettit, G.R.; Herald, C.L.; Shores, R.; Yuspa, S.H. Bryostatin 1, an activator of protein kinase C, inhibits tumor promotion by phorbol esters in SENCAR mouse skin. Carcinogenesis 1987, 8, 1343–1346. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.K.; Alkon, D.L. Bryostatin-1: Pharmacology and therapeutic potential as a CNS drug. CNS Drug Rev. 2006, 12, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.K.; Alkon, D.L. Pharmacology of protein kinase C activators: Cognition-enhancing and antidementic therapeutics. Pharmacol. Ther. 2010, 127, 66–77. [Google Scholar] [CrossRef]
- Nelson, T.J.; Sun, M.K.; Lim, C.; Sen, A.; Khan, T.; Chirila, F.V.; Alkon, D.L. Bryostatin effects on cognitive function and PKCє in Alzheimer’s disease phase IIa and expanded access trials. J. Alzheimer’s Dis. 2017, 58, 521–535. [Google Scholar] [CrossRef] [PubMed]
- Plisson, F.; Huang, X.-C.; Zhang, H.; Khalil, Z.; Capon, R.J. Lamellarins as inhibitors of P-glycoprotein-mediated multidrug resistance in a human colon cancer cell line. Chem-Asian J. 2012, 7, 1616–1623. [Google Scholar] [CrossRef] [PubMed]
- Plisson, F.; Conte, M.; Khalil, Z.; Huang, X.-C.; Piggott, A.M.; Capon, R.J. Kinase inhibitor scaffolds against neurodegenerative diseases from a Southern Australian Ascidian, Didemnum sp. ChemMedChem 2012, 7, 983–990. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Khalil, Z.; Conte, M.M.; Plisson, F.; Capon, R.J. A search for kinase inhibitors and antibacterial agents: Bromopyrrolo-2-aminoimidazoles from a deep-water Great Australian Bight sponge, Axinella sp. Tetrahedron Lett. 2012, 53, 3784–3787. [Google Scholar] [CrossRef]
- Plisson, F.; Prasad, P.; Xiao, X.; Piggott, A.M.; Huang, X.-C.; Khalil, Z.; Capon, R.J. Callyspongisines A–D: Bromopyrrole alkaloids from an Australian marine sponge, Callyspongia sp. Org. Biomol. Chem. 2014, 12, 1579–1584. [Google Scholar] [CrossRef] [PubMed]
- Medina Padilla, M.; Orozco Muñoz, L.; Capon, R.J. Therapeutic Use of Indole-Dihydro-Imidazole Derivatives. European Patent Application No. 12167268.7; Publication No. EP 2 662 081 A1, 13 November 2013. [Google Scholar]
- Skropeta, D.; Pastro, N.; Zivanovic, A. Kinase inhibitors from marine sponges. Mar. Drugs 2011, 9, 2131–2154. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hu, Y.; Waller, D.L.; Wang, J.; Liu, Q. Natural products as kinase inhibitors. Nat. Prod. Rep. 2012, 29, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Pla, D.; Albericio, F.; Álvarez, M. Progress on lamellarins. MedChemComm 2011, 2, 689–697. [Google Scholar] [CrossRef]
- Gao, J.; Hamann, M.T. Chemistry and biology of kahalalides. Chem. Rev. 2011, 111, 3208–3235. [Google Scholar] [CrossRef] [PubMed]
- Bharate, B.S.; Yadav, R.R.; Battula, S.; Vishwakarma, A.R. Meridianins: Marine-derived potent kinase inhibitors. Mini Rev. Med. Chem. 2012, 12, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Bharate, S.B.; Sawant, S.D.; Singh, P.P.; Vishwakarma, R.A. Kinase inhibitors of marine origin. Chem. Rev. 2013, 113, 6761–6815. [Google Scholar] [CrossRef] [PubMed]
- Cecchelli, R.; Berezowski, V.; Lundquist, S.; Culot, M.; Renftel, M.; Dehouck, M.P.; Fenart, L. Modelling of the blood—Brain barrier in drug discovery and development. Nat. Rev. Drug Discovery 2007, 6, 650–661. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Mensch, J.; Oyarzabal, J.; Mackie, C.; Augustijns, P. In vivo, in vitro and in silico methods for small molecule transfer across the BBB. J. Pharm. Sci. 2009, 98, 4429–4468. [Google Scholar] [CrossRef]
- Stanimirovic, D.B.; Bani-Yaghoub, M.; Perkins, M.; Haqqani, A.S. Blood–brain barrier models: In vitro to in vivo translation in preclinical development of CNS-targeting biotherapeutics. Expert Opin. Drug Discovery 2015, 10, 141–155. [Google Scholar] [CrossRef]
- Zhu, L.; Zhao, J.; Zhang, Y.; Zhou, W.; Yin, L.; Wang, Y.; Fan, Y.; Chen, Y.; Liu, H. ADME properties evaluation in drug discovery: In silico prediction of blood–brain partitioning. Mol. Diversity 2018, 979–990. [Google Scholar] [CrossRef]
- Chico, L.K.; Van Eldik, L.J.; Watterson, D.M. Targeting protein kinases in central nervous system disorders. Nat. Rev. Drug Discovery 2009, 8, 892–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghose, A.K.; Herbertz, T.; Hudkins, R.L.; Dorsey, B.D.; Mallamo, J.P. Knowledge-based, central nervous system (CNS) lead selection and lead optimisation for CNS drug discovery. ACS Chem. Neurosci. 2012, 3, 50–68. [Google Scholar] [CrossRef] [PubMed]
- Rankovic, Z. CNS drug design: Balancing physicochemical properties for optimal brain exposure. J. Med. Chem. 2015, 58, 2584–2608. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, T. Harnessing the potential of natural products in drug discovery from a cheminformatics vantage point. Org. Biomol. Chem. 2017, 15, 9275–9282. [Google Scholar] [CrossRef] [PubMed]
- Saldívar-González, F.I.; Pilón-Jiménez, B.A.; Medina-Franco, J.L. Chemical space of naturally occurring compounds. Phys. Sci. Rev. 2018, 0, 1–14. [Google Scholar] [CrossRef]
- Landrum, G. RDKit: Open-Source Cheminformatics Software. Available online: https://www.rdkit.org/ (accessed on 10 June 2018).
- Baunbæk, D.; Trinkler, N.; Ferandin, Y.; Lozach, O.; Ploypradith, P.; Rucirawat, S.; Ishibashi, F.; Iwao, M.; Meijer, L. Anticancer alkaloid lamellarins inhibit protein kinases. Mar. Drugs 2008, 6, 514–527. [Google Scholar] [CrossRef] [PubMed]
- Gompel, M.; Leost, M.; Bal De Kier Joffe, E.; Puricelli, L.; Hernandez Franco, L.; Palermo, J.; Meijer, L. Meridianins, a new family of protein kinase inhibitors isolated from the Ascidian Aplidium meridianum. Bioorg. Med. Chem. Lett. 2004, 14, 1703–1707. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, E.; Debiton, E.; Fabbro, D.; Moreau, P.; Prudhomme, M.; Anizon, F. In-vitro antiproliferative activities and kinase inhibitory potencies of meridianin derivatives. Anti-Cancer Drug 2008, 19, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Akue-Gedu, R.; Debiton, E.; Ferandin, Y.; Meijer, L.; Prudhomme, M.; Anizon, F.; Moreau, P. Synthesis and biological activities of aminopyrimidyl-indoles structurally related to meridianins. Bioorg. Med. Chem. 2009, 17, 4420–4424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giraud, F.; Alves, G.; Debiton, E.; Nauton, L.; Théry, V.; Durieu, E.; Ferandin, Y.; Lozach, O.; Meijer, L.; Anizon, F.; et al. Synthesis, protein kinase inhibitory potencies, and in vitro antiproliferative activities of meridianin derivatives. J. Med. Chem. 2011, 54, 4474–4489. [Google Scholar] [CrossRef] [PubMed]
- Bettayeb, K.; Tirado, O.M.; Marionneau-Lambot, S.; Ferandin, Y.; Lozach, O.; Morris, J.C.; Mateo-Lozano, S.; Drueckes, P.; Schächtele, C.; Kubbutat, M.H.G.; et al. Meriolins, a new class of cell death-inducing kinase inhibitors with enhanced selectivity for cyclin-dependent kinases. Cancer Res. 2007, 67, 8325–8334. [Google Scholar] [CrossRef] [PubMed]
- Feher, M.; Schmidt, J.M. Property distributions: Differences between drugs, natural products, and molecules from combinatorial chemistry. J. Chem. Inf. Comp. Sci. 2003, 43, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, K.; Schneider, G. Properties and architecture of drugs and natural products revisited. Curr. Chem. Biol. 2007, 1, 115–127. [Google Scholar] [CrossRef]
- Li, H.; Yap, C.W.; Ung, C.Y.; Xue, Y.; Cao, Z.W.; Chen, Y.Z. Effect of selection of molecular descriptors on the prediction of blood-brain barrier penetrating and nonpenetrating agents by statistical learning methods. J. Chem. Inf. Model. 2005, 45, 1376–1384. [Google Scholar] [CrossRef] [PubMed]
- Crivori, P.; Cruciani, G.; Carrupt, P.-A.; Testa, B. Predicting blood-brain barrier permeation from three-dimensional molecular structure. J. Med. Chem. 2000, 43, 2204–2216. [Google Scholar] [CrossRef] [PubMed]
- Doniger, S.; Hofmann, T.; Yeh, J. Predicting CNS permeability of drug molecules. J. Comput. Biol. 2002, 9, 849–864. [Google Scholar] [CrossRef] [PubMed]
- Muresan, S.; Hopkins, A.L.; Bickerton, G.R.; Paolini, G.V. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4. [Google Scholar] [CrossRef]
- Hall, L.H.; Kier, L.B. Electrotopological state indices for atom types: A novel combination of electronic, topological, and valence state information. J. Chem. Inf. Comput. Sci. 1995, 35, 1039–1045. [Google Scholar] [CrossRef]
- Kier, L.B.; Hall, L.H. Molecular Structure Description: The Electrotopological State; Elsevier Science (Academic Press): San Diego, CA, USA, 1999; ISBN 0-12-406555-4. [Google Scholar]
- Sahigara, F.; Mansouri, K.; Ballabio, D.; Mauri, A.; Consonni, V.; Todeschini, R. Comparison of different approaches to define the applicability domain of QSAR models. Molecules 2012, 17, 4791–4810. [Google Scholar] [CrossRef]
- Mathea, M.; Klingspohn, W.; Baumann, K. Chemoinformatic classification methods and their applicability domain. Mol. Inform. 2016, 35, 160–180. [Google Scholar] [CrossRef]
- Sushko, I.; Novotarskyi, S.; Ko, R.; Pandey, A.K.; Cherkasov, A.; Liu, H.; Yao, X.; Tomas, O.; Hormozdiari, F.; Dao, P.; et al. Applicability domains for classification problems: Benchmarking of distance to models for ames mutagenicity set. J. Chem. Inf. Model. 2010, 50, 2094–2111. [Google Scholar] [CrossRef] [PubMed]
- Klingspohn, W.; Mathea, M.; Ter Laak, A.; Heinrich, N.; Baumann, K. Efficiency of different measures for defining the applicability domain of classification models. J. Cheminformatics 2017, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Lien, E.A.; Solheim, E.; Ueland, P.M. Distribution of tamoxifen and its metabolites in rat and human tissues during steady-state treatment. Cancer Res. 1991, 51, 4837–4844. [Google Scholar] [PubMed]
- Kimelberg, H.K. Tamoxifen as a powerful neuroprotectant in experimental stroke and implications for human stroke therapy. Recent Pat. CNS Drug Discov. 2008, 3, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Liao, K.; Chang, C. Tamoxifen usage correlates with increased risk of Parkinson’s disease in older women with breast cancer: A case—control study in Taiwan. Eur. J. Clin. Pharmacol. 2018, 74, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Latourelle, J.C.; Dybdahl, M.; Destefano, A.; Myers, R.H.; Lash, T.L. Risk of Parkinson’ s disease after tamoxifen treatment. BMC Neurol. 2010, 10, 2–8. [Google Scholar] [CrossRef]
- Russo, P.; Kisialiou, A.; Lamonaca, P.; Moroni, R.; Prinzi, G.; Fini, M. New drugs from marine organisms in Alzheimer’s disease. Mar. Drugs 2016, 14, 5. [Google Scholar] [CrossRef]
- Sánchez, J.A.; Alfonso, A.; Rodriguez, I.; Alonso, E.; Cifuentes, J.M.; Bermudez, R.; Rateb, M.E.; Jaspars, M.; Houssen, W.E.; Ebel, R.; et al. Spongionella Secondary metabolites, promising modulators of immune response through CD147 receptor modulation. Front. Immunol. 2016, 7, 452. [Google Scholar] [CrossRef]
- Tahtouh, T.; Elkins, J.M.; Filippakopoulos, P.; Soundararajan, M.; Burgy, G.; Durieu, E.; Cochet, C.; Schmid, R.S.; Lo, D.C.; Delhommel, F.; et al. Selectivity, cocrystal structures, and neuroprotective properties of leucettines, a family of protein kinase inhibitors derived from the marine sponge alkaloid Leucettamine B. J. Med. Chem. 2012, 55, 9312–9330. [Google Scholar] [CrossRef]
- Nguyen, T.L.; Duchon, A.; Manousopoulou, A.; Loaëc, N.; Villiers, B.; Pani, G.; Karatas, M.; Mechling, A.E.; Harsan, L.-A.; Linaton, E.; et al. Correction of cognitive deficits in mouse models of down syndrome by a pharmacological inhibitor of DYRK1A. Dis. Mod. Mech. 2018, 11. [Google Scholar] [CrossRef]
- Riniker, S.; Landrum, G.A. Better informed distance geometry: Using what we know to improve conformation generation. J. Chem. Inf. Model. 2015, 55, 2562–2574. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.E. Rapid calculation of polar molecular surface area and its application to the prediction of transport phenomena. Abstr. Pap. Am. Chem. Soc. 1999, 217, U696. [Google Scholar] [CrossRef]
- Platts, J.A.; Abraham, M.H.; Zhao, Y.H.; Hersey, A.; Ijaz, L.; Butina, D. Correlation and prediction of a large blood-brain distribution data set—An LFER study. Eur. J. Chem. 2001, 36, 719–730. [Google Scholar] [CrossRef]
- Efron, B.; Hastie, T.; Johnstone, I.; Tibshirani, R.J. Least angle regression. Ann. Stat. 2004, 32, 407–499. [Google Scholar] [CrossRef] [Green Version]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef] [Green Version]
- Kortagere, S.; Chekmarev, D.; Welsh, W.J.; Ekins, S. New predictive models for blood-brain barrier permeability of drug-like molecules. Pharm. Res. 2008, 25, 1836–1845. [Google Scholar] [CrossRef] [PubMed]
- Ghorbanzad’e, M.; Fatemi, M.H. Classification of central nervous system agents by least squares support vector machine based on their structural descriptors: A comparative study. Chemometr. Intell. Lab. 2012, 110, 102–107. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model Name | Number Variables | Model Accuracy | External Test Accuracy |
|---|---|---|---|
| Logistic Regression | 200 | 0.81 ± 0.06 | 0.68 ± 0.20 |
| Decision Tree | 200 | 0.73 ± 0.07 | 0.73 ± 0.21 |
| Random Forest | 200 | 0.81 ± 0.08 | 0.80 ± 0.22 |
| Gradient Boosting | 200 | 0.76 ± 0.08 | 0.73 ± 0.24 |
| K-Nearest Neighbour | 200 | 0.78 ± 0.09 | 0.63 ± 0.28 |
| Linear Discriminant Analysis | 200 | 0.70 ± 0.06 | 0.65 ± 0.23 |
| Quadratic Discriminant Analysis | 200 | 0.62 ± 0.03 | 0.60 ± 0.20 |
| Naïve BAYES | 200 | 0.59 ± 0.09 | 0.53 ± 0.18 |
| Support Vector Machine | 200 | 0.63 ± 0.01 | 0.68 ± 0.20 |
| Logistic Regression | 161 | 0.81 ± 0.05 | 0.75 ± 0.22 |
| Decision Tree | 161 | 0.71 ± 0.06 | 0.67 ± 0.19 |
| Random Forest | 161 | 0.79 ± 0.08 | 0.80 ± 0.22 |
| Gradient Boosting | 161 | 0.77 ± 0.08 | 0.70 ± 0.22 |
| K-Nearest Neighbour | 161 | 0.76 ± 0.10 | 0.60 ± 0.30 |
| Linear Discriminant Analysis | 161 | 0.75 ± 0.06 | 0.70 ± 0.25 |
| Quadratic Discriminant Analysis | 161 | 0.62 ± 0.05 | 0.58 ± 0.23 |
| Naïve Bayes | 161 | 0.56 ± 0.08 | 0.55 ± 0.15 |
| Support Vector Machine | 161 | 0.63 ± 0.01 | 0.75 ± 0.22 |
| Model Name | Number Variables | Hyperparameters | Model Accuracy | External Test Accuracy |
|---|---|---|---|---|
| After Recursive Feature Elimination with Cross-Validation (RFECV) | ||||
| logistic regression | 135 | default | 0.82 | 0.66 |
| decision tree | 200 | default | 0.75 | 0.69 |
| random forest | 154 | default | 0.82 | 0.78 |
| gradient boosting | 160 | default | 0.78 | 0.72 |
| After Tuning Hyperparameters | ||||
| logistic regression 1 | 200 | l1, 1 | 0.81 ± 0.06 | 0.63 ± 0.18 |
| decision tree 2 | 200 | auto, 5, 1 | 0.70 ± 0.05 | 0.56 ± 0.20 |
| random forest 3 | 200 | sqrt, 400, 5, 2 | 0.80 ± 0.07 | 0.75 ± 0.20 |
| gradient boosting 4 | 200 | log2, 200, 10, 4 | 0.78 ± 0.07 | 0.69 ± 0.20 |
| After Tuning Hyperparameters and RFECV | ||||
| logistic regression 1 | 18 | l1, 1 | 0.82 | 0.63 |
| decision tree 2 | 156 | auto, 5, 1 | 0.77 | 0.63 |
| random forest 3 | 150 | sqrt, 400, 5, 2 | 0.82 | 0.72 |
| gradient boosting 4 | 162 | log2, 200, 10, 4 | 0.82 | 0.75 |
| After Multicollinearity and RFECV | ||||
| logistic regression | 88 | default | 0.82 | 0.66 |
| decision tree | 2 | default | 0.70 | 0.63 |
| random forest | 127 | default | 0.79 | 0.78 |
| gradient boosting | 69 | default | 0.78 | 0.69 |
| After Multicollinearity and Tuning Hyperparameters | ||||
| logistic regression1 | 161 | l2, 10 | 0.81 ± 0.06 | 0.78 ± 0.18 |
| decision tree 2 | 161 | auto, 6, 1 | 0.74 ± 0.04 | 0.60 ± 0.20 |
| random forest3 | 161 | log2, 700, 5, 2 | 0.80 ± 0.07 | 0.80 ± 0.20 |
| gradient boosting4 | 161 | log2, 300, 15, 4 | 0.80 ± 0.07 | 0.80 ± 0.20 |
| After Multicollinearity, Tuning Hyperparameters and RFECV | ||||
| logistic regression 1 | 99 | l2, 10 | 0.83 | 0.72 |
| decision tree 2 | 85 | auto, 6, 1 | 0.76 | 0.63 |
| random forest 3 | 150 | log2, 700, 5, 2 | 0.82 | 0.72 |
| gradient boosting 4 | 161 | log2, 300, 15, 4 | 0.80 | 0.78 |
| Property | CPSMs | CPSM+ | CPSM− | KDs | MDKIs | Outliers |
|---|---|---|---|---|---|---|
| MolWt | 312 ± 127 | 296 ± 97 | 326 ± 129 | 460 ± 83 | 474 ± 246 | 1343 ± 265 |
| TPSA | 55 ± 34 | 36 ± 26 | 68 ± 28 | 96 ± 42 | 114 ± 83 | 399 ± 162 |
| MolLogP | 2.9 ± 1.7 | 3.4 ± 1.2 | 2.7 ± 1.9 | 4.2 ± 1.4 | 3.2 ± 2.6 | 1.8 ± 6.3 |
| NumHDonors | 1.2 ± 1.0 | 0.8 ± 0.8 | 1.5 ± 1.1 | 2.3 ± 1.9 | 3.0 ± 2.6 | 10.1 ± 5.5 |
| NumHAcceptors | 3.8 ± 2.2 | 3.1 ± 1.8 | 4.3 ± 2.0 | 6.4 ± 1.8 | 6.5 ± 5.0 | 23.1 ± 10.2 |
| NOCount | 4.4 ± 2.4 | 3.2 ± 1.9 | 5.2 ± 2.1 | 7.5 ± 2.4 | 7.6 ± 5.5 | 26.5 ± 10.6 |
| MolMR | 86 ± 36 | 84 ± 27 | 89 ± 36 | 123 ± 22 | 122 ± 59 | 325 ± 61 |
| NumRotatableBonds | 4.1 ± 3.0 | 3.5 ± 2.6 | 4.7 ± 3.3 | 5.9 ± 2.8 | 5.5 ± 6.9 | 23.1 ± 12.0 |
| Hall–Kier Alpha | −1.9 ± 1.0 | −1.7 ± 0.9 | −2.1 ± 1.0 | −3.4 ± 0.8 | −2.6 ± 1.6 | −4.2 ± 3.3 |
| BertzCT | 670 ± 367 | 619 ± 301 | 714 ± 375 | 1248 ± 257 | 1142 ± 669 | 2985 ± 1102 |
| qed | 0.7 ± 0.2 | 0.7 ± 0.2 | 0.6 ± 0.2 | 0.5 ± 0.2 | 0.4 ± 0.2 | 0.06 ± 0.03 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Plisson, F.; Piggott, A.M. Predicting Blood–Brain Barrier Permeability of Marine-Derived Kinase Inhibitors Using Ensemble Classifiers Reveals Potential Hits for Neurodegenerative Disorders. Mar. Drugs 2019, 17, 81. https://doi.org/10.3390/md17020081
Plisson F, Piggott AM. Predicting Blood–Brain Barrier Permeability of Marine-Derived Kinase Inhibitors Using Ensemble Classifiers Reveals Potential Hits for Neurodegenerative Disorders. Marine Drugs. 2019; 17(2):81. https://doi.org/10.3390/md17020081
Chicago/Turabian StylePlisson, Fabien, and Andrew M. Piggott. 2019. "Predicting Blood–Brain Barrier Permeability of Marine-Derived Kinase Inhibitors Using Ensemble Classifiers Reveals Potential Hits for Neurodegenerative Disorders" Marine Drugs 17, no. 2: 81. https://doi.org/10.3390/md17020081