Characterization of the Hydrolysis Kinetics of Fucosylated Glycosaminoglycan in Mild Acid and Structures of the Resulting Oligosaccharides

Abstract
:1. Introduction
2. Results and Discussion
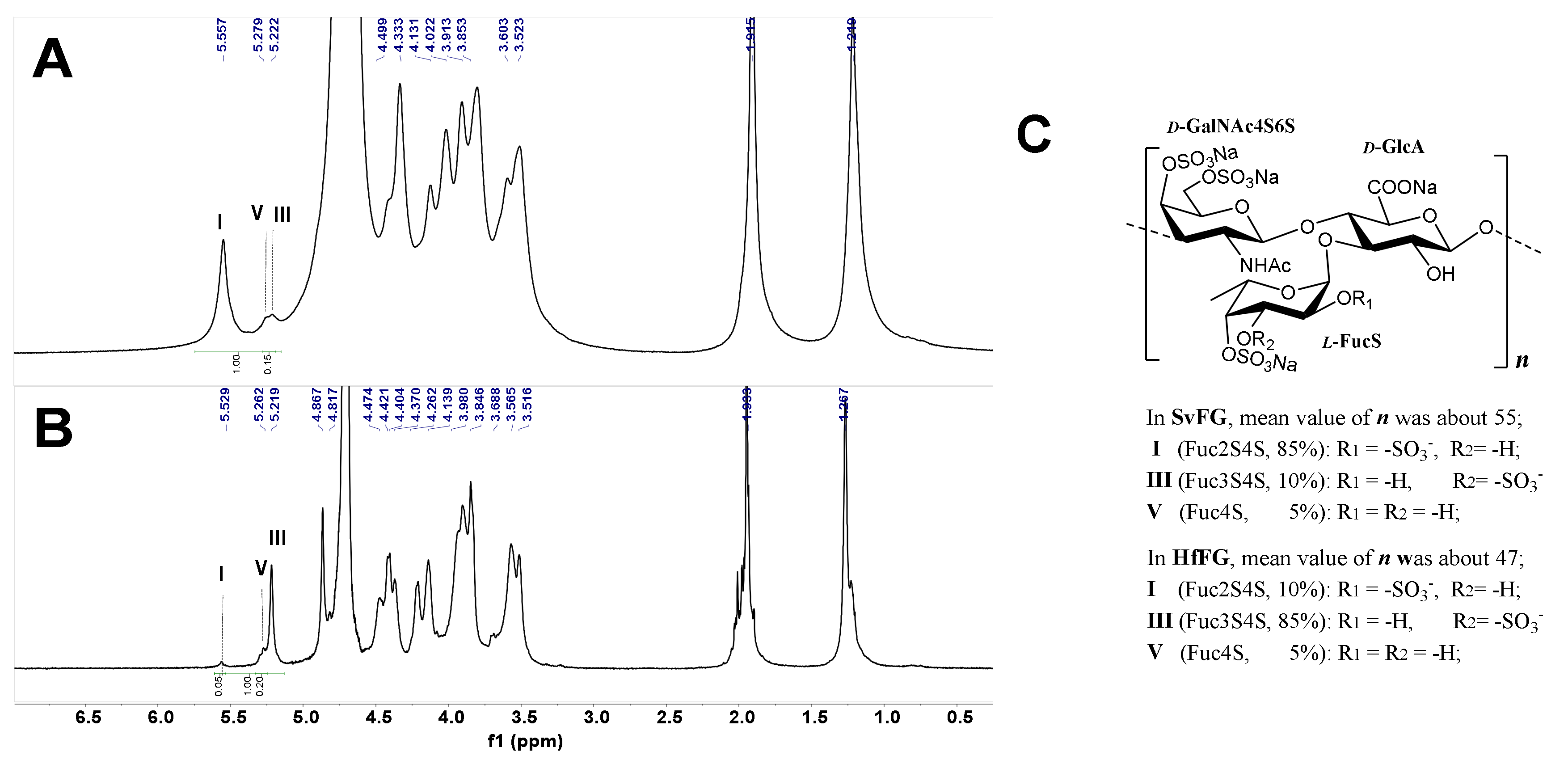
2.1. Structures of SvFG and HfFG
2.2. Mild Acid Hydrolysis of SvFG and HfFG
2.3. Analysis of Defucosylation of the Partial Fucosylated Products by 1H NMR
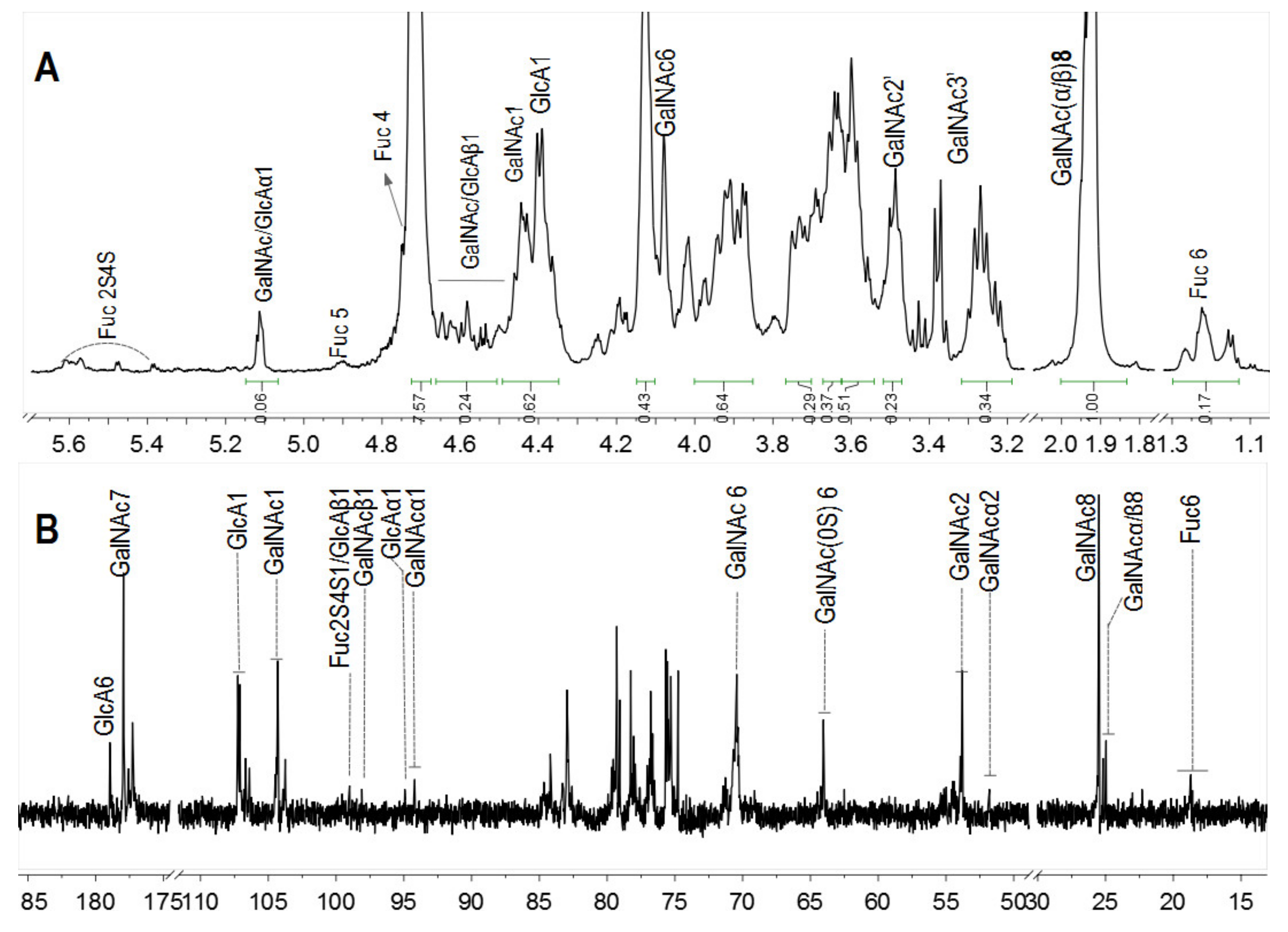
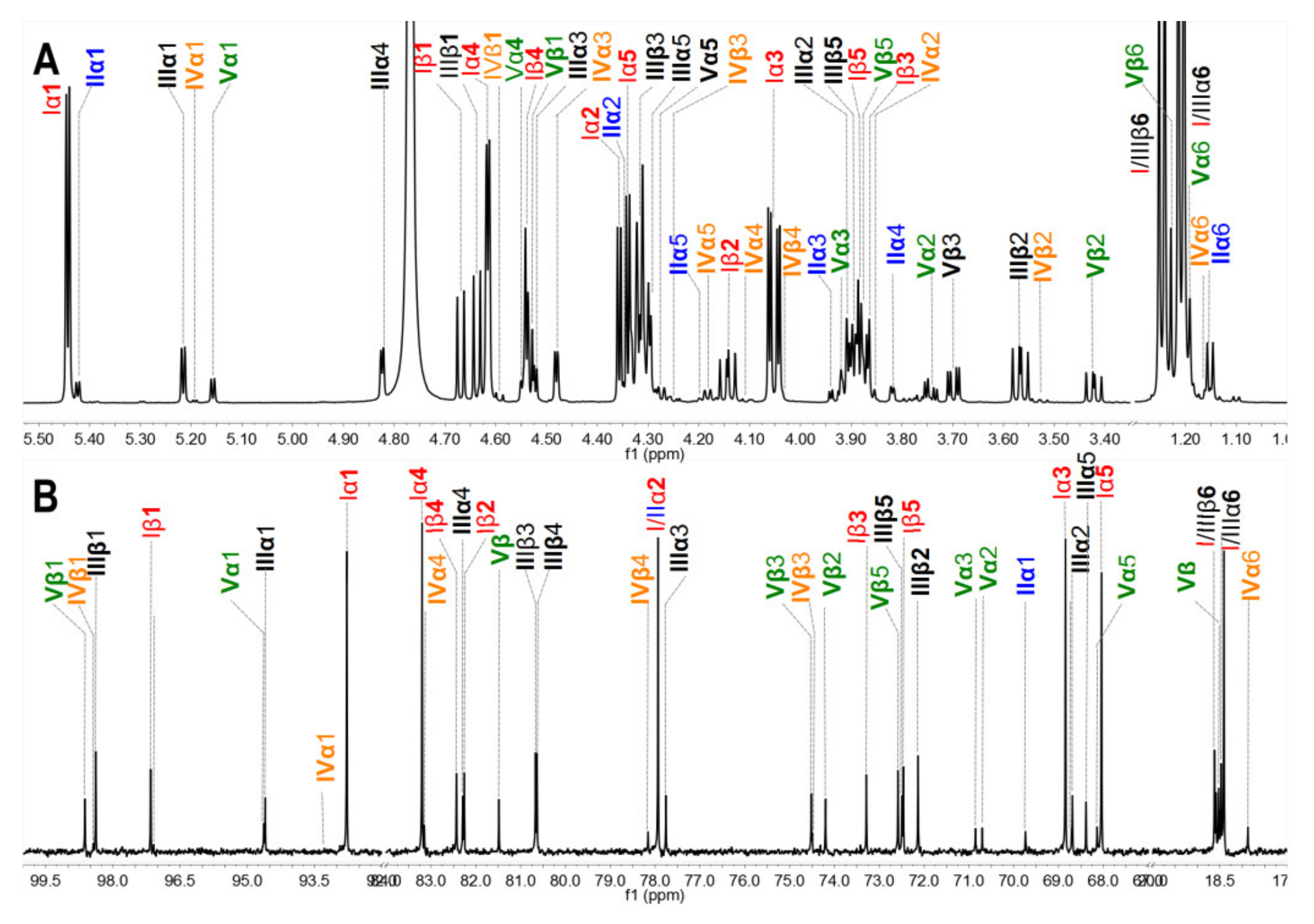
2.4. Physicochemical Properties and Structures of Hydrolysis Products
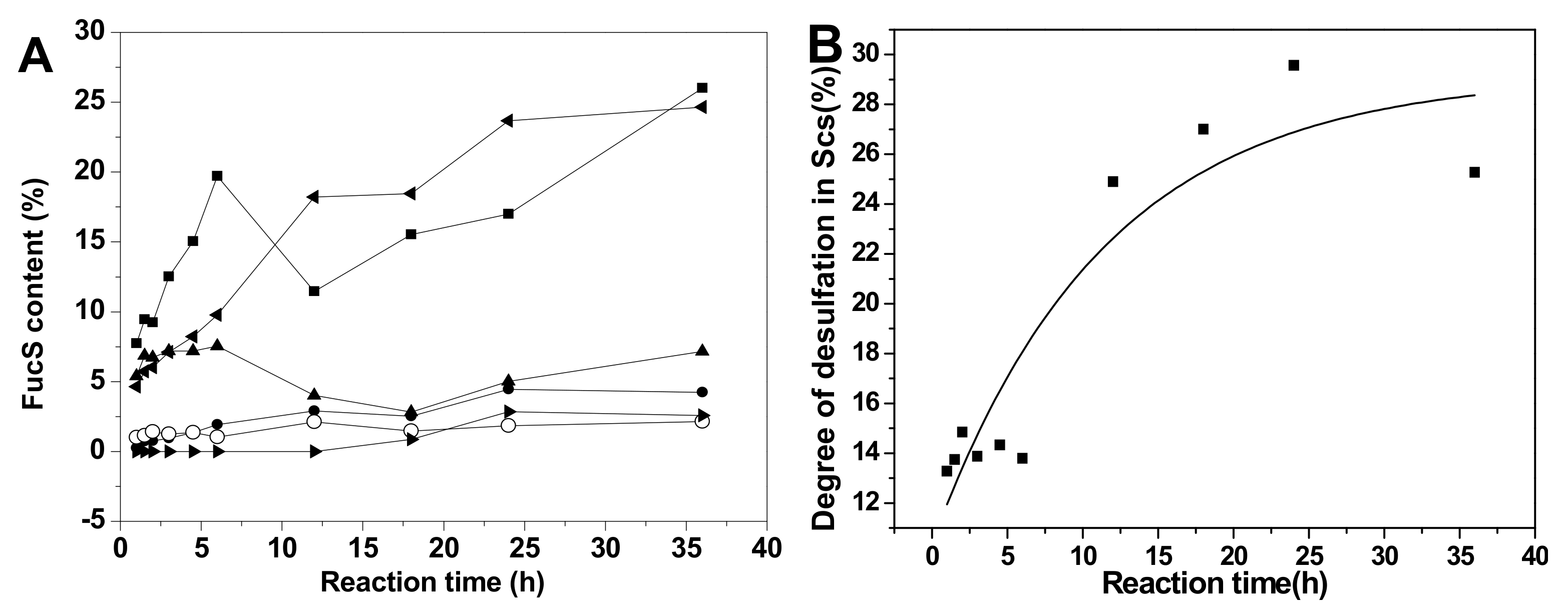
2.5. Chemical Structures and Compositions of Scs
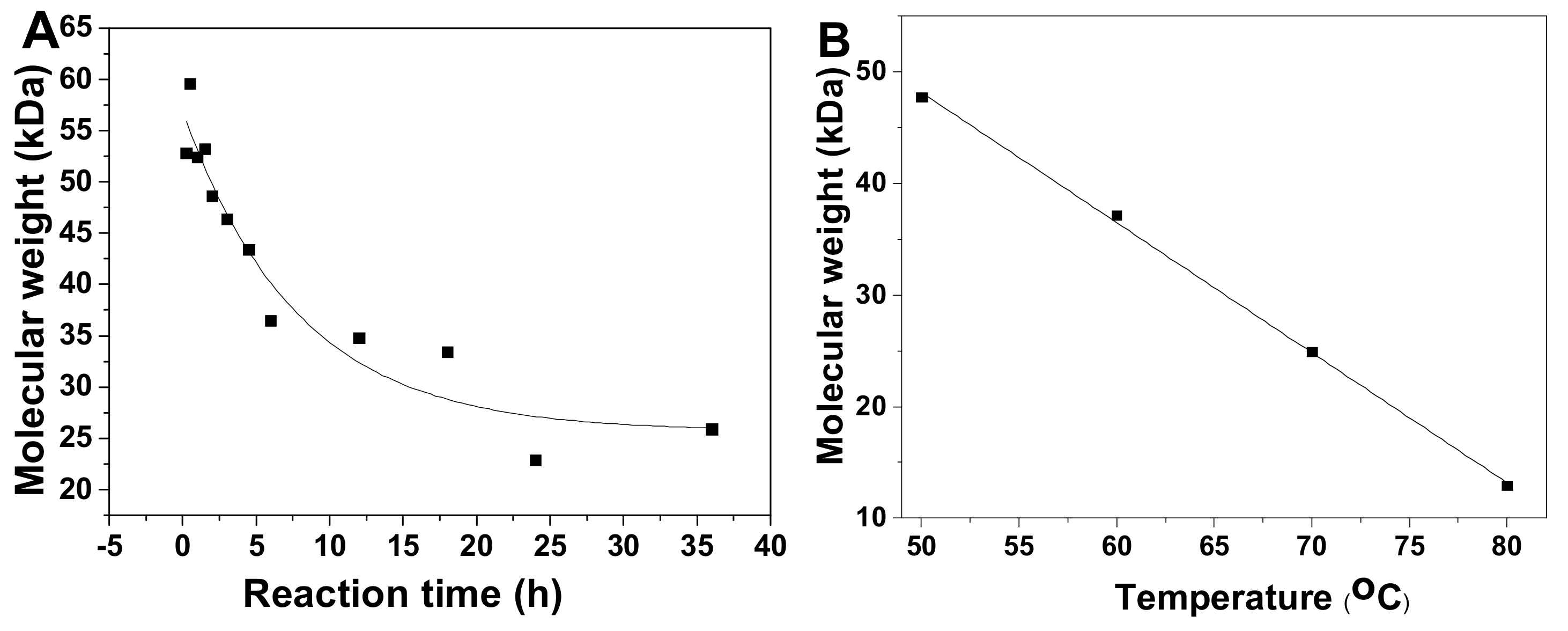
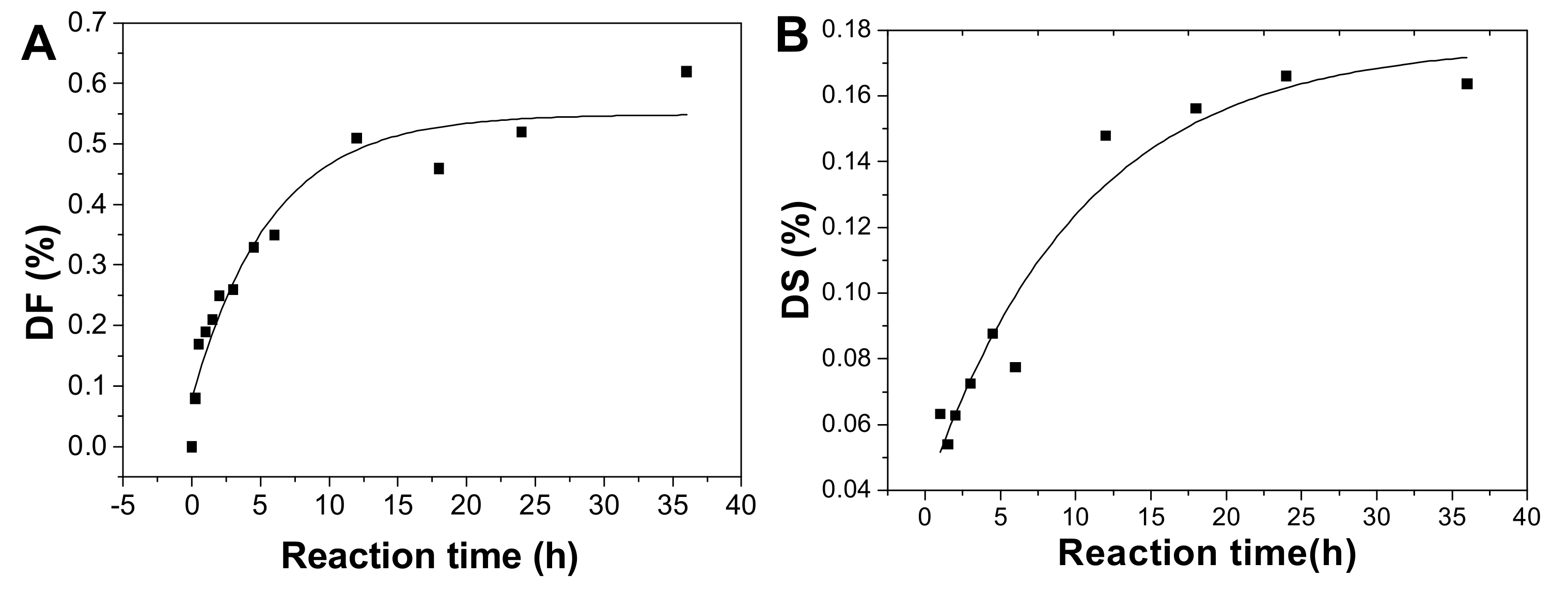
2.6. Kinetics Process of SvFG in Mild Acid Hydrolysis
2.7. Effects of DF, Mw, and SO3- Content on APTT Prolongation Activity in Vitro
3. Materials and Methods
3.1. Materials
3.2. Isolation of Fucosylated Glycosaminoglycan from the Body Wall of Sea Cucumber
3.3. Mild Acid Hydrolysis of Fucosylated Glycosaminoglycan
3.4. Purification of the Acid-released Fucose Fragments
3.5. Determination of Physicochemical Properties
3.6. NMR Method
3.7. Determination of the Kinetic Parameters
3.8. Determination of the APTT Activities in Vitro
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vieira, R.P.; Mourão, P. Occurrence of a unique fucose-branched chondroitin sulfate in the body wall of a sea cucumber. J. Biol. Chem. 1988, 263, 18176–18183. [Google Scholar]
- Santos, G.R.; Glauser, B.F.; Parreiras, L.A.; Vilanova, E.; Mourão, P.A. Distinct structures of the α-fucose branches in fucosylated chondroitin sulfates do not affect their anticoagulant activity. Glycobiology 2015, 25, 1043–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mourão, P.A.; Pereira, M.S.; Pavão, M.S.; Mulloy, B.; Tollefsen, D.M.; Mowinckel, M.C.; Abildgaard, U. Structure and anticoagulant activity of a fucosylated chondroitin sulfate from echinoderm. Sulfated fucose branches on the polysaccharide account for its high anticoagulant action. J. Biol. Chem. 1996, 271, 23973–23984. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Wang, Y.; Jiang, T.; Lv, Z. Novel branch patterns and anticoagulant activity of glycosaminoglycan from sea cucumber Apostichopus japonicus. Int. J. Biol. Macromol. 2015, 72, 911–918. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Wu, M.; Xiao, C.; Yang, L.; Zhou, L.; Gao, N.; Li, Z.; Chen, J.; Chen, J.; Liu, J. Discovery of an intrinsic tenase complex inhibitor: Pure nonasaccharide from fucosylated glycosaminoglycan. Proc. Natl. Acad. Sci. USA 2015, 112, 8284–8289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.; Xu, S.; Zhao, J.; Kang, H.; Ding, H. Free-radical depolymerization of glycosaminoglycan from sea cucumber Thelenata ananas by hydrogen peroxide and copper ions. Carbohydr. Polym. 2010, 80, 1116–1124. [Google Scholar] [CrossRef]
- Kariya, Y.; Watabe, S.; Hashimoto, K.; Yoshida, K. Occurrence of chondroitin sulfate E in glycosaminoglycan isolated from the body wall of sea cucumber Stichopus japonicus. J. Biol. Chem. 1990, 265, 5081–5085. [Google Scholar] [PubMed]
- Tømmeraas, K.; Melander, C. Kinetics of hyaluronan hydrolysis in acidic solution at various pH values. Biomacromolecules 2008, 9, 1535–1540. [Google Scholar]
- Mourão, P.A.; Bastos, I.G. Highly acidic glycans from sea cucumbers: Isolation and fractionation of fucose—rich sulfated polysaccharides from the body wall of Ludwigothurea grisea. Eur. J. Biochem. 1987, 166, 639–645. [Google Scholar]
- Vieira, R.P.; Mulloy, B.; Mourão, P. Structure of a fucose-branched chondroitin sulfate from sea cucumber. Evidence for the presence of 3-O-sulfo-beta-D-glucuronosyl residues. J. Biol. Chem. 1991, 266, 13530–13536. [Google Scholar]
- Borsig, L.; Wang, L.; Cavalcante, M.C.; Cardilo-Reis, L.; Ferreira, P.L.; Mourão, P.A.; Esko, J.D.; Pavao, M.S. Selectin blocking activity of a fucosylated chondroitin sulfate glycosaminoglycan from sea cucumber. Effect on tumor metastasis and neutrophil recruitment. J. Biol. Chem. 2007, 282, 14984–14991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, K.; Minami, Y.; Nemoto, H.; Numata, K.; Yamanaka, E. Structure of DHG, a depolymerized glycosaminoglycan from sea cucumber, Stichopus japonicus. Tetrahedron Lett. 1992, 33, 4959–4962. [Google Scholar] [CrossRef]
- Gao, R.; Wu, N.; Li, Y.; Chen, S. Structures and anticoagulant activities of the partially mild acidic hydrolysis products of the fucosylated chondroitin sulfate from sea cucumber Pearsonothuria graeffei. J. Carbohydr. Chem. 2014, 33, 471–488. [Google Scholar] [CrossRef]
- Wu, M.; Huang, R.; Wen, D.; Gao, N.; He, J.; Li, Z.; Zhao, J. Structure and effect of sulfated fucose branches on anticoagulant activity of the fucosylated chondroitin sulfate from sea cucumber Thelenata ananas. Carbohydr. Polym. 2012, 87, 862–868. [Google Scholar]
- Santos, G.R.; Porto, A.C.; Soares, P.A.; Vilanova, E.; Mourão, P.A. Exploring the structure of fucosylated chondroitin sulfate through bottom-up nuclear magnetic resonance and electrospray ionization-high-resolution mass spectrometry approaches. Glycobiology 2017, 27, 625–634. [Google Scholar] [CrossRef] [Green Version]
- Lian, W.; Wu, M.; Huang, N.; Gao, N.; Xiao, C.; Li, Z.; Zhang, Z.; Zheng, Y.; Peng, W.; Zhao, J. Anti-HIV-1 activity and structure-activity-relationship study of a fucosylated glycosaminoglycan from an echinoderm by targeting the conserved CD4 induced epitope. Biochim. Biophys. Acta 2013, 1830, 4681–4691. [Google Scholar]
- Li, X.; Luo, L.; Cai, Y.; Yang, W.; Lin, L.; Li, Z.; Gao, N.; Purcell, S.W.; Wu, M.; Zhao, J. Structural elucidation and biological activity of a highly regular fucosylated glycosaminoglycan from the edible sea cucumber Stichopus herrmanni. J. Agric. Food. Chem. 2017, 65, 9315–9323. [Google Scholar] [CrossRef]
- Xu, L.; Gao, N.; Xiao, C.; Lin, L.; Purcell, S.W.; Wu, M.; Zhao, J. Modulating the degree of fucosylation of fucosylated chondroitin sulfate enhances heparin cofactor II-dependent thrombin inhibition. Eur. J. Med. Chem. 2018, 154, 133–143. [Google Scholar]
- Nagase, H.; Enjyoji, K.; Minamiguchi, K.; Kitazato, K.T.; Kitazato, K.; Saito, H.; Kato, H. Depolymerized holothurian glycosaminoglycan with novel anticoagulant actions: Antithrombin III-and heparin cofactor II-independent inhibition of factor X activation by factor IXa-factor VIIIa complex and heparin cofactor II-dependent inhibition of thrombin. Blood 1995, 85, 1527–1534. [Google Scholar]
- Nagase, H.; Kitazato, K.T.; Sasaki, E.; Hattori, M.; Kitazato, K.; Saito, H. Antithrombin III-independent effect of depolymerized holothurian glycosaminoglycan (DHG) on acute thromboembolism in mice. Thromb. Haemost. 1997, 77, 399–402. [Google Scholar] [CrossRef]
- Sheehan, J.P.; Walke, E.N. Depolymerized holothurian glycosaminoglycan and heparin inhibit the intrinsic tenase complex by a common antithrombin-independent mechanism. Blood 2006, 107, 3876–3882. [Google Scholar] [PubMed]
- Li, J.H.; Li, S.; Zhi, Z.J.; Yan, L.F.; Ye, X.Q.; Ding, T.; Yan, L.; Linhardt, R.J.; Chen, S.G. Depolymerization of fucosylated chondroitin sulfate with a modified fenton-system and anticoagulant activity of the resulting fragments. Mar Drugs 2016, 14, 170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, L.; Wu, M.; Xu, L.; Lian, W.; Xiang, J.; Lu, F.; Gao, N.; Xiao, C.; Wang, S.; Zhao, J. Comparison of physicochemical characteristics and anticoagulant activities of polysaccharides from three sea cucumbers. Mar Drugs 2013, 11, 399–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Gao, N.; Sun, H.; Xiao, C.; Yang, L.; Lin, L.; Yin, R.; Li, Z.; Zhang, H.; Ji, X.; et al. Effects of native fucosylated glycosaminoglycan, its depolymerized derivatives on intrinsic factor Xase, coagulation, thrombosis, and hemorrhagic risk. Thromb. Haemst. 2020, 120, 607–619. [Google Scholar]
- Xu, C.; Pranovich, A.; Vähäsalo, L.; Hemming, J.; Holmbom, B.; Schols, H.A.; Willför, S. Kinetics of acid hydrolysis of water-soluble spruce O-acetyl galactoglucomannans. J. Agric. Food. Chem. 2008, 56, 2429–2435. [Google Scholar] [CrossRef]
- Simha, R. Kinetics of degradation and size distribution of long chain polymers. J. Appl. Phys. 1941, 12, 569–578. [Google Scholar]
- Yan, J.K.; Wu, J.Y.; Jin, B.; Cui, H.Y. Molecular weight changes and kinetics of acid hydrolysis of exopolysaccharides isolated from cordyceps sinensis. Food Sci. 2012, 33, 82–85. [Google Scholar]
- Guan, R.; Peng, Y.; Zhou, L.; Zheng, W.; Liu, X.; Wang, P.; Yuan, Q.; Gao, N.; Zhao, L.; Zhao, J. Precise Structure and anticoagulant activity of fucosylated glycosaminoglycan from Apostichopus japonicus: Analysis of its depolymerized fragments. Mar. Drugs 2019, 17, 195. [Google Scholar] [CrossRef] [Green Version]
- Honda, S.; Chiba, H.; Kakehi, K. Selective determination of pentoses, hexoses and 6-deoxyhexoses by dual-wavelength spectrophotometry with the cysteine—phenol—sulfuric acid system. Anal. Chim. Acta 1981, 131, 205–211. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samp. | Treat. (°C × h) | a Yield (%) | b RT (min) | c Mwt (kDa) | d PDI | -SO3− (%) | DF (%) | DS (%) | DH (%) |
|---|---|---|---|---|---|---|---|---|---|
| SvFG | -- | 100 | 14.287 | 52.77 | 1.48 | ND | 0 | -- | -- |
| pF1 | 60 × 0.25 | 99.45 | 14.405 | 52.45 | 1.85 | 36 | 8 | -- | -- |
| pF2 | 60 × 0.5 | 89.32 | 14.414 | 53.16 | 1.64 | 36 | 17 | -- | -- |
| pF3 | 60 × 1.0 | 77.76 | 14.465 | 48.56 | 1.9 | 30 | 19 | 6.32 | -0.17 |
| pF4 | 60 × 1.5 | 85.73 | 14.56 | 46.44 | 1.81 | 32 | 22 | 5.39 | -0.02 |
| pF5 | 60 × 2.0 | 93.97 | 14.656 | 43.44 | 1.96 | 28 | 25 | 6.28 | 0.03 |
| pF6 | 60 × 3.0 | 89.79 | 14.869 | 36.52 | 1.98 | 28 | 26 | 7.25 | 0.14 |
| pF7 | 60 × 4.0 | 86.86 | 15.073 | 34.79 | 1.91 | 28 | 33 | 8.75 | 0.43 |
| pF8 | 60 × 6.0 | 83.31 | 15.142 | 33.43 | 1.92 | 26 | 35 | 7.75 | 0.53 |
| pF9 | 60 × 12.0 | 73.09 | 15.964 | 22.93 | 1.95 | 22 | 52 | 14.78 | 0.40 |
| pF10 | 60 × 18.0 | 71.03 | 15.698 | 25.85 | 1.83 | 22 | 46 | 15.62 | 1.48 |
| pF11 | 60 × 24.0 | 67.06 | 16.216 | 19.83 | 1.80 | 26 | 52 | 16.61 | 1.01 |
| pF12 | 60 × 36.0 | 72.65 | 16.949 | 12.54 | 1.80 | 22 | 63 | 16.36 | 1.71 |
| pF13 | 50 × 5.0 | 96.16 | 14.481 | 47.72 | 1.72 | 22 | 17 | 1.00 | 0.07 |
| pF14 | 60 × 5.0 | 86.64 | 14.914 | 37.06 | 1.90 | 26 | 29 | 6.17 | 0.46 |
| pF15 | 70 × 5.0 | 48.64 | 15.778 | 24.94 | 1.78 | 20 | 47 | 17.49 | 1.17 |
| pF16 | 80 × 5.0 | 50.99 | 16.949 | 12.91 | 1.71 | 24 | 63 | 23.50 | 3.42 |
| pF17 | 100 × 0.5 | -- | 17.120 | 5.33 | 1.52 | 20 | 81 | 18.65 | 10.02 |
| pF18 | 100 × 2.0 | -- | 18.794 | 3.50 | 1.47 | 20 | 87 | 34.56 | 14.60 |
| pF19 | 60 × 12.0 | -- | 15.67 | 27.66 | 2.03 | 26 | 62 | 8.91 | 4.753 |
| pF20 | 100 × 2.0 | -- | 19.035 | 3.57 | 1.74 | 24 | 91 | 55.12 | 12.36 |
| Resource | Sugar | JH1,H2 (Hz) | Chemical Shift (ppm) | ||||
|---|---|---|---|---|---|---|---|
| H/C-2 | H/C-3 | H/C-4 | H/C-5 | H/C-6 | |||
| S. variegatus (Sc8) | α-Fuc2S4S (I) | 3.6 | 4.40/77.92 | 4.11/68.82 | 4.67/83.18 | 4.38/68.02 | 1.25/18.39 |
| α-Fuc2S (II) | 4.2 | 4.40/78.13 | 3.40/69.72 | 3.88/74.48 | 4.25/68.70 | 1.21/17.85 | |
| α-Fuc3,4S (III) | 3.6 | 3.96/68.69 | 4.58/77.76 | 4.88/81.48 | 4.36/68.37 | 1.25/18.57 | |
| α-Fuc3S (IV) | 3.6 | 3.91/NDa | 4.53/ND | 4.16/ND | 4.25/ND | 1.22/ND | |
| α-Fuc4S (V) | 4.2 | 3.81/70.69 | 3.97/70.84 | 4.61/83.13 | 4.33/68.14 | 1.26/18.50 | |
| β-Fuc2,4S (I’) | 7.8 | 4.20/82.27 | 3.93/73.28 | 4.60/82.42 | 3.94/72.45 | 1.02/18.45 | |
| β-Fuc3,4S (III’) | 7.8 | 3.62/72.13 | 4.36/80.62 | 4.82/80.66 | 3.96/72.58 | 1.30/18.60 | |
| β-Fuc3S (IV’) | 8.4 | 3.60/ND | 4.31/ND | 4.10/72.01 | 3.85/ND | 1.27/ND | |
| β-Fuc4S (V’) | -- | 3.50/74.20 | 3.76/74.52 | 4.54/82.35 | 3.93/75.48 | 1.29/18.56 | |
| H. fuscopunctata (Sc19) | α-Fuc3,4S (III) | 3.6 | 3.95/68.68 | 4.59/77.77 | 4.88/81.46 | 4.35/68.38 | 1.26/18.37 |
| α-Fuc3S (IV) | 3.6 | 3.92/68.52 | 4.54/80.36 | 4.15/72.62 | 4.25/68.57 | 1.22/18.01 | |
| α-Fuc4S (V) | 4.2 | 3.80/79.71 | 3.97/70.84 | 4.61/83.16 | 4.33/68.15 | 1.26/18.53 | |
| α-Fuc (VI) | 3.6 | 3.73/ND | 3.84/ND | 3.80/73.35 | ND/ND | 1.19/ND | |
| β-Fuc3,4S (III’) | 7.8 | 3.61/72.16 | 4.37/80.63 | 4.82/80.57 | 3.95/72.48 | 1.30/18.70 | |
| β-Fuc3S (IV’) | 7.8 | 3.59/72.09 | 4.31/83.09 | 4.10/72.09 | 3.85/72.98 | 1.25/17.98 | |
| β-Fuc4S (V’) | 7.8 | 3.48/74.21 | 3.76/74.51 | 4.54/82.30 | 3.93/72.49 | 1.29/18.59 | |
| β-Fuc (VI’) | -- | 3.43/74.14 | 3.64/75.36 | 3.73/73.90 | 3.80/ND | 1.23/ND | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Zhang, Z.; Mao, H.; Wang, P.; Zuo, Z.; Gao, L.; Shi, X.; Yin, R.; Gao, N.; Zhao, J. Characterization of the Hydrolysis Kinetics of Fucosylated Glycosaminoglycan in Mild Acid and Structures of the Resulting Oligosaccharides. Mar. Drugs 2020, 18, 286. https://doi.org/10.3390/md18060286
Liu X, Zhang Z, Mao H, Wang P, Zuo Z, Gao L, Shi X, Yin R, Gao N, Zhao J. Characterization of the Hydrolysis Kinetics of Fucosylated Glycosaminoglycan in Mild Acid and Structures of the Resulting Oligosaccharides. Marine Drugs. 2020; 18(6):286. https://doi.org/10.3390/md18060286
Chicago/Turabian StyleLiu, Xixi, Zhexian Zhang, Hui Mao, Pin Wang, Zhichuang Zuo, Li Gao, Xiang Shi, Ronghua Yin, Na Gao, and Jinhua Zhao. 2020. "Characterization of the Hydrolysis Kinetics of Fucosylated Glycosaminoglycan in Mild Acid and Structures of the Resulting Oligosaccharides" Marine Drugs 18, no. 6: 286. https://doi.org/10.3390/md18060286