Using Bioinformatic Approaches to Identify Pathways Targeted by Human Leukemogens

Abstract
:Abbreviations
| AhR | aryl hydrocarbon receptor |
| ALL | acute lymphocytic leukemia |
| AML | acute myeloid leukemia |
| AUC | area-under-the-curve |
| CAS RN | Chemical Abstracts Service Registry Numbers |
| COX | cyclooxygenase |
| FWER | family-wise error rate |
| HOPACH | hierarchical Ordered Partitioning And Collapsing Hybrid |
| HSC | hematopoietic stem cells |
| IARC | International Agency for Research on Cancer |
| LOX | lipoxygenase |
| MAPK | mitogen-activated protein kinase |
| MNCL | mononuclear cell leukemia |
| NTP | National Toxicology Program |
| NTs | Neurotrophins |
| Perc | tetrachloroethylene |
| PTGS2 | prostaglandin-endoperoxide synthase 2 |
| RoC | Report on Carcinogens |
| SEPEA | Structurally Enhanced Pathway Enrichment Analysis |
| SVM | support vector machines |
| TCE | trichloroethylene |
1. Introduction
1.1. Chemical Exposures Associated with Leukemia
1.2. Biological Pathways Involved in Leukemia
1.3. Biological Pathways Targeted by Leukemogens
1.4. Study Aim
2. Results and Discussion
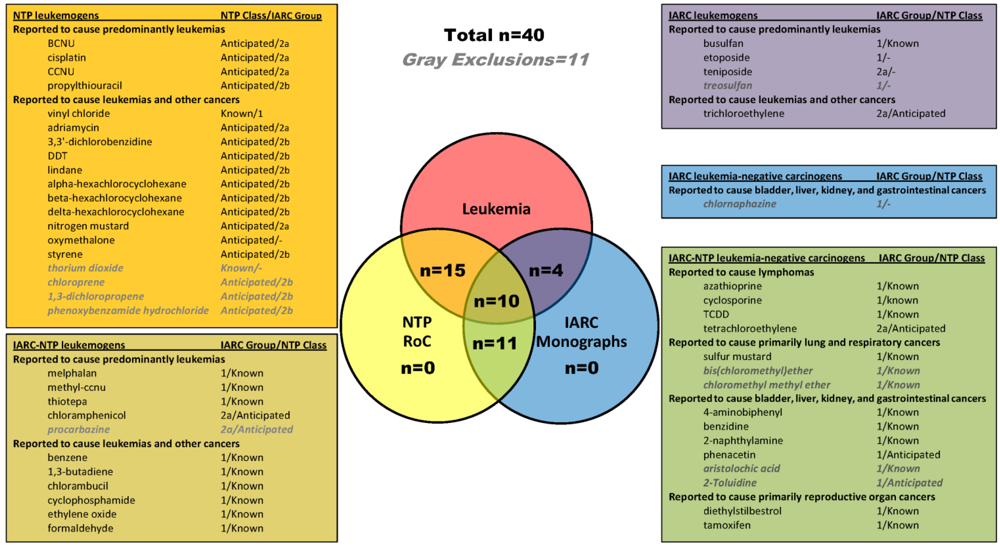
2.1. Identification of Leukemogens and Non-Leukemogenic Carcinogens

2.2. Enrichment of KEGG Pathways in Genes and Proteins Associated with Leukemogens and Non-Leukemogenic Carcinogens


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | No. (%) of Leukemogens | Cluster 0 Probability | Cluster1 Probability |
|---|---|---|---|
| Biological Pathway | |||
| Metabolism_of_xenobiotics_by_cytochrome_P450 | 20 (69) | 0 | 1 |
| Neurotrophin_signaling_pathway | 19 (66) | 0 | 1 |
| Glutathione_metabolism | 18 (62) | 0.02 | 0.98 |
| Apoptosis | 18 (62) | 0.01 | 0.99 |
| MAPK_signaling_pathway | 17 (59) | 0 | 1 |
| Toll-like_receptor_signaling_pathway | 17 (59) | 0 | 1 |
| p53_signaling_pathway | 16 (55) | 0.11 | 0.89 |
| Retinol_metabolism | 15 (52) | 0.02 | 0.98 |
| Bile_secretion | 15 (52) | 0.05 | 0.95 |
| ErbB_signaling_pathway | 15 (52) | 0 | 1 |
| Disease Pathway | |||
| Pathways_in_cancer | 23 (79) | 0 | 1 |
| Prostate_cancer | 20 (69) | 0.14 | 0.86 |
| Colorectal_cancer | 20 (69) | 0 | 1 |
| Bladder_cancer | 19 (66) | 0.1 | 0.91 |
| Melanoma | 19 (66) | 0 | 1 |
| Pancreatic_cancer | 18 (62) | 0.09 | 0.91 |
| Chronic_myeloid_leukemia | 18 (62) | 0.01 | 1 |
| Amyotrophic_lateral_sclerosis_(ALS) | 18 (62) | 0 | 1 |
| Small_cell_lung_cancer | 18 (62) | 0.07 | 0.93 |
| Toxoplasmosis | 17 (59) | 0 | 1 |
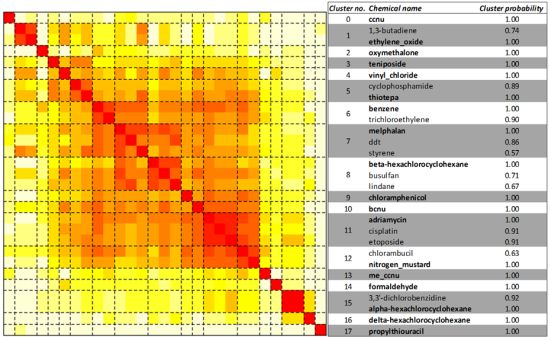
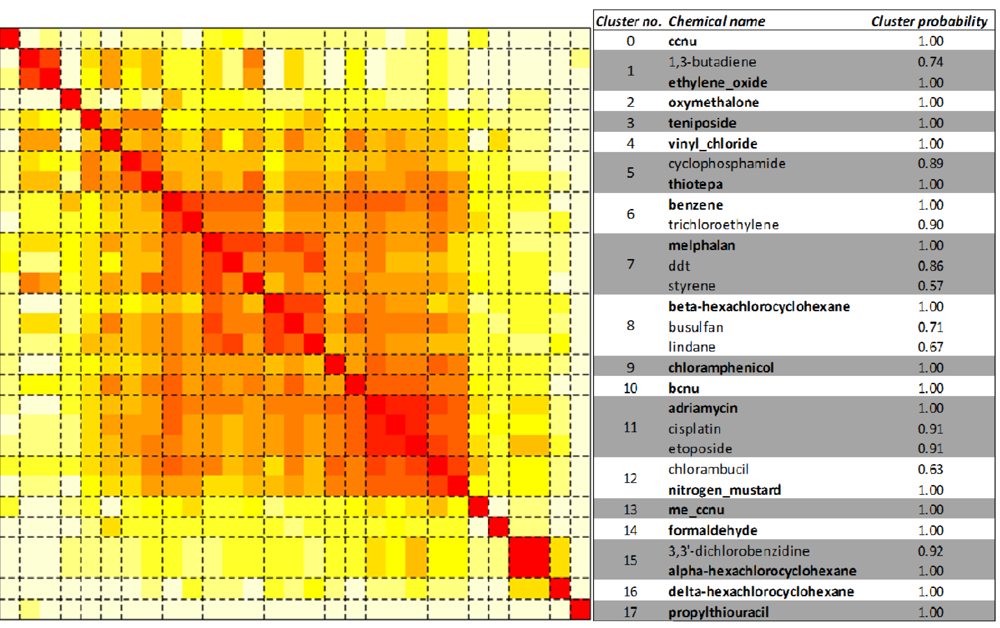
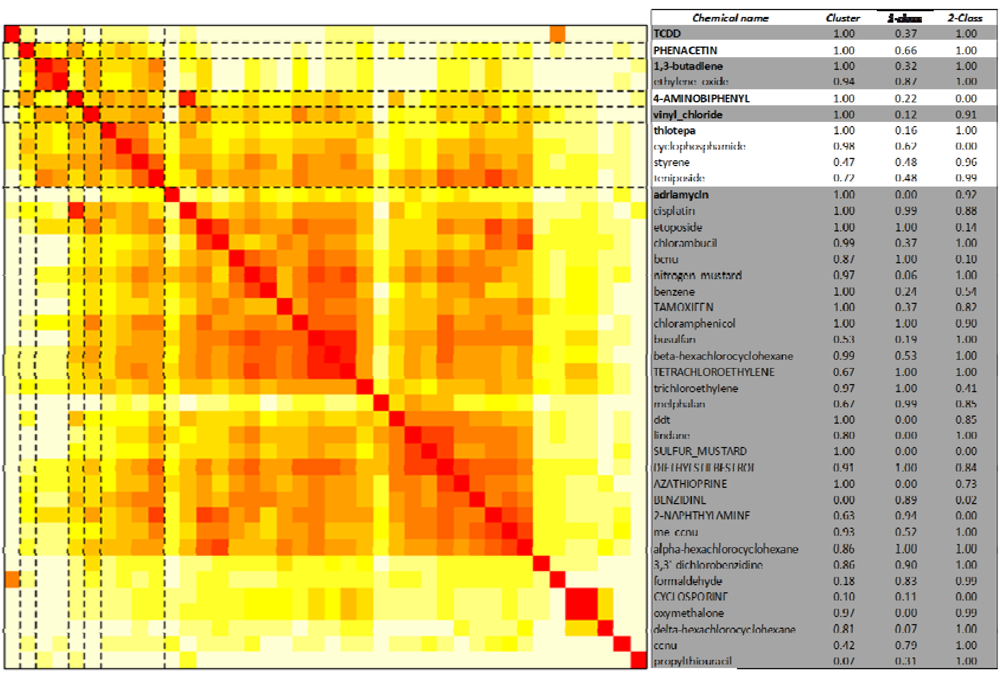
2.3. Unsupervised Clustering of Leukemogens

2.4. Distinguishing Leukemogens and Non-Leukemogenic Carcinogens

2.4.2. One-Class Support Vector Machines
2.4.3. Two-Class Random Forests
| Pathway | No. (%) of Leukemogens | No. (%) of Non-Leukemogens | Cluster 0 Probability | Cluster 1 Probability | Mean Decrease Gini |
|---|---|---|---|---|---|
| Caffeine_metabolism | 3 (10) | 8 (73) | 0.3 | 0.7 | 0.36 |
| Arachidonic_acid_metabolism | 13 (45) | 6 (55) | 0.03 | 0.97 | 0.2 |
| Basal_cell_carcinoma | 9 (31) | 6 (55) | 0.12 | 0.88 | 0.18 |
| Drug_metabolism_other_enzymes | 9 (31) | 5 (45) | 0 | 1 | 0.16 |
| Ribosome | 2 (7) | 5 (45) | 0.62 | 0.38 | 0.15 |
| Retinol_metabolism | 15 (52) | 8 (73) | 0.02 | 0.98 | 0.15 |
| Metabolism_of_xenobiotics_by_cytochrome_P450 | 20 (69) | 7 (64) | 0 | 1 | 0.15 |
| Prostate_cancer | 20 (69) | 7 (64) | 0.14 | 0.86 | 0.14 |
| Pentose_and_glucuronate_inter-conversions | 7 (24) | 5 (45) | 0.89 | 0.11 | 0.14 |
| Renin-angiotensin_system | 2 (7) | 3 (27) | 0.37 | 0.63 | 0.13 |
2.4.4. Challenges in Discriminating Leukemogens and Non-Leukemogenic Carcinogens
2.5. Comparison of Pathway Enrichment in CTD and in Data from a Single, Well-Designed, Toxico-Genomic Study
3. Experimental Section
3.1. Identification of Human Leukemogens and Non-Leukemogenic Carcinogens
3.2. Analysis of Enrichment of KEGG Biochemical Pathways in CTD Data
3.3. Structural Similarity between Chemicals








3.4. Unsupervised Clustering of Chemicals and Pathways






3.5. One-Class Classification of Chemicals







3.6. Two-Class Classification of Chemicals



4. Conclusions
Conflict of Interest
Acknowledgments
References
- Sawyers, C.L.; Denny, C.T.; Witte, O.N. Leukemia and the disruption of normal hematopoiesis. Cell 1991, 64, 337–350. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Vardiman, J.W. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; IARC: Lyon, France, 2008. [Google Scholar]
- Vardiman, J.W. The World Health Organization (WHO) classification of tumors of the hematopoietic and lymphoid tissues: An overview with emphasis on the myeloid neoplasms. Chem. Biol. Interact. 2010, 184, 16–20. [Google Scholar] [CrossRef]
- American Cancer Society. In Cancer Facts & Figures 2012; American Cancer Society: Atlanta, GA, USA, 2012.
- Austin, H.; Delzell, E.; Cole, P. Benzene and leukemia. A review of the literature and a risk assessment. Am. J. Epidemiol. 1988, 127, 419–439. [Google Scholar]
- Khalade, A.; Jaakkola, M.S.; Pukkala, E.; Jaakkola, J.J. Exposure to benzene at work and the risk of leukemia: A systematic review and meta-analysis. Environ. Health 2010, 9. [Google Scholar] [CrossRef] [Green Version]
- Van Maele-Fabry, G.; Duhayon, S.; Lison, D. A systematic review of myeloid leukemias and occupational pesticide exposure. Canc. Causes Contr. 2007, 18, 457–478. [Google Scholar] [CrossRef]
- Goldstein, B.D. Hematological and toxicological evaluation of formaldehyde as a potential cause of human leukemia. Hum. Exp. Toxicol. 2011, 30, 725–735. [Google Scholar] [CrossRef]
- Albin, M.; Bjork, J.; Welinder, H.; Tinnerberg, H.; Mauritzson, N.; Johansson, B.; Billstrom, R.; Stromberg, U.; Mikoczy, Z.; Ahlgren, T.; et al. Acute myeloid leukemia and clonal chromosome aberrations in relation to past exposure to organic solvents. Scand. J. Work Environ. Health 2000, 26, 482–491. [Google Scholar]
- Sandler, D.P.; Shore, D.L.; Anderson, J.R.; Davey, F.R.; Arthur, D.; Mayer, R.J.; Silver, R.T.; Weiss, R.B.; Moore, J.O.; Schiffer, C.A.; et al. Cigarette smoking and risk of acute leukemia: Associations with morphology and cytogenetic abnormalities in bone marrow. J. Natl. Cancer Inst. 1993, 85, 1994–2003. [Google Scholar] [CrossRef]
- Strom, S.S.; Oum, R.; Elhor Gbito, K.Y.; Garcia-Manero, G.; Yamamura, Y. De novo acute myeloid leukemia risk factors: A Texas case-control study. Cancer 2012. [Google Scholar] [CrossRef]
- Cogliano, V.J.; Baan, R.; Straif, K.; Grosse, Y.; Lauby-Secretan, B.; El Ghissassi, F.; Bouvard, V.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; et al. Preventable exposures associated with human cancers. J. Natl. Cancer Inst. 2011, 103, 1827–1839. [Google Scholar]
- Qian, Z.; Joslin, J.M.; Tennant, T.R.; Reshmi, S.C.; Young, D.J.; Stoddart, A.; Larson, R.A.; Le Beau, M.M. Cytogenetic and genetic pathways in therapy-related acute myeloid leukemia. Chem. Biol. Interact. 2010, 184, 50–57. [Google Scholar] [CrossRef]
- Wiemels, J. Perspectives on the causes of childhood leukemia. Chem. Biol. Interact. 2012, 5, 59–67. [Google Scholar] [CrossRef]
- IARC. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. A Review of Human Carcinogens; International Agency for Research on Cancer: Lyon, France, 2011; 100. Available online: http://monographs.iarc.fr/ENG/monographs/PDFs/index.php (accessed on 5 February 2012).
- NTP, 12th Report on Carcinogens; U.S. Department of Health and Human Services, Public Health Service, National Toxicology Program: Research Triangle Park, NC, USA, 2011.
- Rowley, J.D.; Golomb, H.M.; Vardiman, J.W. Nonrandom chromosome abnormalities in acute leukemia and dysmyelopoietic syndromes in patients with previously treated malignant disease. Blood 1981, 58, 759–767. [Google Scholar]
- Smith, S.M.; Le Beau, M.M.; Huo, D.; Karrison, T.; Sobecks, R.M.; Anastasi, J.; Vardiman, J.W.; Rowley, J.D.; Larson, R.A. Clinical-cytogenetic associations in 306 patients with therapy-related myelodysplasia and myeloid leukemia: The University of Chicago series. Blood 2003, 102, 43–52. [Google Scholar]
- Kelly, L.M.; Gilliland, D.G. Genetics of myeloid leukemias. Annu. Rev. Genomics Hum. Genet. 2002, 3, 179–198. [Google Scholar] [CrossRef]
- Pedersen-Bjergaard, J.; Andersen, M.T.; Andersen, M.K. Genetic pathways in the pathogenesis of therapy-related myelodysplasia and acute myeloid leukemia. Hematol. Am. Soc. Hematol. Educ. Program. 2007, 392–397. [Google Scholar] [CrossRef]
- Pedersen-Bjergaard, J.; Christiansen, D.H.; Desta, F.; Andersen, M.K. Alternative genetic pathways and cooperating genetic abnormalities in the pathogenesis of therapy-related myelodysplasia and acute myeloid leukemia. Leukemia 2006, 20, 1943–1949. [Google Scholar] [CrossRef]
- Pedersen-Bjergaard, J.; Andersen, M.K.; Andersen, M.T.; Christiansen, D.H. Genetics of therapy-related myelodysplasia and acute myeloid leukemia. Leukemia 2008, 22, 240–248. [Google Scholar] [CrossRef]
- Takahashi, S. Current findings for recurring mutations in acute myeloid leukemia. J. Hematol. Oncol. 2011, 4. [Google Scholar] [CrossRef]
- Patel, J.P.; Gonen, M.; Figueroa, M.E.; Fernandez, H.; Sun, Z.; Racevskis, J.; van Vlierberghe, P.; Dolgalev, I.; Thomas, S.; Aminova, O.; et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N. Engl. J. Med. 2012, 366, 1079–1089. [Google Scholar]
- Havelange, V.; Stauffer, N.; Heaphy, C.C.; Volinia, S.; Andreeff, M.; Marcucci, G.; Croce, C.M.; Garzon, R. Functional implications of microRNAs in acute myeloid leukemia by integrating microRNA and messenger RNA expression profiling. Cancer 2011, 117, 4696–4706. [Google Scholar] [CrossRef]
- Seca, H.; Almeida, G.M.; Guimaraes, J.E.; Vasconcelos, M.H. miR signatures and the role of miRs in acute myeloid leukaemia. Eur. J. Cancer 2010, 46, 1520–1527. [Google Scholar] [CrossRef]
- Volinia, S.; Galasso, M.; Costinean, S.; Tagliavini, L.; Gamberoni, G.; Drusco, A.; Marchesini, J.; Mascellani, N.; Sana, M.E.; Abu Jarour, R.; et al. Reprogramming of miRNA networks in cancer and leukemia. Genome Res. 2010, 20, 589–599. [Google Scholar] [CrossRef]
- Voso, M.T.; D’Alo, F.; Greco, M.; Fabiani, E.; Criscuolo, M.; Migliara, G.; Pagano, L.; Fianchi, L.; Guidi, F.; Hohaus, S.; Leone, G. Epigenetic changes in therapy-related MDS/AML. Chem. Biol. Interact. 2010, 184, 46–49. [Google Scholar] [CrossRef]
- Theilgaard-Monch, K.; Boultwood, J.; Ferrari, S.; Giannopoulos, K.; Hernandez-Rivas, J.M.; Kohlmann, A.; Morgan, M.; Porse, B.; Tagliafico, E.; Zwaan, C.M.; et al. Gene expression profiling in MDS and AML: Potential and future avenues. Leukemia 2011, 25, 909–920. [Google Scholar]
- Miller, B.G.; Stamatoyannopoulos, J.A. Integrative meta-analysis of differential gene expression in acute myeloid leukemia. PLoS One 2010, 5. [Google Scholar] [CrossRef]
- Mrozek, K.; Radmacher, M.D.; Bloomfield, C.D.; Marcucci, G. Molecular signatures in acute myeloid leukemia. Curr. Opin. Hematol. 2009, 16, 64–69. [Google Scholar] [CrossRef]
- Kornblau, S.M.; Minden, M.D.; Rosen, D.B.; Putta, S.; Cohen, A.; Covey, T.; Spellmeyer, D.C.; Fantl, W.J.; Gayko, U.; Cesano, A. Dynamic single-cell network profiles in acute myelogenous leukemia are associated with patient response to standard induction therapy. Clin. Cancer Res. 2010, 16, 3721–3733. [Google Scholar]
- Cesano, A.; Rosen, D.B.; O’Meara, P.; Putta, S.; Gayko, U.; Spellmeyer, D.C.; Cripe, L.D.; Sun, Z.; Uno, H.; Litzow, M.R.; et al. Functional pathway analysis in acute myeloid leukemia using single cell network profiling assay: Effect of specimen source (bone marrow or peripheral blood) on assay readouts. Cytometry B Clin. Cytom. 2012, 82, 158–172. [Google Scholar]
- Reikvam, H.; Olsnes, A.M.; Gjertsen, B.T.; Ersvar, E.; Bruserud, O. Nuclear factor-kappaB signaling: A contributor in leukemogenesis and a target for pharmacological intervention in human acute myelogenous leukemia. Crit. Rev. Oncog. 2009, 15, 1–41. [Google Scholar]
- Towatari, M.; Iida, H.; Tanimoto, M.; Iwata, H.; Hamaguchi, M.; Saito, H. Constitutive activation of mitogen-activated protein kinase pathway in acute leukemia cells. Leukemia 1997, 11, 479–484. [Google Scholar]
- Simon, M.; Grandage, V.L.; Linch, D.C.; Khwaja, A. Constitutive activation of the Wnt/beta-catenin signalling pathway in acute myeloid leukaemia. Oncogene 2005, 24, 2410–2420. [Google Scholar] [CrossRef]
- Wang, Y.; Krivtsov, A.V.; Sinha, A.U.; North, T.E.; Goessling, W.; Feng, Z.; Zon, L.I.; Armstrong, S.A. The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science 2010, 327, 1650–1653. [Google Scholar]
- Luis, T.C.; Ichii, M.; Brugman, M.H.; Kincade, P.; Staal, F.J. Wnt signaling strength regulates normal hematopoiesis and its deregulation is involved in leukemia development. Leukemia 2012, 26, 414–421. [Google Scholar] [CrossRef]
- Altman, J.K.; Sassano, A.; Platanias, L.C. Targeting mTOR for the treatment of AML. New agents and new directions. Oncotarget 2011, 2, 510–517. [Google Scholar]
- Chung, E.; Kondo, M. Role of Ras/Raf/MEK/ERK signaling in physiological hematopoiesis and leukemia development. Immunol. Res. 2011, 49, 248–268. [Google Scholar] [CrossRef]
- Casado, F.L.; Singh, K.P.; Gasiewicz, T.A. The aryl hydrocarbon receptor: Regulation of hematopoiesis and involvement in the progression of blood diseases. Blood Cells Mol. Dis. 2010, 44, 199–206. [Google Scholar] [CrossRef]
- Shaham, L.; Binder, V.; Gefen, N.; Borkhardt, A.; Izraeli, S. miR-125 in normal and malignant hematopoiesis. Leukemia 2012. [Google Scholar] [CrossRef]
- Smith, M.T. Advances in understanding benzene health effects and susceptibility. Annu. Rev. Public Health 2010, 31, 133–148. [Google Scholar] [CrossRef]
- Lan, Q.; Zhang, L.; Li, G.; Vermeulen, R.; Weinberg, R.S.; Dosemeci, M.; Rappaport, S.M.; Shen, M.; Alter, B.P.; Wu, Y.; et al. Hematotoxicity in workers exposed to low levels of benzene. Science 2004, 306, 1774–1776. [Google Scholar]
- Zhang, L.; Tang, X.; Rothman, N.; Vermeulen, R.; Ji, Z.; Shen, M.; Qiu, C.; Guo, W.; Liu, S.; Reiss, B.; et al. Occupational exposure to formaldehyde, hematotoxicity, and leukemia-specific chromosome changes in cultured myeloid progenitor cells. Cancer Epidemiol. Biomark. Prev. 2010, 19, 80–88. [Google Scholar] [CrossRef]
- McHale, C.M.; Zhang, L.; Smith, M.T. Current understanding of the mechanism of benzene-induced leukemia in humans: Implications for risk assessment. Carcinogenesis 2012, 33, 240–252. [Google Scholar]
- McHale, C.M.; Zhang, L.; Lan, Q.; Vermeulen, R.; Li, G.; Hubbard, A.E.; Porter, K.E.; Thomas, R.; Portier, C.J.; Shen, M.; et al. Global gene expression profiling of a population exposed to a range of benzene levels. Environ. Health Perspect. 2011, 119, 628–634. [Google Scholar]
- Li, L.; Li, M.; Sun, C.; Francisco, L.; Chakraborty, S.; Sabado, M.; McDonald, T.; Gyorffy, J.; Chang, K.; Wang, S.; et al. Altered hematopoietic cell gene expression precedes development of therapy-related myelodysplasia/acute myeloid leukemia and identifies patients at risk. Cancer Cell 2011, 20, 591–605. [Google Scholar] [CrossRef]
- Guyton, K.Z.; Kyle, A.D.; Aubrecht, J.; Cogliano, V.J.; Eastmond, D.A.; Jackson, M.; Keshava, N.; Sandy, M.S.; Sonawane, B.; Zhang, L.; et al. Improving prediction of chemical carcinogenicity by considering multiple mechanisms and applying toxicogenomic approaches. Mutat. Res. 2009, 681, 230–240. [Google Scholar] [CrossRef]
- Mattingly, C.J.; Colby, G.T.; Forrest, J.N.; Boyer, J.L. The Comparative Toxicogenomics Database (CTD). Environ. Health Perspect. 2003, 111, 793–795. [Google Scholar] [CrossRef]
- Gohlke, J.M.; Thomas, R.; Zhang, Y.; Rosenstein, M.C.; Davis, A.P.; Murphy, C.; Becker, K.G.; Mattingly, C.J.; Portier, C.J. Genetic and environmental pathways to complex diseases. BMC Syst. Biol. 2009, 3. [Google Scholar] [CrossRef]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Hattori, M.; Aoki-Kinoshita, K.; Itoh, M.; Kawashima, S.; Katayama, T.; Araki, M.; Hirakawa, M. From genomics to chemical genomics: New developments in KEGG. Nucleic Acids Res. 2006, 34, D354–D357. [Google Scholar] [CrossRef]
- ATSDR. Toxicological Profile For Benzene. U.S. Department of Health And Human Services. Agency for Toxic Substances and Disease Registry: Atlanta, GA, USA, 2007. Available online: http://www.atsdr.cdc.gov/toxprofiles/tp3.html (accessed on 5 February 2012).
- Cronkite, E.P.; Drew, R.T.; Inoue, T.; Hirabayashi, Y.; Bullis, J.E. Hematotoxicity and carcinogenicity of inhaled benzene. Environ. Health Perspect. 1989, 82, 97–108. [Google Scholar] [CrossRef]
- Snyder, C.A.; Goldstein, B.D.; Sellakumar, A.R.; Bromberg, I.; Laskin, S.; Albert, R.E. The inhalation toxicology of benzene: Incidence of hematopoietic neoplasms and hematotoxicity in ARK/J and C57BL/6J mice. Toxicol. Appl. Pharmacol. 1980, 54, 323–331. [Google Scholar] [CrossRef]
- Thomas, J.; Haseman, J.K.; Goodman, J.I.; Ward, J.M.; Loughran, T.P., Jr.; Spencer, P.J. A review of large granular lymphocytic leukemia in Fischer 344 rats as an initial step toward evaluating the implication of the endpoint to human cancer risk assessment. Toxicol. Sci. 2007, 99, 3–19. [Google Scholar] [CrossRef]
- Thomas, R.; Gohlke, J.; Stopper, G.; Parham, F.; Portier, C. Choosing the right path: Enhancement of biologically relevant sets of genes or proteins using pathway structure. Genome Biol. 2009, 10. [Google Scholar] [CrossRef]
- van der Laan, M.J.; Pollard, K.S. A new algorithm for hybrid hierarchical clustering with visualization and the bootstrap. J. Stat. Plan. Inference 2003, 117, 275–303. [Google Scholar] [CrossRef]
- Hole, P.S.; Darley, R.L.; Tonks, A. reactive oxygen species play a role in myeloid leukemias? Blood 2011, 117, 5816–5826. [Google Scholar] [CrossRef]
- Rucker, F.G.; Schlenk, R.F.; Bullinger, L.; Kayser, S.; Teleanu, V.; Kett, H.; Habdank, M.; Kugler, C.M.; Holzmann, K.; Gaidzik, V.I.; et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood 2012, 119, 2114–2121. [Google Scholar]
- Li, Z.; Beutel, G.; Rhein, M.; Meyer, J.; Koenecke, C.; Neumann, T.; Yang, M.; Krauter, J.; von Neuhoff, N.; Heuser, M.; Diedrich, H.; et al. High-affinity neurotrophin receptors and ligands promote leukemogenesis. Blood 2009, 113, 2028–2037. [Google Scholar] [CrossRef]
- Mongan, N.P.; Gudas, L.J. Diverse actions of retinoid receptors in cancer prevention and treatment. Differentiation 2007, 75, 853–870. [Google Scholar] [CrossRef]
- Mi, J. Current treatment strategy of acute promyelocytic leukemia. Front. Med. 2011, 5, 341–347. [Google Scholar] [CrossRef]
- Crivori, P.; Pennella, G.; Magistrelli, M.; Grossi, P.; Giusti, A.M. Predicting myelosuppression of drugs from in silico models. J. Chem. Inf. Model. 2011, 51, 434–445. [Google Scholar] [CrossRef]
- Smith, M.T.; Zhang, L.; McHale, C.M.; Skibola, C.F.; Rappaport, S.M. Benzene, the exposome and future investigations of leukemia etiology. Chem. Biol. Interact. 2011, 192, 155–159. [Google Scholar] [CrossRef]
- Wartenberg, D.; Reyner, D.; Scott, C.S. Trichloroethylene and cancer: Epidemiologic evidence. Environ. Health Perspect. 2000, 108(Suppl 2), 161–176. [Google Scholar]
- Scott, C.S.; Chiu, W.A. Trichloroethylene cancer epidemiology: A consideration of select issues. Environ. Health Perspect. 2006, 114, 1471–1478. [Google Scholar] [CrossRef]
- Aschengrau, A.; Ozonoff, D.; Paulu, C.; Coogan, P.; Vezina, R.; Heeren, T.; Zhang, Y. Cancer risk and tetrachloroethylene-contaminated drinking water in Massachusetts. Arch. Environ. Health 1993, 48, 284–292. [Google Scholar] [CrossRef]
- Ramlow, J.M. Apparent increased risk of leukemia in their highest category of exposure to tetrachloroethylene (PCE) in drinking water. Arch. Environ. Health 1995, 50, 170–173. [Google Scholar] [CrossRef]
- Ishmael, J.; Dugard, P.H. A review of perchloroethylene and rat mononuclear cell leukemia. Regul. Toxicol. Pharmacol. 2006, 45, 178–184. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- La Vecchia, C.; Tavani, A. Coffee and cancer risk: An update. Eur. J. Cancer Prev. 2007, 16, 385–389. [Google Scholar] [CrossRef]
- Tworoger, S.S.; Gertig, D.M.; Gates, M.A.; Hecht, J.L.; Hankinson, S.E. Caffeine, alcohol, smoking, and the risk of incident epithelial ovarian cancer. Cancer 2008, 112, 1169–1177. [Google Scholar] [CrossRef]
- Kuper, H.; Titus-Ernstoff, L.; Harlow, B.L.; Cramer, D.W. Population based study of coffee, alcohol and tobacco use and risk of ovarian cancer. Int. J. Cancer 2000, 88, 313–318. [Google Scholar] [CrossRef]
- Kotsopoulos, J.; Vitonis, A.F.; Terry, K.L.; De Vivo, I.; Cramer, D.W.; Hankinson, S.E.; Tworoger, S.S. Coffee intake, variants in genes involved in caffeine metabolism, and the risk of epithelial ovarian cancer. Canc. Causes Contr. 2009, 20, 335–344. [Google Scholar] [CrossRef]
- Sachse, C.; Brockmoller, J.; Bauer, S.; Roots, I. Functional significance of a C–A polymorphism in intron 1 of the cytochrome P450 CYP1A2 gene tested with caffeine. Br. J. Clin. Pharmacol. 1999, 47, 445–449. [Google Scholar]
- Han, X.M.; Ou-Yang, D.S.; Lu, P.X.; Jiang, C.H.; Shu, Y.; Chen, X.P.; Tan, Z.R.; Zhou, H.H. Plasma caffeine metabolite ratio (17X/137X) in vivo associated with G-2964A and C734A polymorphisms of human CYP1A2. Pharmacogenetics 2001, 11, 429–435. [Google Scholar] [CrossRef]
- Le Marchand, L.; Donlon, T.; Kolonel, L.N.; Henderson, B.E.; Wilkens, L.R. Estrogen metabolism-related genes and breast cancer risk: The multiethnic cohort study. Cancer Epidemiol. Biomark. Prev. 2005, 14, 1998–2003. [Google Scholar] [CrossRef]
- Zevin, S.; Benowitz, N.L. Drug interactions with tobacco smoking. An update. Clin. Pharmacokinet. 1999, 36, 425–438. [Google Scholar] [CrossRef]
- Vistisen, K.; Loft, S.; Poulsen, H.E. Cytochrome P450 IA2 activity in man measured by caffeine metabolism: Effect of smoking, broccoli and exercise. Adv. Exp. Med. Biol. 1991, 283, 407–411. [Google Scholar] [CrossRef]
- Zeldin, D.C. Epoxygenase pathways of arachidonic acid metabolism. J. Biol. Chem. 2001, 276, 36059–36062. [Google Scholar] [CrossRef]
- Wang, D.; Dubois, R.N. Eicosanoids and cancer. Nat. Rev. Cancer 2010, 10, 181–193. [Google Scholar] [CrossRef]
- Greene, E.R.; Huang, S.; Serhan, C.N.; Panigrahy, D. Regulation of inflammation in cancer by eicosanoids. Prostaglandins Other Lipid Mediat. 2011, 96, 27–36. [Google Scholar] [CrossRef]
- Davis, A.P.; King, B.L.; Mockus, S.; Murphy, C.G.; Saraceni-Richards, C.; Rosenstein, M.; Wiegers, T.; Mattingly, C.J. The comparative toxicogenomics database: Update 2011. Nucleic Acids Res. 2011, 39, D1067–D1072. [Google Scholar]
- Bolton, E.E.; Wang, Y.; Thiessen, P.A.; Bryant, S.H. PubChem: Integrated platform of small molecules and biological activities. Annu. Rep. Comput. Chem. 2008, 4, 217–241. [Google Scholar]
- Chen, X.; Reynolds, C.H. Performance of similarity measures in 2D fragment-based similarity searching: Comparison of structural descriptors and similarity coefficients. J. Chem. Inf. Comput. Sci. 2002, 42, 1407–1414. [Google Scholar] [CrossRef]
- Holliday, J.D.; Salim, N.; Whittle, M.; Willett, P. Analysis and display of the size dependence of chemical similarity coefficients. J. Chem. Inf. Comput. Sci. 2003, 43, 819–828. [Google Scholar] [CrossRef]
- Van der Laan, M.; Pollard, K. A new algorithm for hybrid hierarchical clustering with visualization and the bootstrap. J. Stat. Plan. Inference 2003, 117, 275–303. [Google Scholar] [CrossRef]
- R Development Core Team. In R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2009.
- Pollard, K.S.; Wall, G.; van der Laan, M.J. Hopach: Hierarchical Ordered Partitioning and Collapsing Hybrid (HOPACH); R Package Version 2.10.0. 2010. Available online: http://CRAN.R-project.org/package=hopach (accessed on 4 April 2012).
- Schölkopf, B.; Platt, J.C.; Shawe-Taylor, J.; Smola, A.J.; Williamson, R.C. Estimating the support of a high-dimensional distribution. Neural Comput. 2001, 13, 1443–1471. [Google Scholar] [CrossRef]
- Dimitriadou, E.; Hornik, K.; Leisch, F.; Meyer, D.; Weingessel, A. e1071: Misc Functions of the Department of Statistics (e1071),TU Wien; R Package Version 1.5-24. 2010. Available online: http://CRAN.R-project.org/package=e1071 (accessed on 2 November 2010).
- Polley, E.C. SuperLearner: Super Learner Prediction; R Package Version 1.1-18. 2010. Available online: http://www.stat.berkeley.edu/~ecpolley/SL/ (accessed on 2 November 2010).
- Liaw, A.; Wiener, M. Classification and Regression by randomForest. R. News 2002, 2, 18–22. [Google Scholar]
- Sing, T.; Sander, O.; Beerenwinkel, N.; Lengauer, T. Visualizing the Performance of Scoring Classifiers; R Package Version 1.0-4. 2009. Available online: http://CRAN.R-project.org/package=ROCR (accessed on 2 April 2012).
Supplementary Files
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Thomas, R.; Phuong, J.; McHale, C.M.; Zhang, L. Using Bioinformatic Approaches to Identify Pathways Targeted by Human Leukemogens. Int. J. Environ. Res. Public Health 2012, 9, 2479-2503. https://doi.org/10.3390/ijerph9072479
Thomas R, Phuong J, McHale CM, Zhang L. Using Bioinformatic Approaches to Identify Pathways Targeted by Human Leukemogens. International Journal of Environmental Research and Public Health. 2012; 9(7):2479-2503. https://doi.org/10.3390/ijerph9072479
Chicago/Turabian StyleThomas, Reuben, Jimmy Phuong, Cliona M. McHale, and Luoping Zhang. 2012. "Using Bioinformatic Approaches to Identify Pathways Targeted by Human Leukemogens" International Journal of Environmental Research and Public Health 9, no. 7: 2479-2503. https://doi.org/10.3390/ijerph9072479
APA StyleThomas, R., Phuong, J., McHale, C. M., & Zhang, L. (2012). Using Bioinformatic Approaches to Identify Pathways Targeted by Human Leukemogens. International Journal of Environmental Research and Public Health, 9(7), 2479-2503. https://doi.org/10.3390/ijerph9072479