Bromopyrrole Alkaloids as Lead Compounds against Protozoan Parasites

 ,
,  ,
,
Abstract
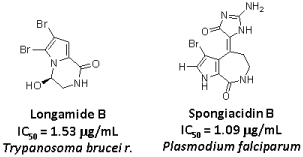
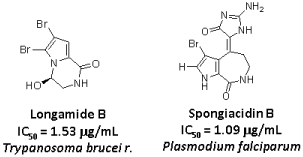
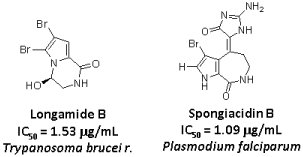
:
1. Introduction
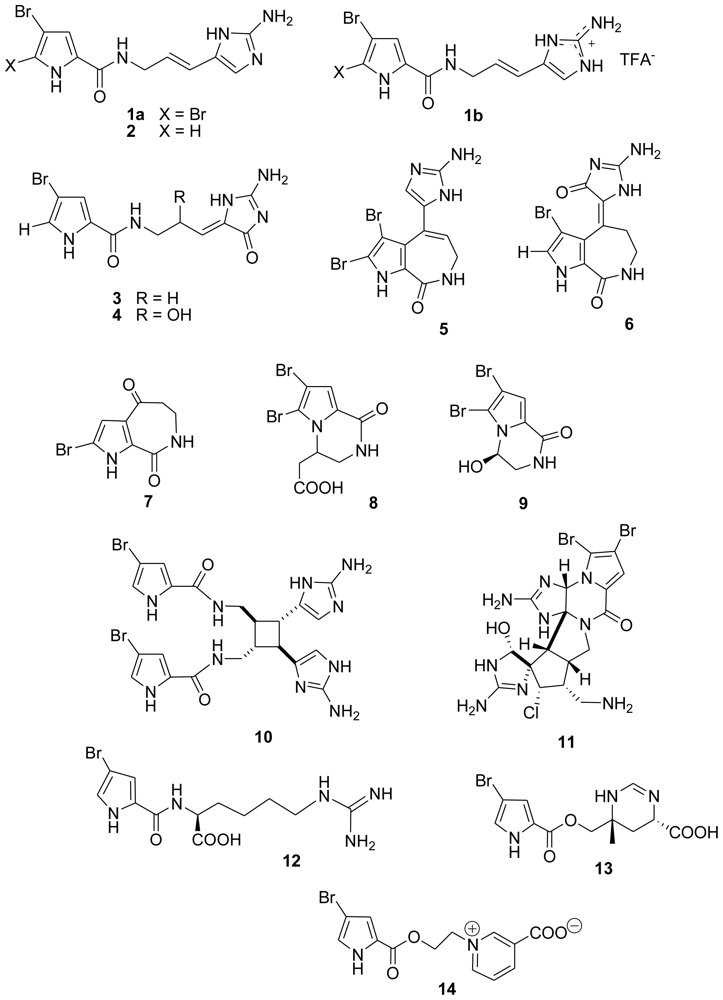
2. Results and Discussion
3. Experimental Section
3.1. Isolation of compounds 2–14
3.2. Activity against Plasmodium falciparum
3.3. Activity against Trypanosoma brucei rhodesiense
3.4. Activity against Trypanosoma cruzi
3.5. Activity against Leishmania donovani
3.6. Cytotoxicity against L6-cells
3.7. PfFAS-II enzyme inhibition assays
4. Conclusions
Acknowledgements
- Samples Availability: Available from the authors.
References
- Data taken from Malaria Foundation International. http://www.malaria.org and linked sites (accessed on 3 May 2010).
- Brun, R; Blum, J; Chappuis, F; Burri, C. Human African trypanosomiasis. Lancet 2010, 375, 148–159. [Google Scholar] [Green Version]
- Bern, C; Maguire, JH; Alvar, J. Complexities of Assessing the Disease Burden Attributable to Leishmaniasis. PLoS Neglect. Trop. D 2008, 2, e313. [Google Scholar]
- Reithinger, R. Leishmaniases’ burden of disease: ways forward for getting from speculation to reality. Plos Neglect. Trop. D 2008, 2, e285. [Google Scholar]
- Fattorusso, E; Taglialatela-Scafati, O. Marine Antimalarials. Mar. Drugs 2009, 7, 130–152. [Google Scholar]
- Mayer, AM; Rodriguez, AD; Berlinck, RG; Hamann, MT. Marine pharmacology in 2005–6: Marine compounds with anthelmintic, antibacterial, anticoagulant, antifungal, anti-inflammatory, antimalarial, antiprotozoal, antituberculosis, and antiviral activities; affecting the cardiovascular, immune, and nervous systems and other miscellaneous mechanisms of action. Biochim. Biophys. Acta 2009, 1790, 283–308. [Google Scholar]
- Tasdemir, D; Topaloglu, B; Perozzo, R; Brun, R; O’Neill, R; Carballeira, NM; Zhang, X; Tonge, PJ; Linden, A; Ruedi, P. Marine natural products from the Turkish sponge Agelas oroides that inhibit the enoyl reductases from Plasmodium falciparum, Mycobacterium tubercolosis and Escherichia coli. Bioorg. Med. Chem 2007, 15, 6834–6845. [Google Scholar]
- Waller, RF; Keeling, PJ; Donald, RG; Striepen, B; Handman, E; Lang-Unnasch, N; Cowman, AF; Besra, GS; Roos, DS; McFadden, GI. Nuclear-encoded proteins target to the plastid in Toxoplasma gondii and Plasmodium falciparum. Proc. Natl. Acad. Sci. USA 1998, 95, 12352–12357. [Google Scholar]
- Surolia, N; Surolia, A. Triclosan offers protection against blood stages of malaria by inhibiting enoyl-ACP reductase of Plasmodium falciparum. Nat. Med 2001, 7, 167–173. [Google Scholar]
- Vaughan, AM; O’Neill, MT; Tarun, AS; Camargo, N; Phuong, TM; Aly, AS; Cowman, AF; Kappe, SH. Type II fatty acid synthesis is essential only for malaria parasite late liver stage development. Cell. Microbiol 2009, 11, 506–520. [Google Scholar]
- Yu, M; Kumar, TR; Nkrumah, LJ; Coppi, A; Retzlaff, S; Li, CD; Kelly, BJ; Moura, PA; Lakshmanan, V; Freundlich, JS; Valderramos, JC; Vilcheze, C; Siedner, M; Tsai, JH; Falkard, B; Sidhu, AB; Purcell, LA; Gratraud, P; Kremer, L; Waters, AP; Schiehser, G; Jacobus, DP; Janse, CJ; Ager, A; Jacobs, WR, Jr; Sacchettini, JC; Heussler, V; Sinnis, P; Fidock, DA. The fatty acid biosynthesis enzyme FabI plays a key role in the development of liver-stage malarial parasites. Cell Host Microbe 2008, 4, 567–578. [Google Scholar]
- Aiello, A; Fattorusso, E; Menna, M; Taglialatela-Scafati, O. Fattorusso, E, Taglialatela-Scafati, O, Eds.; Modern Alkaloids; Wiley-VCH: Weinheim, Germany, 2007; pp. 271–304. [Google Scholar]
- Kobayashi, J; Suzuki, M; Tsuda, M. Konbu’acidin A, a new bromopyrrole alkaloid with cdk4 inhibitory activity from Hymeniacidon sponge. Tetrahedron 1997, 53, 15681–15684. [Google Scholar]
- Cafieri, F; Fattorusso, E; Mangoni, A; Taglialatela-Scafati, O. Dispacamides, anti-histamine alkaloids from Caribbean Agelas sponges. Tetrahedron Lett 1996, 37, 3587–3590. [Google Scholar]
- Cafieri, F; Carnuccio, R; Fattorusso, E; Taglialatela-Scafati, O; Vallefuoco, T. Anti-histaminic activity of bromopyrrole alkaloids isolated from Caribbean Agelas sponges. Bioorg. Med. Chem. Lett 1997, 7, 2283–2288. [Google Scholar]
- Kobayashi, J; Ohizumi, Y; Nakamura, H; Hirata, Y. A novel antagonist of serotonergic receptors, hymenidin, isolated from the Okinawan marine sponge Hymeniacidon species. Experientia 1986, 42, 1176–1177. [Google Scholar]
- Cafieri, F; Fattorusso, E; Mangoni, A; Taglialatela-Scafati, O. A novel bromopyrrole alkaloid from the sponge Agelas longissima with antiserotonergic activity. Bioorg. Med. Chem. Lett 1995, 5, 799–804. [Google Scholar]
- Kinnel, RB; Gehrken, HP; Scheuer, PJ. Palau’amine: a cytotoxic and immunosuppressive hexacyclic bisguanidine antibiotic from the sponge Stylotella agminata. J. Am. Chem. Soc 1993, 115, 3376–3377. [Google Scholar]
- Walker, RP; Faulkner, DJ; van Engen, D; Clardy, J. Sceptrin, an antimicrobial agent from the sponge Agelas sceptrum. J. Am. Chem. Soc 1981, 103, 6772–6773. [Google Scholar]
- Cipres, A; O’Malley, DP; Li, K; Finlay, D; Baran, PS; Vuori, K. Sceptrin, a Marine Natural Compound, Inhibits Cell Motility in a Variety of Cancer Cell Lines. ACS Chem. Biol 2010, 5, 195–202. [Google Scholar]
- Aiello, A; D’Esposito, M; Fattorusso, E; Menna, M; Mueller, WEG; Perovic-Ottstadt, S; Schroeder, HC. Novel bioactive bromopyrrole alkaloids from the Mediterranean sponge Axinella verrucosa. Bioorg. Med. Chem 2006, 14, 17–24. [Google Scholar]
- Cafieri, F; Fattorusso, E; Mangoni, A; Taglialatela-Scafati, O. Longamide and 3,7-dimethylisoguanine, two novel alkaloids from the marine sponge Agelas longissima. Tetrahedron Lett 1995, 36, 7893–7896. [Google Scholar]
- Cafieri, F; Fattorusso, E; Taglialatela-Scafati, O. Novel bromopyrrole alkaloids from the sponge Agelas dispar. J. Nat. Prod 1998, 61, 122–125. [Google Scholar]
- Matile, H; Pink, JRL. Lefkovits, I, Pernis, B, Eds.; Immunological Methods; Academic Press: San Diego, CA, USA, 1990; pp. 221–234. [Google Scholar]
- Baltz, T; Baltz, D; Giroud, C; Crockett, J. Cultivation in a semi-defined medium of animal infective forms of Trypanosoma brucei, T. equiperdum, T. evansi, T. rhodesiense and T. gambiense. EMBO J 1985, 4, 1273–1277. [Google Scholar]
- Thuita, JK; Karanja, SM; Wenzler, T; Mdachi, RE; Ngotho, JM; Kagira, JM; Tidwell, R; Brun, R. Efficacy of the diamidine DB75 and its prodrug DB289, against murine models of human African trypanosomiasis. Acta Trop 2008, 108, 6–10. [Google Scholar]
- Buckner, FS; Verlinde, CL; La Flamme, AC; van Voorhis, WC. Efficient technique for screening drugs for activity against Trypanosoma cruzi using parasites expressing betagalactosidase. Antimicrob. Agents Chemother 1996, 40, 2592–2597. [Google Scholar]
- Mikus, J; Steverding, D. A simple colorimetric method to screen drug cytotoxicity against Leishmania using the dye Alamar Blue. Parasitol. Int 2000, 48, 265–269. [Google Scholar]
- Tasdemir, D; Lack, G; Brun, R; Rüedi, P; Scapozza, L; Perozzo, R. Inhibition of Plasmodium falciparum fatty acid biosynthesis: evaluation of FabG, FabZ, and FabI as drug targets for flavonoids. J. Med. Chem 2006, 49, 3345–3353. [Google Scholar]
- Fresneda, PM; Molina, P; Sanz, MA. A convergent approach to midpacamide and dispacamide pyrrole-imidazole marine alkaloids. Tetrahedron Lett 2001, 42, 851–854. [Google Scholar]
- Sun, X-T; Chen, A. Total synthesis of rac- longamide B. Tetrahedron Lett 2007, 48, 3459–3461. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
| Compound | T. b. rhodesiense | T. cruzi | L. donovani | P. falciparum | Cytotoxicity L6 cells |
|---|---|---|---|---|---|
| 1a | 17.30 1 | >30 1 | >30 1 | 3.90 1 | 88.60 1 |
| 1b | 12.20 1 | >30 1 | 15.40 1 | 7.90 1 | 76.40 1 |
| 2 | 77.64 | 73.10 | 29.87 | 12.54 | 75.73 |
| 3 | 40.12 | >90 | >90 | 1.34 | >90 |
| 4 | 68.25 | >90 | 53.75 | >20 | >90 |
| 5 | 25.34 | >90 | 75.86 | 4.88 | >90 |
| 6 | 13.48 | 72.25 | 41.59 | 1.09 | 35.61 |
| 7 | 61.48 | >90 | >90 | >20 | >90 |
| 8 | 1.53 | 33.03 | 3.85 | 7.46 | 9.94 |
| 9 | >90 | >90 | >90 | >20 | >90 |
| 10 | 9.71 | 60.08 | 51.58 | 11.08 | >90 |
| 11 | 0.46 | 68.88 | 1.09 | 1.48 | 4.46 |
| 12 | 67.13 | >90 | 34.49 | >20 | 62.32 |
| 13 | 73.76 | >90 | 75.83 | >20 | >90 |
| 14 | 49.96 | >90 | 43.22 | 11.18 | >90 |
| Standards | 0.004 a | 0.312 b | 0.206 c | 0.065 d | 0.005 e |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Scala, F.; Fattorusso, E.; Menna, M.; Taglialatela-Scafati, O.; Tierney, M.; Kaiser, M.; Tasdemir, D. Bromopyrrole Alkaloids as Lead Compounds against Protozoan Parasites. Mar. Drugs 2010, 8, 2162-2174. https://doi.org/10.3390/md8072162
Scala F, Fattorusso E, Menna M, Taglialatela-Scafati O, Tierney M, Kaiser M, Tasdemir D. Bromopyrrole Alkaloids as Lead Compounds against Protozoan Parasites. Marine Drugs. 2010; 8(7):2162-2174. https://doi.org/10.3390/md8072162
Chicago/Turabian StyleScala, Fernando, Ernesto Fattorusso, Marialuisa Menna, Orazio Taglialatela-Scafati, Michelle Tierney, Marcel Kaiser, and Deniz Tasdemir. 2010. "Bromopyrrole Alkaloids as Lead Compounds against Protozoan Parasites" Marine Drugs 8, no. 7: 2162-2174. https://doi.org/10.3390/md8072162