
First-Principles Study of Vacancies in Ti3SiC2 and Ti3AlC2

 ,
,
Abstract
:
1. Introduction
2. Theoretical Method
3. Results and Discussion
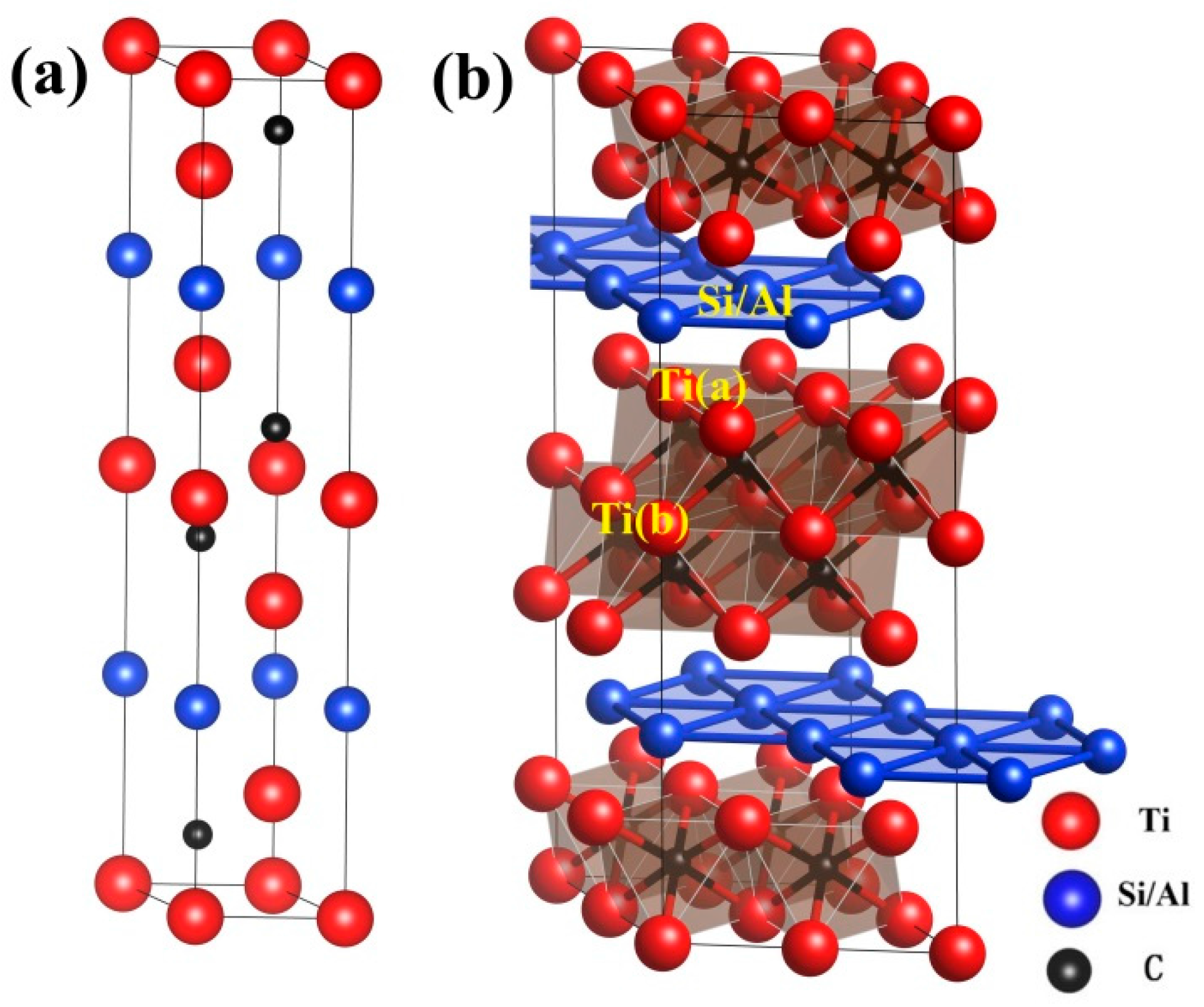
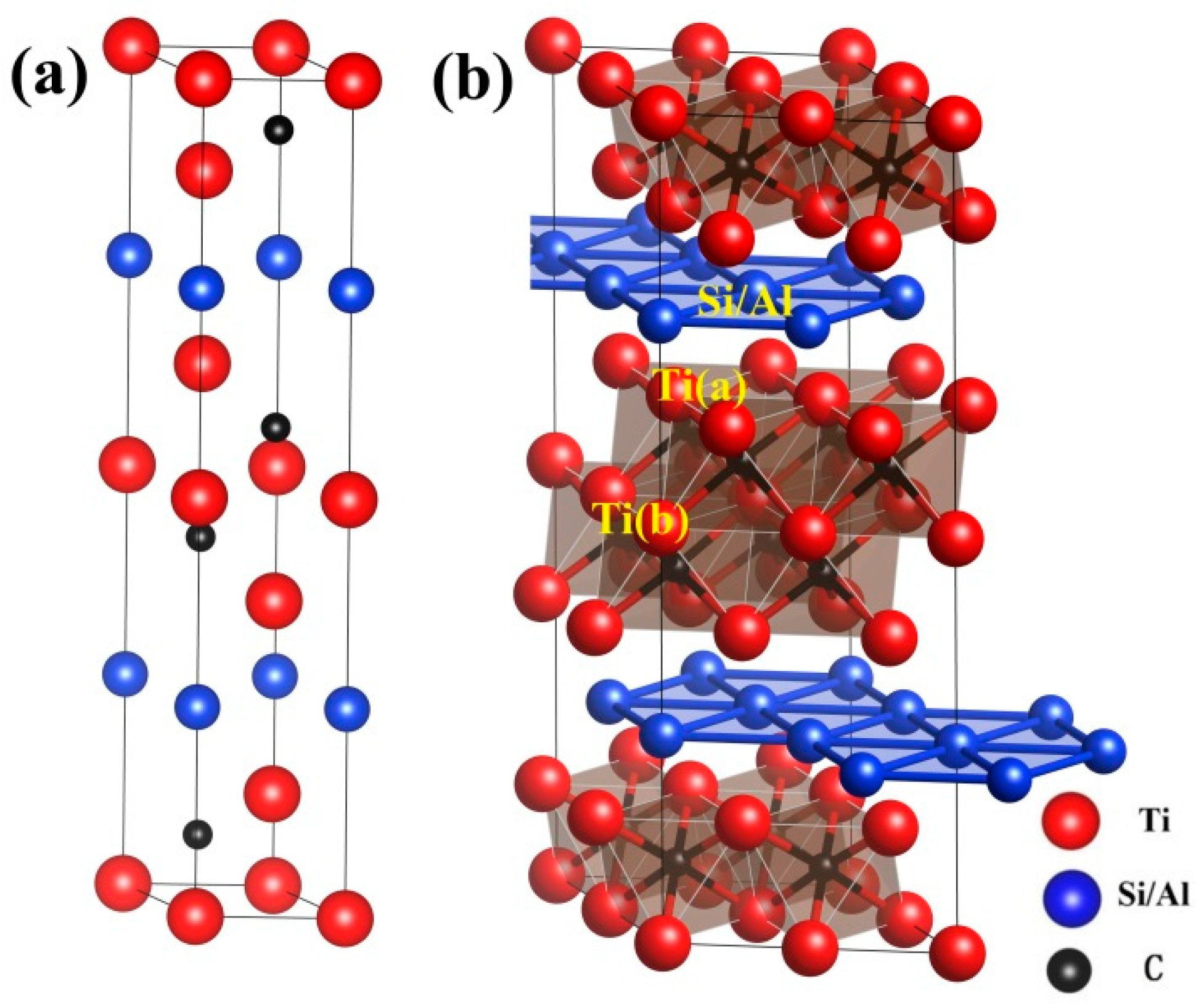
3.1. Properties of Perfect Ti3SiC2 and Ti3AlC2
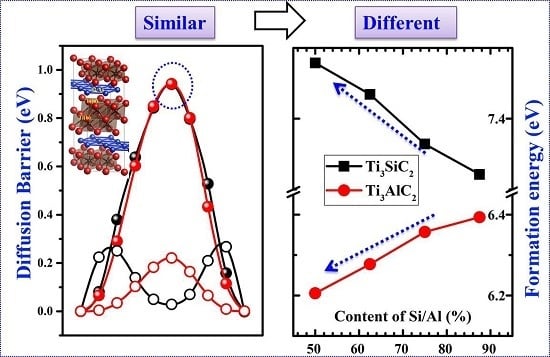
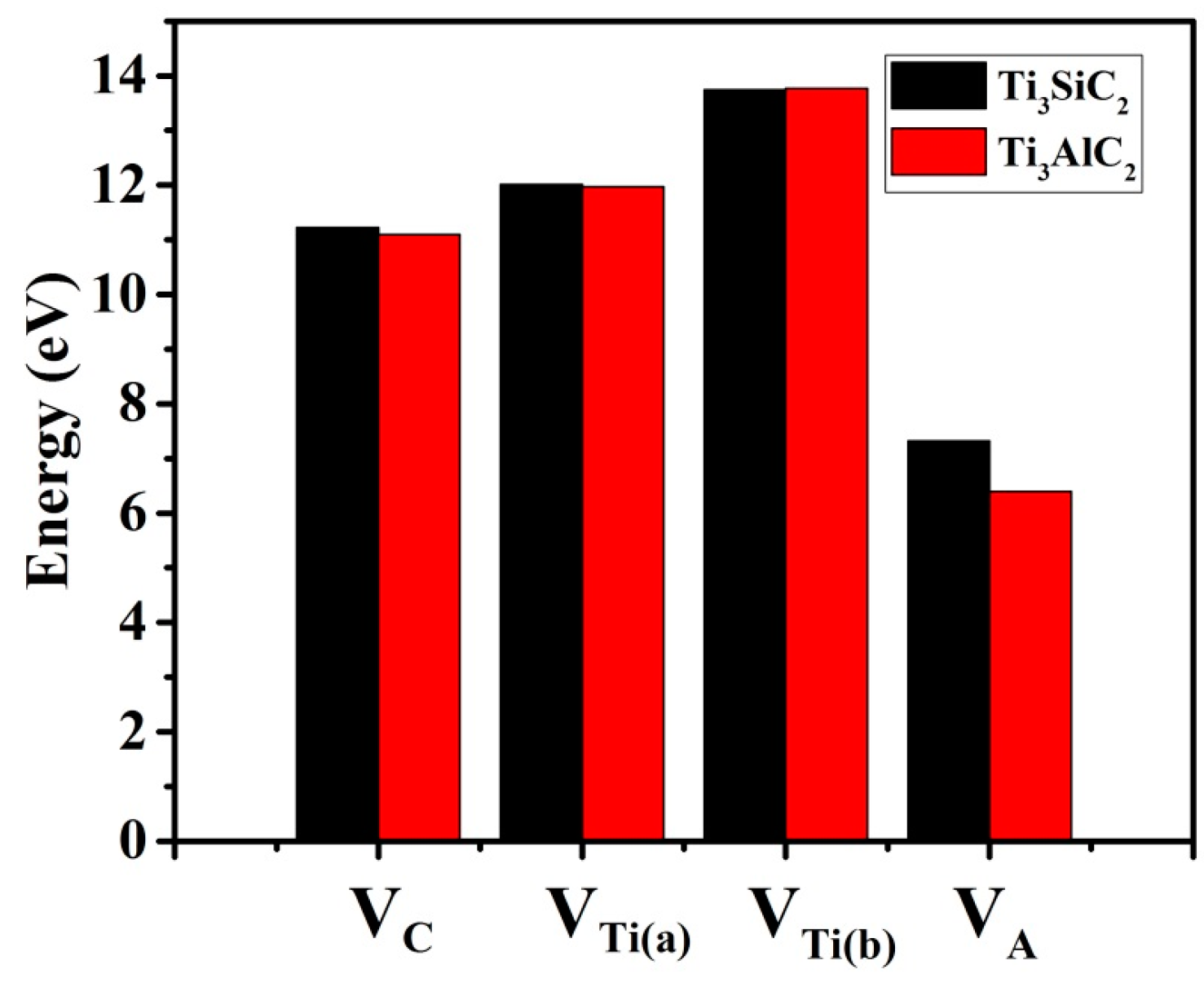
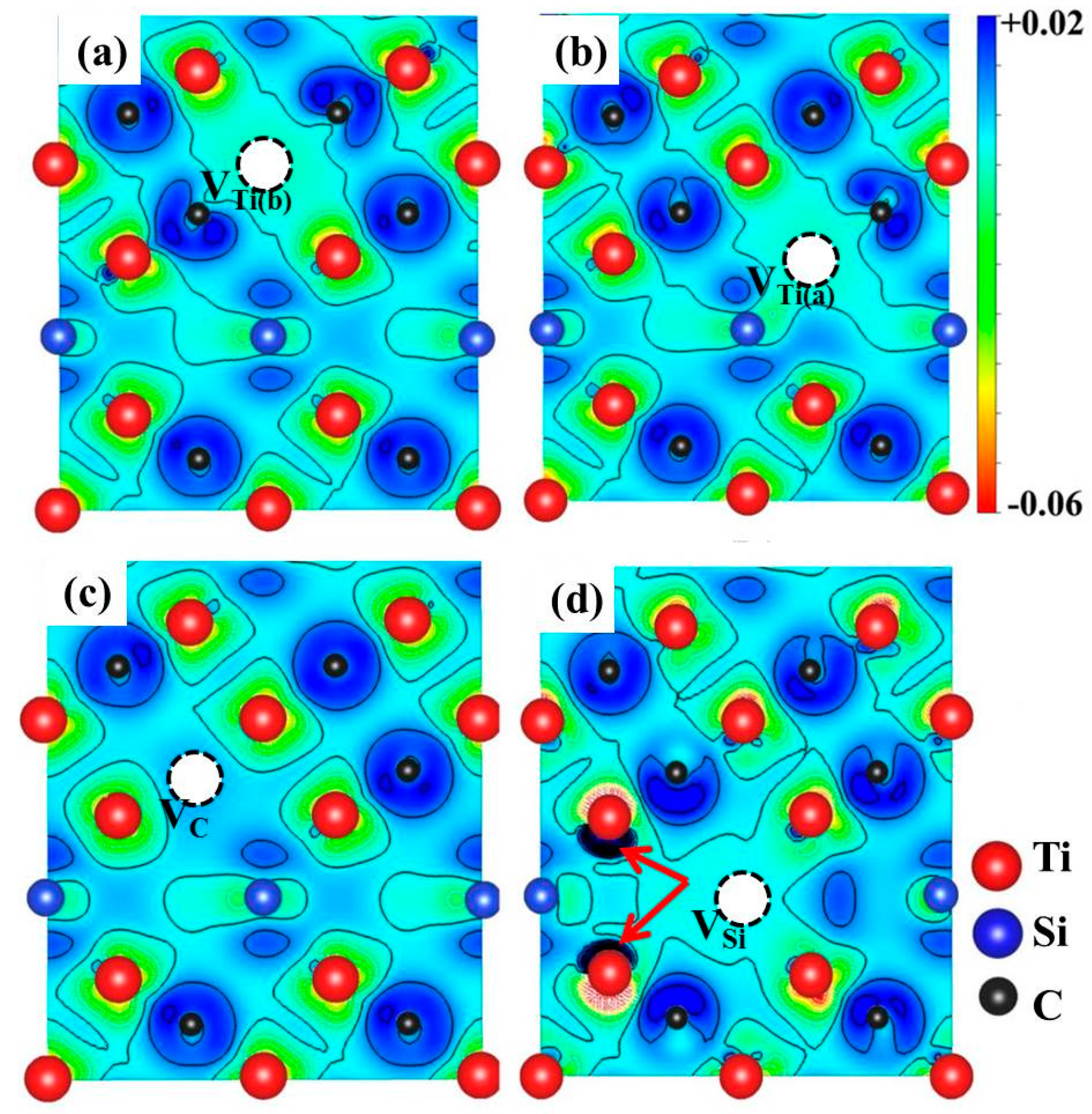
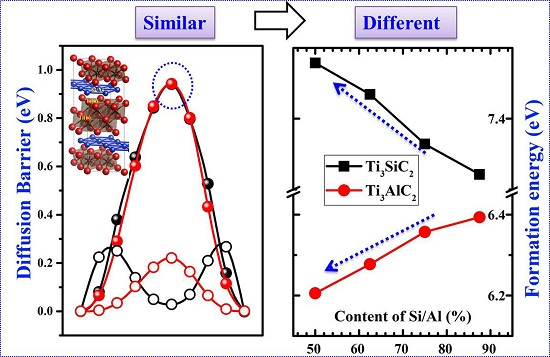
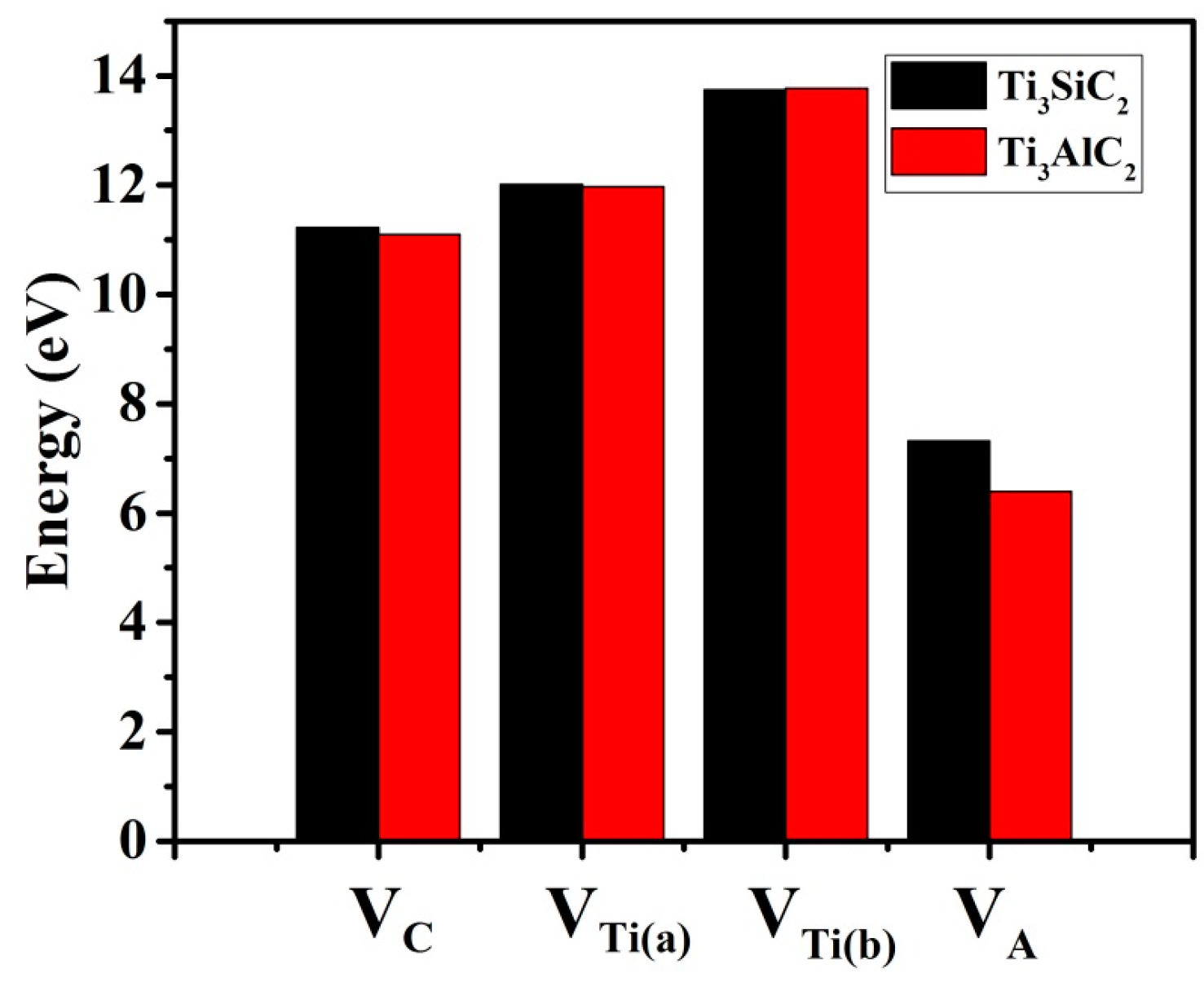
3.2. Formation Energies of Vacancies in Ti3SiC2 and Ti3AlC2
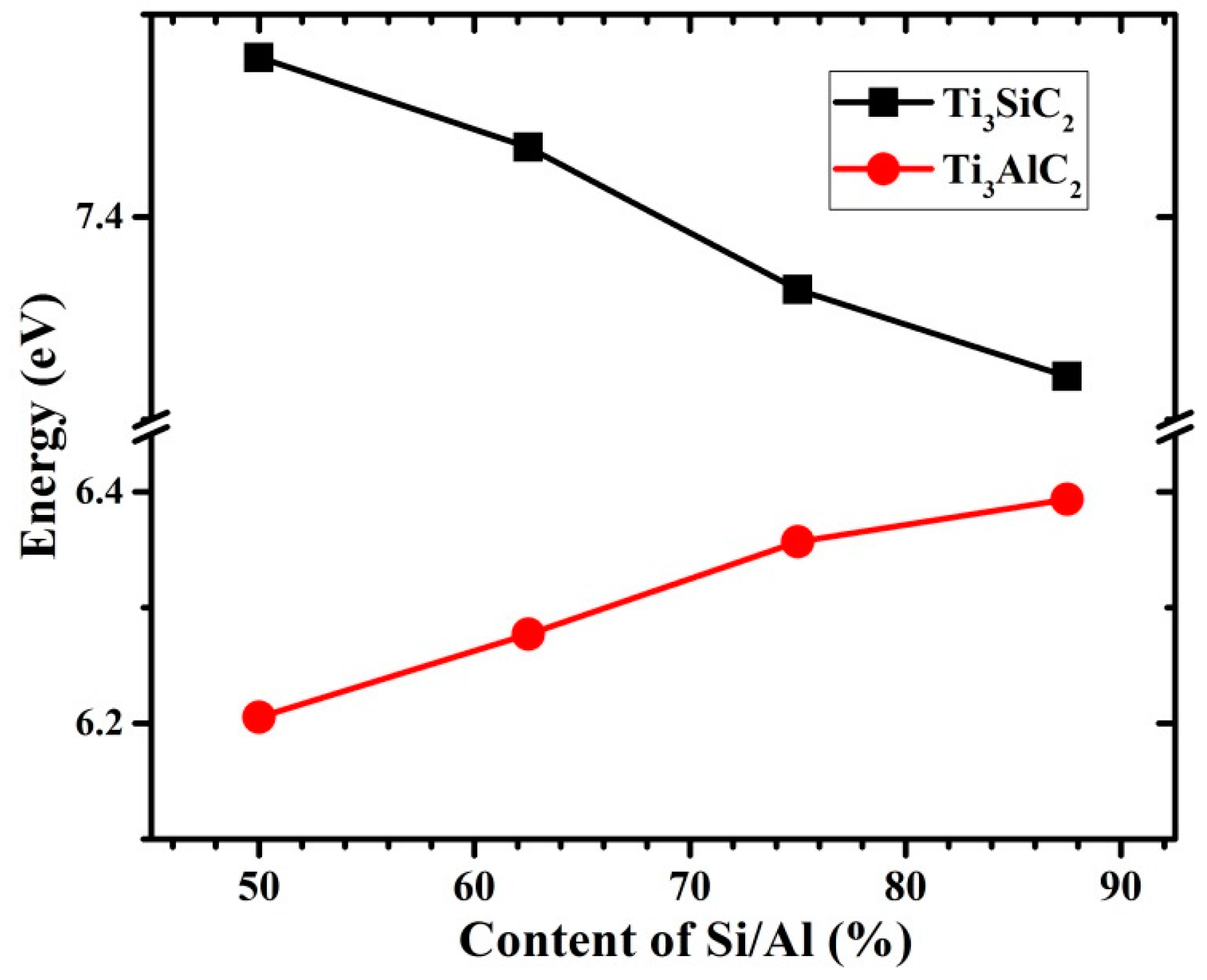
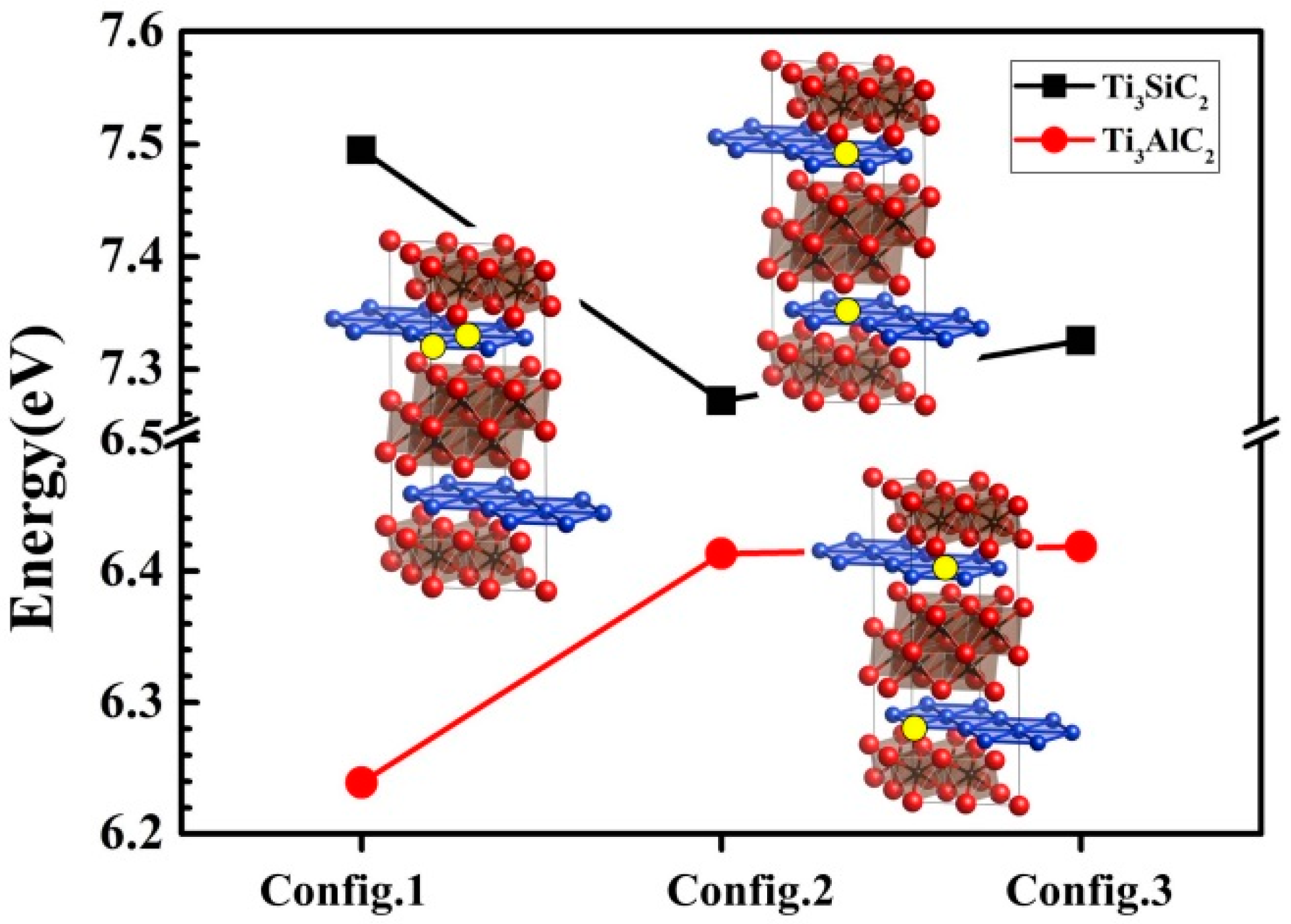
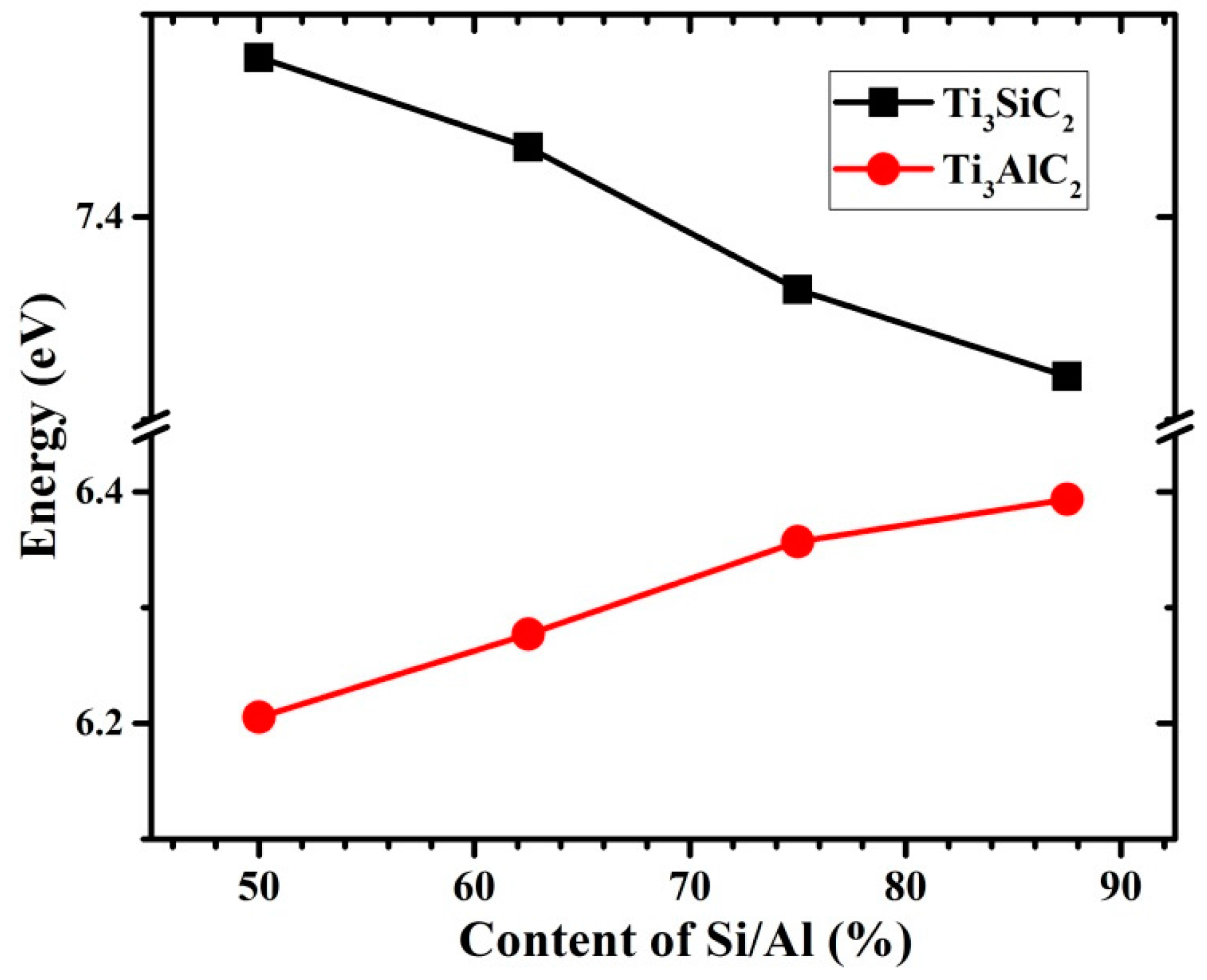
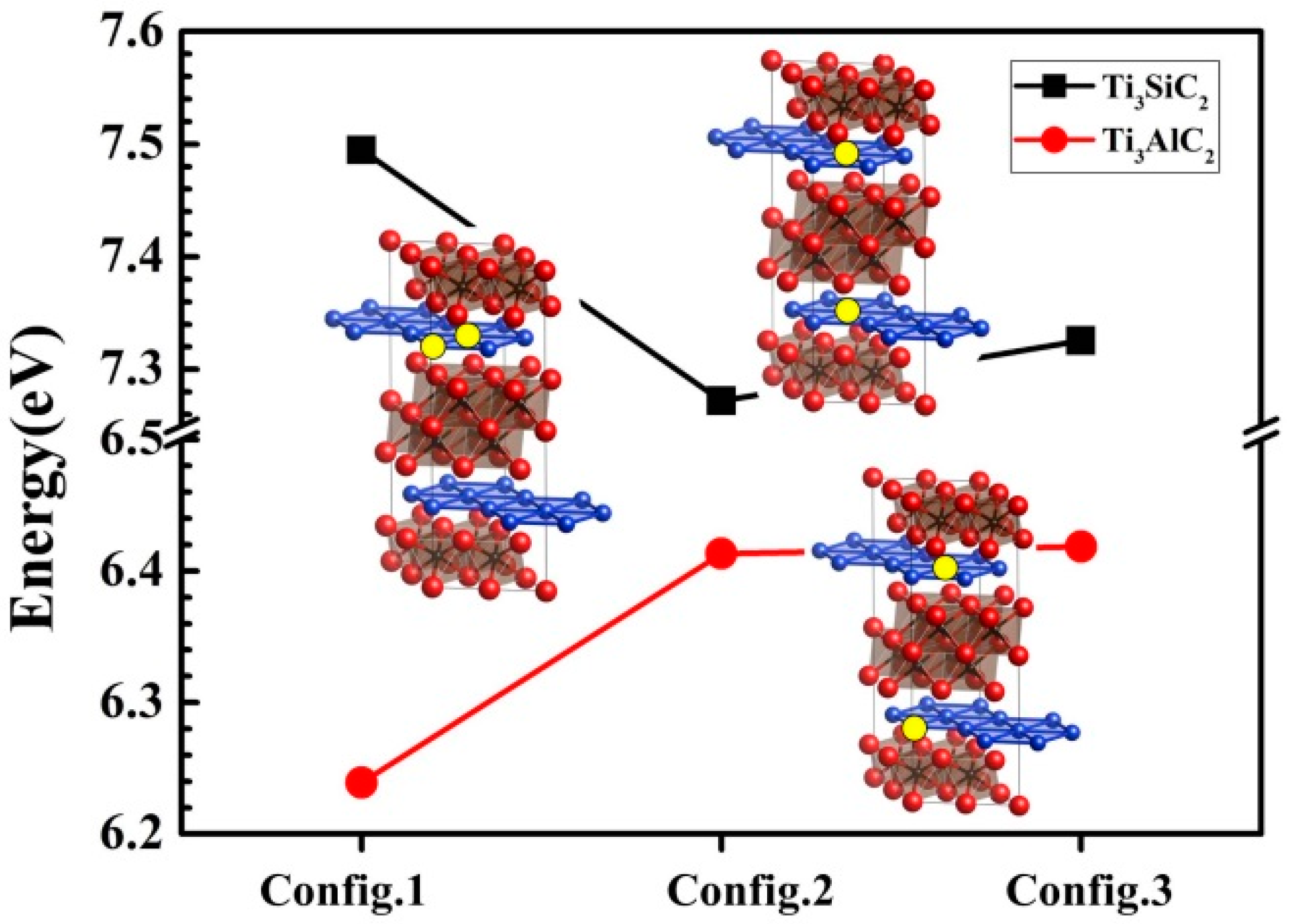
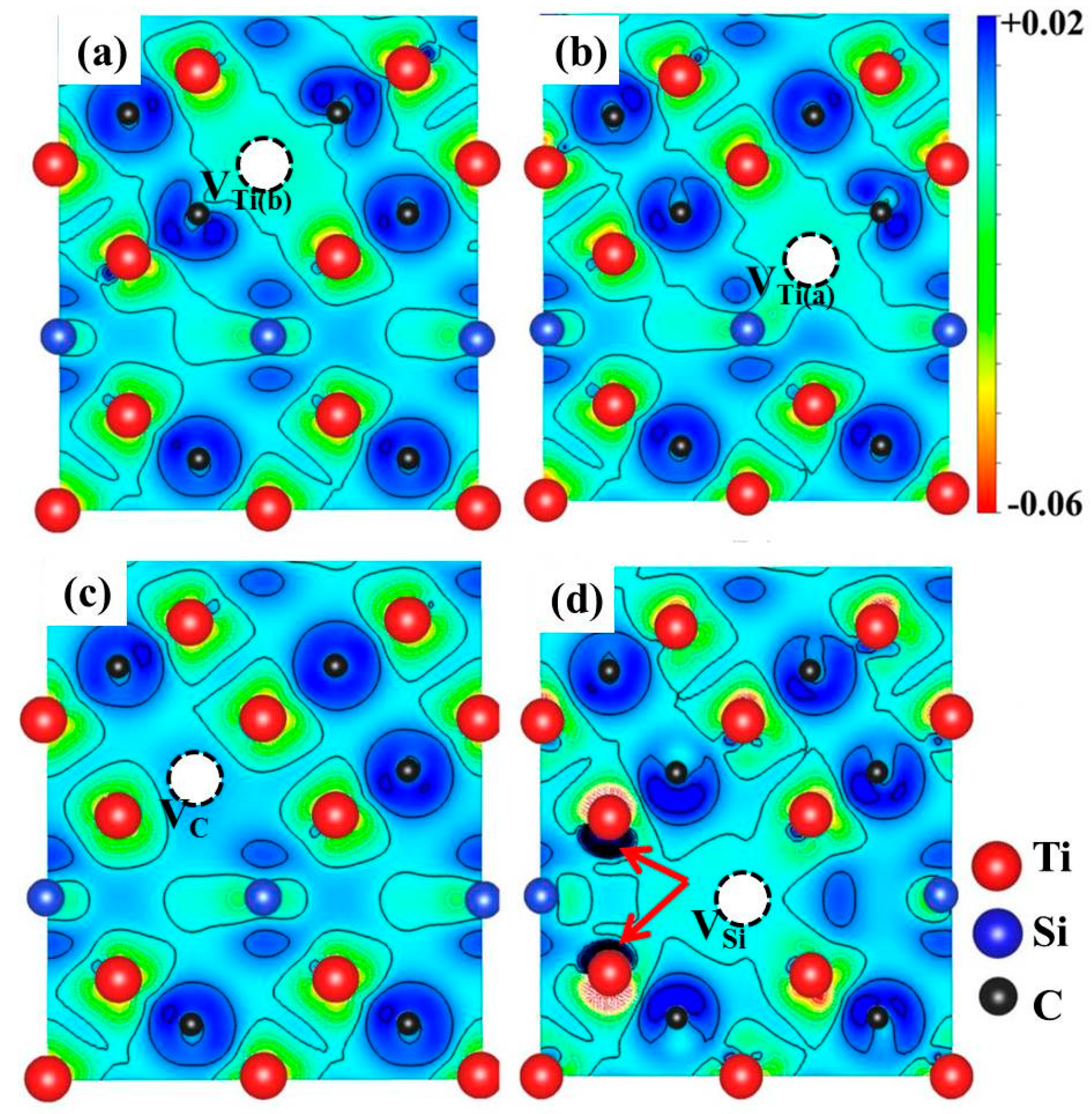
3.3. Vacancy–Vacancy Interactions of VA
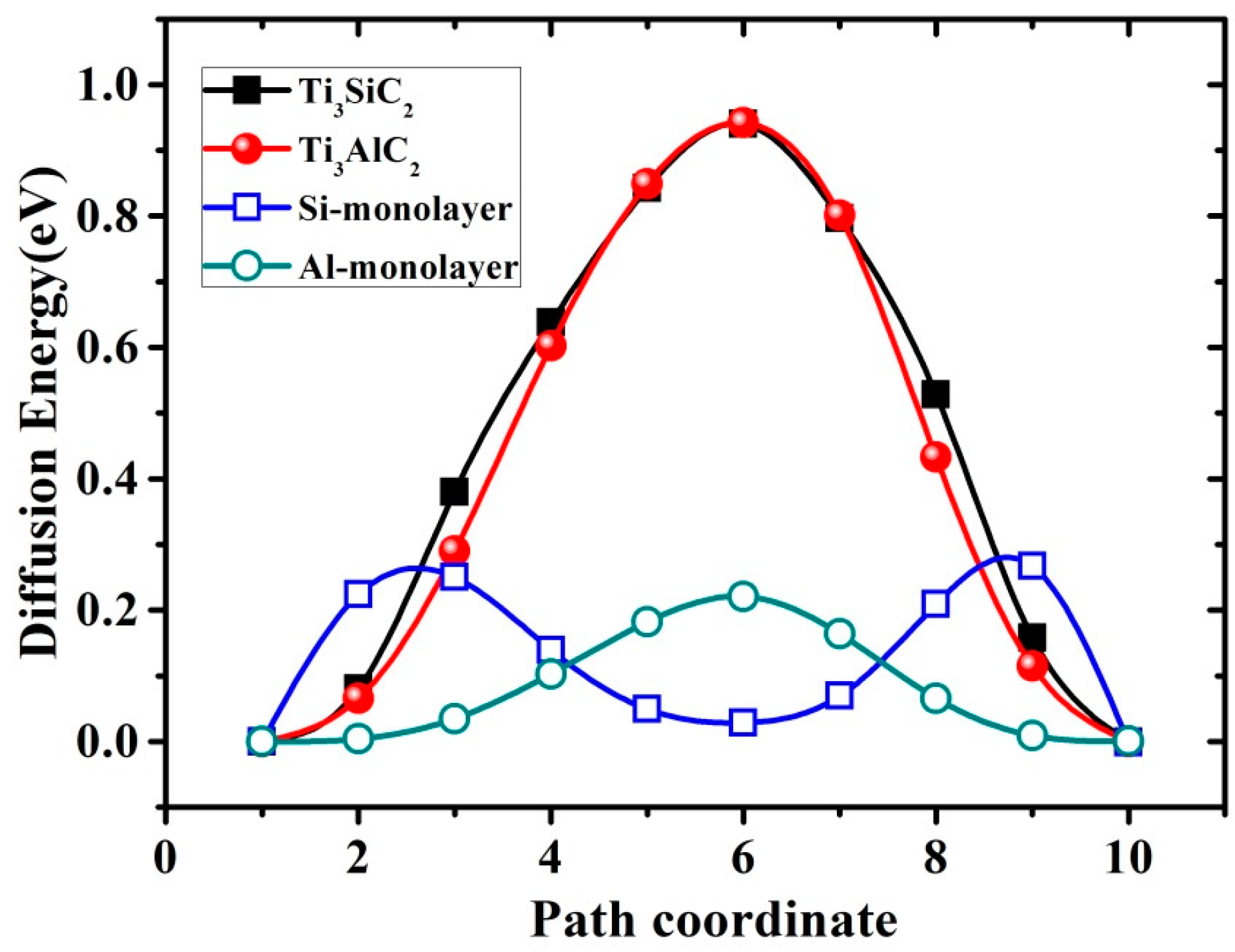
3.4. Diffusion of VA Vacancies
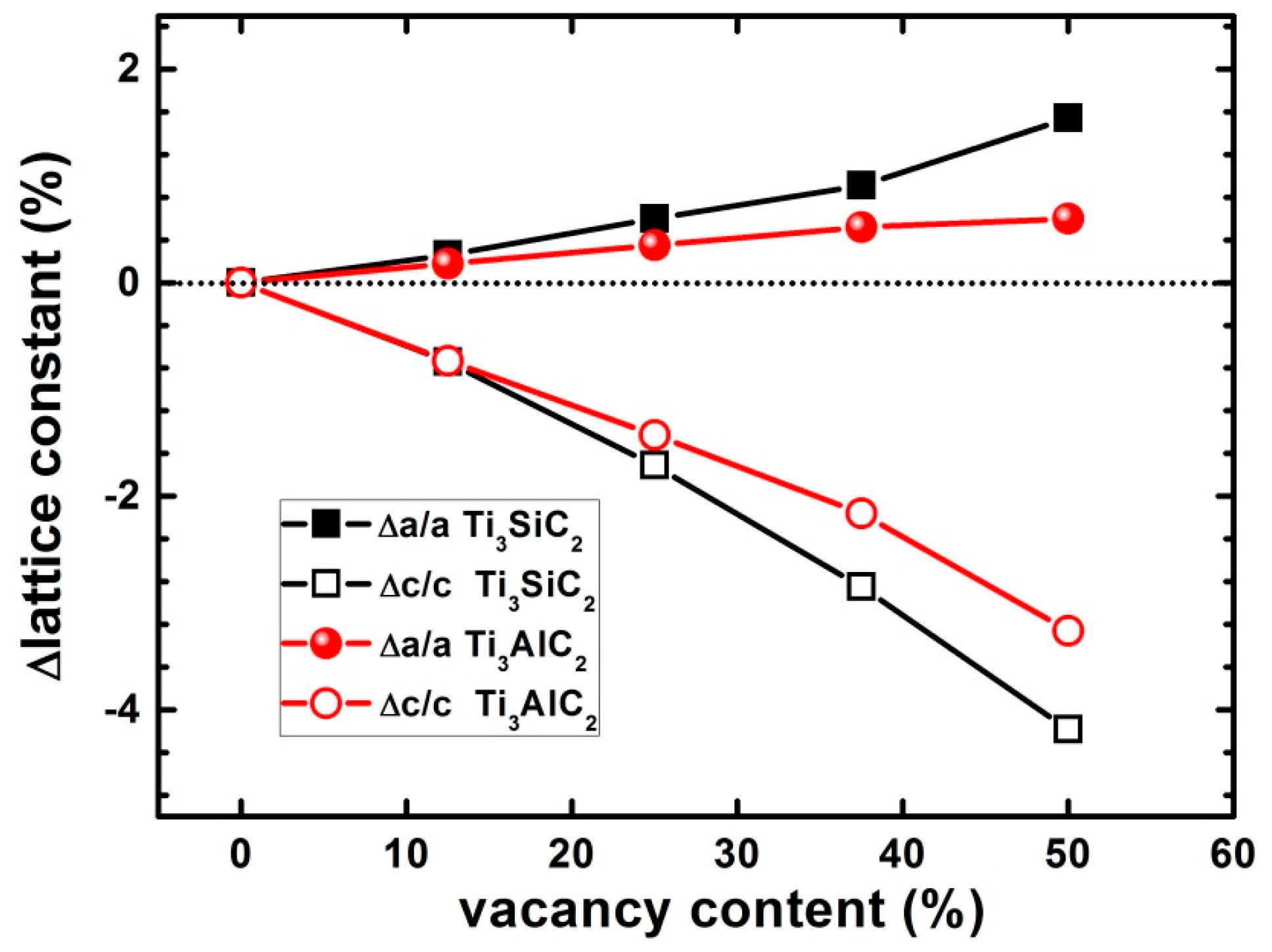
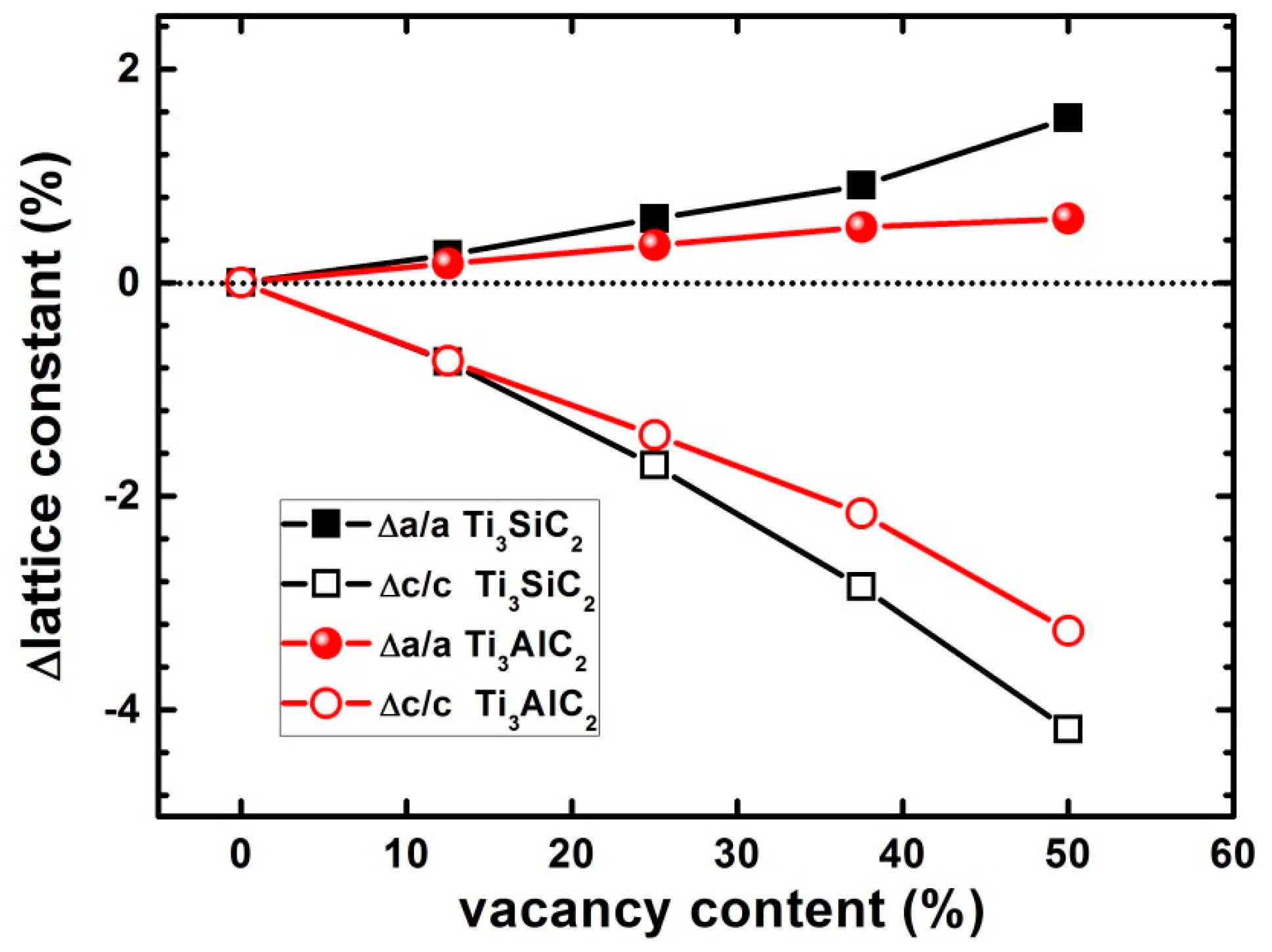
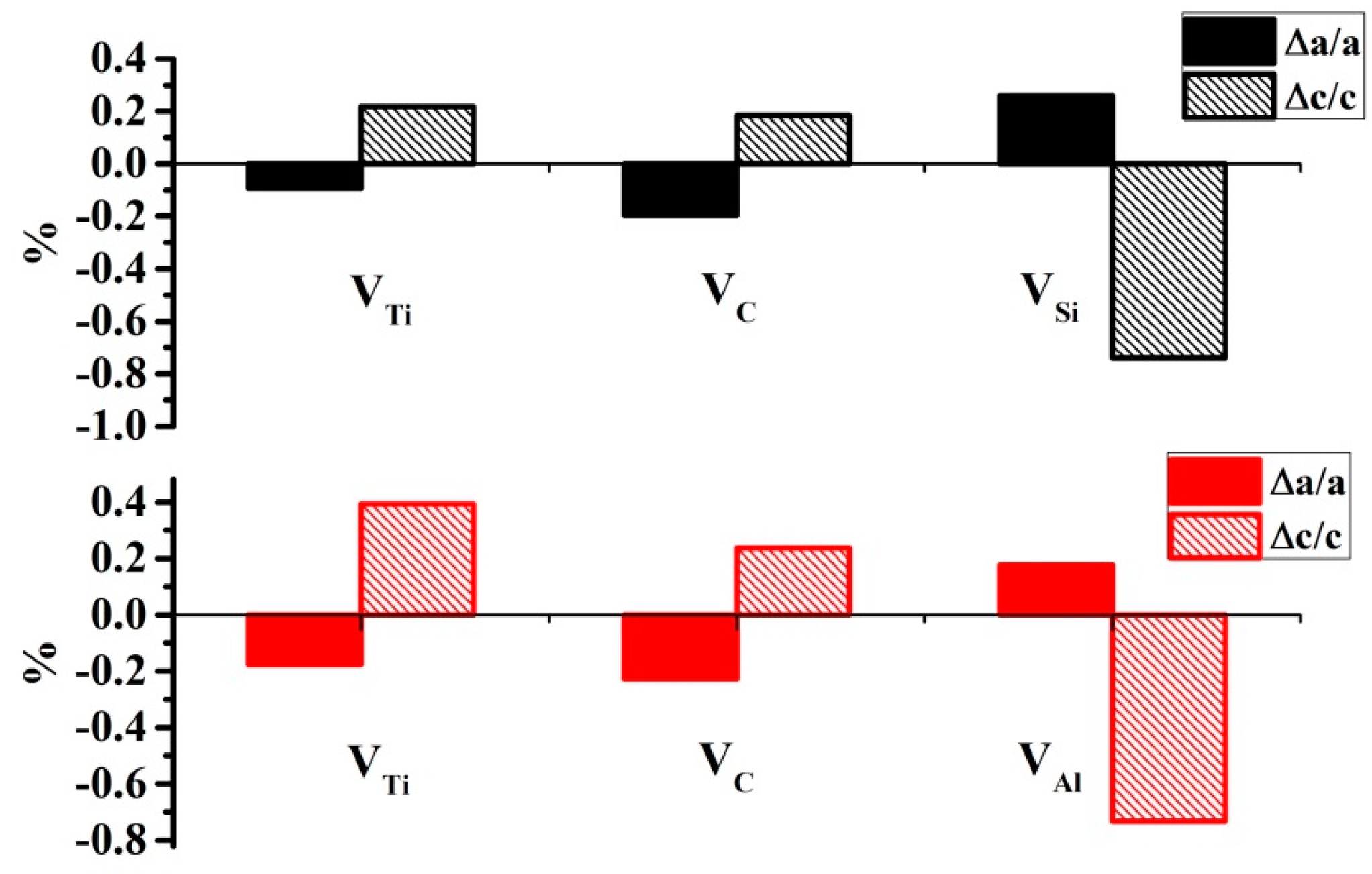
3.5. Effects of Vacancies on Lattice Constants
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ghidiu, M.; Lukatskaya, M.R.; Zhao, M.-Q.; Gogotsi, Y.; Barsoum, M.W. Conductive two-dimensional titanium carbide ‘clay’ with high volumetric capacitance. Nature 2014, 516, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Naguib, M.; Mochalin, V.N.; Barsoum, M.W.; Gogotsi, Y. 25th Anniversary Article: Mxenes: A New Family of Two-Dimensional Materials. Adv. Mater. 2014, 26, 992–1005. [Google Scholar] [CrossRef] [PubMed]
- Radovic, M.; Barsoum, M.W. Max phases: Bridging the gap between metals and ceramics. Am. Ceram. Soc. Bull. 2013, 92, 20–27. [Google Scholar]
- Barsoum, M.W.; El-Raghy, T. Synthesis and Characterization of a Remarkable Ceramic: Ti3SiC2. J. Am. Ceram. Soc. 1996, 79, 1953–1956. [Google Scholar] [CrossRef]
- Barsoum, M.W. The MN+1AXN Phases: A New Class of Solids: Thermodynamically stable nanolaminates. Prog. Solid State Chem. 2000, 28, 201–281. [Google Scholar] [CrossRef]
- Barsoum, M.W.; El-Raghy, T.; Rawn, C.J.; Porter, W.D.; Wang, H.; Payzant, E.A.; Hubbard, C.R. Thermal properties of Ti3SiC2. J. Phys. Chem. Solids 1999, 60, 429–439. [Google Scholar] [CrossRef]
- Kisi, E.H.; Crossley, J.A.A.; Myhra, S.; Barsoum, M.W. Structure and Crystal Chemistry of Ti3SiC2. J. Phys. Chem. Solids 1998, 59, 1437–1443. [Google Scholar] [CrossRef]
- Zhang, H.; Bao, Y.; Zhou, Y. Current Status in Layered Ternary Carbide Ti3SiC2, a Review. J. Mater. Sci. Technol. 2009, 25, 1–38. [Google Scholar]
- Farber, L.; Levin, I.; Barsoum, M.W.; El-Raghy, T.; Tzenov, T. High-resolution transmission electron microscopy of some Tin+1AXn compounds (n = 1, 2; A = Al or Si; X = C or N). J. Appl. Phys. 1999, 86, 2540–2543. [Google Scholar] [CrossRef]
- Wang, J.-Y.; Zhou, Y.-C. Polymorphism of Ti3SiC2 ceramic: First-principles investigations. Phys. Rev. B 2004, 69, 144108. [Google Scholar] [CrossRef]
- Nappé, J.C.; Monnet, I.; Grosseau, P.; Audubert, F.; Guilhot, B.; Beauvy, M.; Benabdesselam, M.; Thomé, L. Structural changes induced by heavy ion irradiation in titanium silicon carbide. J. Nucl. Mater. 2011, 409, 53–61. [Google Scholar] [CrossRef]
- Liu, X.M.; Le Flem, M.; Béchade, J.L.; Monnet, I. Nanoindentation investigation of heavy ion irradiated Ti3(Si,Al)C2. J. Nucl. Mater. 2010, 401, 149–153. [Google Scholar] [CrossRef]
- Whittle, K.R.; Blackford, M.G.; Aughterson, R.D.; Moricca, S.; Lumpkin, G.R.; Riley, D.P.; Zaluzec, N.J. Radiation tolerance of Mn+1AXn phases, Ti3AlC2 and Ti3SiC2. Acta Mater. 2010, 58, 4362–4368. [Google Scholar] [CrossRef]
- Hoffman, E.N.; Vinson, D.W.; Sindelar, R.L.; Tallman, D.J.; Kohse, G.; Barsoum, M.W. MAX phase carbides and nitrides: Properties for future nuclear power plant in-core applications and neutron transmutation analysis. Nucl. Eng. Des. 2012, 244, 17–24. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, Y.; Liao, T.; Zhang, J.; Lin, Z. A first-principles investigation of the phase stability of Ti2AlC with Al vacancies. Scr. Mater. 2008, 58, 227–230. [Google Scholar] [CrossRef]
- Liao, T.; Wang, J.; Zhou, Y. Ab initio modeling of the formation and migration of monovacancies in Ti2AlC. Scr. Mater. 2008, 59, 854–857. [Google Scholar] [CrossRef]
- Liao, T.; Wang, J.; Zhou, Y. First-principles investigation of intrinsic defects and (N, O) impurity atom stimulated Al vacancy in Ti2AlC. Appl. Phys. Lett. 2008, 93, 261911. [Google Scholar] [CrossRef]
- Music, D.; Ahuja, R.; Schneider, J.M. Theoretical study of nitrogen vacancies in Ti4AlN3. Appl. Phys. Lett. 2005, 86, 031911. [Google Scholar] [CrossRef]
- Jie, T.; Han, H.; Darshana, W.; Wenguan, L.; Mingwen, Z.; Ping, H. A comparative first-principles study of the electronic, mechanical, defect and acoustic properties of Ti2AlC and Ti3AlC. J. Phys. D Appl. Phys. 2014, 47, 215301. [Google Scholar]
- Du, Y.L.; Sun, Z.M.; Hashimoto, H.; Tian, W.B. First-Principles Study of Carbon Vacancy in Ta4AlC3. Mater. Trans. 2008, 49, 1934–1936. [Google Scholar] [CrossRef]
- Han, H.; Wickramaratne, D.; Huang, Q.; Dai, J.; Li, T.; Wang, H.; Zhang, W.; Huai, P. A first-principles study on the defective properties of MAX phase Cr2AlC: The magnetic ordering and strong correlation effect. RSC Adv. 2016, 6, 84262–84268. [Google Scholar] [CrossRef]
- Medvedeva, N.I.; Novikov, D.L.; Ivanovsky, A.L.; Kuznetsov, M.V.; Freeman, A.J. Electronic properties of Ti3SiC2-based solid solutions. Phys. Rev. B 1998, 58, 16042–16050. [Google Scholar] [CrossRef]
- Zhao, S.; Xue, J.; Wang, Y.; Huang, Q. Ab initio study of irradiation tolerance for different Mn+1AXn phases: Ti3SiC2 and Ti3AlC2. J. Appl. Phys. 2014, 115, 023503. [Google Scholar] [CrossRef]
- Liu, X.; Le Flem, M.; Béchade, J.-L.; Onimus, F.; Cozzika, T.; Monnet, I. XRD investigation of ion irradiated Ti3Si0.90Al0.10C2. Nucl. Instrum. Methods B 2010, 268, 506–512. [Google Scholar] [CrossRef]
- Nappé, J.C.; Monnet, I.; Audubert, F.; Grosseau, P.; Beauvy, M.; Benabdesselam, M. Formation of nanosized hills on Ti3SiC2 oxide layer irradiated with swift heavy ions. Nucl. Instrum. Methods B 2012, 270, 36–43. [Google Scholar] [CrossRef]
- Marion, L.F.; Monnet, I. Saturation of irradiation damage in (Ti,Zr)3(Si,Al)C2 compounds. J. Nucl. Mater. 2013, 433, 534–537. [Google Scholar] [CrossRef]
- Song, P.; Sun, J.; Wang, Z.; Cui, M.; Shen, T.; Li, Y.; Pang, L.; Zhu, Y.; Huang, Q.; Lü, J. Irradiation resistance properties studies on helium ions irradiated MAX phase Ti3AlC2. Nucl. Instrum. Methods B 2014, 326, 332–336. [Google Scholar] [CrossRef]
- Zhang, L.; Qi, Q.; Shi, L.Q.; O’Connor, D.J.; King, B.V.; Kisi, E.H.; Venkatachalam, D.K. Damage tolerance of Ti3SiC2 to high energy iodine irradiation. Appl. Surf. Sci. 2012, 258, 6281–6287. [Google Scholar] [CrossRef]
- Huang, Q.; Liu, R.; Lei, G.; Huang, H.; Li, J.; He, S.; Li, D.; Yan, L.; Zhou, J.; Huang, Q. Irradiation resistance of MAX phases Ti3SiC2 and Ti3AlC2: Characterization and comparison. J. Nucl. Mater. 2015, 465, 640–647. [Google Scholar] [CrossRef]
- Wang, C.; Yang, T.; Kong, S.; Xiao, J.; Xue, J.; Wang, Q.; Hu, C.; Huang, Q.; Wang, Y. Effects of He irradiation on Ti3AlC2: Damage evolution and behavior of He bubbles. J. Nucl. Mater. 2013, 440, 606–611. [Google Scholar] [CrossRef]
- Yang, T.; Wang, C.; Taylor, C.A.; Huang, X.; Huang, Q.; Li, F.; Shen, L.; Zhou, X.; Xue, J.; Yan, S.; et al. The structural transitions of Ti3AlC2 induced by ion irradiation. Acta Mater. 2014, 65, 351–359. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Henkelman, G.; Arnaldsson, A.; Jónsson, H. A fast and robust algorithm for bader decomposition of charge density. Comp. Mater. Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]
- Huang, Q.; Han, H.; Liu, R.; Lei, G.; Yan, L.; Zhou, J.; Huang, Q. Saturation of ion irradiation effects in MAX phase Cr2AlC. Acta Mater. 2016, 110, 1–7. [Google Scholar] [CrossRef]
- Pack, J.D.; Monkhorst, H.J. “Special points for Brillouin-zone integrations”—A reply. Phys. Rev. B 1977, 16, 1748–1749. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- Henkelman, G.; Jónsson, H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. J. Chem. Phys. 2000, 113, 9978–9985. [Google Scholar] [CrossRef]
- Onodera, A.; Hirano, H.; Yuasa, T.; Gao, N.F.; Miyamoto, Y. Static compression of Ti3SiC2 to 61 GPa. Appl. Phys. Lett. 1999, 74, 3782–3784. [Google Scholar] [CrossRef]
- Hill, R. The Elastic Behaviour of a Crystalline Aggregate. Proc. Phys. Soc. A 1952, 65, 349. [Google Scholar] [CrossRef]
- Han, H.; Cheng, C.; Xiong, X.-G.; Su, J.; Dai, J.-X.; Wang, H.; Yin, G.; Huai, P. Piezoelectric, Mechanical and Acoustic Properties of KNaNbOF5 from First-Principles Calculations. Materials 2015, 8, 8578–8589. [Google Scholar] [CrossRef]
- Han, H.; Yin, G.; Wickramaratne, D. A first-principles investigation of the electronic, elastic, piezoelectric and acoustic properties of K3B6O10Cl. Comput. Mater. Sci. 2013, 69, 81–86. [Google Scholar] [CrossRef]
- Pietzka, M.A.; Schuster, J.C. Summary of Constitutional Data on the Aluminum-Carbon-Titanium System. J. Phase Equilib. 1994, 15, 392–400. [Google Scholar] [CrossRef]
- Kornblit, L.; Pelleg, J.; Rabinovitch, A. Self-diffusion calculation for fcc metals. Phys. Rev. B 1977, 16, 1164–1167. [Google Scholar] [CrossRef]
- García Ortega, M.; Ramos de Debiaggi, S.; Monti, A. Self-Diffusion in FCC Metals: Static and Dynamic Simulations in Aluminium and Nickel. Phys. Stat. Sol. (b) 2002, 234, 506–521. [Google Scholar] [CrossRef]
- Atkinson, A.; Taylor, R.I. The diffusion of Ni in the bulk and along dislocations in NiO single crystals. Philos. Mag. A 1979, 39, 581–595. [Google Scholar] [CrossRef]
- Debiaggi, S.; Decorte, P.; Monti, A. Diffusion by Vacancy Mechanism in Ni, Al, and Ni3Al: Calculation Based on Many-Body Potentials. Phys. Stat. Sol. (b) 1996, 195, 37–54. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | Method | a (Å) | c (Å) | c/a | zTi | zC |
|---|---|---|---|---|---|---|
| Ti3SiC2 | Cal. | 3.075 | 17.734 | 5.767 | 0.135 | 0.572 |
| Exp. [42] | 3.07 | 17.67 | 5.76 | 0.135 | 0.568 | |
| Ti3AlC2 | Cal. | 3.082 | 18.642 | 0.648 | 0.127 | 0.569 |
| Exp. [6] | 3.075 | 18.578 | 0.641 | 0.128 | 0.564 |
| Properties | B (GPa) | G (GPa) | E (GPa) | v |
| Cal. | 200.3 | 132.3 | 325.2 | 0.23 |
| Exp. | 187 | 142 | 339 | 0.2 |
| Properties | Vl (Km/s) | Vt (Km/s) | Va (Km/s) | ΘD (K) |
| Cal. | 9.17 | 5.43 | 6.0 | 780 |
| Exp. | 9.14 | 5.61 | 6.2 | 804 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Han, H.; Yin, G.; Wang, C.-Y.; Hou, Y.-Y.; Tang, J.; Dai, J.-X.; Ren, C.-L.; Zhang, W.; Huai, P. First-Principles Study of Vacancies in Ti3SiC2 and Ti3AlC2. Materials 2017, 10, 103. https://doi.org/10.3390/ma10020103
Wang H, Han H, Yin G, Wang C-Y, Hou Y-Y, Tang J, Dai J-X, Ren C-L, Zhang W, Huai P. First-Principles Study of Vacancies in Ti3SiC2 and Ti3AlC2. Materials. 2017; 10(2):103. https://doi.org/10.3390/ma10020103
Chicago/Turabian StyleWang, Hui, Han Han, Gen Yin, Chang-Ying Wang, Yu-Yang Hou, Jun Tang, Jian-Xing Dai, Cui-Lan Ren, Wei Zhang, and Ping Huai. 2017. "First-Principles Study of Vacancies in Ti3SiC2 and Ti3AlC2" Materials 10, no. 2: 103. https://doi.org/10.3390/ma10020103
APA StyleWang, H., Han, H., Yin, G., Wang, C.-Y., Hou, Y.-Y., Tang, J., Dai, J.-X., Ren, C.-L., Zhang, W., & Huai, P. (2017). First-Principles Study of Vacancies in Ti3SiC2 and Ti3AlC2. Materials, 10(2), 103. https://doi.org/10.3390/ma10020103