Application of Operando X-ray Diffractometry in Various Aspects of the Investigations of Lithium/Sodium-Ion Batteries




Abstract
:1. Introduction
2. Operando X-ray Diffraction Technique
3. The Operando XRD Studies in Li+/Na+ Batteries
3.1. Reaction Mechanisms
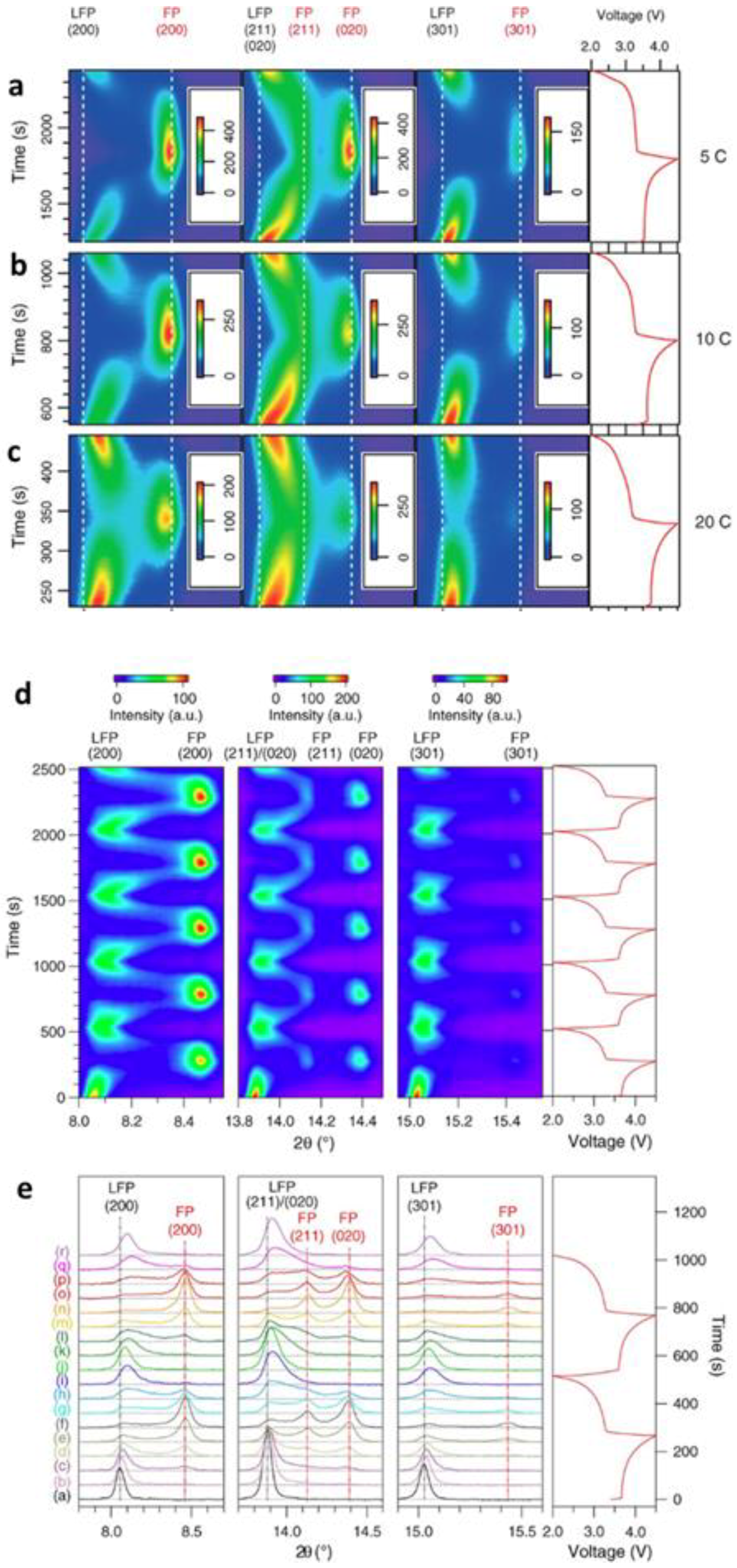
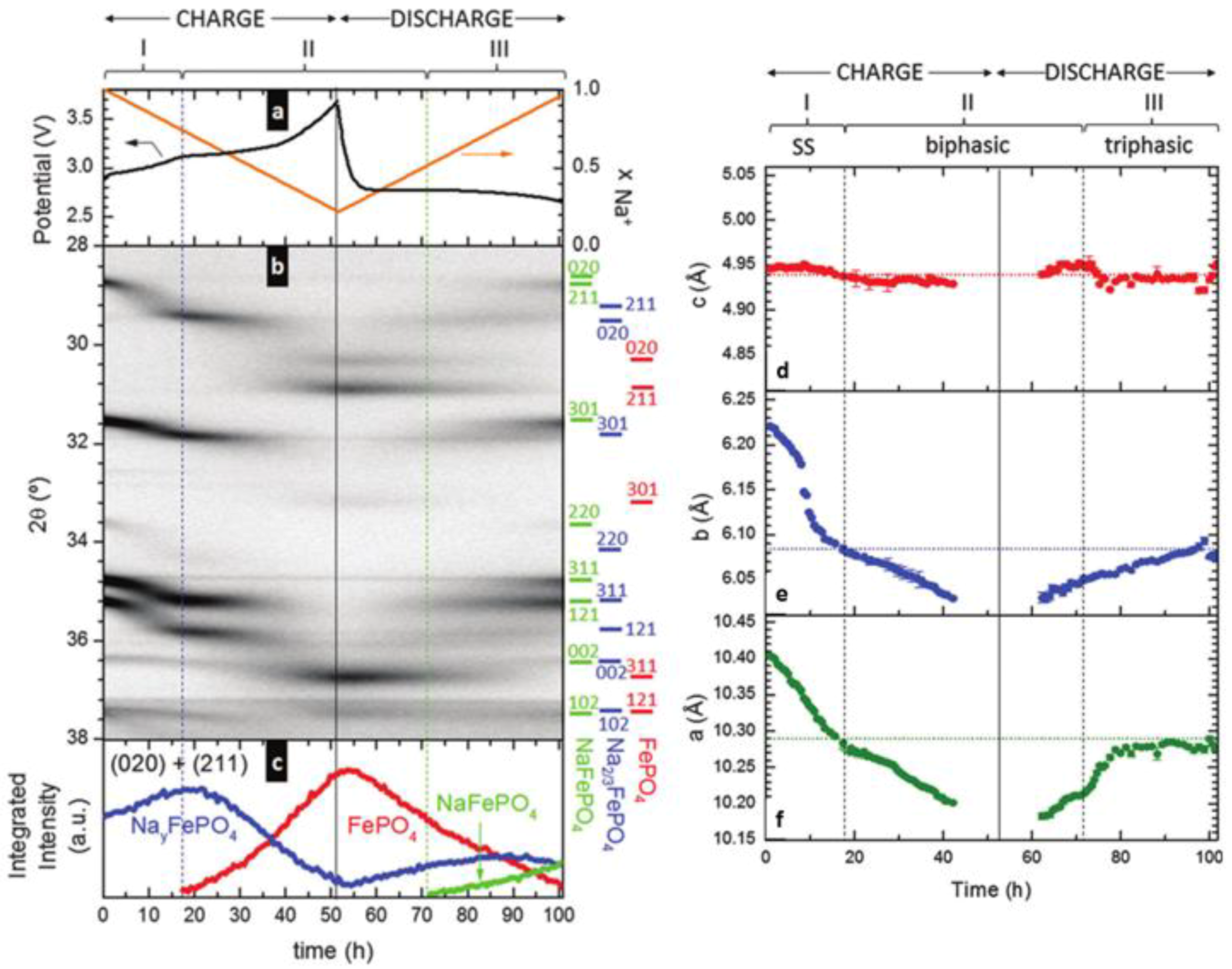
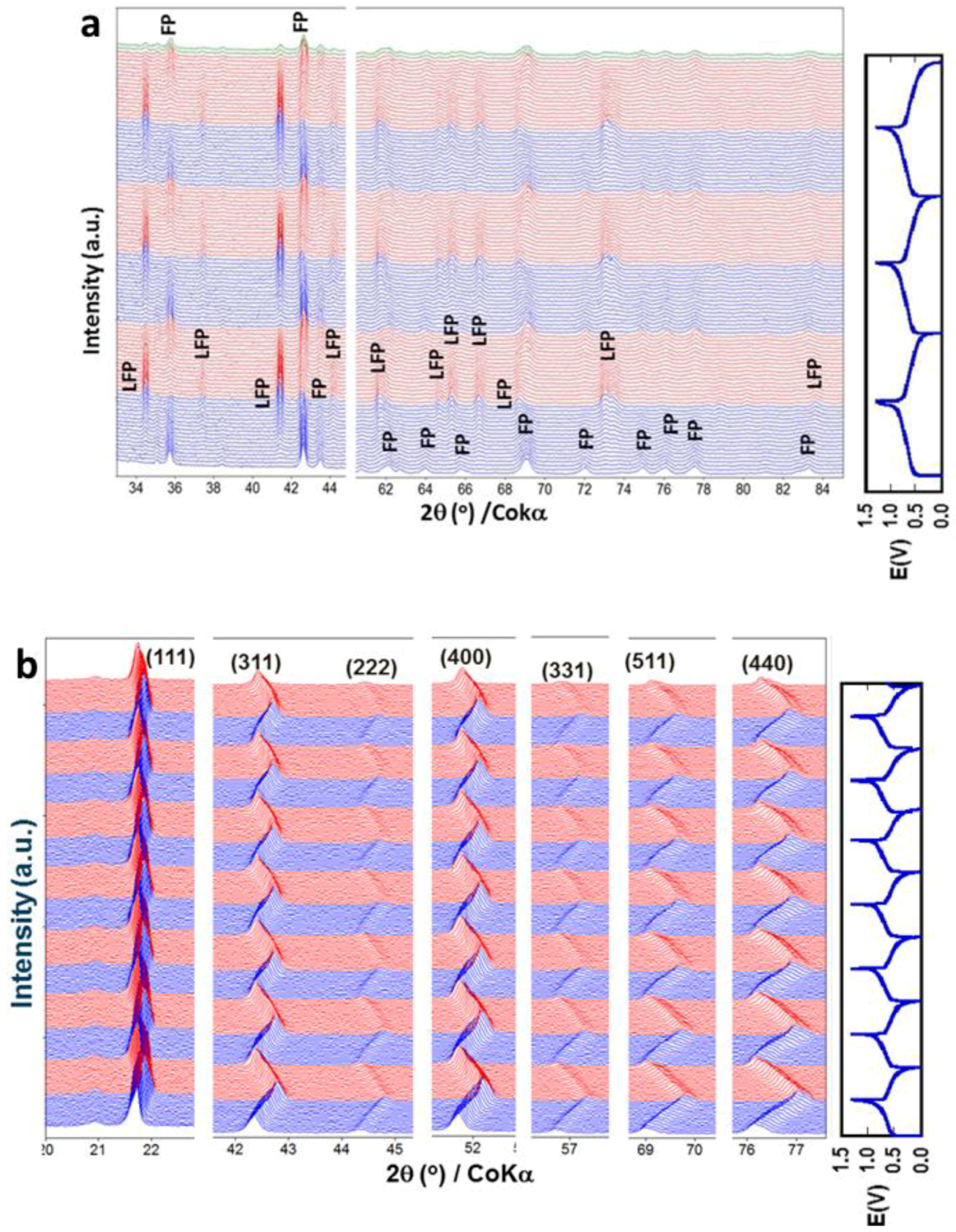
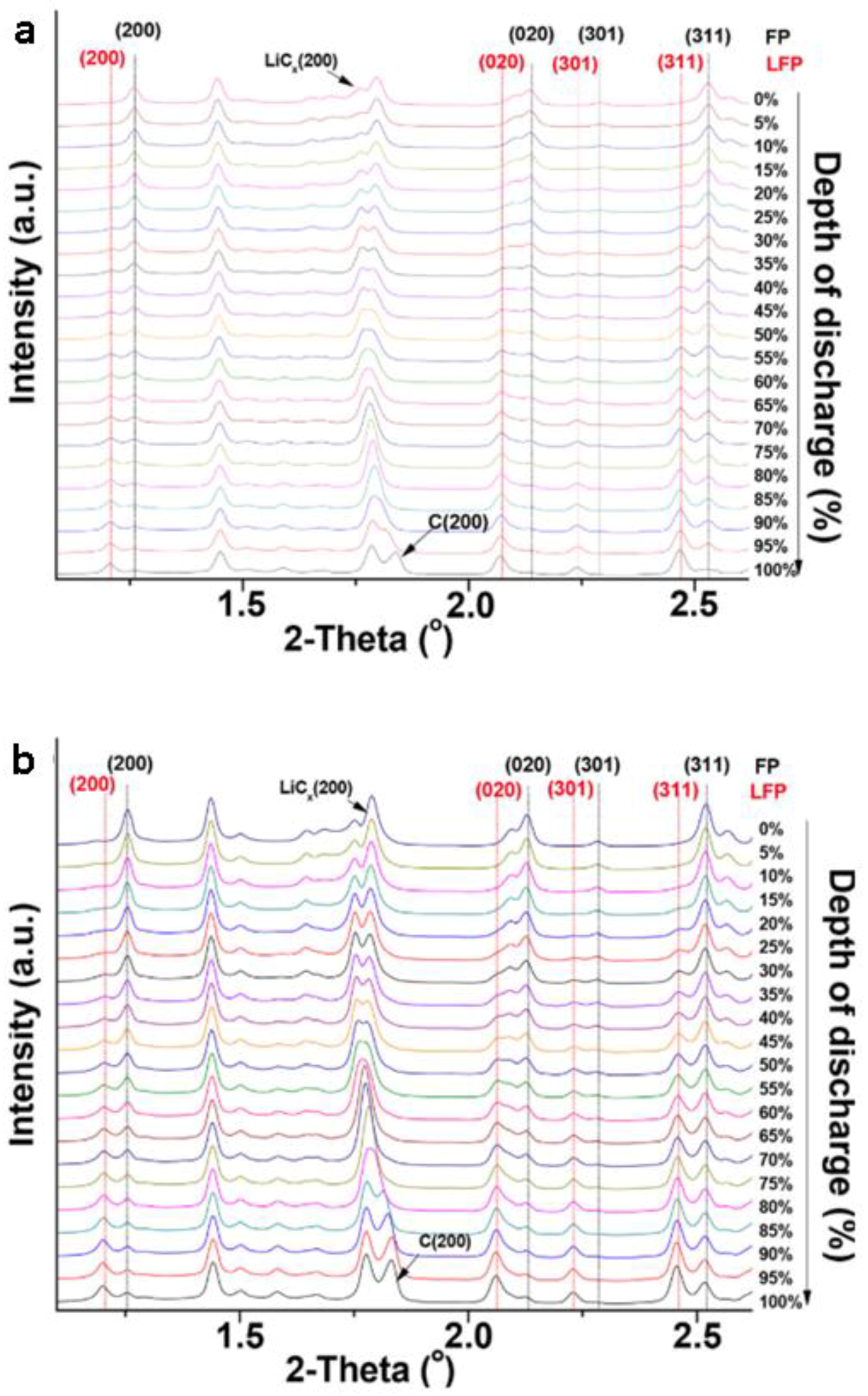
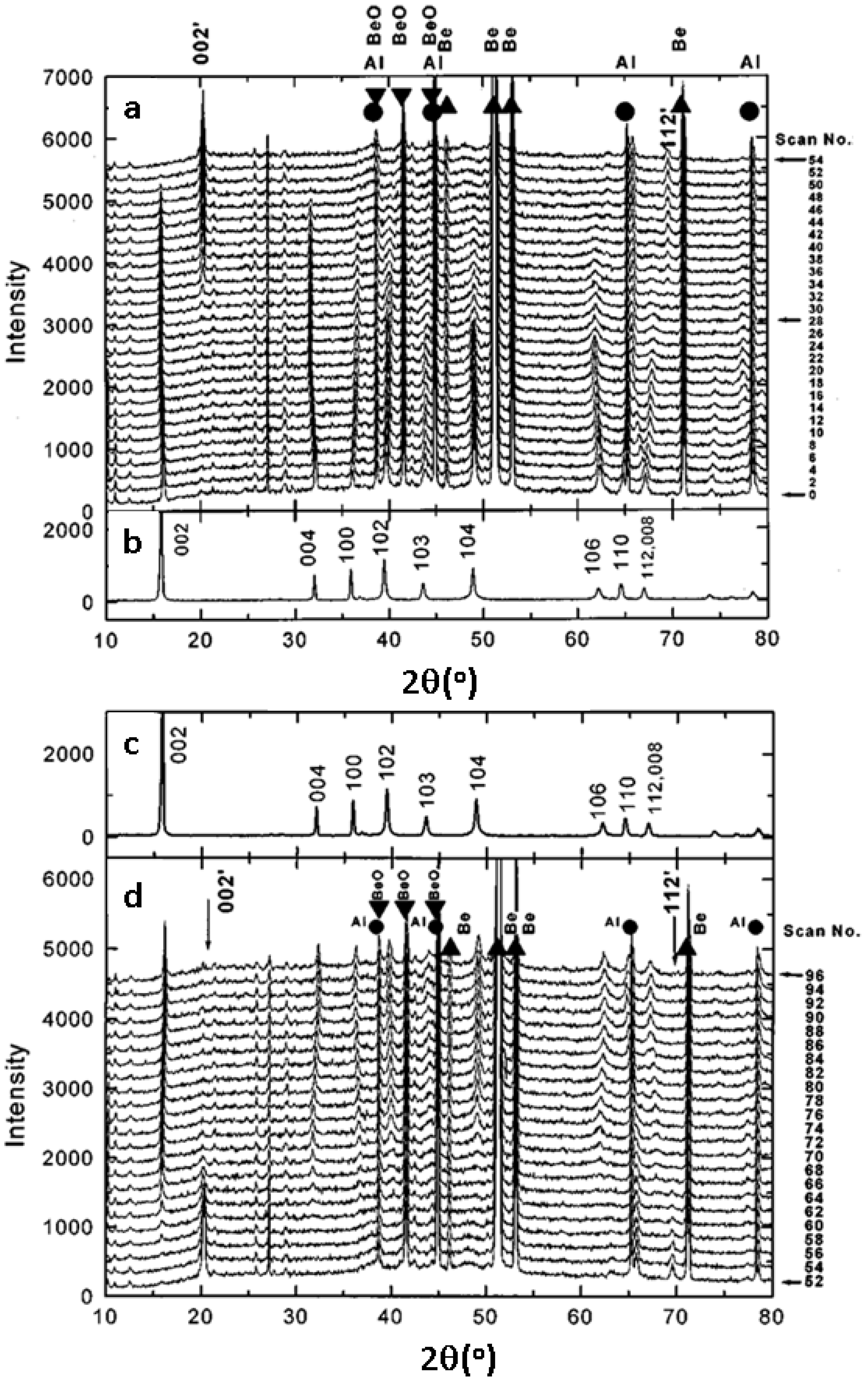
3.1.1. Olivine Structured Cathodes of LiFePO4 and NaFePO4
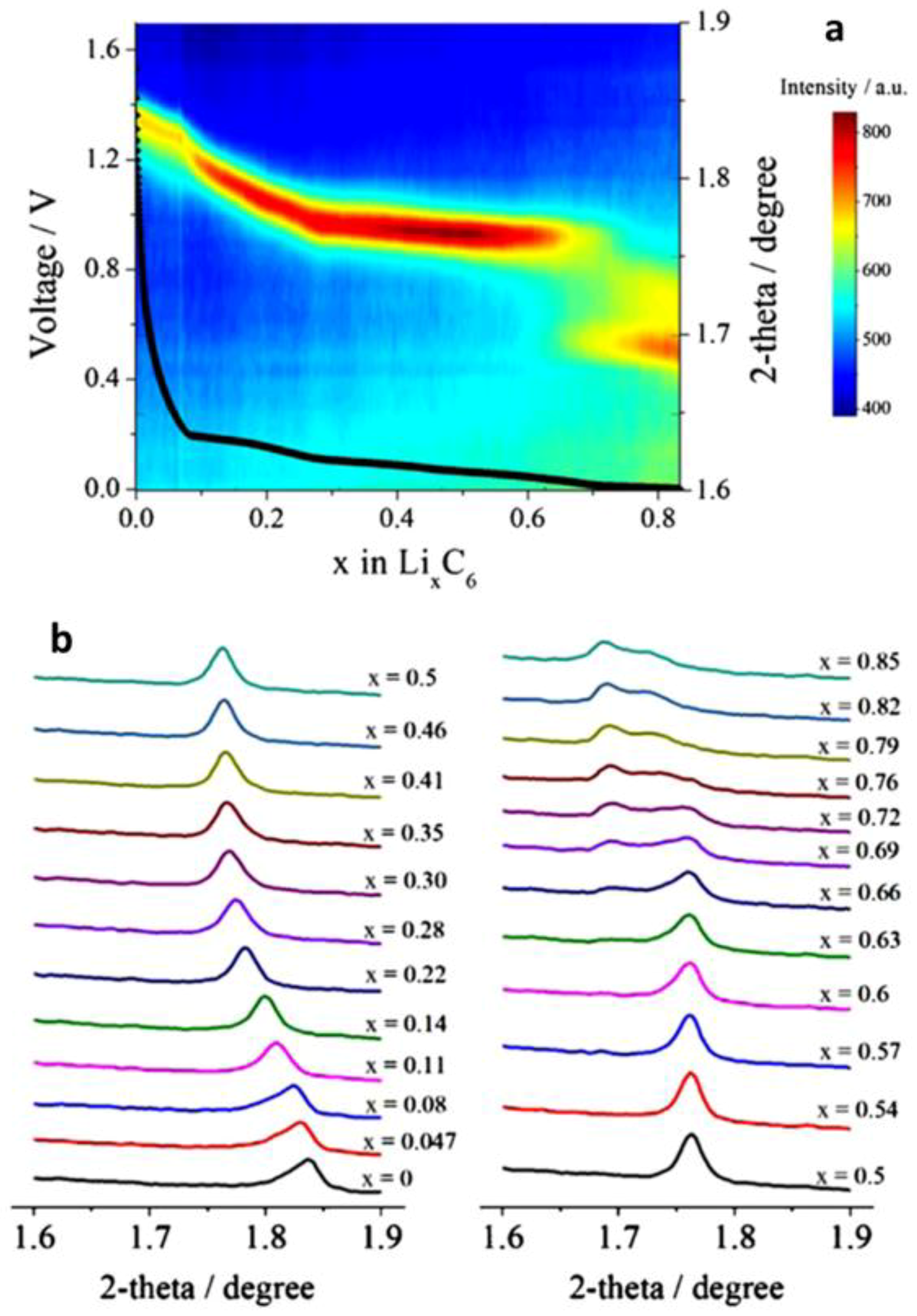
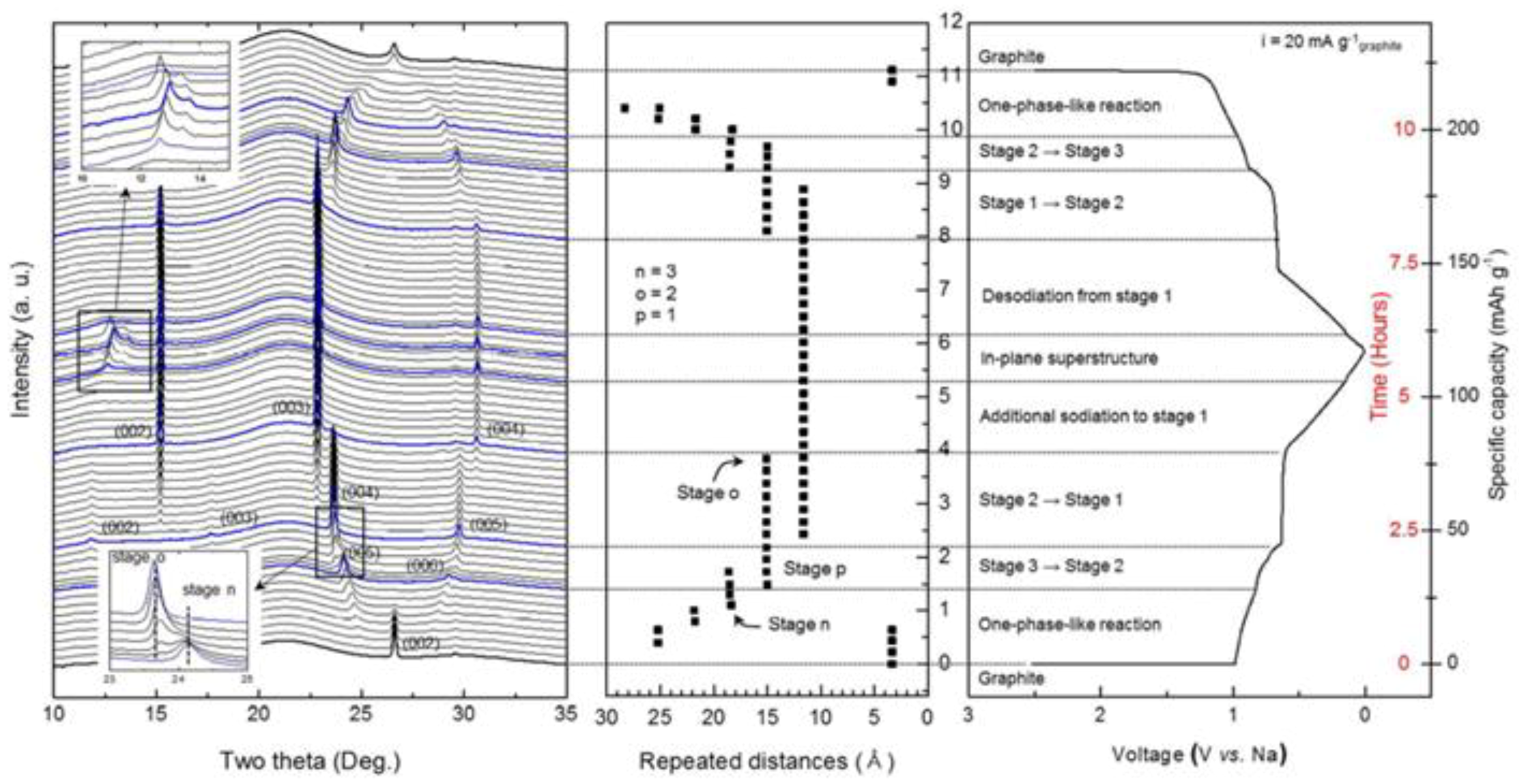
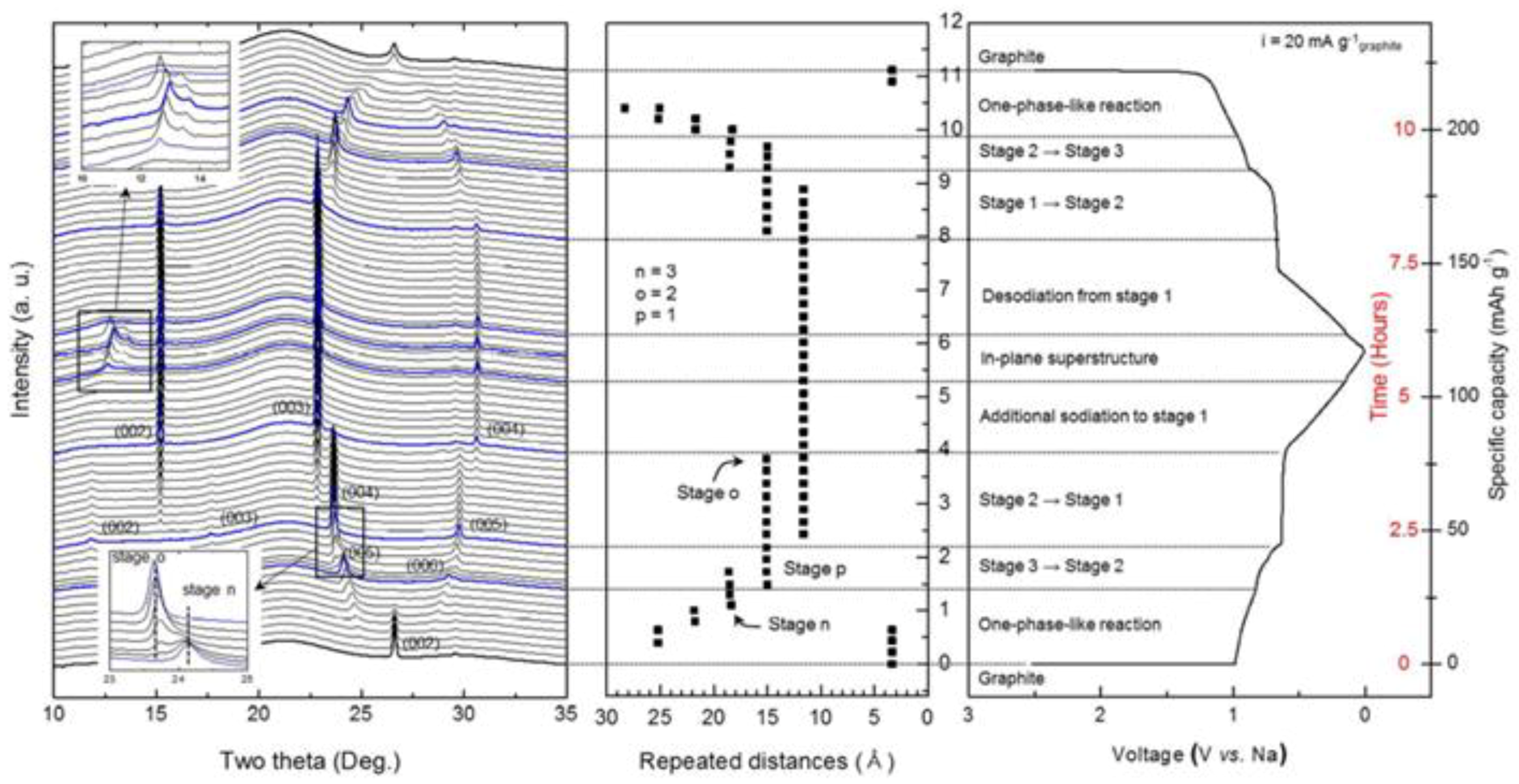
3.1.2. Layer Structured Li2MoO3, Nax(NiMn)O2 and Graphite
- (1)
- 1.5 < Li+ < 2 (3 < V < 3.6 V), (003) and (110) diffractions shift to the lower angles continuously, indicating a solid-solution reaction with increasing c and a lattice parameters of the phase I, but the percentage increase of a is greater than c, implying that Li ions are mainly removed from the LiMo2 layers [163];
- (2)
- 1.0 < Li+ < 1.5 (3.6 < V < 3.7 V), phase II, with the same layered structure as phase I, forms and grows at the expense of phase I. The lattice parameters of phase II are larger than those of phase I;
- (3)
- 0.53 < Li+ < 1.0 (3.7 < V < 4.8 V), phase I is completely consumed and phase II grows via a solid solution route with a continuous increase of lattice parameters a and c.
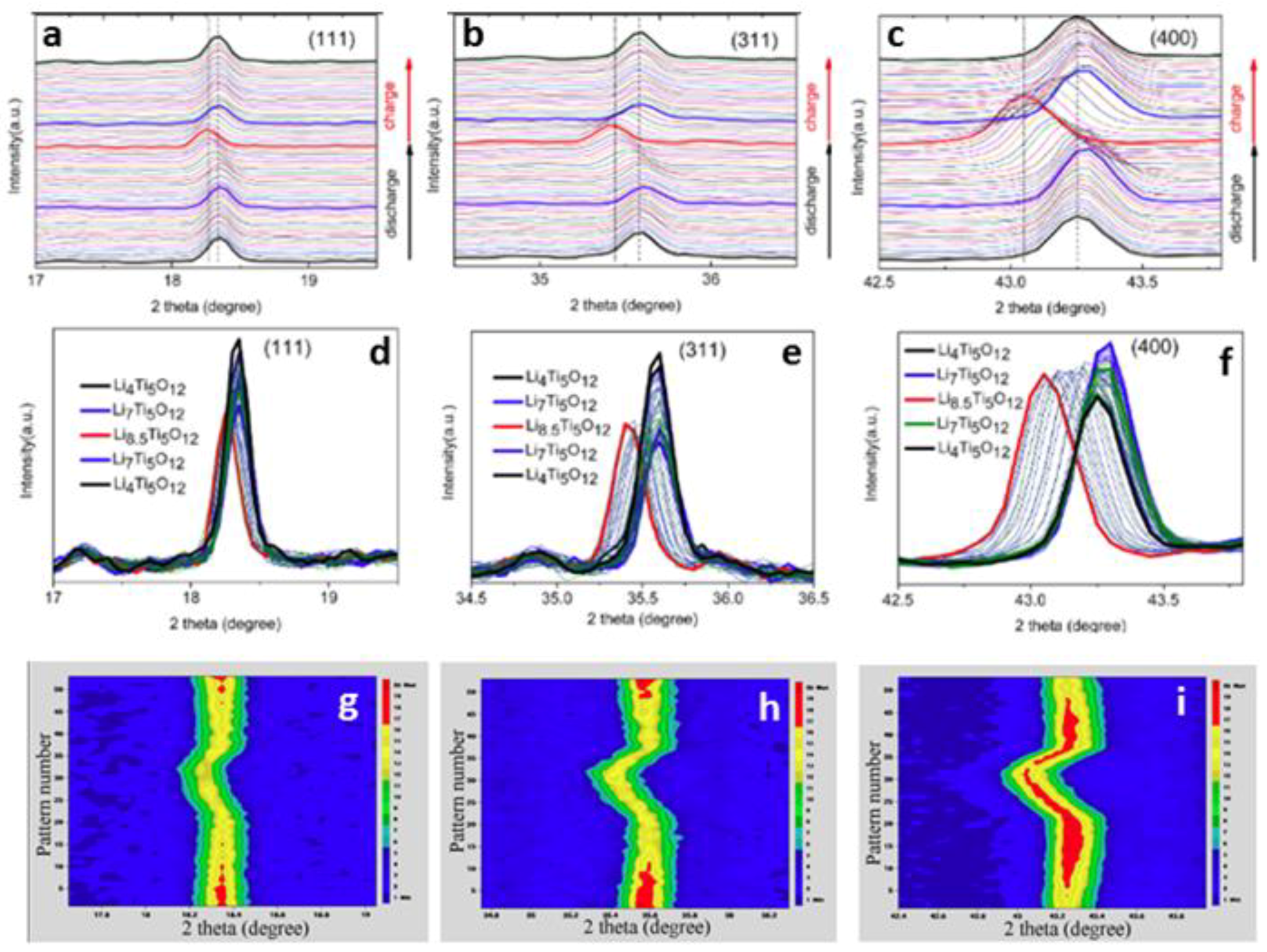
3.1.3. Spinel-Li4Ti5O12
3.1.4. Electrodes for Aqueous Battery
3.2. Degradation Mechanisms
3.3. Thermal Stability
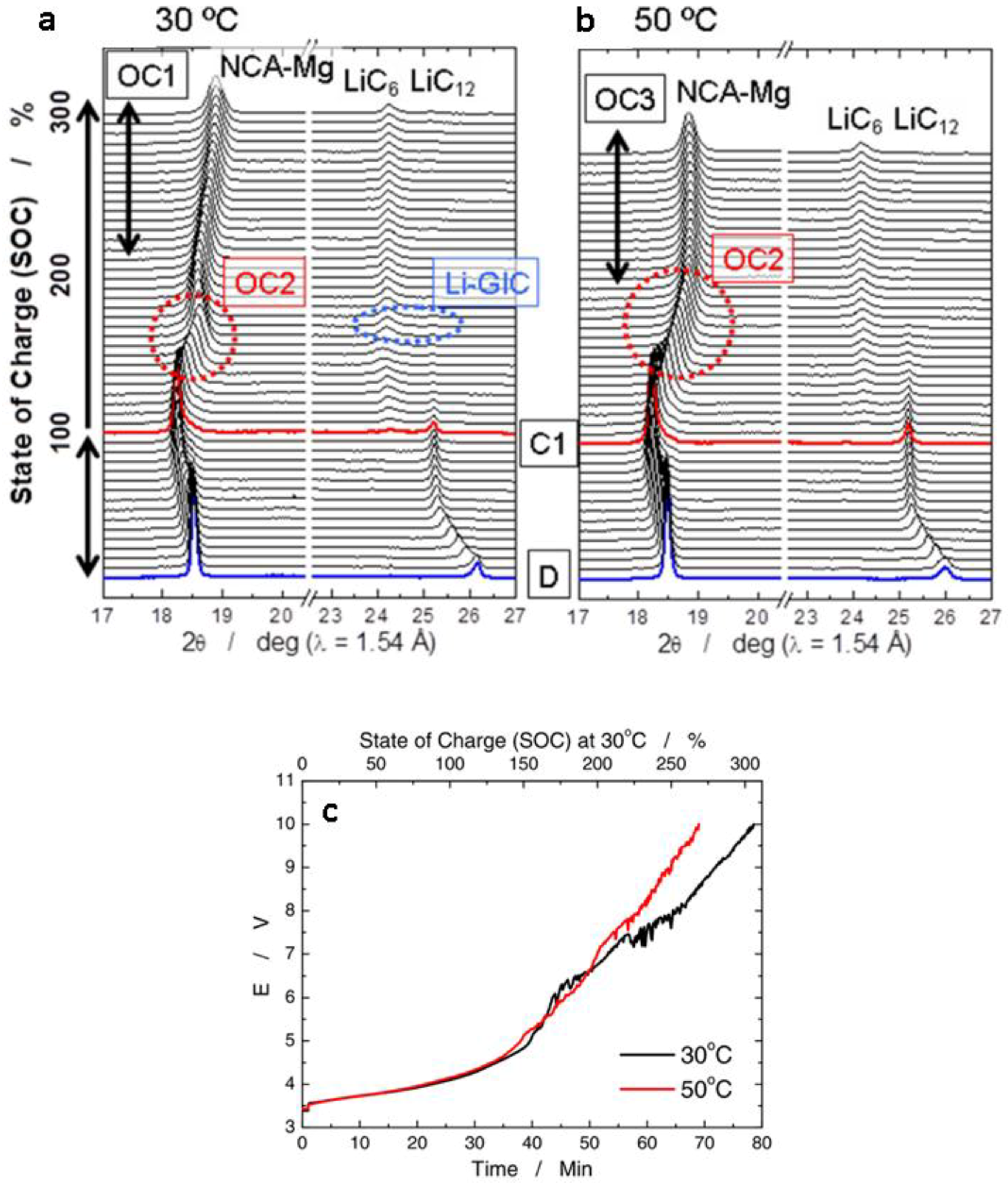
- (1)
- Low voltage region (0 ≤ SOC ≤ 100%, V ≤ 4.1V). The peak (003)NCA-Mg shifts to lower diffraction angle owing to the expansion of distance between transition metal layers induced by lithium de-intercalation. This distance reaches maximum when the cells are charged at 100% SOC (C1 in Figure 20a,b) at both 30 and 50 °C, implying the side reactions at NCA-Mg electrodes are negligible in this region;
- (2)
- Mid voltage region (100% < SOC < ~ 200%, V ≤ 6.6 V). The (003)NCA-Mg peaks shift back to higher angle caused by the contraction of the interlayer distance. The shift of (003)NCA-Mg is slower at 50 °C than at 30 °C in the region marked as OC2, suggesting more side reactions at 50 °C than at 30 °C. The X-ray absorption results indicate that the oxidation of nickel and cobalt in NCA-Mg reached the maximum values at cell voltage of 6.6 V;
- (3)
- High voltage region (~200% < SOC, V > 6.6 V). The rate of (003)NCA-Mg peak shift slows down in the case of 30 °C, suggesting that side reactions accelerated at the NCA-Mg cathode in the high voltage region, labeled as OC1 in Figure 20a. The cell at 50 °C exhibits no (003)NCA-Mg peak shift in the overcharge region with voltage > 8 V (OC3), indicating that the charging current is fully consumed by side reactions at NCA-Mg positive-electrode. On the anode side, lithium ions intercalate into graphite during charge. At 30 °C, fully-intercalated graphite, LiC6, is formed at the overcharge state, marked with “Li-GIC”. Graphite cannot accommodate more Li+ in the structure with charging the cell beyond this point, which results in lithium plating on the graphite anode [195,196]. In the cell at 50 °C, graphite is not fully intercalated with Li+ even at 10 V owing to the increased side-reactions on the graphite at this temperature. Based on the operando XRD and SAFS results, the authors proposed different side reaction mechanisms for the overcharged cells at 30 and 50 °C, which can help researchers to understand the overcharge effect on both cathode and anode, and to design batteries that can reduce and optimize the side reactions for the long life and safe batteries [197].
3.4. Heat Treatment
3.4.1. Phase evolution of LiFePO4 Precursor
3.4.2. Phase Evolution of LiFePO4 Precursor
- 30–100 °C: Li-Mn-Fe-PO4/GO precursor → (Mn/Fe)Cl2 + (Mn/Fe)PO4 + Li containing phase
- 100–280 °C: (Mn/Fe)Cl2 +(Mn/Fe)PO4 + Li containing phase → Li3PO4 + (Mn/Fe)PO4
- 280–450 °C: Li3PO4 + (Mn/Fe)PO4 → Li(Mn/Fe)PO4
- 450–650 °C: (Mn/Fe)PO4 + Li containing phase → Li(Mn/Fe)PO4 (single phase)
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Etacheri, V.; Marom, R.; Elazari, R.; Salitraa, G.; Aurbach, D. Challenges in the development of advanced Li-ion batteries: a review. Energy Environ. Sci. 2011, 4, 3243–3262. [Google Scholar] [CrossRef]
- Wen, J.; Yu, Y.; Chen, C. A Review on Lithium-Ion Batteries Safety Issues: Existing Problems and Possible Solutions. Mater. Express 2012, 2, 197–212. [Google Scholar] [CrossRef]
- Armand, M.; Tarascon, J.-M. Building better batteries. Nature 2008, 451, 652–657. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lu, Y.; Zhao, C.; Hu, Y.-S.; Titirici, M.-M.; Li, H.; Huang, X.; Chen, L. Recent advances of electrode materials for low-cost sodium-ion batteries towards practical application for grid energy storage. Energy Storage Mater. 2017, 7, 130–151. [Google Scholar] [CrossRef]
- Nitta, N.; Wu, F.; Lee, J.T.; Yushin, G. Li-ion battery materials: present and future. Mater. Today 2015, 18, 252–264. [Google Scholar] [CrossRef]
- Meng, J.; Guo, H.; Niu, C.; Zhao, Y.; Xu, L.; Li, Q.; Mai, L. Advances in Structure and Property Optimizations of Battery Electrode Materials. Joule 2017, 1, 522–547. [Google Scholar] [CrossRef]
- Ding, Y.; Wang, R.; Wang, L.; Cheng, K.; Zhao, Z.; Mu, D.; Wu, B. A Short Review on Layered LiNi0.8Co0.1Mn0.1O2 Positive Electrode Material for Lithium-ion Batteries. Energy Procedia 2017, 105, 2941–2952. [Google Scholar] [CrossRef]
- Ding, Y.; Mu, D.; Wu, B.; Wang, R.; Zhao, Z.; Wu, F. Recent progresses on nickel-rich layered oxide positive electrode materials used in lithium-ion batteries for electric vehicles. Appl. Energy 2017, 195, 586–599. [Google Scholar] [CrossRef]
- Manivannan, V.; Chennabasappa, M.; Garrett, J. Optimization and Characterization of Lithium Ion Cathode Materials in the System (1 – x – y)LiNi0.8Co0.2O2 • xLi2MnO3 • yLiCoO2. Energies 2010, 3, 847–866. [Google Scholar] [CrossRef]
- Rozier, P.; Tarascon, J.M. Review—Li-Rich Layered Oxide Cathodes for Next-Generation Li-Ion Batteries: Chances and Challenges. J. Electrochem. Soc. 2015, 162, A2490–A2499. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, S.; Tan, H.; Yang, Z.; Zeng, J. Preparation and Doping Mode of Doped LiMn2O4 for Li-Ion Batteries. Energies 2013, 6, 1718–1730. [Google Scholar] [CrossRef]
- Zhu, W.; Liu, D.; Trottier, J.; Gagnon, C.; Guerfi, A.; Julien, C.M.; Mauger, A.; Zaghib, K. Comparative studies of the phase evolution in M-doped LixMn1.5Ni0.5O4 (M = Co, Al, Cu and Mg) by in-situ X-ray diffraction. J. Power Sources 2014, 264, 290–298. [Google Scholar] [CrossRef]
- Yang, J.; Han, X.; Zhang, X.; Cheng, F.; Chen, J. Spinel LiNi0.5Mn1.5O4 cathode for rechargeable lithiumion batteries: Nano vs micro, ordered phase (P4332) vs disordered phase (Fd3-m). Nano Res. 2013, 6, 679–687. [Google Scholar] [CrossRef]
- Liu, D.; Lu, Y.; Goodenough, J.B. Rate Properties and Elevated-Temperature Performances of LiNi0.5−xCr2xMn 1.5−xO4 (0 ≤ 2x ≤ 0.8) as 5 V Cathode Materials for Lithium-Ion Batteries. J. Electrochem. Soc. 2010, 157, A1269. [Google Scholar] [CrossRef]
- Dimesso, L.; Forster, C.; Jaegermann, W.; Khanderi, J.P.; Tempel, H.; Popp, A.; Engstler, J.; Schneider, J.J.; Sarapulova, A.; Mikhailova, D.; et al. Developments in nanostructured LiMPO4 (M = Fe, Co, Ni, Mn) composites based on three dimensional carbon architecture. Chem. Soc. Rev. 2012, 41, 5068–5680. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.C.; Nam, K.W.; Chang, W.Y.; Cho, B.W.; Yoon, W.-S.; Yang, X.-Q.; Chung, K.Y. Comparative studies on C-coated and uncoated LiFePO4 cycling at various rates and temperatures using synchrotron based in situ X-ray diffraction. Electrochim. Acta 2011, 56, 1182–1189. [Google Scholar] [CrossRef]
- Joachin, H.; Kaun, T.D.; Zaghib, K.; Prakasha, J. Electrochemical and Thermal Studies of Carbon-Coated LiFePO4 Cathode. J. Electrochem. Soc. 2009, 156, A401–A406. [Google Scholar] [CrossRef]
- Kim, M.-S.; Kim, H.-K.; Lee, S.-W.; Kim, D.-H.; Ruan, D.; Chung, K.Y.; Lee, S.H.; Roh, K.C.; Kim, K.-B. Synthesis of Reduced Graphene Oxide-Modified LiMn0.75Fe0.25PO4 Microspheres by Salt-Assisted Spray Drying for High-Performance Lithium-Ion Batteries. Sci. Rep. 2016, 6, 26686. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zhu, W.; Kim, C.; Cho, M.; Guerfi, A.; Delp, S.A.; Allen, J.L.; Jow, T.R.; Zaghib, K. High-energy lithium-ion battery using substituted LiCoPO4: From coin type to 1 Ah cell. J. Power Sources 2018, 388, 52–56. [Google Scholar] [CrossRef]
- Vijaya Babu, K.; Seeta Devi, L.; Veeraiah, V.; Anand, K. Structural and dielectric studies of LiNiPO4 and LiNi0.5Co0.5PO4 cathode materials for lithium-ion batteries. J. Asian Ceram. Soc. 2016, 4, 269–276. [Google Scholar] [CrossRef]
- Qi, X.; Wang, Y.; Jiang, L.; Mu, L.; Zhao, C.; Liu, L.; Hu, Y.-S.; Chen, L.; Huang, X. Sodium-Deficient O3-Na0.9[Ni0.4MnxTi0.6−x]O2 Layered-Oxide Cathode Materials for Sodium-Ion Batteries. Part. Part. Syst. Charact. 2015, 33, 538–544. [Google Scholar] [CrossRef]
- Mu, L.; Xu, S.; Li, Y.; Hu, Y.-S.; Li, H.; Chen, L.; Huang, X. Prototype Sodium-Ion Batteries Using an Air-Stable and Co/Ni-Free O3-Layered Metal Oxide Cathode. Adv. Mater. 2015, 27, 6928–6933. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Seo, D.-H.; Kim, H.; Park, I.; Yoo, J.-K.; Jung, S.-K.; Park, Y.-U.; Goddard, W.A., III; Kang, K. Unexpected discovery of low-cost maricite NaFePO4 as a high-performance electrode for Na-ion batteries. Energy Environ. Sci. 2015, 8, 540–545. [Google Scholar] [CrossRef]
- Jian, Z.; Han, W.; Lu, X.; Yang, H.; Hu, Y.-S.; Zhou, J.; Zhou, Z.; Li, J.; Chen, W.; Chen, D.; Chen, L. Superior Electrochemical Performance and Storage Mechanism of Na3V2(PO4)3 Cathode for Room-Temperature Sodium-Ion Batteries. Adv. Energy Mater. 2012, 3, 156–160. [Google Scholar] [CrossRef]
- Zhou, W.; Xue, L.; Lü, X.; Gao, H.; Li, Y.; Xin, S.; Fu, G.; Cui, Z.; Zhu, Y.; Goodenough, J.B. NaxMV(PO4)3 (M = Mn, Fe, Ni) Structure and Properties for Sodium Extraction. Nano Lett. 2016, 16, 7836–7841. [Google Scholar] [CrossRef] [PubMed]
- Aurbach, D.; Markovsky, B.; Weissman, I.; Levi, E.; Ein-Eli, Y. On the correlation between surface chemistry and performance of graphite negative electrodes for Li ion batteries. Electrochim. Acta 1999, 45, 67–86. [Google Scholar] [CrossRef]
- Wang, Y.; Feng, Z.; Zhu, W.; Gariépy, V.; Gagnon, C.; Provencher, M.; Laul, D.; Veillette, R.; Trudeau, M.; Guerfi, A.; et al. High Capacity and High Efficiency Maple Tree-Biomass-Derived Hard Carbon as an Anode Material for Sodium-Ion Batteries. Materials 2018, 11, 1294. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Jin, Y.; Sun, S.; Huang, Y.; Peng, J.; Luo, J.; Zhang, Q.; Qiu, Y.; Fang, C.; Han, J. Low-Cost and High-Performance Hard Carbon Anode Materials for Sodium-Ion Batteries. ACS Omega 2017, 2, 1687–1695. [Google Scholar] [CrossRef]
- He, Y.-B.; Li, B.; Liu, M.; Zhang, C.; Lv, W.; Yang, C.; Li, J.; Du, H.; Zhang, B.; Yang, Q.-H.; et al. Gassing in Li4Ti5O12-based batteries and its remedy. Sci. Rep. 2012, 2, 913. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Ouyang, M.; Lu, L.; Li, J. Cycle Life of Commercial Lithium-Ion Batteries with Lithium Titanium Oxide Anodes in Electric Vehicles. Energies 2014, 7, 4896–4909. [Google Scholar] [CrossRef]
- Wen, Y.; He, K.; Zhu, Y.; Han, F.; Xu, Y.; Matsuda, I.; Ishii, Y.; Cumings, J.; Wang, C. Expanded graphite as superior anode for sodium-ion batteries. Nat. Commun. 2014, 5, 4033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Lv, W.; Tao, Y.; He, Y.-B.; Wang, D.-W.; You, C.-H.; Li, B.; Kang, F.; Yang, Q.-H. Ultrafast high-volumetric sodium storage of folded-graphene electrodes through surface-induced redox reactions. Energy Storage Mater. 2015, 1, 112–118. [Google Scholar] [CrossRef]
- Liu, S.; Lei, W.; Liu, Y.; Zhang, W.-H. Uniform ReS2 porous nanospheres assembled from curly ReS2 few-layers with an expanded interlayer spacing for high-performance lithium-ion batteries. Chem. Eng. J. 2019, 357, 112–119. [Google Scholar] [CrossRef]
- Bella, F.; Muñoz-García, A.B.; Colò, F.; Meligrana, G.; Lamberti, A.; Destro, M.; Pavone, M.; Gerbaldi, C. Combined Structural, Chemometric, and Electrochemical Investigation of Vertically Aligned TiO2 Nanotubes for Na-ion Batteries. ACS Omega 2018, 3, 8440–8450. [Google Scholar] [CrossRef]
- Wang, H.; Jiang, Y.; Manthiram, A. N-doped Fe3C@C as an efficient polyselenide reservoir for high-performance sodium-selenium batteries. Energy Storage Mater. 2019, 16, 374–382. [Google Scholar] [CrossRef]
- Xu, K. Electrolytes and interphases in Li-ion batteries and beyond. Chem. Rev. 2014, 114, 11503–11618. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Lochala, J.A.; Kwok, A.; Deng, Z.D.; Xiao, J. Research Progress towards Understanding the Unique Interfaces between Concentrated Electrolytes and Electrodes for Energy Storage Applications. Adv. Sci. 2017, 4, 1700032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheers, J.; Fantini, S.; Johansson, P. A review of electrolytes for lithium–sulphur batteries. J. Power Sources 2014, 255, 204–218. [Google Scholar] [CrossRef]
- Erickson, E.M.; Markevich, E.; Salitra, G.; Sharon, D.; Hirshberg, D.; de la Llave, E.; Shterenberg, I.; Rozenman, A.; Frimer, A.; Aurbach, D. Review—Development of Advanced Rechargeable Batteries: A Continuous Challenge in the Choice of Suitable Electrolyte Solutions. J. Electrochem. Soc. 2015, 162, A2424–A2438. [Google Scholar] [CrossRef]
- Cheng, X.B.; Zhang, R.; Zhao, C.Z.; Wei, F.; Zhang, J.G.; Zhang, Q. A Review of Solid Electrolyte Interphases on Lithium Metal Anode. Adv. Sci. 2016, 3, 1500213. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Henderson, W.A.; Appetecchi, G.B.; Montanino, M.; Passerini, S. Physical and Electrochemical Properties of N-Alkyl-N-methylpyrrolidinium Bis(fluorosulfonyl)imide Ionic Liquids: PY13FSI and PY14FSI. J. Phys. Chem. B 2008, 112, 13577–13580. [Google Scholar] [CrossRef] [PubMed]
- Nazet, A.; Sokolov, S.; Sonnleitner, T.; Makino, T.; Kanakubo, M.; Buchner, R. Densities, Viscosities, and Conductivities of the Imidazolium Ionic Liquids [Emim][Ac], [Emim][FAP], [Bmim][BETI], [Bmim][FSI], [Hmim][TFSI], and [Omim][TFSI]. J. Chem. Eng. Data 2015, 60, 2400–2411. [Google Scholar] [CrossRef]
- Moreno, M.; Simonetti, E.; Appetecchi, G.B.; Carewska, M.; Montanino, M.; Kim, G.T.; Loeffler, N.; Passerini, S. Ionic Liquid Electrolytes for Safer Lithium Batteries. J. Electrochem. Soc. 2016, 164, A6026–A6031. [Google Scholar] [CrossRef] [Green Version]
- Marczewski, M.J.; Stanje, B.; Hanzu, I.; Wilkening, M.; Johansson, P. “Ionic liquids-in-salt”—A promising electrolyte concept for high-temperature lithium batteries? Phys. Chem. Chem. Phys. 2014, 16, 12341–12349. [Google Scholar] [CrossRef] [PubMed]
- Colò, F.; Bella, F.; Nair, J.R.; Gerbaldi, C. Light-cured polymer electrolytes for safe, low-cost and sustainable sodium-ion batteries. J. Power Sources 2017, 365, 293–302. [Google Scholar] [CrossRef]
- Edman, L. Ion Association and Ion Solvation Effects at the Crystalline−Amorphous Phase Transition in PEO−LiTFSI. J. Phys. Chem. B 2000, 104, 7254–7258. [Google Scholar] [CrossRef]
- Das, S.; Ghosh, A. Structure, ion transport, and relaxation dynamics of polyethylene oxide/poly (vinylidene fluoride co-hexafluoropropylene)—lithium bis(trifluoromethane sulfonyl) imide blend polymer electrolyte embedded with ionic liquid. J. Appl. Phys. 2016, 119, 095101. [Google Scholar] [CrossRef]
- Chan, C.K.; Yang, T.; Mark Weller, J. Nanostructured Garnet-type Li7La3Zr2O12: Synthesis, Properties, and Opportunities as Electrolytes for Li-ion Batteries. Electrochim. Acta 2017, 253, 268–280. [Google Scholar] [CrossRef]
- Wu, J.-F.; Chen, E.-Y.; Yu, Y.; Liu, L.; Wu, Y.; Pang, W.K.; Peterson, V.K.; Guo, X. Gallium-Doped Li7La3Zr2O12 Garnet-Type Electrolytes with High Lithium-Ion Conductivity. ACS Appl. Mater. Inter. 2017, 9, 1542–1552. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Li, Y.; Goodenough, J.B. Low-temperature synthesis of Li7La3Zr2O12 with cubic garnet-type structure. Mater. Res. Bull. 2012, 47, 1229–1232. [Google Scholar] [CrossRef]
- Ooms, F.G.B.; Wagemaker, M.; van Well, A.A.; Mulder, F.M.; Kelder, E.M.; Schoonman, J. Structure determination of high-voltage LiMgδNi0.5-δMn1.5O4 spinels for Li-ion batteries. Appl. Phys. A 2002, 74, s1089–s1091. [Google Scholar] [CrossRef]
- Fürtauer, S.; Effenberger, H.S.; Flandorfer, H. CuLi2Sn and Cu2LiSn: Characterization by single crystal XRD and structural discussion towards new anode materials for Li-ion batteries. J. Solid State Chem. 2014, 220, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Boivin, E.; Masquelier, C.; Croguennec, L.; Chotard, J.-N. Crystal Structure and Lithium Diffusion Pathways of a Potential Positive Electrode Material for Lithium-Ion Batteries: Li2VIII(H0.5PO4)2. Inorg. Chem. 2017, 56, 6776–6779. [Google Scholar] [CrossRef] [PubMed]
- Reimers, J.N.; Dahn, J.R. Electrochemical and In Situ X-ray Diffraction Studies of Lithium Intercalation in LixCo02. J. Electrochem. Soc. 1992, 139, 2091–2097. [Google Scholar] [CrossRef]
- Whitehead, A.H.; Edstrtim, K.; Rao, N.; Owen, J.R. In situ X-ray diffraction studies of a graphite-based Li-ion battery negative electrode. J. Power Sources 1996, 63, 41–45. [Google Scholar] [CrossRef]
- Yang, X.Q.; McBreen, J.; Yoon, W.-W.; Grey, C.P. Crystal structure changes of LiMn0.5Ni0.5O2 cathode materials during charge and discharge studied by synchrotron based in situ XRD. Electrochem. Commun. 2002, 4, 649–654. [Google Scholar] [CrossRef]
- Zhang, X.; Kuhnel, R.-S.; Schroeder, M.; Balducci, A. Revisiting Li3V2(PO4)3 as an anode—An outstanding negative electrode for high power energy storage devices. J. Mater. Chem. Phys. 2014, 2, 17906–17913. [Google Scholar] [CrossRef]
- Wang, H.; Wang, F. In situ, operando measurements of rechargeable batteries. Curr. Opin. Chem. Eng. 2016, 13, 170–178. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Xiong, T.; He, L.; Yan, M.; Zhao, K.; Xu, X.; Mai, L. Electrochemical in situ X-ray probing in lithium-ion and sodium-ion batteries. J. Mater. Sci. 2017, 52, 3697–3718. [Google Scholar] [CrossRef]
- Sun, Y.; Zhao, L.; Pan, H.; Lu, X.; Gu, L.; Hu, Y.S.; Li, H.; Armand, M.; Ikuhara, Y.; Chen, L.; et al. Direct atomic-scale confirmation of three-phase storage mechanism in Li4Ti5O12 anodes for room-temperature sodium-ion batteries. Nat. Commun. 2013, 4, 1870. [Google Scholar] [CrossRef] [PubMed]
- Bobrikov, I.A.; Samoylova, N.Y.; Sumnikov, S.V.; Ivanshina, O.Y.; Vasin, R.N.; Beskrovnyi, A.I.; Balagurov, A.M. In-situ time-of-flight neutron diffraction study of the structure evolution of electrode materials in a commercial battery with LiNi0.8Co0.15Al0.05O2 cathode. J. Power Sources 2017, 372, 74–81. [Google Scholar] [CrossRef]
- Dolotko, O.; Senyshyn, A.; Mühlbauer, M.J.; Nikolowski, K.; Ehrenberg, H. Understanding structural changes in NMC Li-ion cells by in situ neutron diffraction. J. Power Sources 2014, 255, 197–203. [Google Scholar] [CrossRef]
- Li, J.; Petibon, R.; Glazier, S.; Sharma, N.; Pang, W.K.; Peterson, V.K.; Dahn, J.R. In-situ Neutron Diffraction Study of a High Voltage Li(Ni0.42Mn0.42Co0.16)O2/Graphite Pouch Cell. Electrochim. Acta 2015, 180, 234–240. [Google Scholar] [CrossRef]
- Wang, X.L.; An, K.; Cai, L.; Feng, Z.; Nagler, S.E.; Daniel, C.; Rhodes, K.J.; Stoica, A.D.; Skorpenske, H.D.; Liang, C.; et al. Visualizing the chemistry and structure dynamics in lithium-ion batteries by in-situ neutron diffraction. Sci. Rep. 2012, 2, 747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobrikova, I.A.; Hub, C.W.; Lee, C.H.; Chenb, T.Y.; Deleg, S.; Balagurova, D.A. Structural evolution in LiFePO4-based battery materials: In-situ and ex-situ time-of-flight neutron diffraction study. J. Power Sources 2014, 258, 356–364. [Google Scholar] [CrossRef]
- Sottmann, J.; DiMichiel, M.; Fjellvåg, H.; Malavasi, L.; Margadonna, S.; Vajeeston, P.; Vaughan, G.B.M.; Wragg, D.S. Chemical Structures of Specific Sodium Ion Battery Components Determined by Operando Pair Distribution Function and X-ray Diffraction Computed Tomography. Angew. Chem. Int. Edit. 2017, 56, 11385–11389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Chen-Wiegart, Y.-C.K.; Wang, J. In operando tracking phase transformation evolution of lithium iron phosphate with hard X-ray microscopy. Nat. Commun. 2014, 5, 4570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, H.-W.; Duh, J.-G.; Lee, J.-F. Valence change by in situ XAS in surface modified LiMn2O4 for Li-ion battery. Electrochem. Commun. 2006, 8, 1731–1736. [Google Scholar] [CrossRef]
- Dominko, R.; Arčon, I.; Kodre, A.; Hanžel, D.; Gaberšček, M. In-situ XAS study on Li2MnSiO4 and Li2FeSiO4 cathode materials. J. Power Sources 2009, 189, 51–58. [Google Scholar] [CrossRef]
- Fehse, M.; Monconduit, L.; Fischer, F.; Tessier, C.; Stievano, L. Study of the insertion mechanism of lithium into anatase by operando X-ray diffraction and absorption spectroscopy. Solid State Ion. 2014, 268, 252–255. [Google Scholar] [CrossRef]
- Giorgetti, M. A Review on the Structural Studies of Batteries and Host Materials by X-ray Absorption Spectroscopy. ISRN Mater. Sci. 2013, 2013, 1–22. [Google Scholar] [CrossRef]
- Giuliana, A.; Marco, G.; Robert, D.; Lorenzo, S.; Iztok, A.; Nicola, N.; Luca, O. Operando characterization of batteries using X-ray absorption spectroscopy: Advances at the beamline XAFS at synchrotron Elettra. J. Phys. D Appl. Phys. 2017, 50, 074001. [Google Scholar] [CrossRef]
- Menzel, M.; Schlifke, A.; Falk, M.; Janek, J.; Fröba, M.; Fittschen, U.E.A. Surface and in-depth characterization of lithium-ion battery cathodes at different cycle states using confocal micro-X-ray fluorescence-X-ray absorption near edge structure analysis. Spectrochim. Acta B 2013, 85, 62–70. [Google Scholar] [CrossRef]
- Dokko, K.; Mohamedi, M.; Anzue, N.; Itoh, T.; Uchida, I. In situ Raman spectroscopic studies of LiNixMn2−xO4thin film cathode materials for lithium ion secondary batteries. J. Mater. Chem. 2002, 12, 3688–3693. [Google Scholar] [CrossRef]
- Hardwick, L.J.; Ruch, P.W.; Hahn, M.; Scheifele, W.; Kötz, R.; Novák, P. In situ Raman spectroscopy of insertion electrodes for lithium-ion batteries and supercapacitors: First cycle effects. J. Phys. Chem. Solids 2008, 69, 1232–1237. [Google Scholar] [CrossRef]
- Wu, J.; Dathar, G.K.; Sun, C.; Theivanayagam, M.G.; Applestone, D.; Dylla, A.G.; Manthiram, A.; Henkelman, G.; Goodenough, J.B.; Stevenson, K.J. In situ Raman spectroscopy of LiFePO4: size and morphology dependence during charge and self-discharge. Nanotechnology 2013, 24, 9. [Google Scholar] [CrossRef] [PubMed]
- Membreno, N.; Xiao, P.; Park, K.S.; Goodenough, J.B.; Henkelman, G.; Stevenso, K.J. In Situ Raman Study of Phase Stability of α-Li3V2(PO4)3 upon Thermal and Laser Heating. J. Phys. Chem. C 2013, 117, 11994–12002. [Google Scholar] [CrossRef]
- Stancovski, V.; Badilescu, S. In situ Raman spectroscopic–electrochemical studies of lithium-ion battery materials: a historical overview. J. Appl. Electrochem. 2014, 44, 23–43. [Google Scholar] [CrossRef]
- Aurbach, D.; Chusid, O. The use of in situ Fourier-transform infrared spectroscopy for the study of surface phenomena on electrodes in selected lithium battery electrolyte solutions. J. Power Sources 1997, 68, 463–470. [Google Scholar] [CrossRef]
- Cheng, H.; Zhu, C.B.; Lu, M.; Yang, Y. In situ micro-FTIR study of the solid–solid interface between lithium electrode and polymer electrolytes. J. Power Sources 2007, 174, 1027–1031. [Google Scholar] [CrossRef]
- Chusid, O.; Gofer, Y.; Aurbach, D.; Watanabe, M.; Momma, T.; Osaka, T. Studies of the interface between lithium electrodes and polymeric electrolyte systems using in situ FTIR spectroscopy. J. Power Sources 2001, 97–98, 632–636. [Google Scholar] [CrossRef]
- Ye, J.-Y.; Jiang, Y.-X.; Sheng, T.; Sun, S.-G. In-situ FTIR spectroscopic studies of electrocatalytic reactions and processes. Nano Energy 2016, 29, 414–427. [Google Scholar] [CrossRef]
- Hovington, P.; Dontigny, M.; Guerfi, A.; Trottier, J.; Lagacé, M.; Mauger, A.; Julien, C.M.; Zaghib, K. In situ Scanning electron microscope study and microstructural evolution of nano silicon anode for high energy Li-ion batteries. J. Power Sources 2014, 248, 457–464. [Google Scholar] [CrossRef]
- Marceau, H.; Kim, C.-S.; Paolella, A.; Ladouceur, S.; Lagacé, M.; Chaker, M.; Vijh, A.; Guerfi, A.; Julien, C.M.; Mauger, A.; et al. In operando scanning electron microscopy and ultraviolet–visible spectroscopy studies of lithium/sulfur cells using all solid-state polymer electrolyte. J. Power Sources 2016, 319, 247–254. [Google Scholar] [CrossRef]
- Miller, D.J.; Proff, C.; Wen, J.G.; Abraham, D.P.; Bareño, J. Observation of Microstructural Evolution in Li Battery Cathode Oxide Particles by In Situ Electron Microscopy. Adv. Energy Mater. 2013, 3, 1098–1103. [Google Scholar] [CrossRef]
- Liu, S.; Xie, J.; Su, Q.; Du, G.; Zhang, S.; Cao, G.; Zhu, T.; Zhao, X. Understanding Li-storage mechanism and performance of MnFe2O4 by in situ TEM observation on its electrochemical process in nano lithium battery. Nano Energy 2014, 8, 84–94. [Google Scholar] [CrossRef]
- Chen, K.; Cao, K.; Xing, C.; Hu, Y.; Liu, J.; He, Y.; Wang, J.; Li, A.; Qin, H. In-situ TEM study of the lithiation and delithiation of FeS nanosheets. J. Alloy. Compd. 2016, 688, 946–952. [Google Scholar] [CrossRef]
- Janish, M.T.; Carter, C.B. In situ TEM observations of the lithiation of molybdenum disulfide. Scr. Mater. 2015, 107, 22–25. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.M. In situ transmission electron microscopy and spectroscopy studies of rechargeable batteries under dynamic operating conditions: A retrospective and perspective view. J. Mater. Res. 2015, 30, 326–339. [Google Scholar] [CrossRef]
- Xu, Z.-L.; Cao, K.; Abouali, S.; Akbari Garakani, M.; Huang, J.; Huang, J.-Q.; Kamali Heidari, E.; Wang, H.; Kim, J.-K. Study of lithiation mechanisms of high performance carbon-coated Si anodes by in-situ microscopy. Energy Storage Mater. 2016, 3, 45–54. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, J.W.; Liu, Y.; Liu, X.; Kushima, A.; Liu, Y.; Xu, Y.; Mao, S.X.; Li, J.; Wang, C.; et al. In Situ Atomic-Scale Imaging of Phase Boundary Migration in FePO4 Microparticles During Electrochemical Lithiation. Adv. Mater. 2013, 25, 5461–5466. [Google Scholar] [CrossRef] [PubMed]
- Orikasa, Y.; Maeda, T.; Koyama, Y.; Murayama, H.; Fukuda, K.; Tanida, H.; Arai, H.; Matsubara, E.; Uchimoto, Y.; Ogumi, Z. Direct Observation of a Metastable Crystal Phase of LixFePO4 under Electrochemical Phase Transition. J. Am. Chem. Soc. 2013, 135, 5497–5500. [Google Scholar] [CrossRef] [PubMed]
- Bak, S.-M.; Hu, E.; Zhou, Y.; Yu, X.; Senanayake, S.D.; Cho, S.-J.; Kim, K.-B.; Chung, K.Y.; Yang, X.-Q.; Nam, K.-W. Structural Changes and Thermal Stability of Charged LiNixMnyCozO2 Cathode Materials Studied by Combined In Situ Time-Resolved XRD and Mass Spectroscopy. ACS Appl. Mater. Interfaces 2014, 6, 22594–22601. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Shunmugasundaram, R.; Doig, R.; Dahn, J.R. In Situ X-ray Diffraction Study of Layered Li–Ni–Mn–Co Oxides: Effect of Particle Size and Structural Stability of Core–Shell Materials. Chem. Mater. 2016, 28, 162–171. [Google Scholar] [CrossRef]
- Kim, H.; Hong, J.; Yoon, G.; Kim, H.; Park, K.-Y.; Park, M.-S.; Yoon, W.-S.; Kang, K. Sodium intercalation chemistry in graphite. Energy Environ. Sci. 2015, 8, 2963–2969. [Google Scholar] [CrossRef]
- Wang, P.-F.; Yao, H.-R.; Liu, X.-Y.; Yin, Y.-X.; Zhang, J.-N.; Wen, Y.; Yu, X.; Gu, L.; Guo, Y.-G. Na+/vacancy disordering promises high-rate Na-ion batteries. Sci. Adv. 2018, 4, eaar6018. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Xiao, L.; Sushko, M.L.; Han, K.S.; Shao, Y.; Yan, M.; Liang, X.; Mai, L.; Feng, F.; Cao, Y.; et al. Manipulating Adsorption–Insertion Mechanisms in Nanostructured Carbon Materials for High-Efficiency Sodium Ion Storage. Adv. Energy Mater. 2017, 7, 1700403. [Google Scholar] [CrossRef]
- Bock, D.C.; Pelliccione, C.J.; Zhang, W.; Timoshenko, J.; Knehr, K.W.; West, A.C.; Wang, F.; Li, Y.; Frenkel, A.I.; Takeuchi, E.S.; et al. Size dependent behavior of Fe3O4 crystals during electrochemical (de)lithiation: An in situ X-ray diffraction, ex situ X-ray absorption spectroscopy, transmission electron microscopy and theoretical investigation. Phys. Chem. Chem. Phys. 2017, 19, 20867–20880. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Pan, H.; Wan, W.; Ma, M.; Bai, J.; Meng, Q.; Ehrlich, S.N.; Hu, Y.S.; Yang, X.Q. A Size-Dependent Sodium Storage Mechanism in Li4Ti5O12 Investigated by a Novel Characterization Technique Combining in Situ X-ray Diffraction and Chemical Sodiation. Nano Lett. 2013, 13, 4721–4727. [Google Scholar] [CrossRef] [PubMed]
- Orikasa, Y.; Maeda, T.; Koyama, Y.; Murayama, H.; Fukuda, K.; Tanida, H.; Arai, H.; Matsubara, E.; Uchimoto, Y.; Ogumi, Z. Transient Phase Change in Two Phase Reaction between LiFePO4 and FePO4 under Battery Operation. Chem. Mater. 2013, 25, 1032–1039. [Google Scholar] [CrossRef]
- Fell, C.R.; Chi, M.; Meng, Y.S.; Jones, J.L. In situ X-ray diffraction study of the lithium excess layered oxide compound Li[Li0.2Ni0.2Mn0.6]O2 during electrochemical cycling. Solid State Ion. 2012, 207, 44–49. [Google Scholar] [CrossRef]
- Muhammad, S.; Lee, S.; Kim, H.; Yoon, J.; Jang, D.; Yoon, J.; Park, J.-H.; Yoon, W.-S. Deciphering the thermal behavior of lithium rich cathode material by in situ X-ray diffraction technique. J. Power Sources 2015, 285, 156–160. [Google Scholar] [CrossRef]
- Yao, K.P.C.; Kwabi, D.G.; Quinlan, R.A.; Mansour, A.N.; Grimaud, A.; Lee, Y.-L.; Lu, Y.-C.; Shao-Horn, Y. Thermal Stability of Li2O2 and Li2O for Li-Air Batteries: In Situ XRD and XPS Studies. J. Electrochem. Soc. 2013, 160, A824–A831. [Google Scholar] [CrossRef]
- Cañas, N.A.; Einsiedel, P.; Freitag, O.T.; Heim, C.; Steinhauer, M.; Park, D.-W.; Friedrich, K.A. Operando X-ray diffraction during battery cycling at elevated temperatures: A quantitative analysis of lithium-graphite intercalation compounds. Carbon 2017, 116, 255–263. [Google Scholar] [CrossRef]
- Kim, U.-H.; Myung, S.-T.; Yoon, C.S.; Sun, Y.-K. Extending the Battery Life Using an Al-Doped Li[Ni0.76Co0.09Mn0.15]O2 Cathode with Concentration Gradients for Lithium Ion Batteries. ACS Energy Lett. 2017, 2, 1848–1854. [Google Scholar] [CrossRef]
- Ma, H.; Su, H.; Amine, K.; Liu, X.; Jaffer, S.; Shang, T.; Gu, L.; Yu, H. Triphase electrode performance adjustment for rechargeable ion batteries. Nano Energy 2018, 43, 1–10. [Google Scholar] [CrossRef]
- Krumeich, F.; Waser, O.; Pratsinis, S.E. Thermal annealing dynamics of carbon-coated LiFePO4 nanoparticles studied by in-situ analysis. J. Solid State Chem. 2016, 242, 96–102. [Google Scholar] [CrossRef]
- Bleith, P.; Kaiser, H.; Novák, P.; Villevieille, C. In situ X-ray diffraction characterisation of Fe0.5TiOPO4 and Cu0.5TiOPO4 as electrode material for sodium-ion batteries. Electrochim. Acta 2015, 176, 18–21. [Google Scholar] [CrossRef]
- Zhu, W.; Liu, D.; Trottier, J.; Gagnon, C.; Mauger, A.; Julien, C.M.; Zaghib, K. In-situ X-ray diffraction study of the phase evolution in undoped and Cr-doped LixMn1.5Ni0.5O4 (0.1 < x < 1.0) 5-V cathode materials. J. Power Sources 2013, 242, 236–243. [Google Scholar] [CrossRef]
- Roberts, M.R.; Madsen, A.; Nicklin, C.; Rawle, J.; Palmer, M.G.; Owen, J.R.; Hector, A.L. Direct Observation of Active Material Concentration Gradients and Crystallinity Breakdown in LiFePO4 Electrodes During Charge/Discharge Cycling of Lithium Batteries. J. Phys. Chem. C 2014, 118, 6548–6557. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Reimers, J.N.; Dahn, J.R. In situ X-ray diffraction and electrochemical studies of Li1-xNiO2. Solid State Ion. 1993, 67, 123–130. [Google Scholar] [CrossRef]
- Leriche, J.B.; Hamelet, S.; Shu, J.; Morcrette, M.; Masquelier, C.; Ouvrard, G.; Zerrouki, M.; Soudan, P.; Belin, S.; Elkaïm, E.; et al. An Electrochemical Cell for Operando Study of Lithium Batteries Using Synchrotron Radiation. J. Electrochem. Soc. 2010, 157, A606. [Google Scholar] [CrossRef]
- Lowe, M.A.; Gao, J.; Abruña, H.D. Mechanistic insights into operational lithium–sulfur batteries by in situ X-ray diffraction and absorption spectroscopy. RSC Adv. 2014, 4, 18347. [Google Scholar] [CrossRef]
- Sasaki, T.; Villevieill, C.; Takeuchi, Y.; Novák, P. Understanding Inhomogeneous Reactions in Li-Ion Batteries: Operando Synchrotron X-ray Diffraction on Two-Layer Electrodes. Adv. Sci. 2015, 1500083. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Shao, L.; Wu, K.; Shui, M.; Wang, D.; Pan, J.; Long, N.; Ren, Y.; Shu, J. Comparison of LiVPO4F to Li4Ti5O12 as anode materials for lithium-ion batteries. ACS Appl. Mater. Interfaces 2013, 5, 8615–8627. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Einarsrud, M.-A.; Vullum-Bruer, F. In situ X-ray diffraction and electrochemical impedance spectroscopy of a nanoporous Li2FeSiO4/C cathode during the initial charge/discharge cycle of a Li-ion battery. J. Power Sources 2013, 238, 478–484. [Google Scholar] [CrossRef]
- Mohanty, D.; Kalnaus, S.; Meisner, R.A.; Rhodes, K.J.; Li, J.; Payzant, E.A.; Wood, D.L.; Daniel, C. Structural transformation of a lithium-rich Li1.2Co0.1Mn0.55Ni0.15O2 cathode during high voltage cycling resolved by in situ X-ray diffraction. J. Power Sources 2013, 229, 239–248. [Google Scholar] [CrossRef]
- Morcrette, M.; Chabre, Y.; Vaughan, G.; Amatucci, G.; Leriche, J.B.; Patoux, S.; Masquelier, C.; Tarascon, J.M. In situ X-ray diffraction techniques as a powerful tool to study battery electrode materials. Electrochim. Acta 2002, 47, 3137–3149. [Google Scholar] [CrossRef]
- He, H.; Huang, C.; Luo, C.-W.; Liu, J.-J.; Chao, Z.-S. Dynamic study of Li intercalation into graphite by in situ high energy synchrotron XRD. Electrochim. Acta 2013, 92, 148–152. [Google Scholar] [CrossRef]
- Harks, P.P.R.M.L.; Mulder, F.M.; Notten, P.H.L. In situ methods for Li-ion battery research: A review of recent developments. J. Power Sources 2015, 288, 92–105. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; He, H.; Liu, Y.; Dun, C.; Ren, Y.; Liu, Q.; Wang, M.-X.; Xie, J. Failure Investigation of LiFePO4 Cells under Overcharge Conditions. J. Electrochem. Soc. 2012, 159, A678–A687. [Google Scholar] [CrossRef]
- Liu, Q.; Liu, Y.; Yang, F.; He, H.; Xiao, X.; Ren, Y.; Lu, W.; Stach, E.; Xie, J. Capacity Fading Mechanism of the Commercial 18650 LiFePO4-Based Lithium-Ion Batteries: An in Situ Time-Resolved High-Energy Synchrotron XRD Study. ACS Appl. Mater. Interfaces 2018, 10, 4622–4629. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Mitra, S. Carbon Coated Submicron sized-LiFePO4: Improved High Rate Performance Lithium Battery Cathode. Energy Procedia 2014, 54, 718–724. [Google Scholar] [CrossRef]
- Zhao, N.; Li, Y.; Zhi, X.; Wang, L.; Zhao, X.; Wang, Y.; Liang, G. Effect of Ce3+ doping on the properties of LiFePO4 cathode material. J. Rare Earths 2016, 34, 174–180. [Google Scholar] [CrossRef]
- MacNeil, D.D.; Lu, Z.; Chen, Z.; Dahn, J.R. A comparison of the electrode/electrolyte reaction at elevated temperatures for various Li-ion battery cathodes. J. Power Sources 2002, 108, 8–14. [Google Scholar] [CrossRef]
- Islam, M.S.; Driscoll, D.J.; Fisher, C.A.J.; Slater, P.R. Atomic-Scale Investigation of Defects, Dopants, and Lithium Transport in the LiFePO4 Olivine-Type Battery Material. Chem. Mater. 2005, 17, 5085–5092. [Google Scholar] [CrossRef]
- Allen, J.L.; Jow, T.R.; Wolfenstine, J. Kinetic Study of the Electrochemical FePO4 to LiFePO4 Phase Transition. Chem. Mater. 2007, 19, 2108–2111. [Google Scholar] [CrossRef]
- Yamada, A.; Koizumi, H.; Sonoyama, N.; Kanno, R. Phase Change in LixFePO4. Electrochem. Solid-State Lett. 2005, 8, A409–A413. [Google Scholar] [CrossRef]
- Sharma, N.; Guo, X.; Du, G.; Guo, Z.; Wang, J.; Wang, Z.; Peterson, V.K. Direct Evidence of Concurrent Solid-Solution and Two-Phase Reactions and the Nonequilibrium Structural Evolution of LiFePO4. J. Am. Chem. Soc. 2012, 134, 7867–7873. [Google Scholar] [CrossRef] [PubMed]
- Yamada, A.; Koizumi, H.; Nishimura, S.-I.; Sonoyama, N.; Kanno, R.; Yonemura, M.; Nakamura, T.; Kobayashi, Y. Room-temperature miscibility gap in LixFePO4. Nat. Mater. 2006, 5, 357. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Song, X.; Richardson, T.J. Electron Microscopy Study of the LiFePO4 to FePO4 Phase Transition. Electrochem. Solid-State Lett. 2006, 9, A295–A298. [Google Scholar] [CrossRef]
- Laffont, L.; Delacourt, C.; Gibot, P.; Wu, M.Y.; Kooyman, P.; Masquelier, C.; Tarascon, J.M. Study of the LiFePO4/FePO4 Two-Phase System by High-Resolution Electron Energy Loss Spectroscopy. Chem. Mater. 2006, 18, 5520–5529. [Google Scholar] [CrossRef]
- Andersson, A.S.; Kalska, B.; Häggström, L.; Thomas, J.O. Lithium extraction/insertion in LiFePO4: An X-ray diffraction and Mössbauer spectroscopy study. Solid State Ion. 2000, 130, 41–52. [Google Scholar] [CrossRef]
- Shin, H.C.; Chung, K.Y.; Min, W.S.; Byun, D.J.; Jang, H.; Cho, B.W. Asymmetry between charge and discharge during high rate cycling in LiFePO4—In Situ X-ray diffraction study. Electrochem. Commun. 2008, 10, 536–540. [Google Scholar] [CrossRef]
- Tan, H.J.; Dodd, J.L.; Fultz, B. Thermodynamic and Kinetic Stability of the Solid Solution Phase in Nanocrystalline LixFePO4. J. Phys. Chem. C 2009, 113, 20527–20530. [Google Scholar] [CrossRef]
- Li, D.; Zhou, H. Two-phase transition of Li-intercalation compounds in Li-ion batteries. Mater. Today 2014, 17, 451–463. [Google Scholar] [CrossRef]
- Li, Z.; Yang, J.; Li, C.; Wang, S.; Zhang, L.; Zhu, K.; Wang, X. Orientation-Dependent Lithium Miscibility Gap in LiFePO4. Chem. Mater. 2018, 30, 874–878. [Google Scholar] [CrossRef]
- Wagemaker, M.; Singh, D.P.; Borghols, W.J.H.; Lafont, U.; Haverkate, L.; Peterson, V.K.; Mulder, F.M. Dynamic Solubility Limits in Nanosized Olivine LiFePO4. J. Am. Chem. Soc. 2011, 133, 10222–10228. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, G.; Nishimura, S.-I.; Park, M.-S.; Kanno, R.; Yashima, M.; Ida, T.; Yamada, A. Isolation of Solid Solution Phases in Size-Controlled LixFePO4 at Room Temperature. Adv. Funct. Mater. 2008, 19, 395–403. [Google Scholar] [CrossRef]
- Takahashi, I.; Mori, T.; Yoshinari, T.; Orikasa, Y.; Koyama, Y.; Murayama, H.; Fukuda, K.; Hatano, M.; Arai, H.; Uchimoto, Y.; et al. Irreversible phase transition between LiFePO4 and FePO4 during high-rate charge-discharge reaction by operando X-ray diffraction. J. Power Sources 2016, 309, 122–126. [Google Scholar] [CrossRef]
- Liu, H.; Strobridge, F.C.; Borkiewicz, O.J.; Wiaderek, K.M.; Chapman, K.W.; Chupas, P.J.; Grey, C.P. Capturing metastable structures during high-rate cycling of LiFePO4 nanoparticle electrodes. Science 2014, 344, 1252817. [Google Scholar] [CrossRef] [PubMed]
- Cogswell, D.A.; Bazant, M.Z. Coherency Strain and the Kinetics of Phase Separation in LiFePO4 Nanoparticles. ACS Nano 2012, 6, 2215–2225. [Google Scholar] [CrossRef] [PubMed]
- Van der Ven, A.; Garikipati, K.; Kim, S.; Wagemaker, M. The Role of Coherency Strains on Phase Stability in LixFePO4: Needle Crystallites Minimize Coherency Strain and Overpotential. J. Electrochem. Soc. 2009, 156, A949–A957. [Google Scholar] [CrossRef]
- Meethong, N.; Huang, H.-Y.S.; Carter, W.C.; Chiang, Y.-M. Size-Dependent Lithium Miscibility Gap in Nanoscale Li1−x FePO4. Electrochem. Solid-State Lett. 2007, 10, A134–A138. [Google Scholar] [CrossRef]
- Bai, P.; Cogswell, D.A.; Bazant, M.Z. Suppression of Phase Separation in LiFePO4 Nanoparticles During Battery Discharge. Nano Lett. 2011, 11, 4890–4896. [Google Scholar] [CrossRef] [PubMed]
- Malik, R.; Zhou, F.; Ceder, G. Kinetics of non-equilibrium lithium incorporation in LiFePO4. Nat. Mater. 2011, 10, 587. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Zhang, G.; Wei, Q.; Tian, X.; Zhao, K.; An, Q.; Zhou, L.; Zhao, Y.; Niu, C.; Ren, W.; et al. In operando observation of temperature-dependent phase evolution in lithium-incorporation olivine cathode. Nano Energy 2016, 22, 406–413. [Google Scholar] [CrossRef]
- Galceran, M.; Saurel, D.; Acebedo, B.; Roddatis, V.V.; Martin, E.; Rojo, T.; Casas-Cabanas, M. The mechanism of NaFePO4 (de)sodiation determined by in situ X-ray diffraction. Phys. Chem. Chem. Phys. 2014, 16, 8837–8842. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Guyomard, D.; Gaubicher, J.; Boucher, F. Structure and Stability of Sodium Intercalated Phases in Olivine FePO4. Chem. Mater. 2010, 22, 4126–4128. [Google Scholar] [CrossRef]
- Lu, J.; Chung, S.C.; Nishimura, S.-I.; Yamada, A. Phase Diagram of Olivine NaxFePO4 (0 < x < 1). Chem. Mater. 2013, 25, 4557–4565. [Google Scholar] [CrossRef]
- Chung, K.Y.; Yoon, W.-S.; Lee, H.S.; McBreen, J.; Yang, X.-Q.; Oh, S.H.; Ryu, W.H.; Lee, J.L.; Cho, W.I.; Cho, B.W. In situ XRD studies of the structural changes of ZrO2-coated LiCoO2 during cycling and their effects on capacity retention in lithium batteries. J. Power Sources 2006, 163, 185–190. [Google Scholar] [CrossRef]
- Chung, K.Y.; Yoon, W.-S.; McBreen, J.; Yang, X.Q.; Oh, S.Y.; Shin, H.C.; Cho, W.I.; Cho, B.W. In situ X-ray diffraction studies on the mechanism of capacity retention improvement by coating at the surface of LiCoO2. J. Power Sources 2007, 174, 619–623. [Google Scholar] [CrossRef]
- Sun, X.; Yang, X.Q.; McBreen, J.; Gao, Y.; Yakovleva, M.V.; Xing, X.K.; Daroux, M.L. New phases and phase transitions observed in over-charged states of LiCoO2-based cathode materials. J. Power Sources 2001, 97–98, 274–276. [Google Scholar] [CrossRef]
- Chen, Z.; Dahn, J.R. Methods to obtain excellent capacity retention in LiCoO2 cycled to 4.5 V. Electrochim. Acta 2004, 49, 1079–1090. [Google Scholar] [CrossRef]
- Ohzuku, T.; Ueda, A. Solid-State Redox Reactions of LiCoO (R3m) for 4 Volt Secondary Lithium Cells. J. Electrochem. Soc. 1994, 141, 2972–2977. [Google Scholar] [CrossRef]
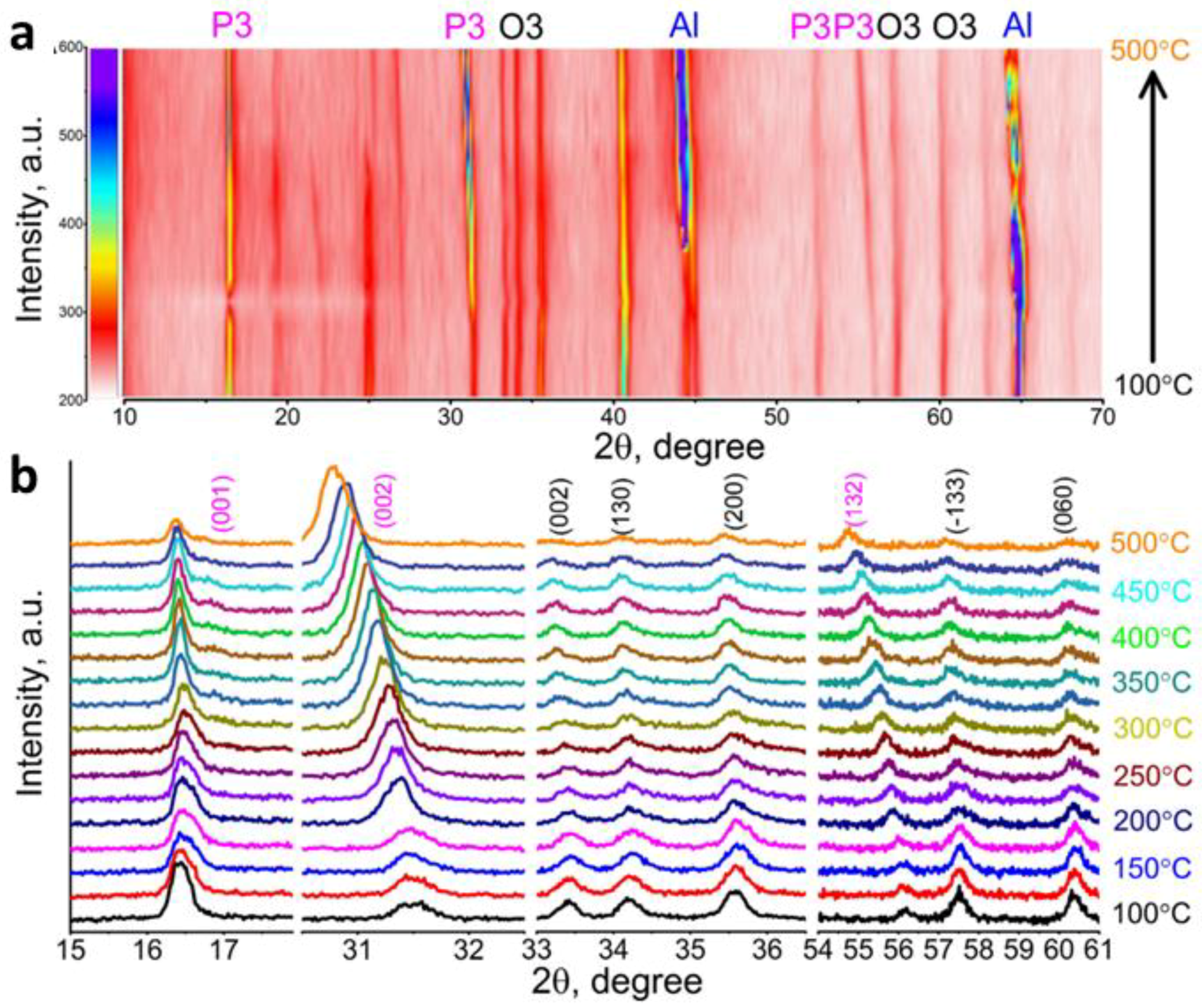
- Wang, P.-F.; You, Y.; Yin, Y.-X.; Guo, Y.-G. Layered Oxide Cathodes for Sodium-Ion Batteries: Phase Transition, Air Stability, and Performance. Adv. Energy Mater. 2018, 8, 1701912. [Google Scholar] [CrossRef]
- Delmas, C.; Fouassier, C.; Hagenmuller, P. Structural classification and properties of the layered oxides. Physica B+C 1980, 99, 81–85. [Google Scholar] [CrossRef]
- Yabuuchi, N.; Komaba, S. Recent research progress on iron- and manganese-based positive electrode materials for rechargeable sodium batteries. Sci. Technol. Adv. Mater. 2014, 15, 043501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Chen, Z.; Schwarz, B.; Sigel, F.; Ehrenberg, H.; An, K.; Zhang, Z.; Zhang, Q.; Li, Y.; Li, J. Kinetic characteristics up to 4.8 V of layered LiNi1/3Co1/3Mn1/3O2 cathode materials for high voltage lithium-ion batteries. Electrochim. Acta 2017, 227, 152–161. [Google Scholar] [CrossRef]
- Yoon, W.-S.; Chung, K.Y.; McBreen, J.; Yang, X.-Q. A comparative study on structural changes of LiCo1/3Ni1/3Mn1/3O2 and LiNi0.8Co0.15Al0.05O2 during first charge using in situ XRD. Electrochem. Commun. 2006, 8, 1257–1262. [Google Scholar] [CrossRef]
- Godbole, V.A.; Colin, J.-F.; Novak, P. Study of Overcharge Behavior of Li1+x(Ni1/3Mn1/3Co1/3)1−xO2 Using In Situ and Ex Situ X-ray Synchrotron Diffraction. J. Electrochem. Soc. 2011, 158, A1005–A1010. [Google Scholar] [CrossRef]
- Zhou, Y.-N.; Ma, J.; Hu, E.; Yu, X.; Gu, L.; Nam, K.-W.; Chen, L.; Wang, Z.; Yang, X.-Q. Tuning charge–discharge induced unit cell breathing in layer-structured cathode materials for lithium-ion batteries. Nat. Commun. 2014, 5, 5381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sathiya, M.; Rousse, G.; Ramesha, K.; Laisa, C.P.; Vezin, H.; Sougrati, M.T.; Doublet, M.L.; Foix, D.; Gonbeau, D.; Walker, W.; et al. Reversible anionic redox chemistry in high-capacity layered-oxide electrodes. Nat. Mater. 2013, 12, 827. [Google Scholar] [CrossRef] [PubMed]
- James, A.C.W.P.; Goodenough, J.B. Structure and bonding in Li2MoO3 and Li2−xMoO3 (0 ≤ x ≤ 1.7). J. Solid State Chem. 1988, 76, 87–96. [Google Scholar] [CrossRef]
- Lu, Z.; Dahn, J.R. Can All the Lithium be Removed from T2 Li2 / 3[ Ni1 / 3Mn2 / 3 ] O 2 ? J. Electrochem. Soc. 2001, 148, A710–A715. [Google Scholar] [CrossRef]
- Berthelot, R.; Carlier, D.; Delmas, C. Electrochemical investigation of the P2–NaxCoO2 phase diagram. Nat. Mater. 2011, 10, 74. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Dahn, J.R. In Situ X-ray Diffraction Study of P2 Na2 / 3[ Ni1 / 3Mn2 / 3 ] O2. J. Electrochem. Soc. 2001, 148, A1225–A1229. [Google Scholar] [CrossRef]
- Wang, P.-F.; You, Y.; Yin, Y.-X.; Wang, Y.-S.; Wan, L.-J.; Gu, L.; Guo, Y.-G. Suppressing the P2–O2 Phase Transition of Na0.67Mn0.67Ni0.33O2 by Magnesium Substitution for Improved Sodium-Ion Batteries. Angew. Chem. Int. Ed. 2016, 55, 7445–7449. [Google Scholar] [CrossRef] [PubMed]
- Han, M.H.; Gonzalo, E.; Sharma, N.; López del Amo, J.M.; Armand, M.; Avdeev, M.; Saiz Garitaonandia, J.J.; Rojo, T. High-Performance P2-Phase Na2/3Mn0.8Fe0.1Ti0.1O2 Cathode Material for Ambient-Temperature Sodium-Ion Batteries. Chem. Mater. 2016, 28, 106–116. [Google Scholar] [CrossRef]
- Xu, J.; Lee, D.H.; Clément, R.J.; Yu, X.; Leskes, M.; Pell, A.J.; Pintacuda, G.; Yang, X.Q.; Grey, C.P.; Meng, Y.S. Identifying the Critical Role of Li Substitution in P2− Nax[LiyNizMn1−y−z]O2 (0 < x, y, z < 1) Intercalation Cathode Materials for High-Energy Na-Ion Batteries. Chem. Mater. 2014, 26, 1260–1269. [Google Scholar] [CrossRef]
- Lu, Z.; Dahn, J.R. Effects of Stacking Fault Defects on the X-ray Diffraction Patterns of T2, O2, and O6 Structure Li2/3[CoxNi1/3−xMn2/3]O2. Chem. Mater. 2001, 13, 2078–2083. [Google Scholar] [CrossRef]
- Singh, G.; Tapia-Ruiz, N.; Lopez del Amo, J.M.; Maitra, U.; Somerville, J.W.; Armstrong, A.R.; Martinez de Ilarduya, J.; Rojo, T.; Bruce, P.G. High Voltage Mg-Doped Na0.67Ni0.3–xMgxMn0.7O2 (x = 0.05, 0.1) Na-Ion Cathodes with Enhanced Stability and Rate Capability. Chem. Mater. 2016, 28, 5087–5094. [Google Scholar] [CrossRef]
- Billaud, J.; Singh, G.; Armstrong, A.R.; Gonzalo, E.; Roddatis, V.; Armand, M.; Rojo, T.; Bruce, P.G. Na0.67Mn1−xMgxO2 (0 ≤ x ≤ 0.2): A high capacity cathode for sodium-ion batteries. Energy Environ. Sci. 2014, 7, 1387–1391. [Google Scholar] [CrossRef]
- Boehm, R.C.; Banerjee, A. Theoretical study of lithium intercalated graphite. J. Chem. Phys. 1992, 96, 1150–1157. [Google Scholar] [CrossRef]
- Ge, H.; Li, N.; Li, D.; Dai, C.; Wang, D. Study on the Theoretical Capacity of Spinel Lithium Titanate Induced by Low-Potential Intercalation. J. Phys. Chem. C 2009, 113, 6324–6326. [Google Scholar] [CrossRef]
- Ge, H.; Li, N.; Li, D.; Dai, C.; Wang, D. Electrochemical characteristics of spinel Li4Ti5O12 discharged to 0.01 V. Electrochem. Commun. 2008, 10, 719–722. [Google Scholar] [CrossRef]
- Venkateswarlu, M.; Chen, C.H.; Do, J.S.; Lin, C.W.; Chou, T.C.; Hwang, B.J. Electrochemical properties of nano-sized Li4Ti5O12 powders synthesized by a sol–gel process and characterized by X-ray absorption spectroscopy. J. Power Sources 2005, 146, 204–208. [Google Scholar] [CrossRef]
- Shu, J. Electrochemical behavior and stability of Li4Ti5O12 in a broad voltage window. J. Solid State Electrochem. 2009, 13, 1535–1539. [Google Scholar] [CrossRef]
- Zhao, L.; Pan, H.-L.; Hu, Y.-S.; Li, H.; Chen, L.-Q. Spinel lithium titanate (Li4Ti5O12) as novel anode material for room-temperature sodium-ion battery. Chin. Phys. B 2012, 21, 028201. [Google Scholar] [CrossRef]
- Yang, L.Y.; Li, H.Z.; Liu, J.; Tang, S.S.; Lu, Y.K.; Li, S.T.; Min, J.; Yan, N.; Lei, M. Li4Ti5O12 nanosheets as high-rate and long-life anode materials for sodium-ion batteries. J. Mater. Chem. A 2015, 3, 24446–24452. [Google Scholar] [CrossRef]
- Kitta, M.; Akita, T.; Tanaka, S.; Kohyama, M. Two-phase separation in a lithiated spinel Li4Ti5O12 crystal as confirmed by electron energy-loss spectroscopy. J. Power Sources 2014, 257, 120–125. [Google Scholar] [CrossRef]
- Kitta, M.; Kuratani, K.; Tabuchi, M.; Takeichi, N.; Akita, T.; Kiyobayashi, T.; Kohyama, M. Irreversible structural change of a spinel Li4Ti5O12 particle via Na insertion-extraction cycles of a sodium-ion battery. Electrochim. Acta 2014, 148, 175–179. [Google Scholar] [CrossRef]
- Li, W.; Dahn, J.R.; Wainwright, D.S. Rechargeable Lithium Batteries with Aqueous Electrolytes. Science 1994, 264, 1115–1118. [Google Scholar] [CrossRef] [PubMed]
- Pasta, M.; Wessells, C.D.; Huggins, R.A.; Cui, Y. A high-rate and long cycle life aqueous electrolyte battery for grid-scale energy storage. Nat. Commun. 2012, 3, 1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Xu, C.; Chen, Z.; Ni, S.; Shen, Z.X. Progress in aqueous rechargeable batteries. Green Energy Environ. 2018, 3, 20–41. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Lee, B.; Qiao, R.; Yang, Z.; Xu, S.; Yu, X.; Gu, L.; Hu, Y.-S.; Yang, W.; et al. Ti-substituted tunnel-type Na0.44MnO2 oxide as a negative electrode for aqueous sodium-ion batteries. Nat. Commun. 2015, 6, 6401. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yu, X.; Xu, S.; Bai, J.; Xiao, R.; Hu, Y.-S.; Li, H.; Yang, X.-Q.; Chen, L.; Huang, X. A zero-strain layered metal oxide as the negative electrode for long-life sodium-ion batteries. Nat. Commun. 2013, 4, 2365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köhler, J.; Makihara, H.; Uegaito, H.; Inoue, H.; Toki, M. LiV3O8: Characterization as anode material for an aqueous rechargeable Li-ion battery system. Electrochim. Acta 2000, 46, 59–65. [Google Scholar] [CrossRef]
- Wu, J.; Gao, X.; Yu, H.; Ding, T.; Yan, Y.; Yao, B.; Yao, X.; Chen, D.; Liu, M.; Huang, L. A Scalable Free-Standing V2O5/CNT Film Electrode for Supercapacitors with a Wide Operation Voltage (1.6 V) in an Aqueous Electrolyte. Adv. Funct. Mater. 2016, 26, 6114–6120. [Google Scholar] [CrossRef]
- Yamada, Y.; Usui, K.; Sodeyama, K.; Ko, S.; Tateyama, Y.; Yamada, A. Hydrate-melt electrolytes for high-energy-density aqueous batteries. Nat. Energy 2016, 1, 16129. [Google Scholar] [CrossRef]
- Wang, H.; Huang, K.; Zeng, Y.; Yang, S.; Chen, L. Electrochemical properties of TiP2O7 and LiTi2(PO4)3 as anode material for lithium ion battery with aqueous solution electrolyte. Electrochim. Acta 2007, 52, 3280–3285. [Google Scholar] [CrossRef]
- Wu, M.-S.; Wang, M.-J.; Jow, J.-J.; Yang, W.-D.; Hsieh, C.-Y.; Tsai, H.-M. Electrochemical fabrication of anatase TiO2 nanostructure as an anode material for aqueous lithium-ion batteries. J. Power Sources 2008, 185, 1420–1424. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, S.-Z.; You, Y.; Feng, Z.; Zhu, W.; Gariépy, V.; Xia, J.; Commarieu, B.; Darwiche, A.; Guerfi, A.; Zaghib, K. High-Capacity and Long-Cycle Life Aqueous Rechargeable Lithium-Ion Battery with the FePO4 Anode. ACS Appl. Mater. Interfaces 2018, 10, 7061–7068. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Yang, C.; Ou, X.; Liang, X.; Xue, H.; Wang, W.; Xu, G. Unravelling the electrochemical properties and thermal behavior of NaNi2/3Sb1/3O2 cathode for sodium-ion batteries by in situ X-ray diffraction investigation. Electrochim. Acta 2017, 257, 146–154. [Google Scholar] [CrossRef]
- Same, A.; Park, J.W.; Battaglia, V.; Tang, H.Y. In situ neutron radiography analysis of graphite/NCA lithium-ion battery during overcharge. J. Appl. Electrochem. 2012, 42, 1–9. [Google Scholar] [CrossRef]
- Lu, W.; López, C.M.; Liu, N.; Vaughey, J.T.; Jansen, A.; Dennis, W.D. Overcharge Effect on Morphology and Structure of Carbon Electrodes for Lithium-Ion Batteries. J. Electrochem. Soc. 2012, 159, A566–A570. [Google Scholar] [CrossRef]
- Makimura, Y.; Sasaki, T.; Oka, H.; Okuda, C.; Nonaka, T.; Nishimura, Y.F.; Kawauchi, S.; Takeuchi, Y. Studying the Charging Process of a Lithium-Ion Battery toward 10 V by In Situ X-ray Absorption and Diffraction: Lithium Insertion/Extraction with Side Reactions at Positive and Negative Electrodes. J. Electrochem. Soc. 2016, 163, A1450–A1456. [Google Scholar] [CrossRef]
- Patterson, A.L. The Scherrer Formula for X-ray Particle Size Determination. Phys. Rev. 1939, 56, 978–982. [Google Scholar] [CrossRef]
- Ziolkowska, D.A.; Jasinski, J.B.; Hamankiewicz, B.; Korona, K.P.; Wu, S.-H.; Czerwinski, A. In Situ XRD and TEM Studies of Sol-Gel-Based Synthesis of LiFePO4. Cryst. Growth Des. 2016, 16, 5006–5013. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pristine Cathode | LiNi0.4Mn0.3Co0.3O2 (NMC433) | LiNi0.5Mn0.3Co0.2O2 (NMC532) | LiNi0.6Mn0.2Co0.2O2 (NMC622) | LiNi0.8Mn0.1Co0.1O2 (NMC811) |
|---|---|---|---|---|
| After charge | Li = 0.34 | Li = 0.29 | Li = 0.27 | Li = 0.22 |
| Phase at 25 °C | R | R | R | R |
| T0–Tf (°C) | 245–325 | 235–315 | 185–245 | T0 = 135 |
| NMC→S1 | ΔT = 80 °C | ΔT = 80 °C | ΔT = 60 °C | |
| T0/Tf (°C) | T0 = 450 | T0 = 400 | T0 = 310 | Tf = 155 |
| S1→S2 | ||||
| T0/Tf (°C) | no RS up to 600 °C | no RS up to 600 °C | T0 = 550 | Tf = 365 |
| S2→RS | ||||
| Phases at 600 o C | S1 + S2 | S2 | RS | RS + Ni (minor) |
| Tpeak, O2 (°C) | ~230 | ~150 | ||
| (from MS) | ||||
| Peak shape | Broad/low ~ 220–320 °C | Broad/low ~ 220–320 °C | Broad/medium ~ 170–340 °C | sharp/high ~ 130–260 °C |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, W.; Wang, Y.; Liu, D.; Gariépy, V.; Gagnon, C.; Vijh, A.; Trudeau, M.L.; Zaghib, K. Application of Operando X-ray Diffractometry in Various Aspects of the Investigations of Lithium/Sodium-Ion Batteries. Energies 2018, 11, 2963. https://doi.org/10.3390/en11112963
Zhu W, Wang Y, Liu D, Gariépy V, Gagnon C, Vijh A, Trudeau ML, Zaghib K. Application of Operando X-ray Diffractometry in Various Aspects of the Investigations of Lithium/Sodium-Ion Batteries. Energies. 2018; 11(11):2963. https://doi.org/10.3390/en11112963
Chicago/Turabian StyleZhu, Wen, Yuesheng Wang, Dongqiang Liu, Vincent Gariépy, Catherine Gagnon, Ashok Vijh, Michel L. Trudeau, and Karim Zaghib. 2018. "Application of Operando X-ray Diffractometry in Various Aspects of the Investigations of Lithium/Sodium-Ion Batteries" Energies 11, no. 11: 2963. https://doi.org/10.3390/en11112963