
Crystallization of Polymers Investigated by Temperature-Modulated DSC


{kind=link}
{kind=link}
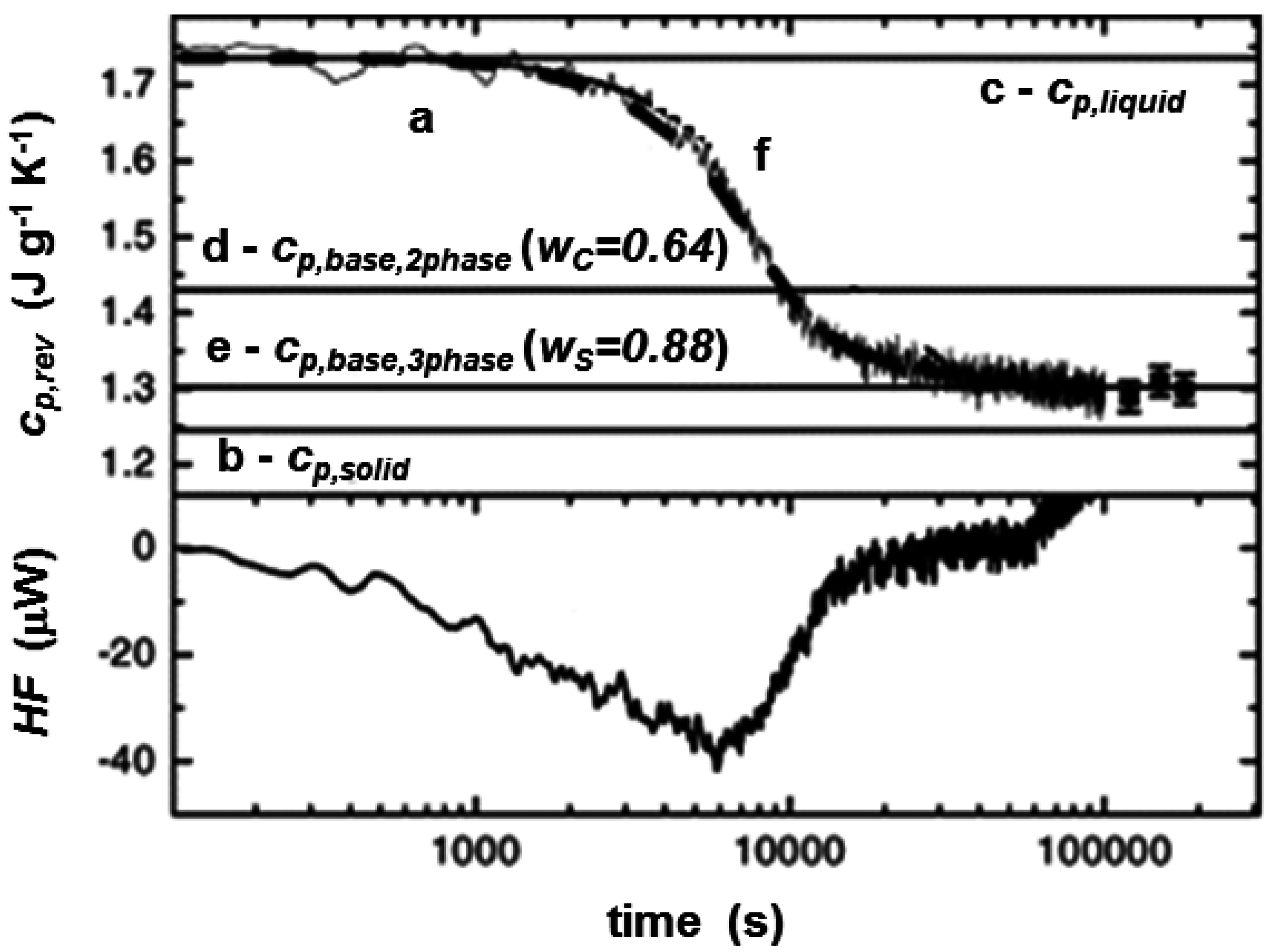{kind=link}
{kind=link}
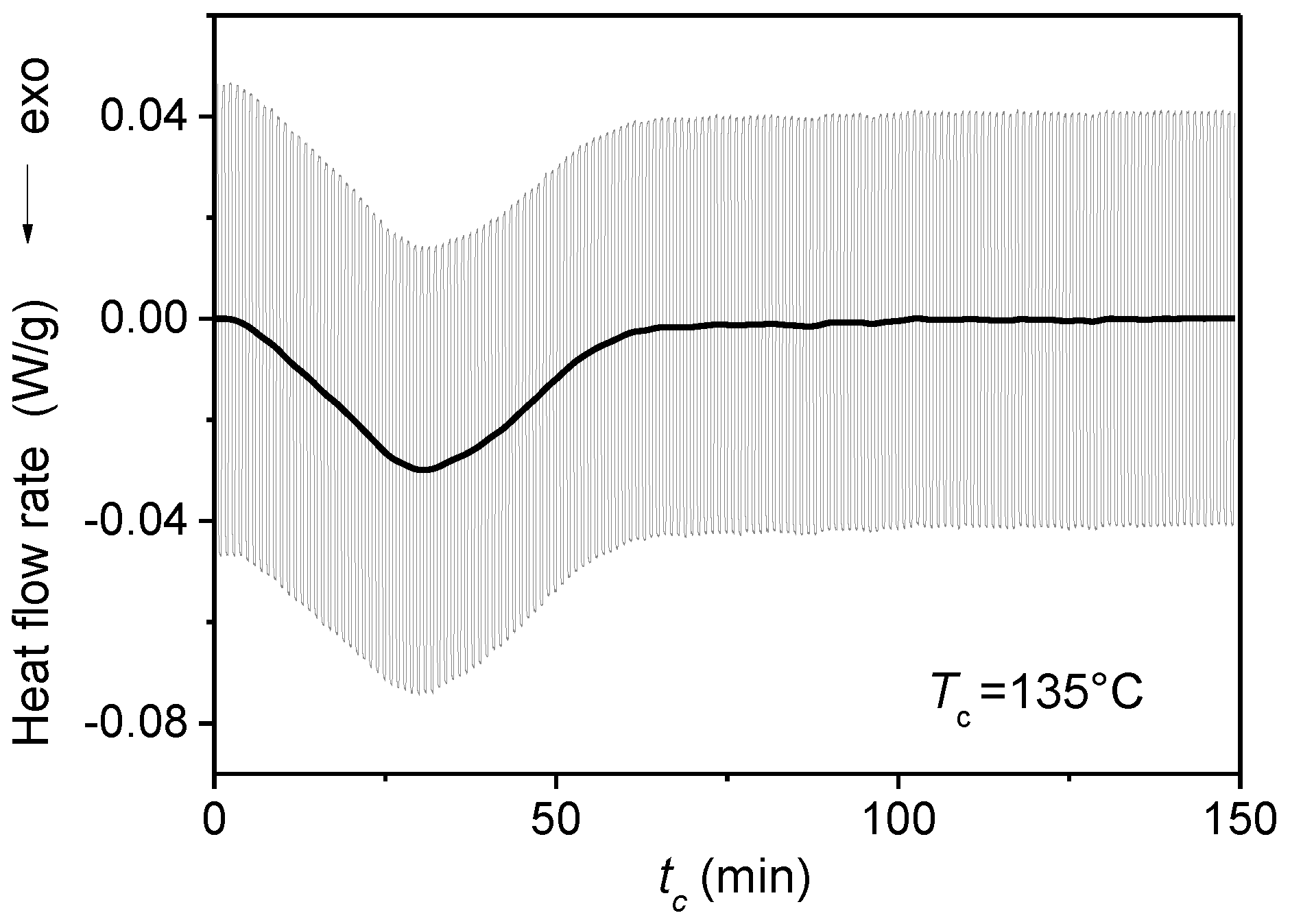{kind=link}
{kind=link}
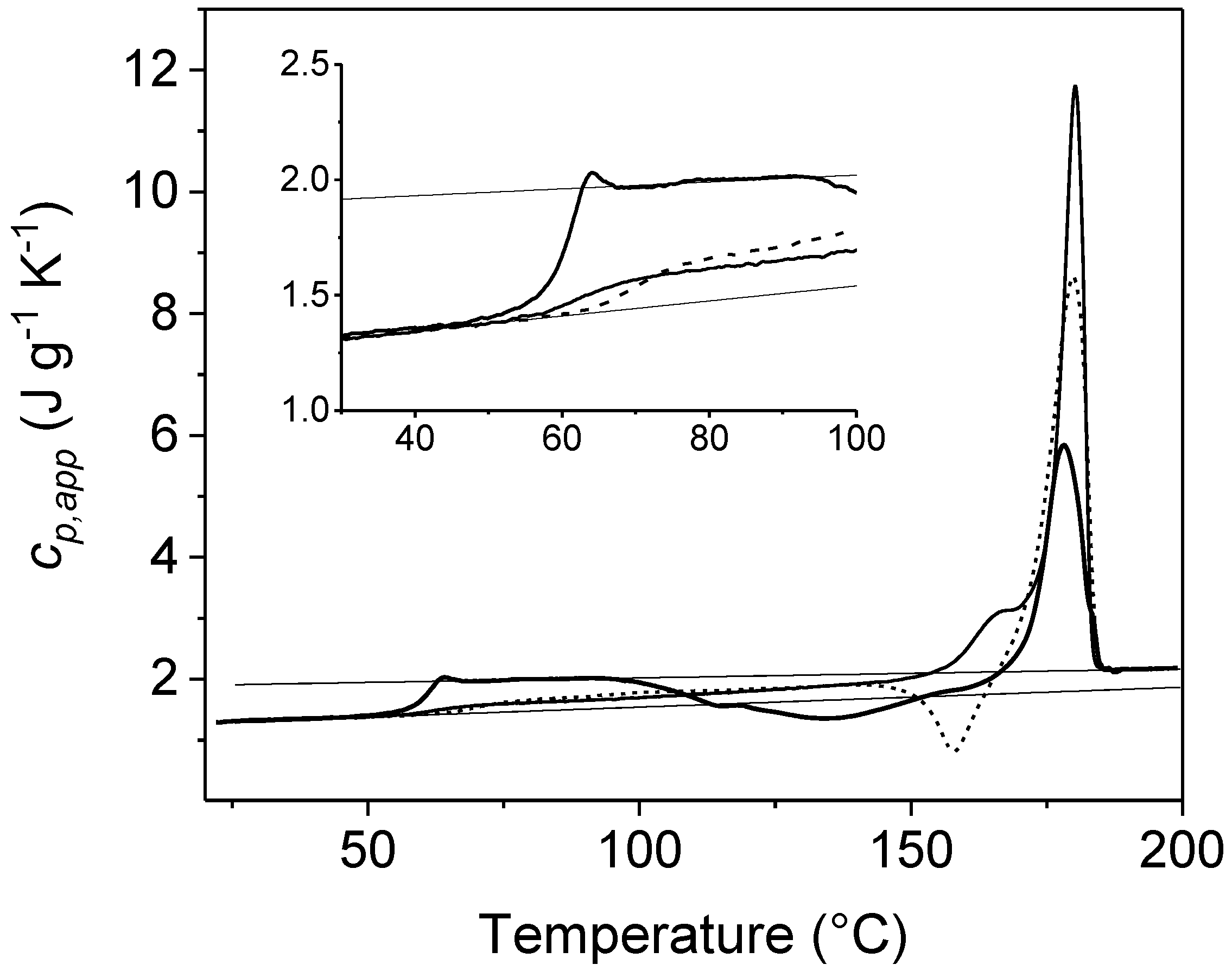{kind=link}
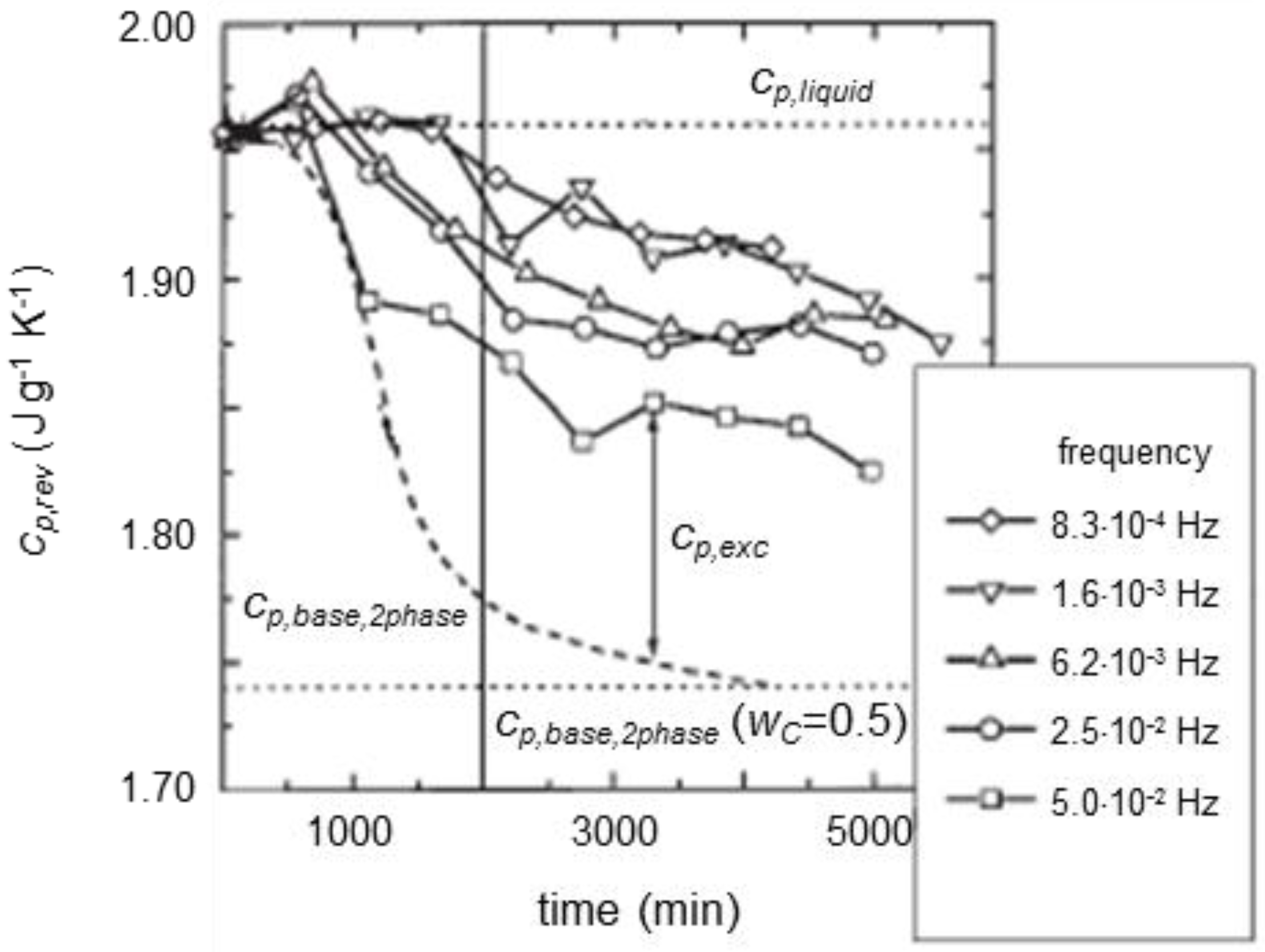{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
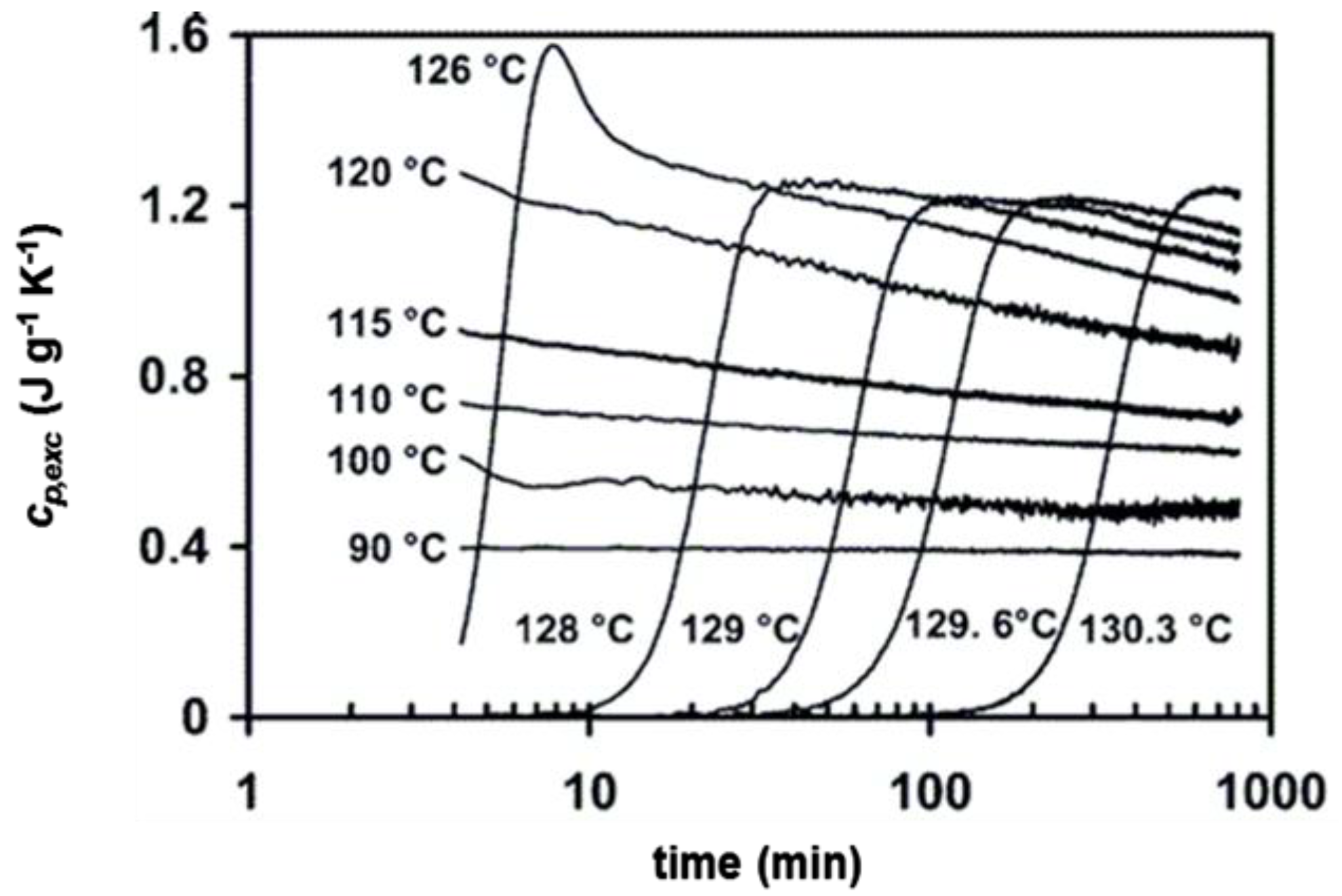
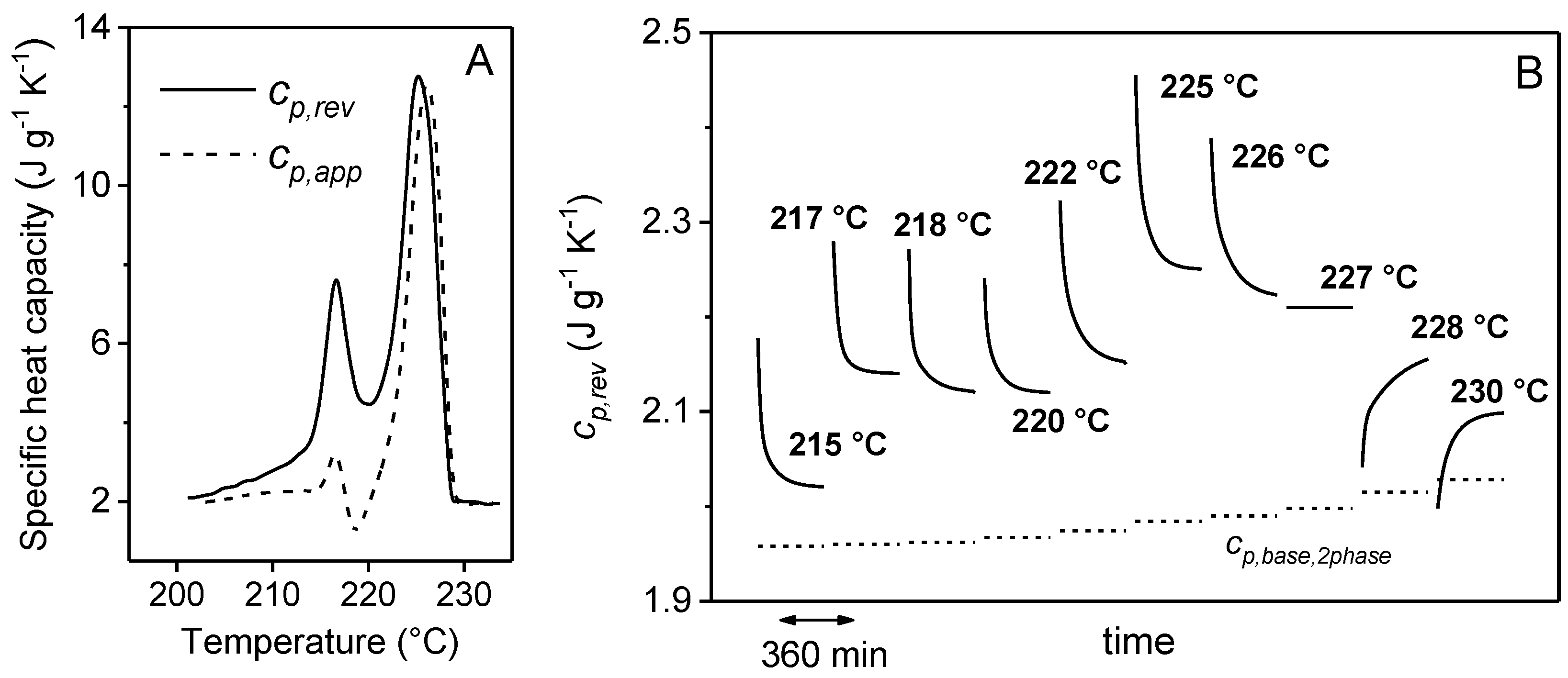
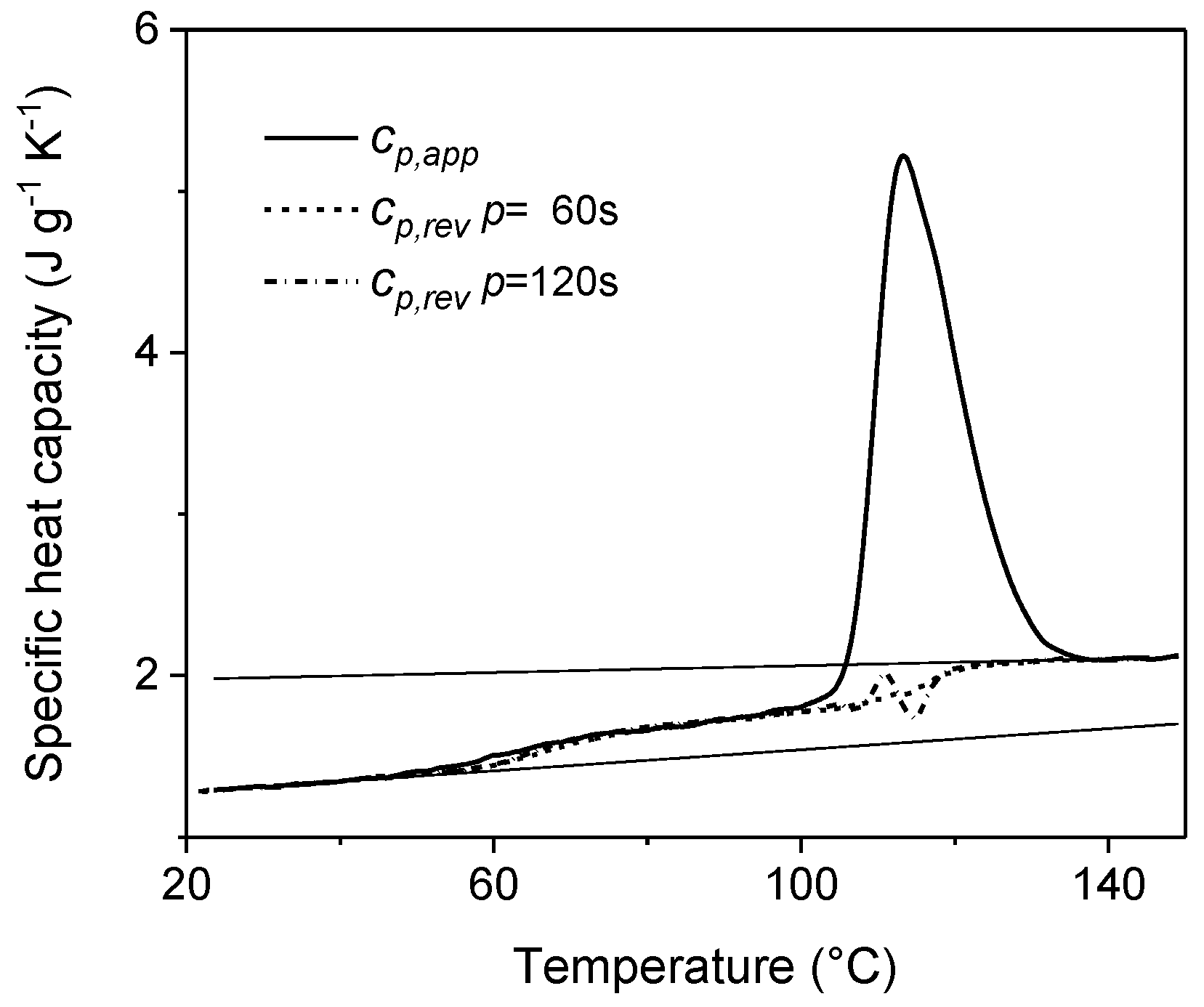
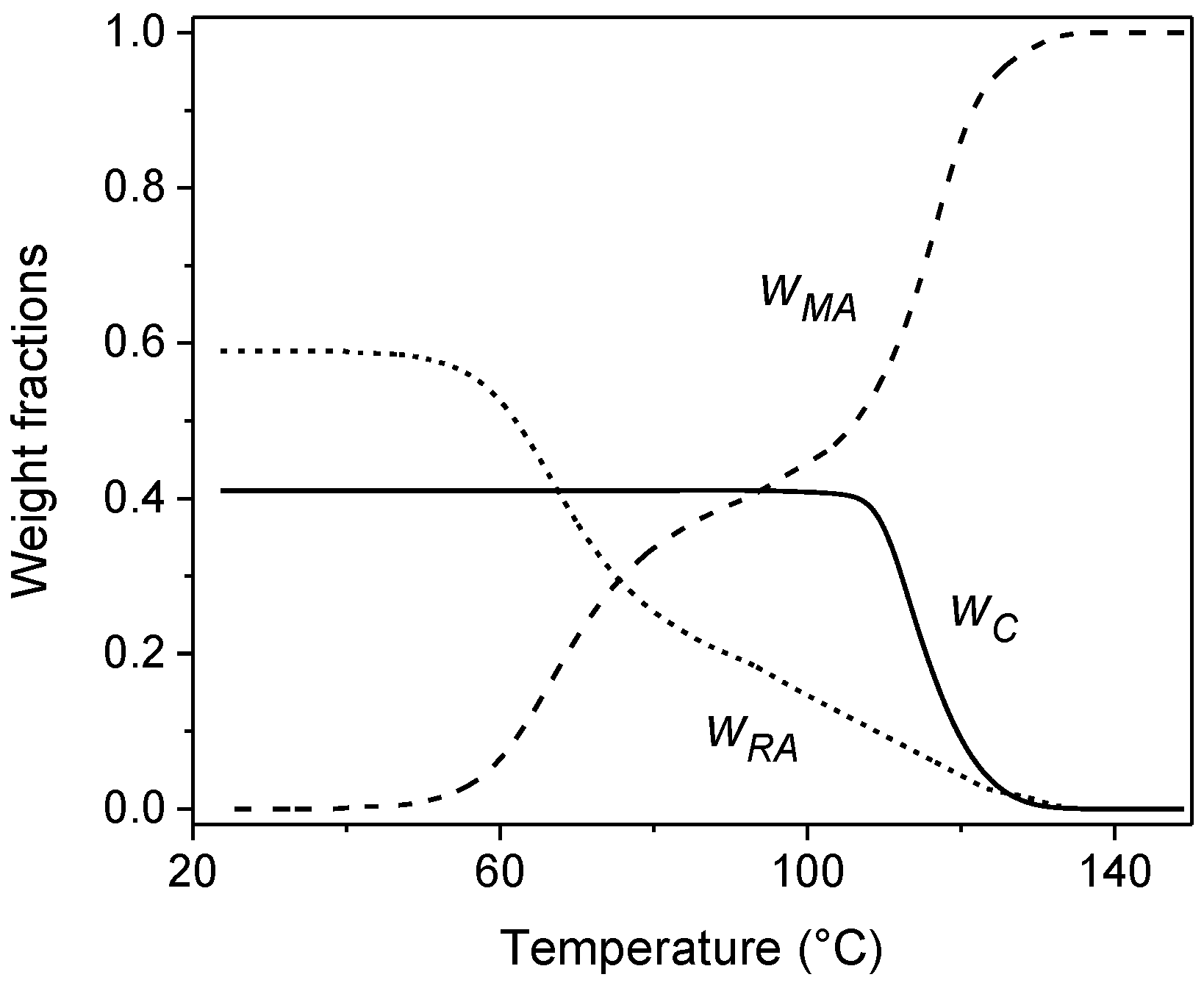
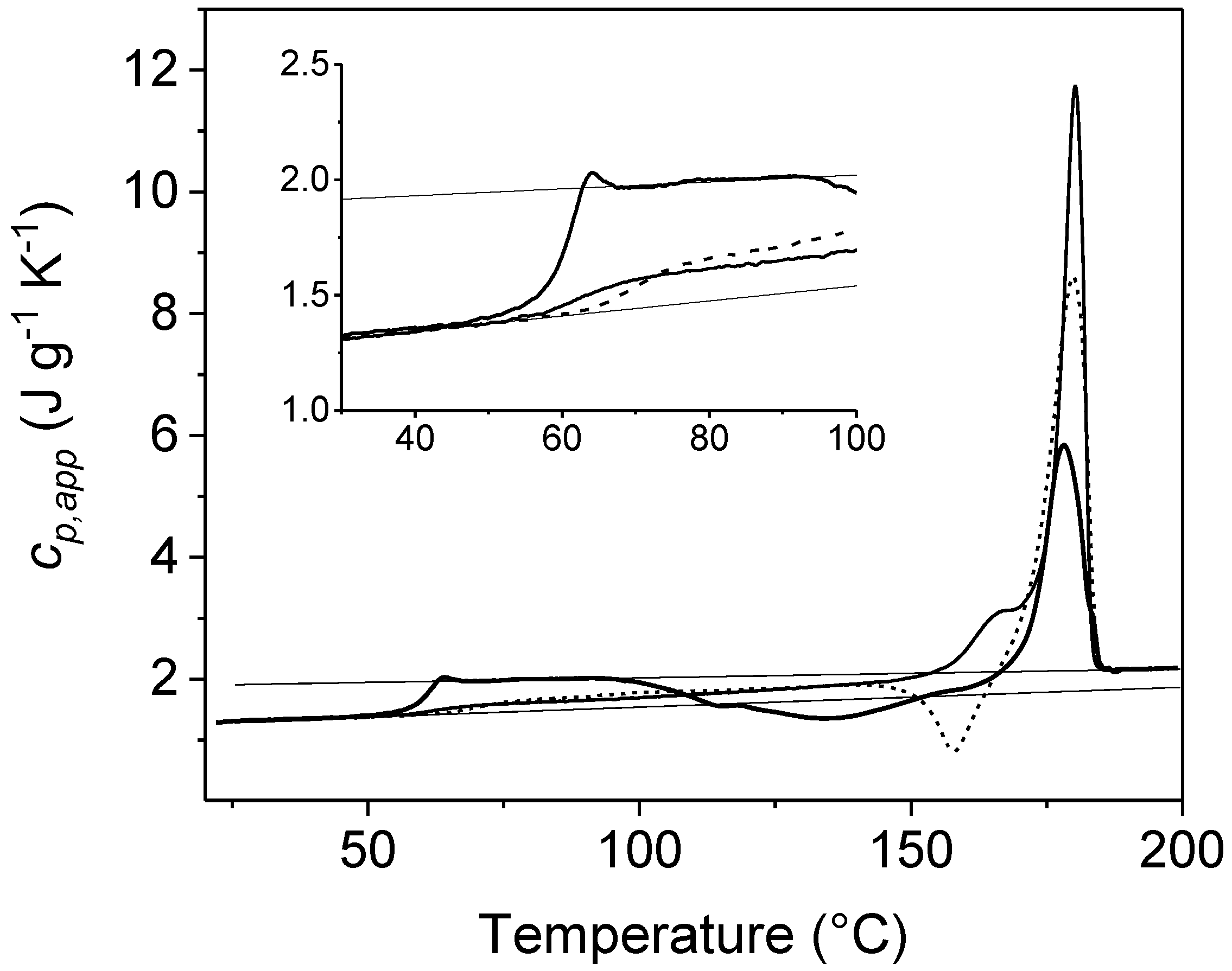
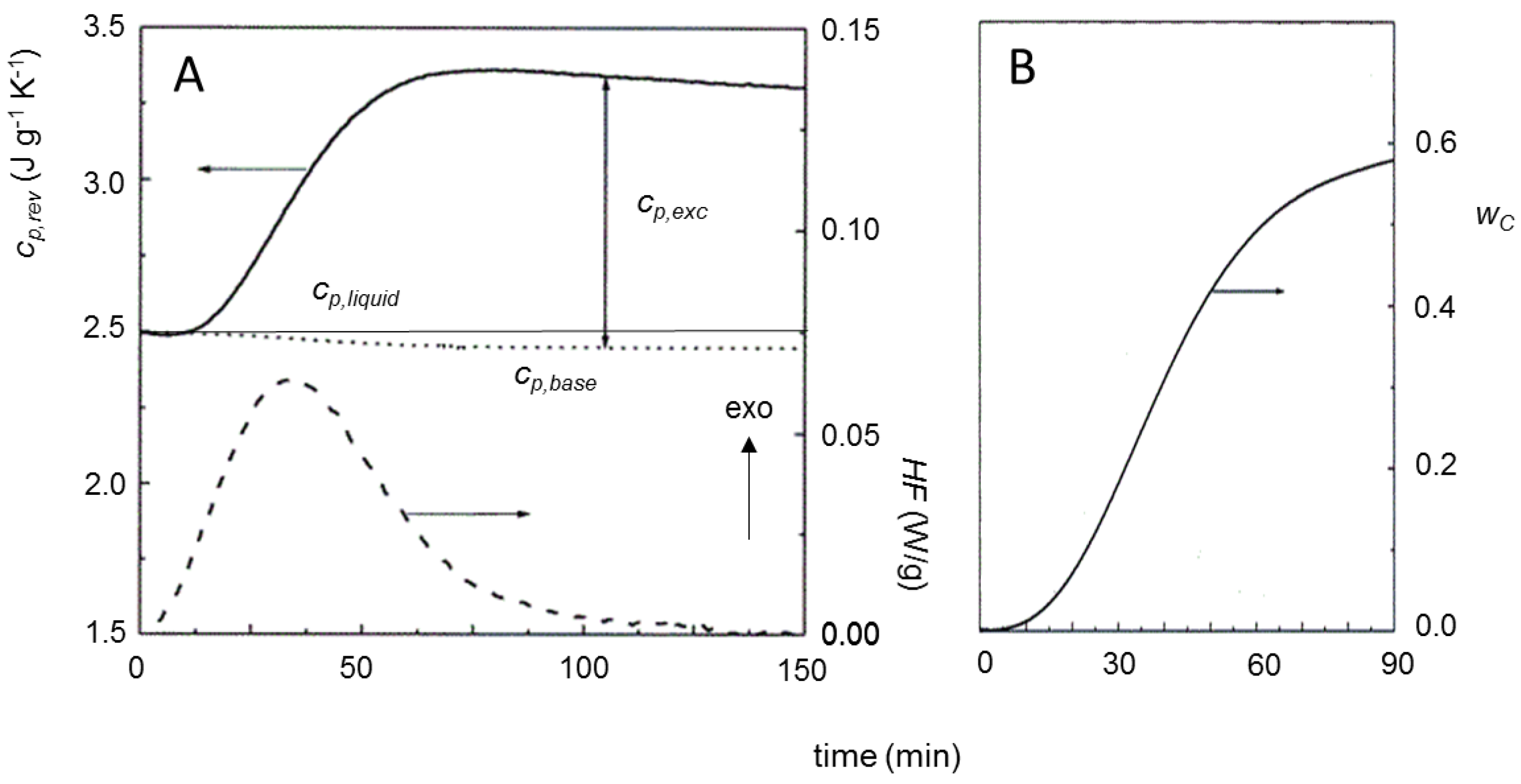
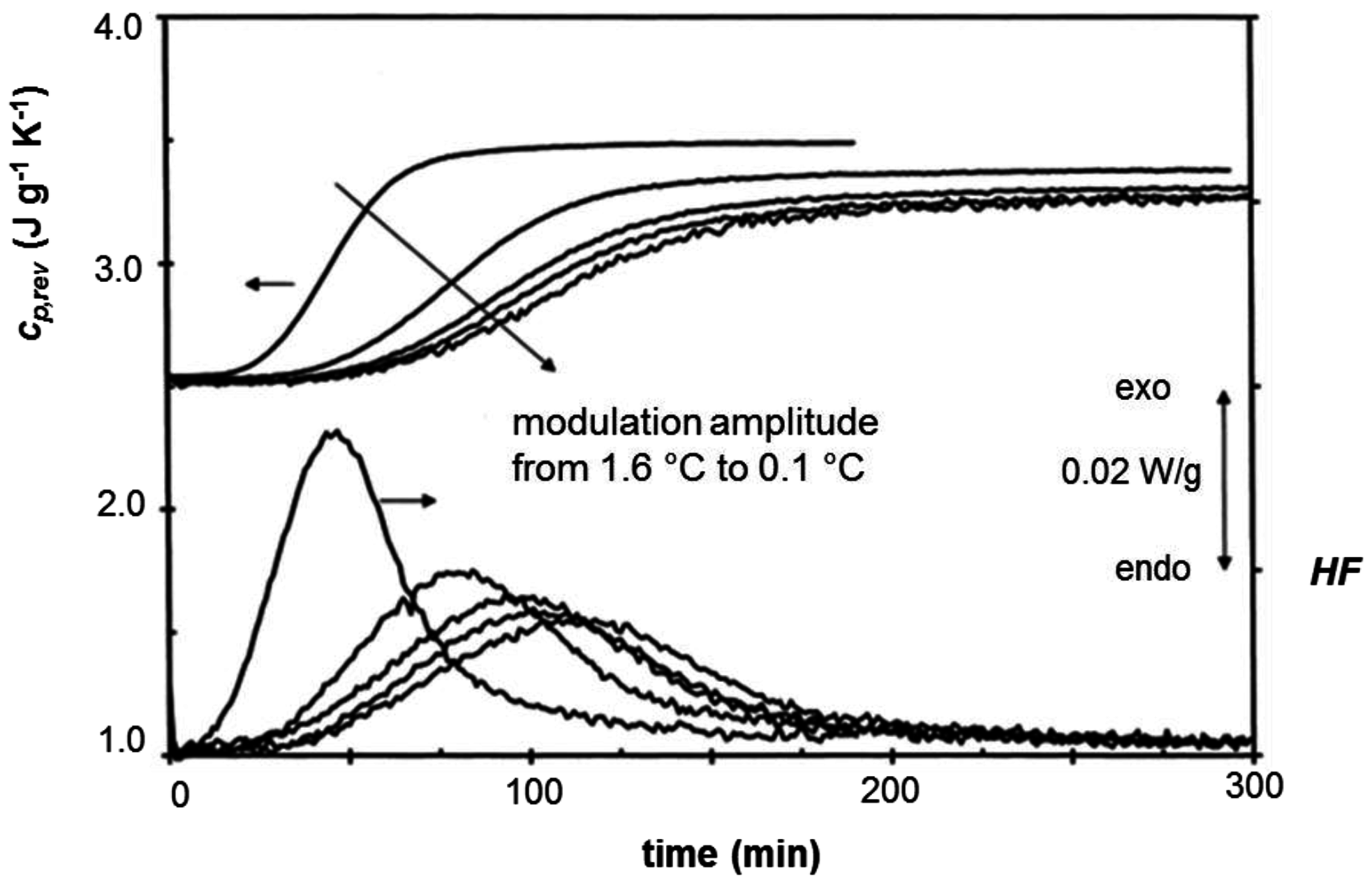
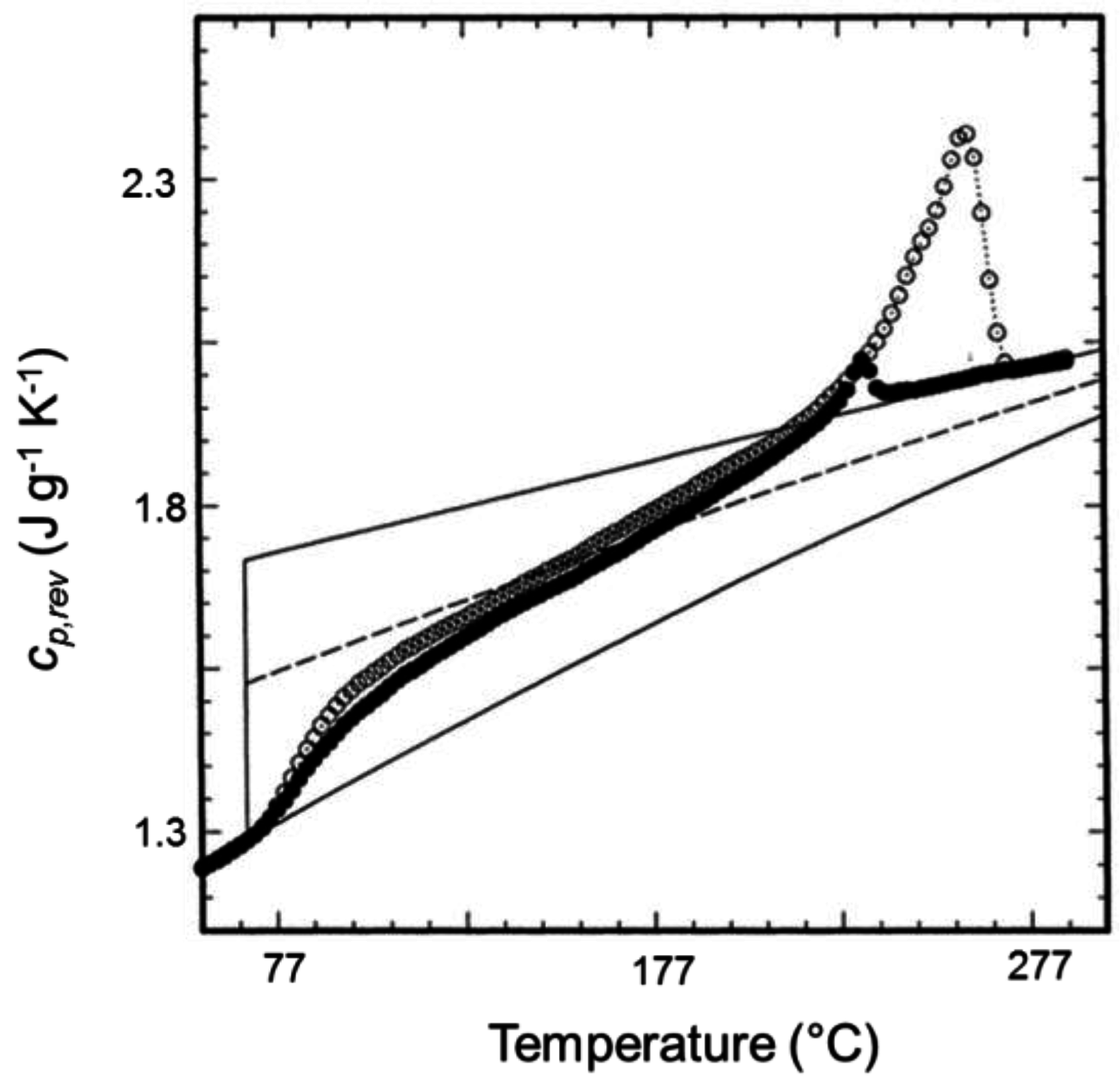
2. Non-Isothermal Crystallization and Stepwise Quasi-Isothermal Crystallization upon Heating
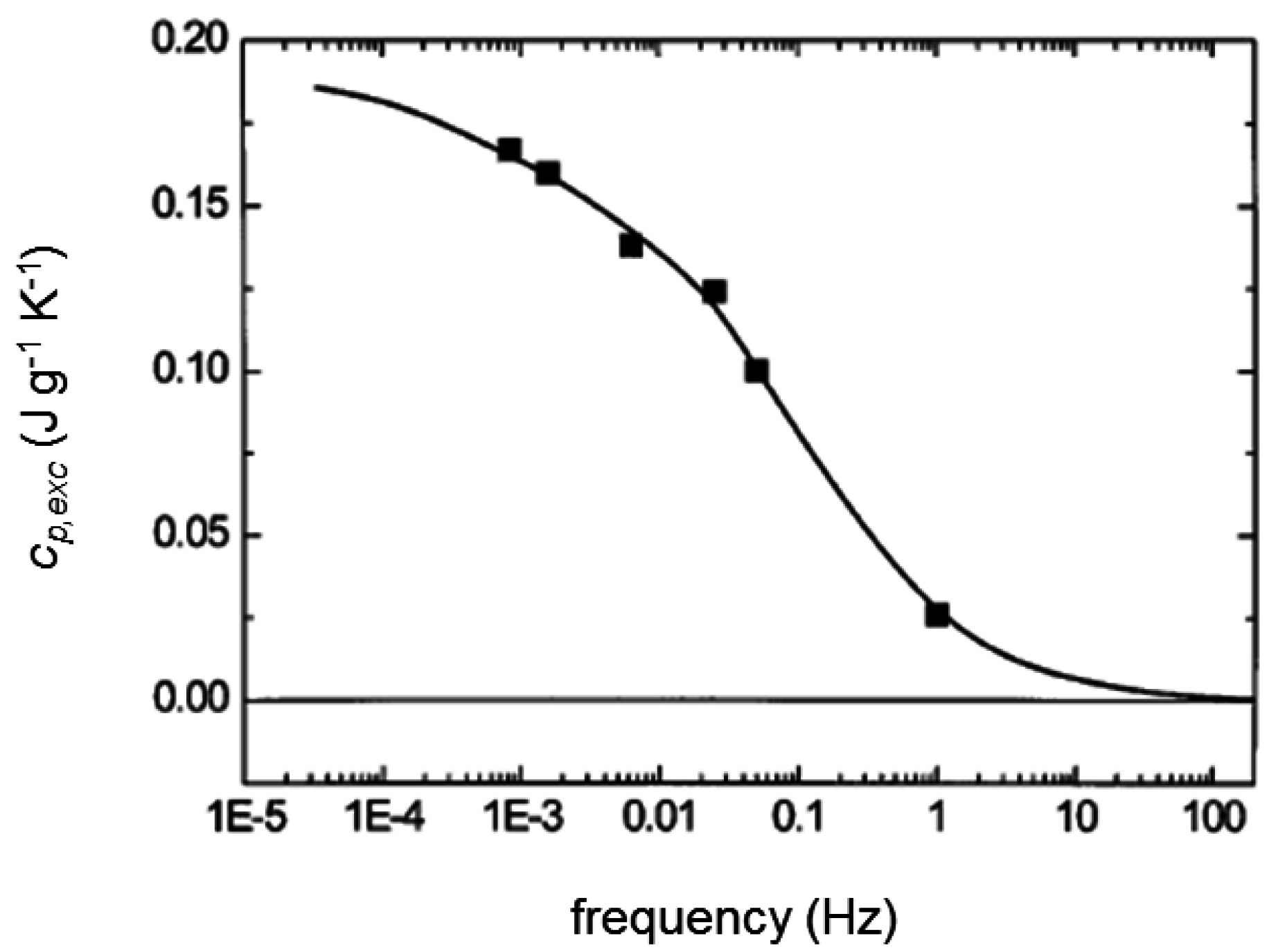
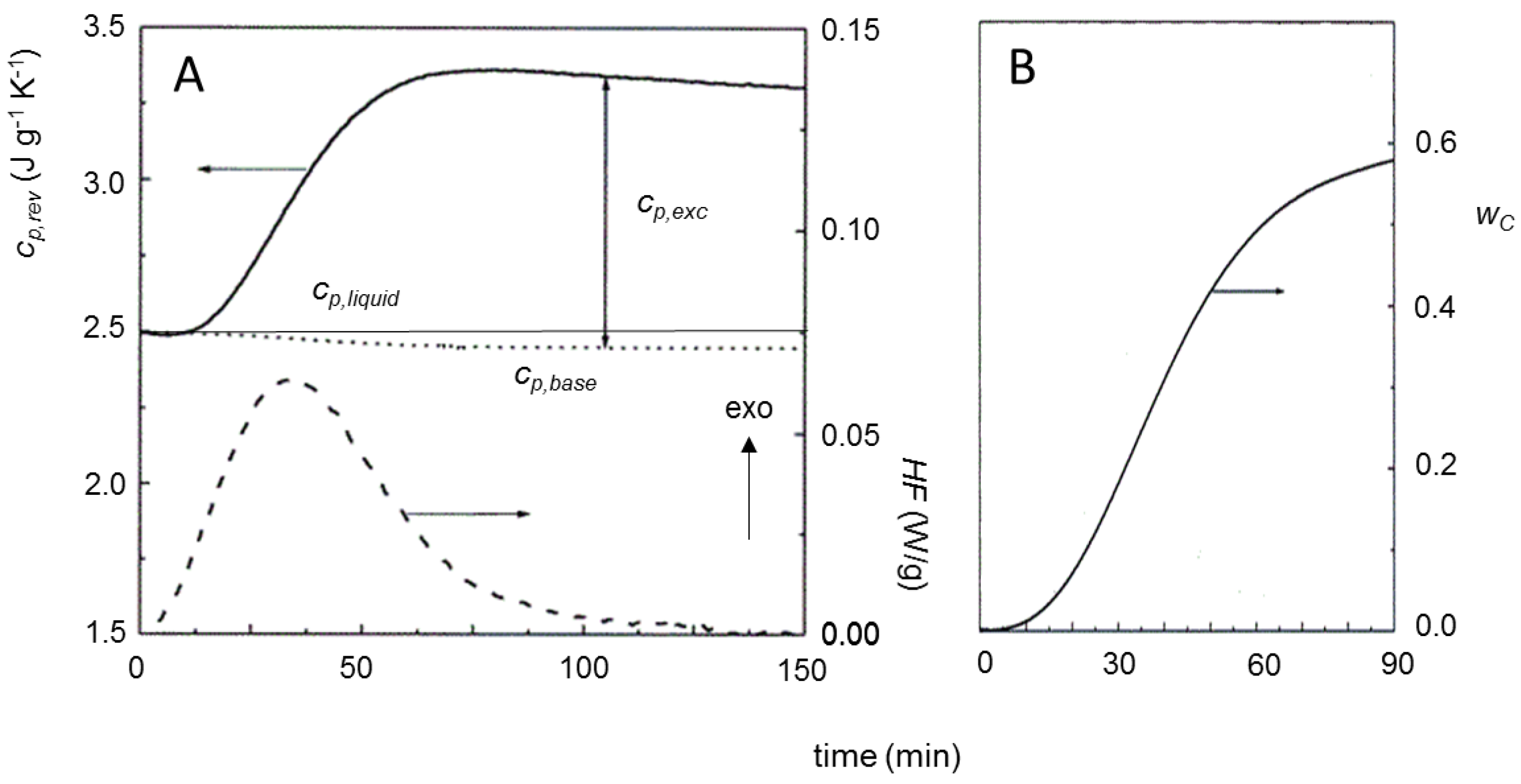
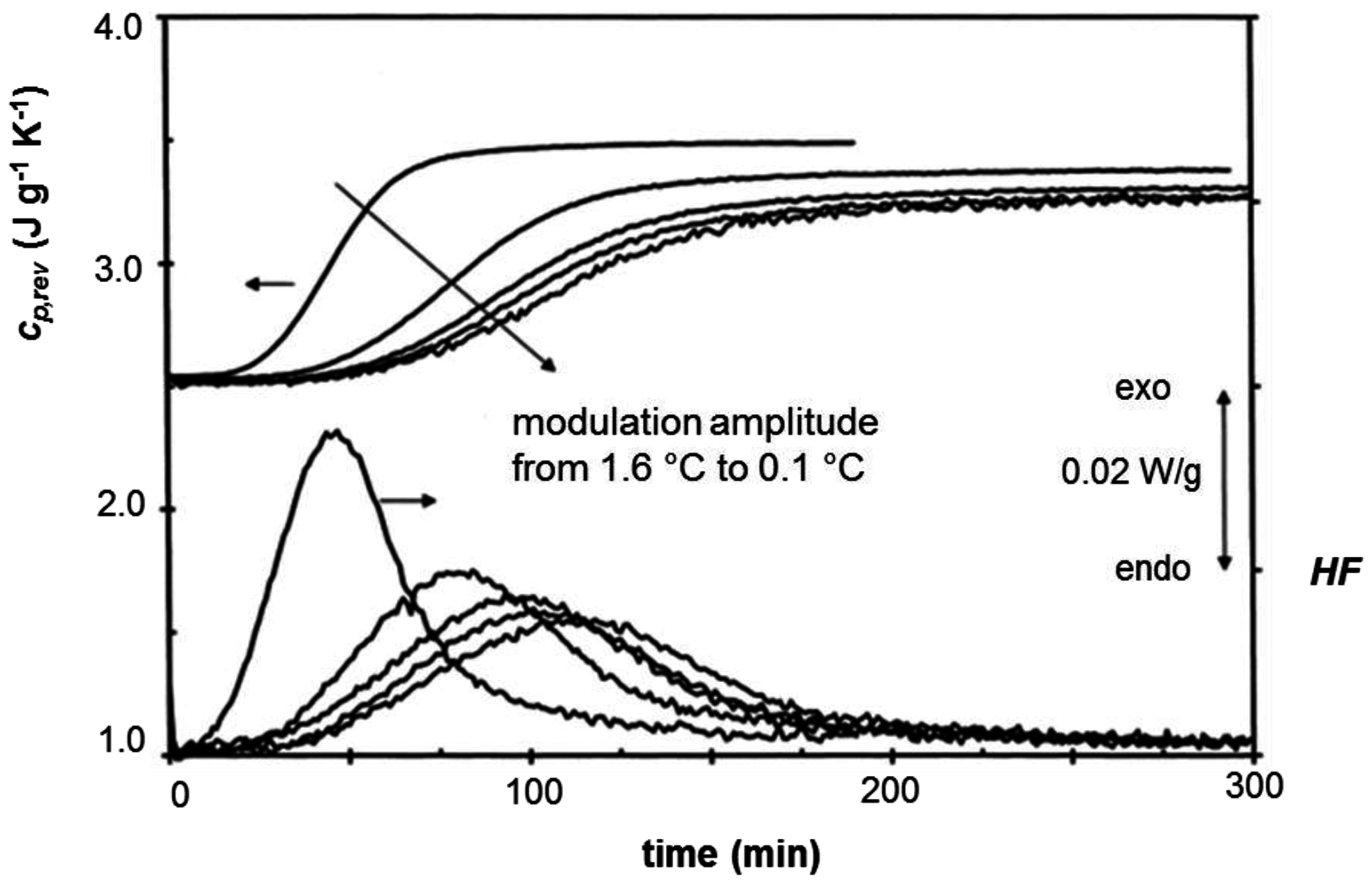
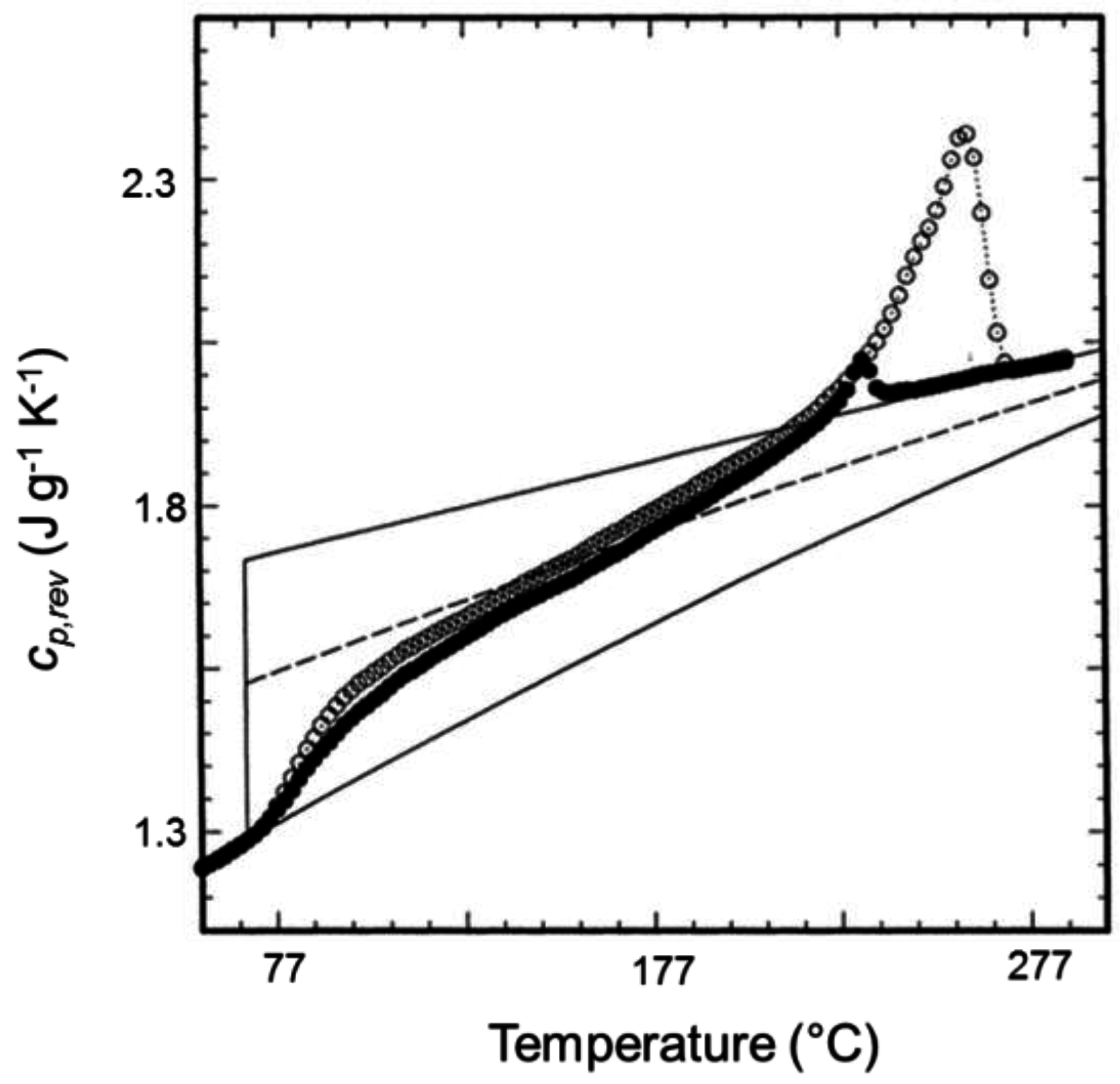
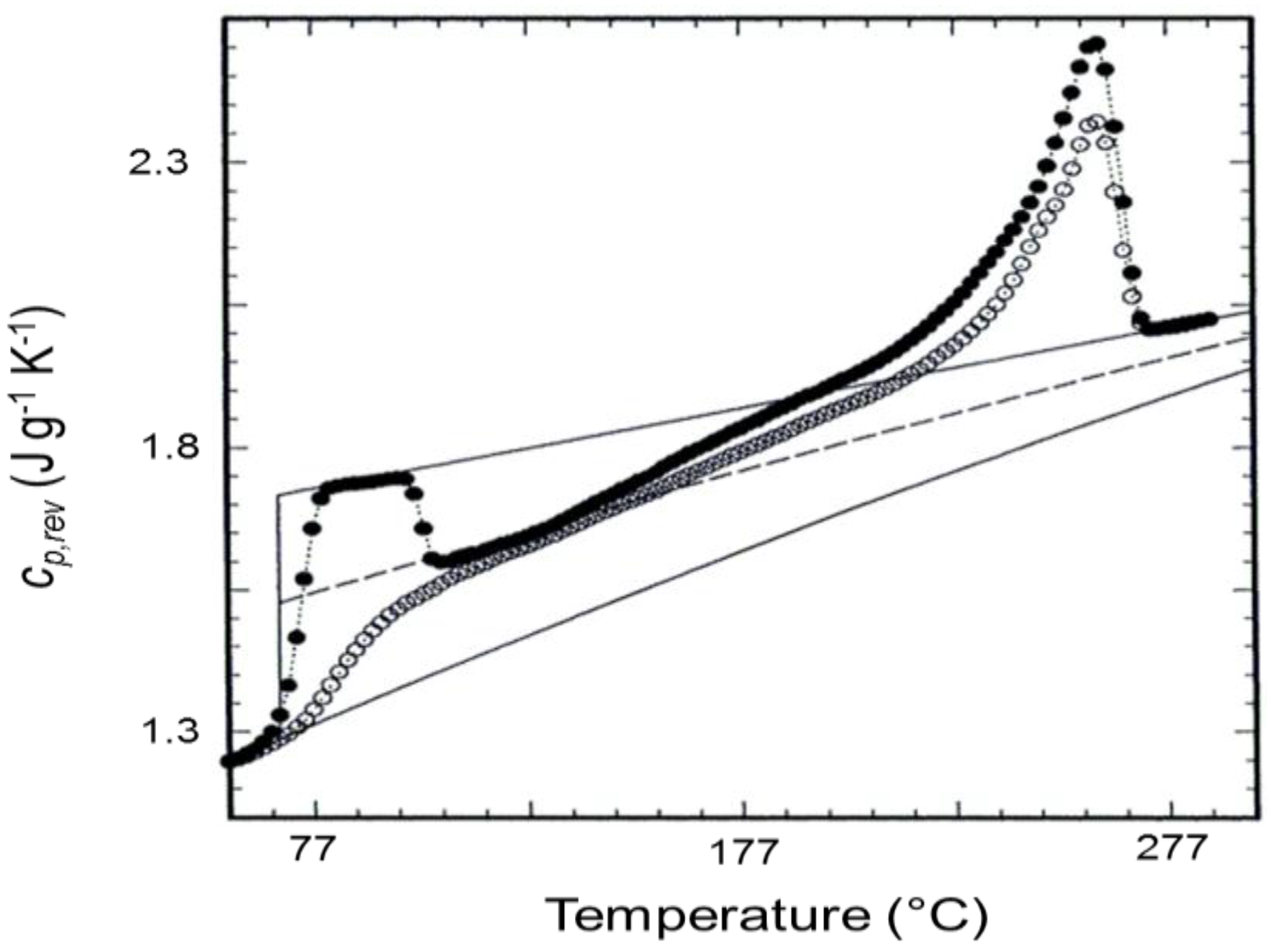
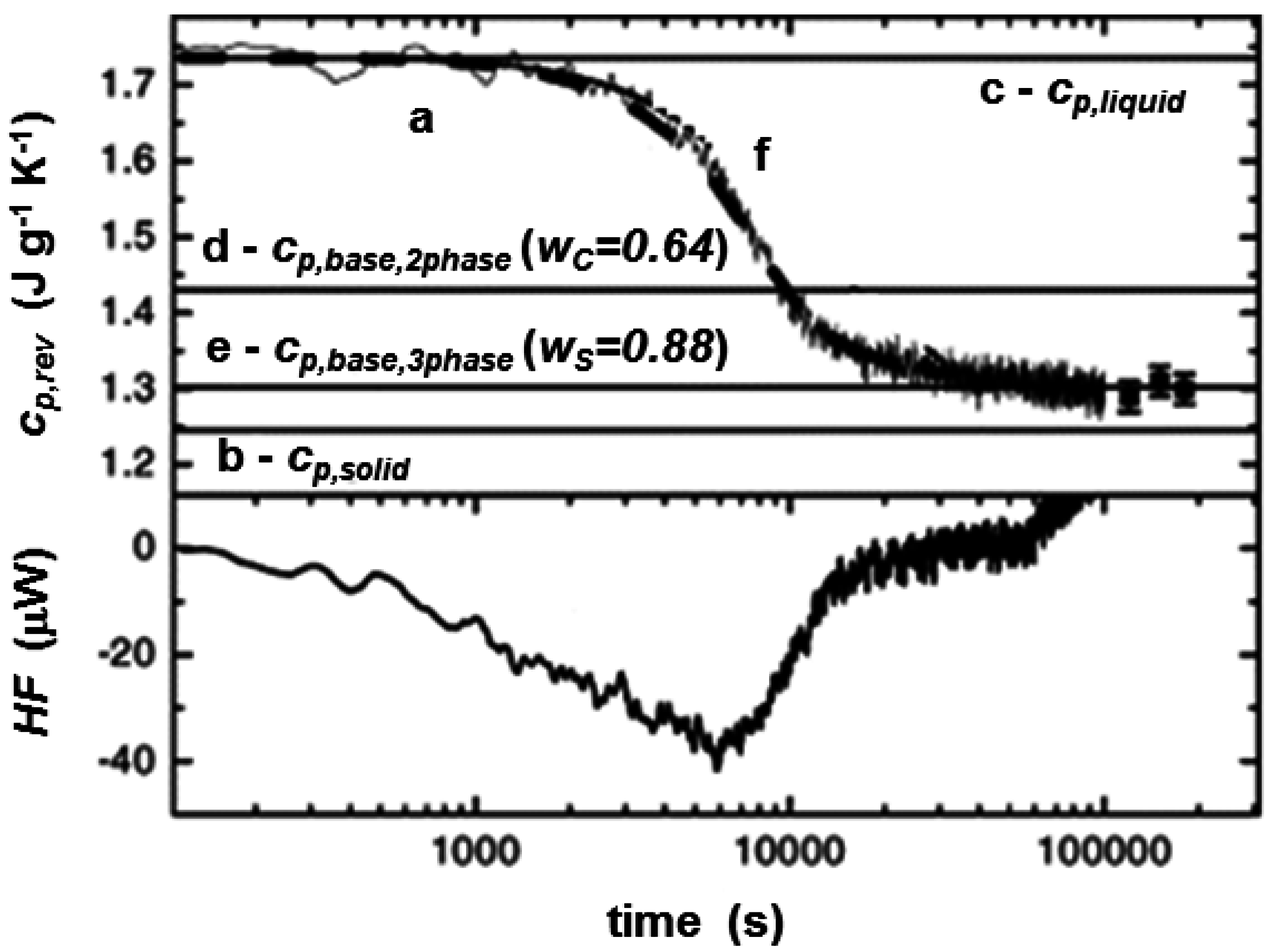
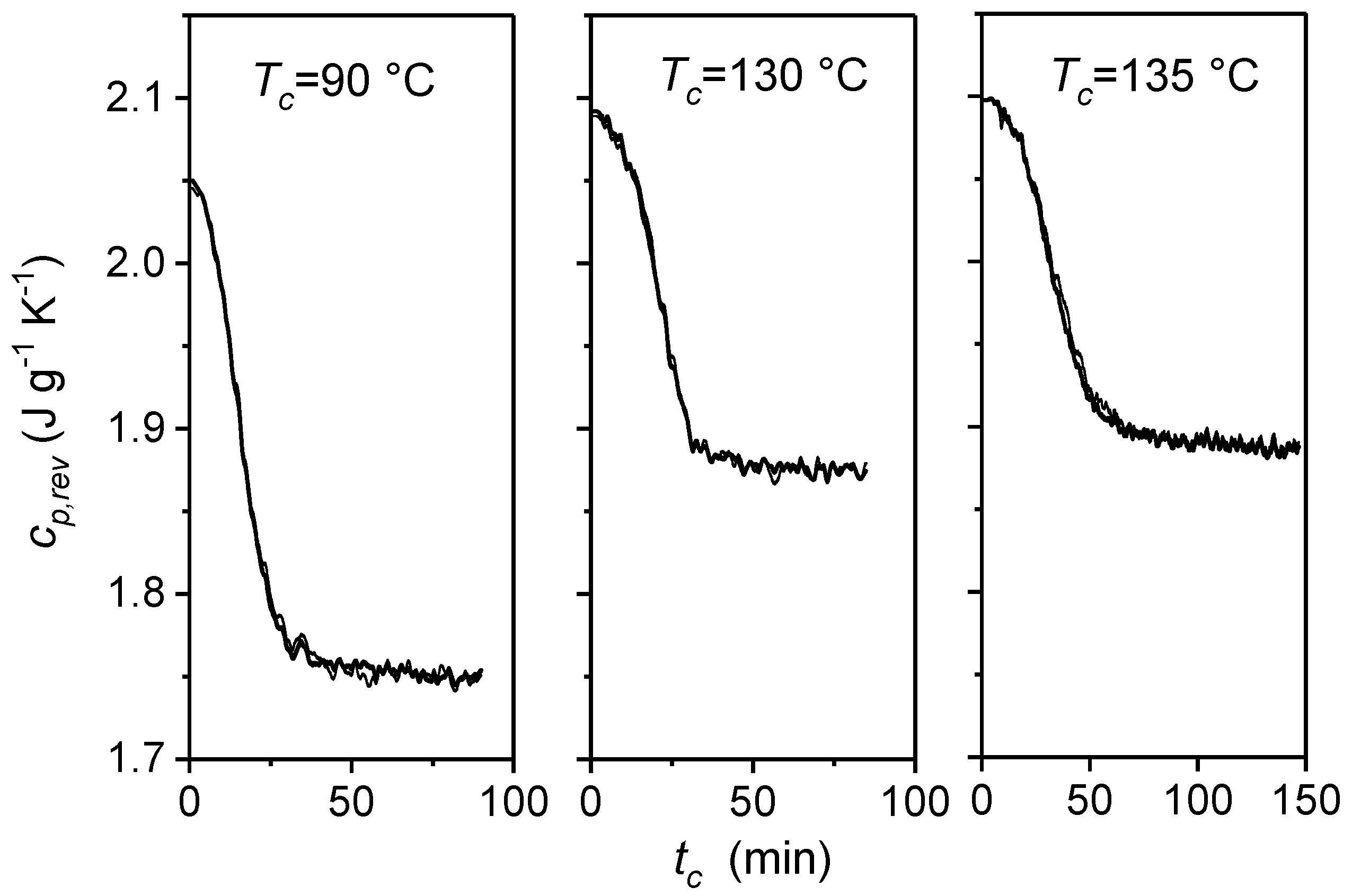
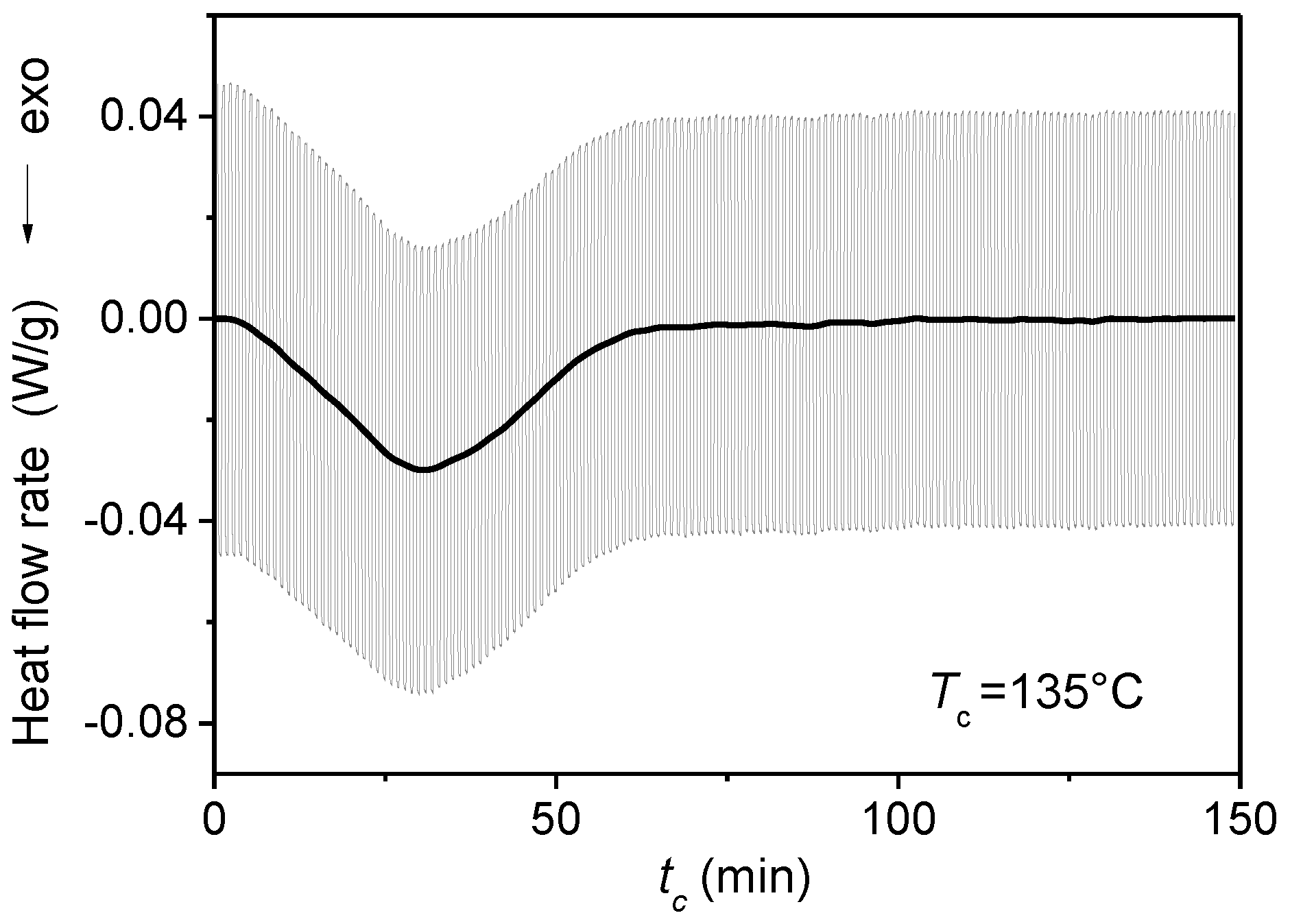
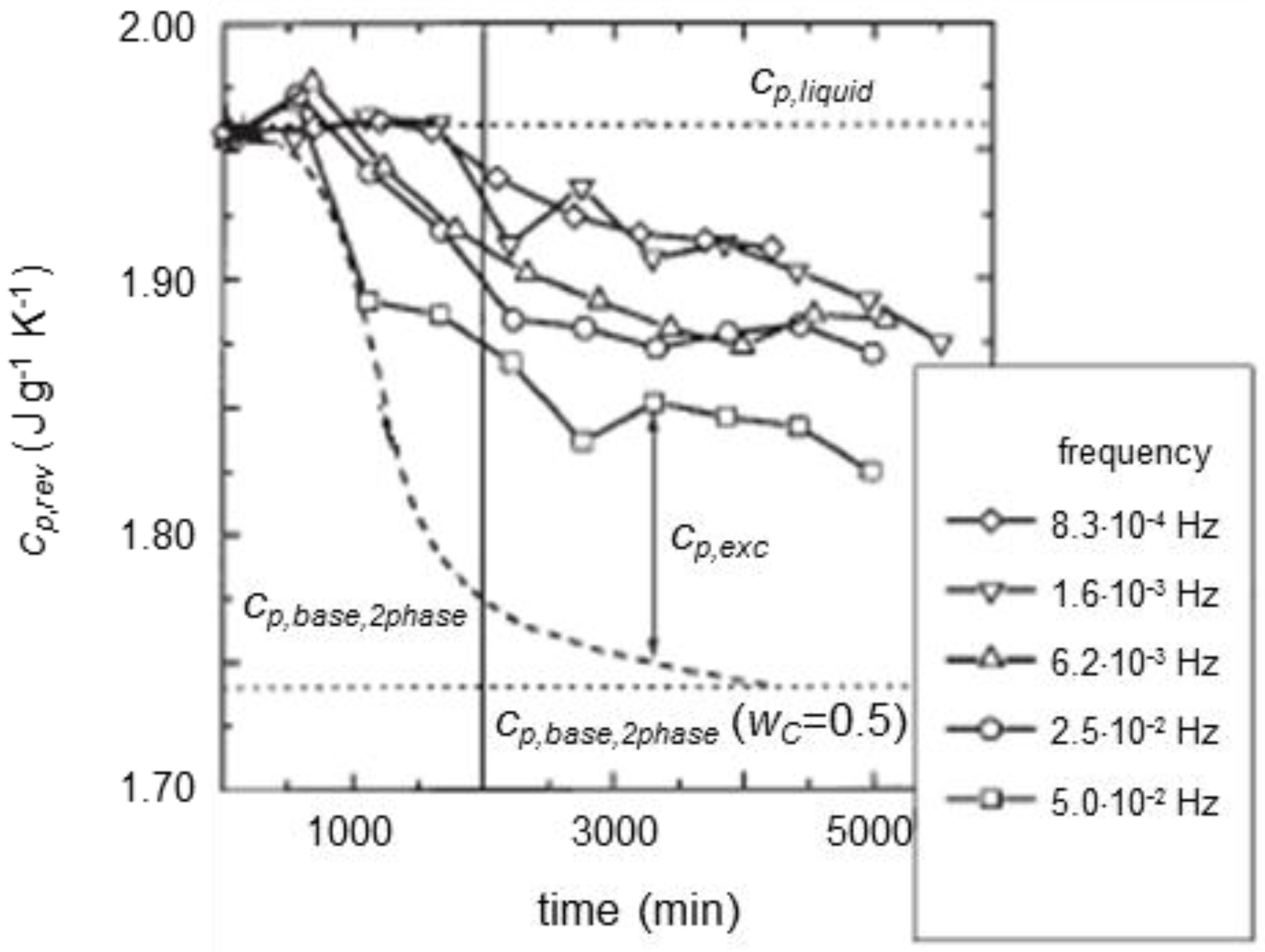
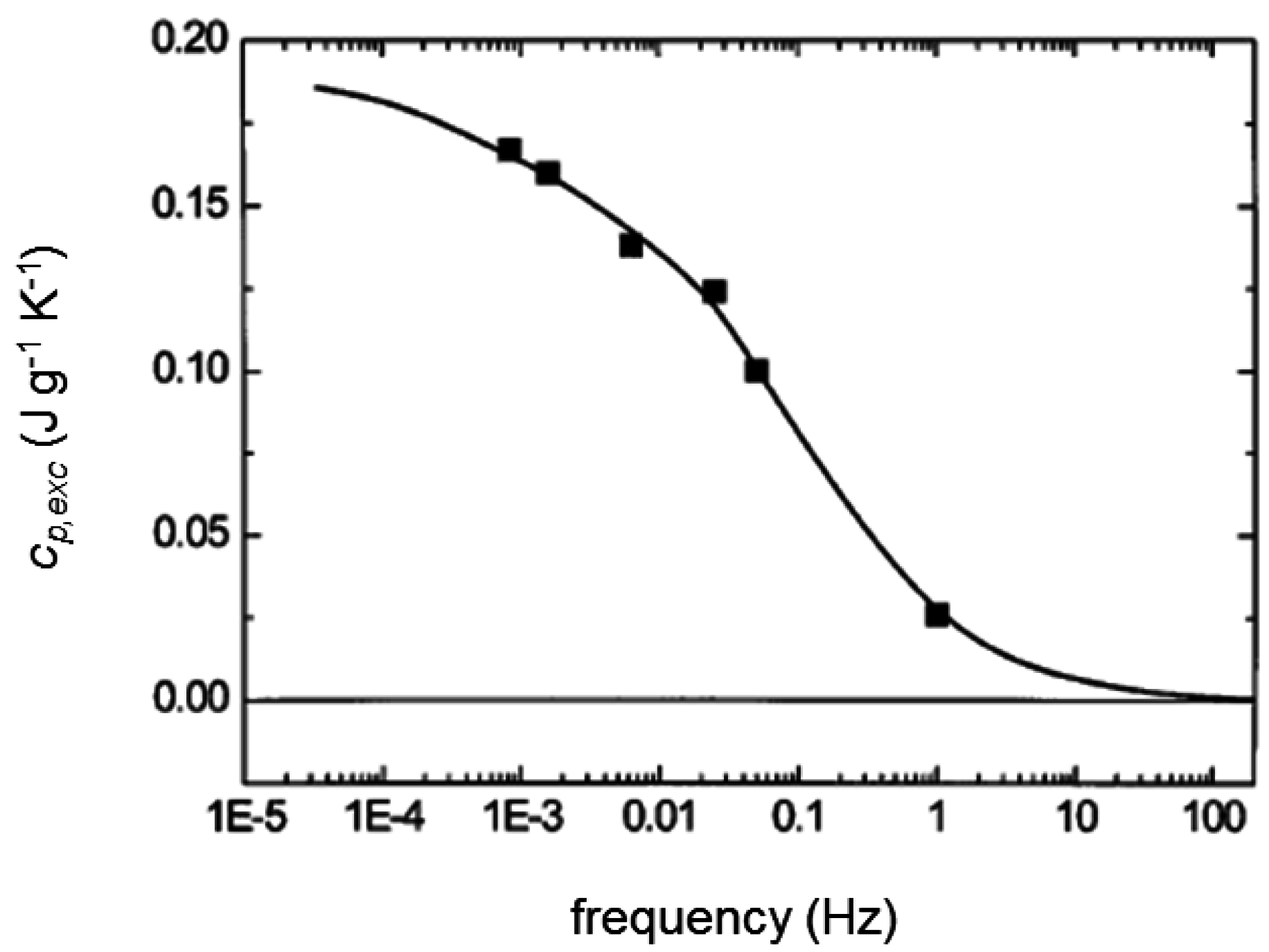
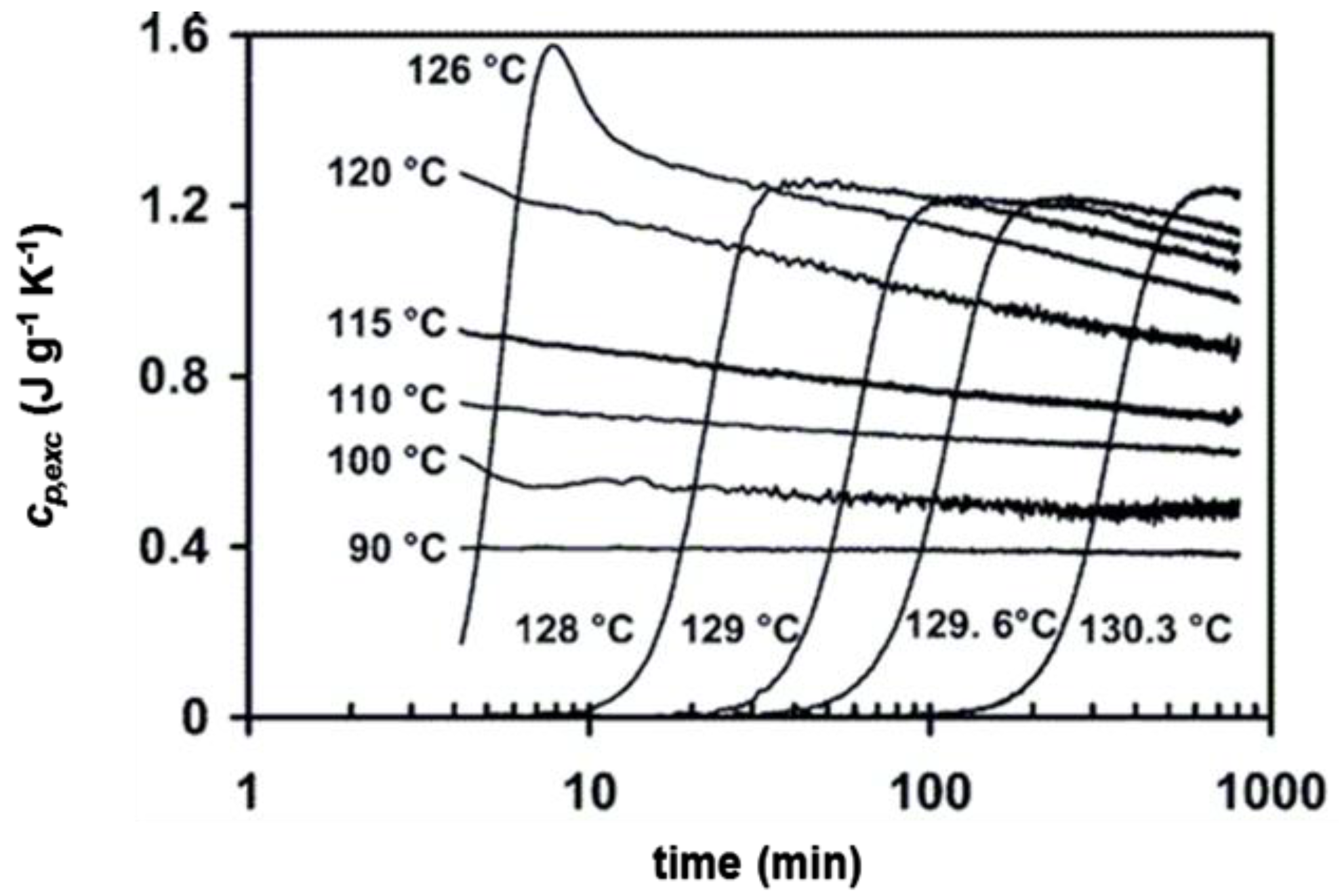
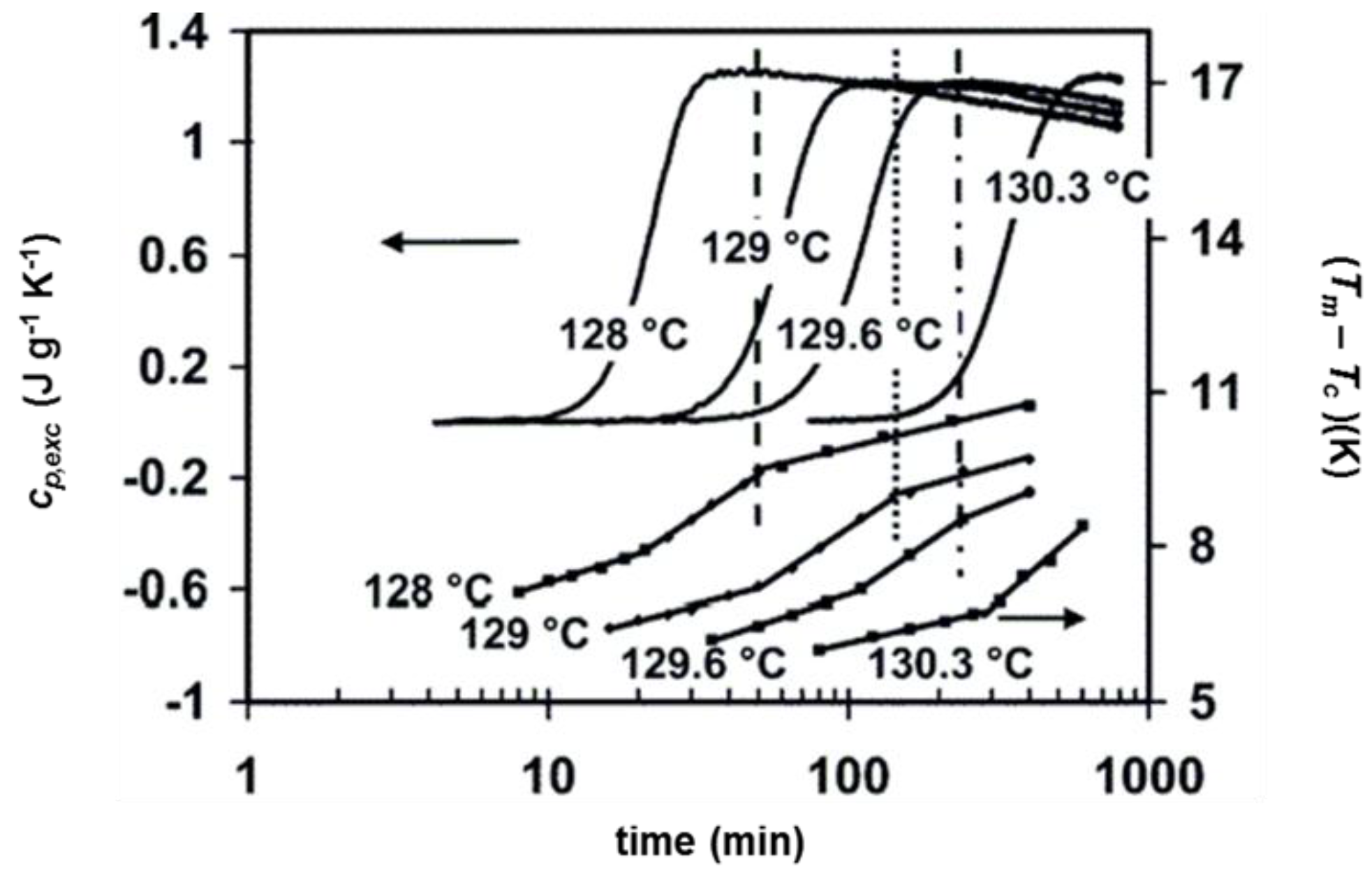
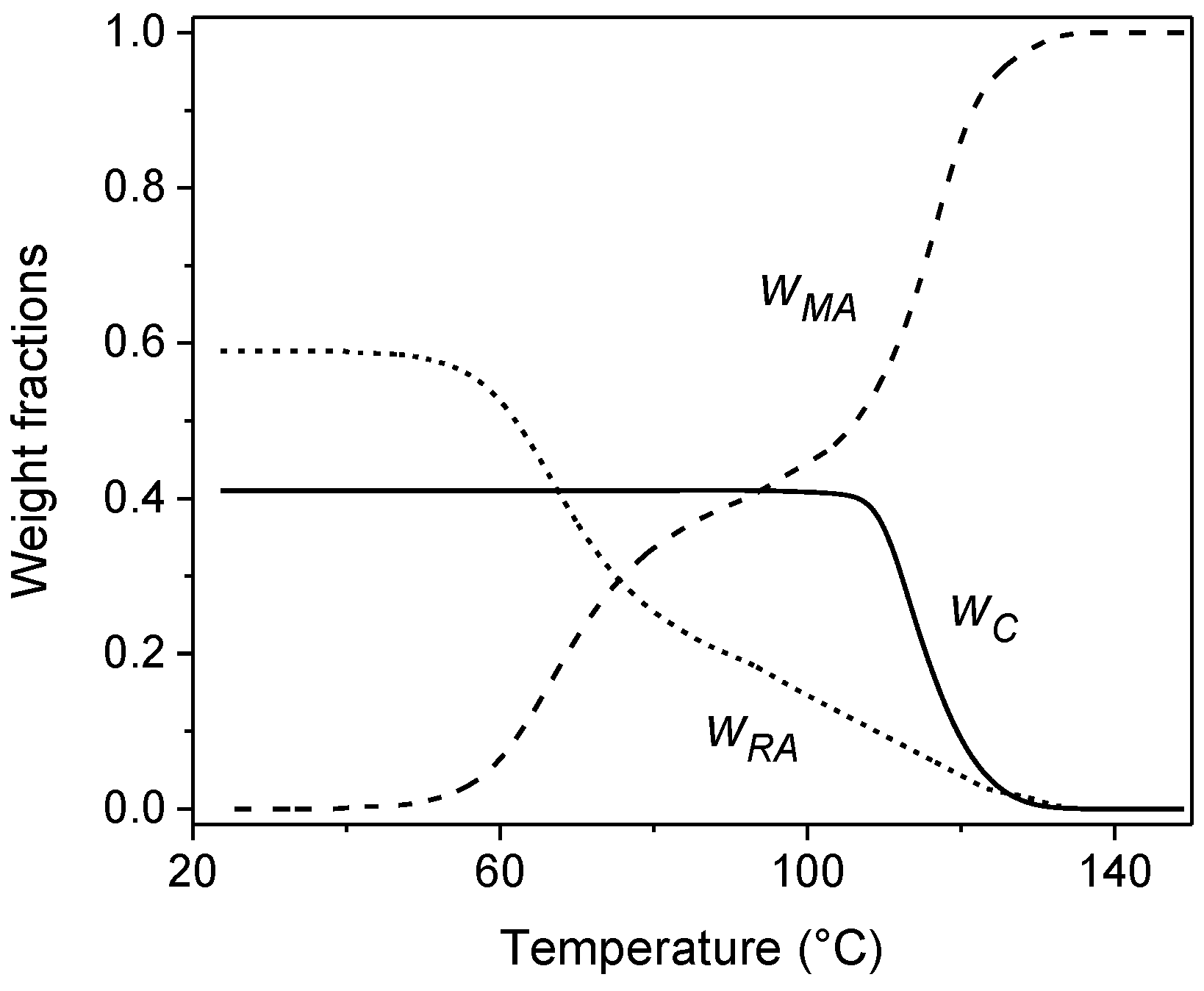
3. Quasi-Isothermal Crystallization
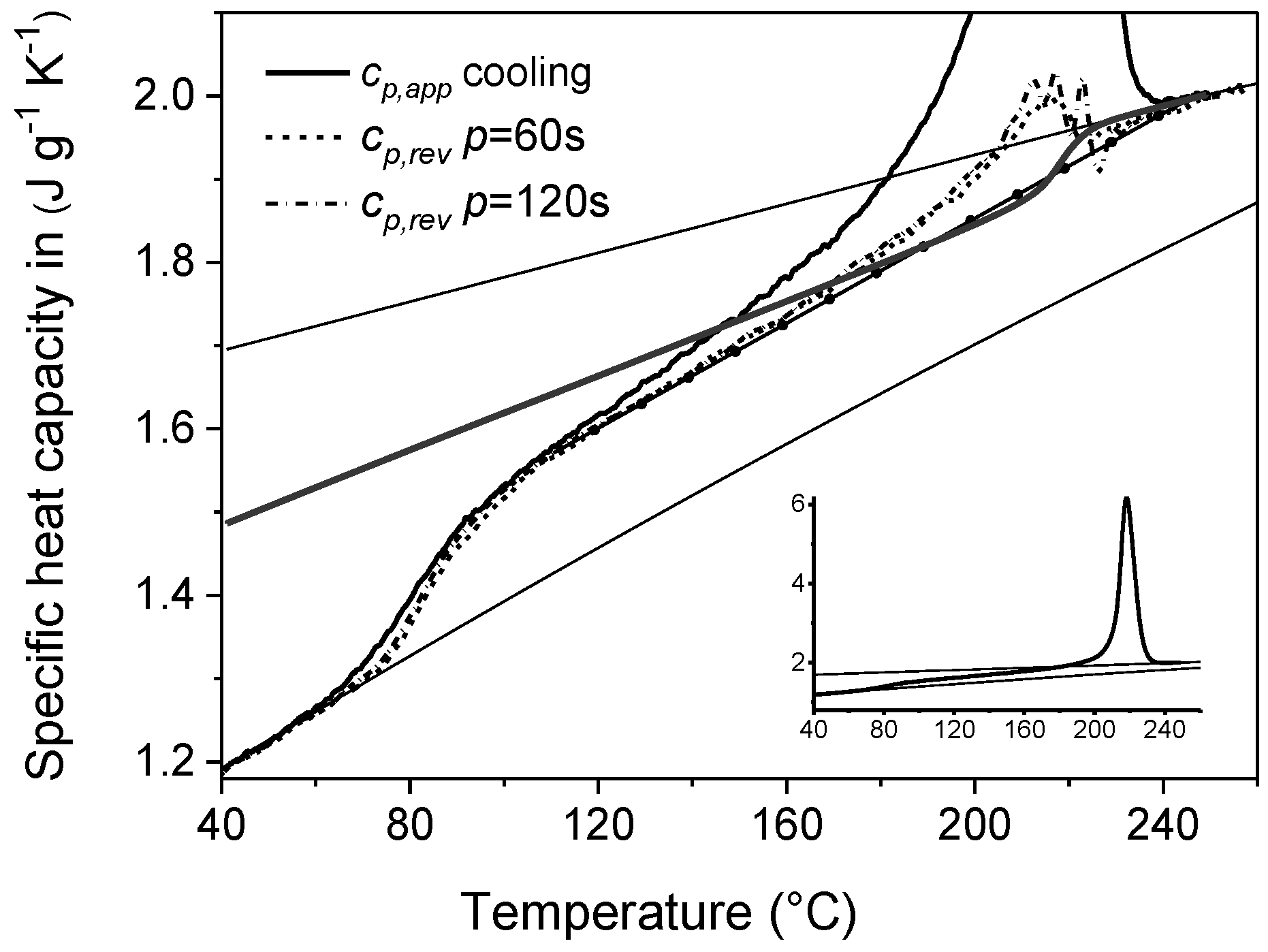
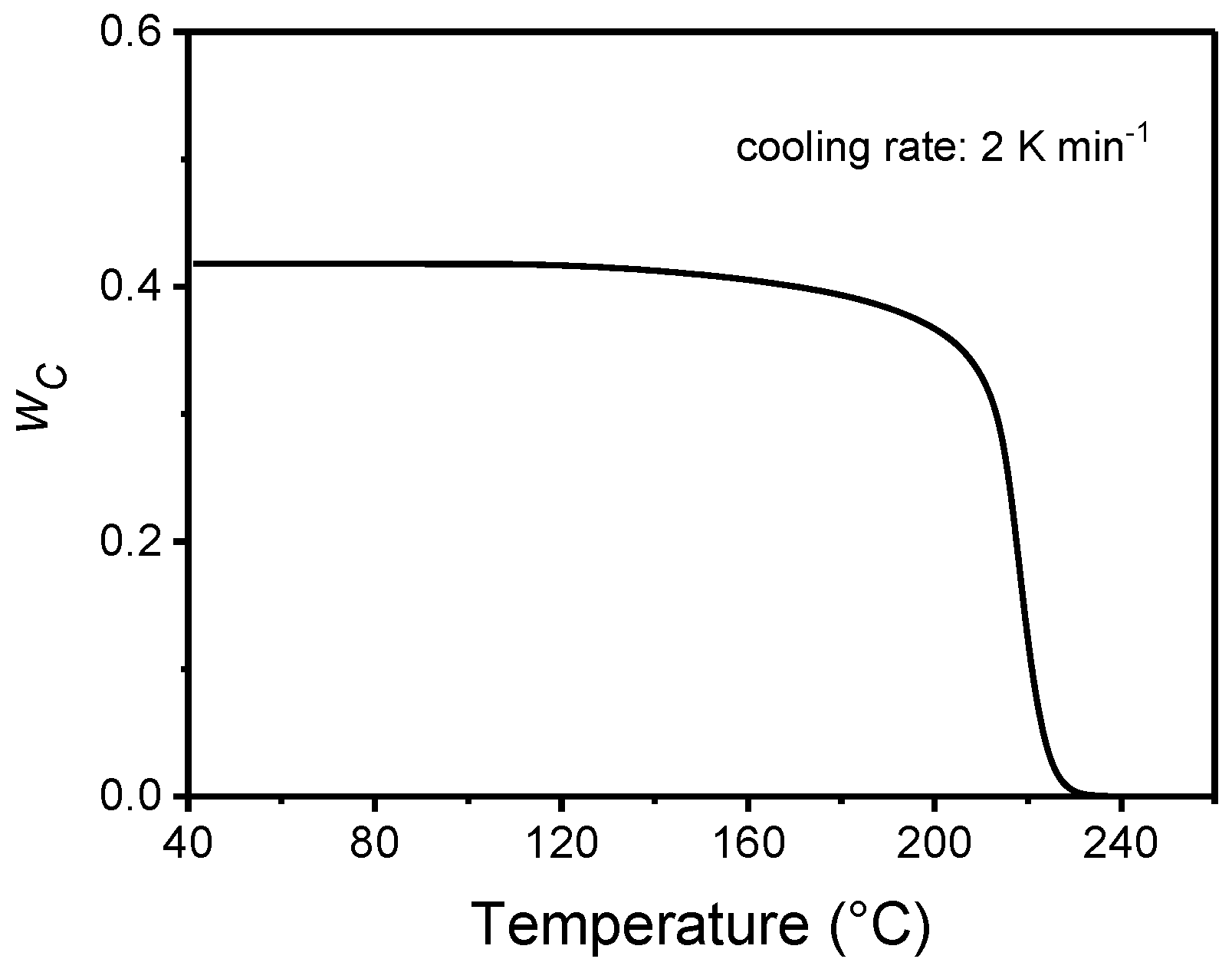
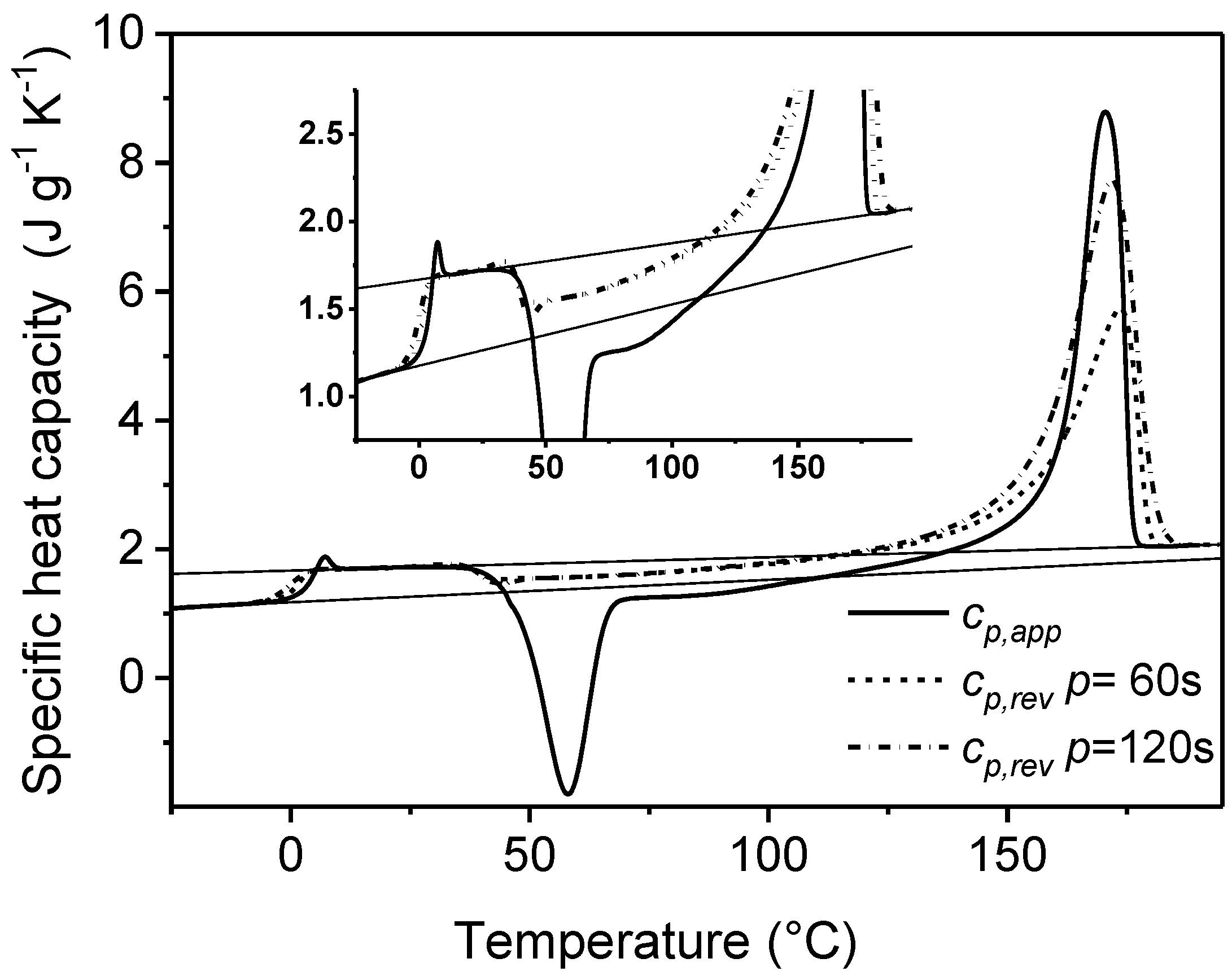
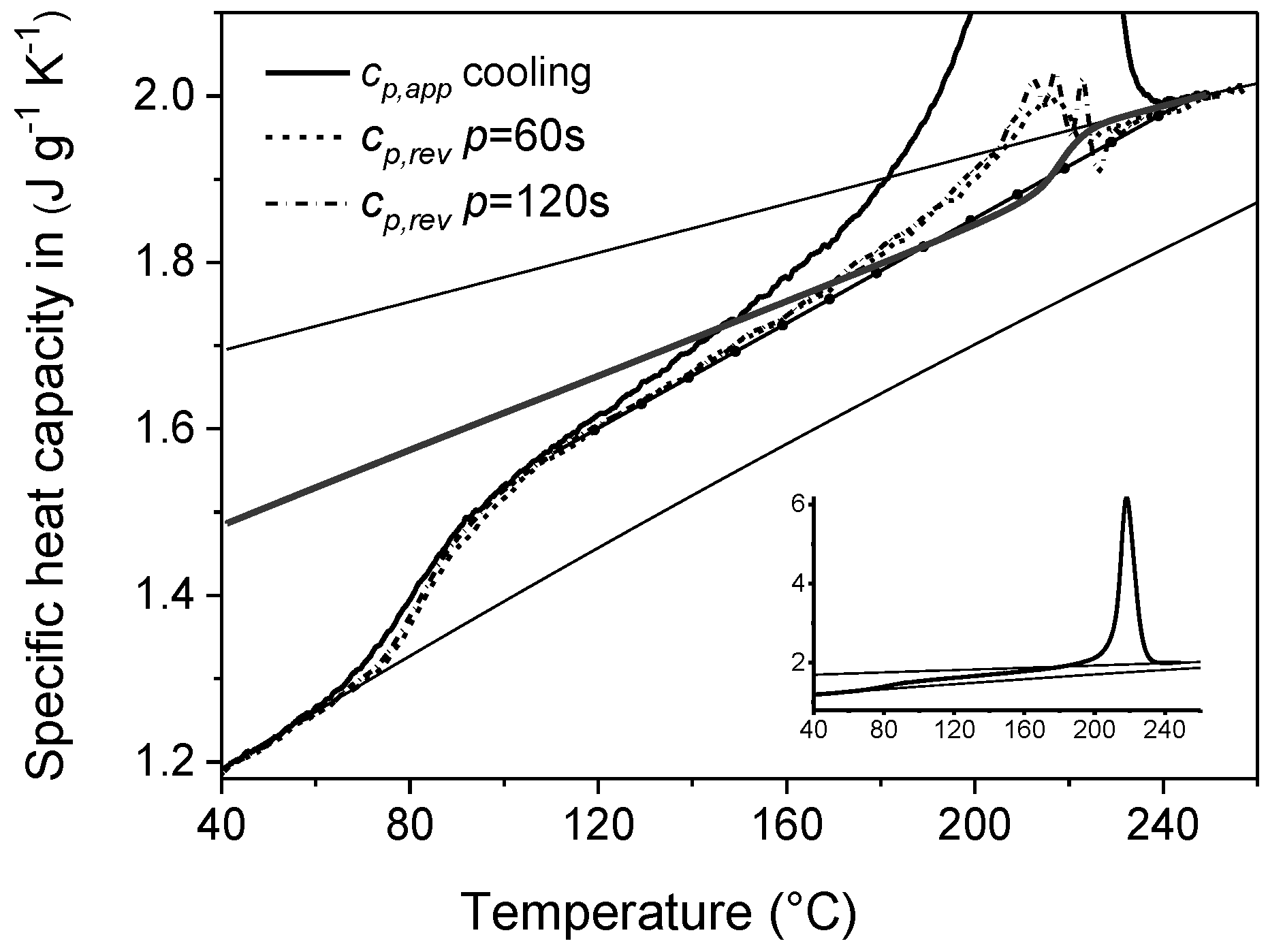
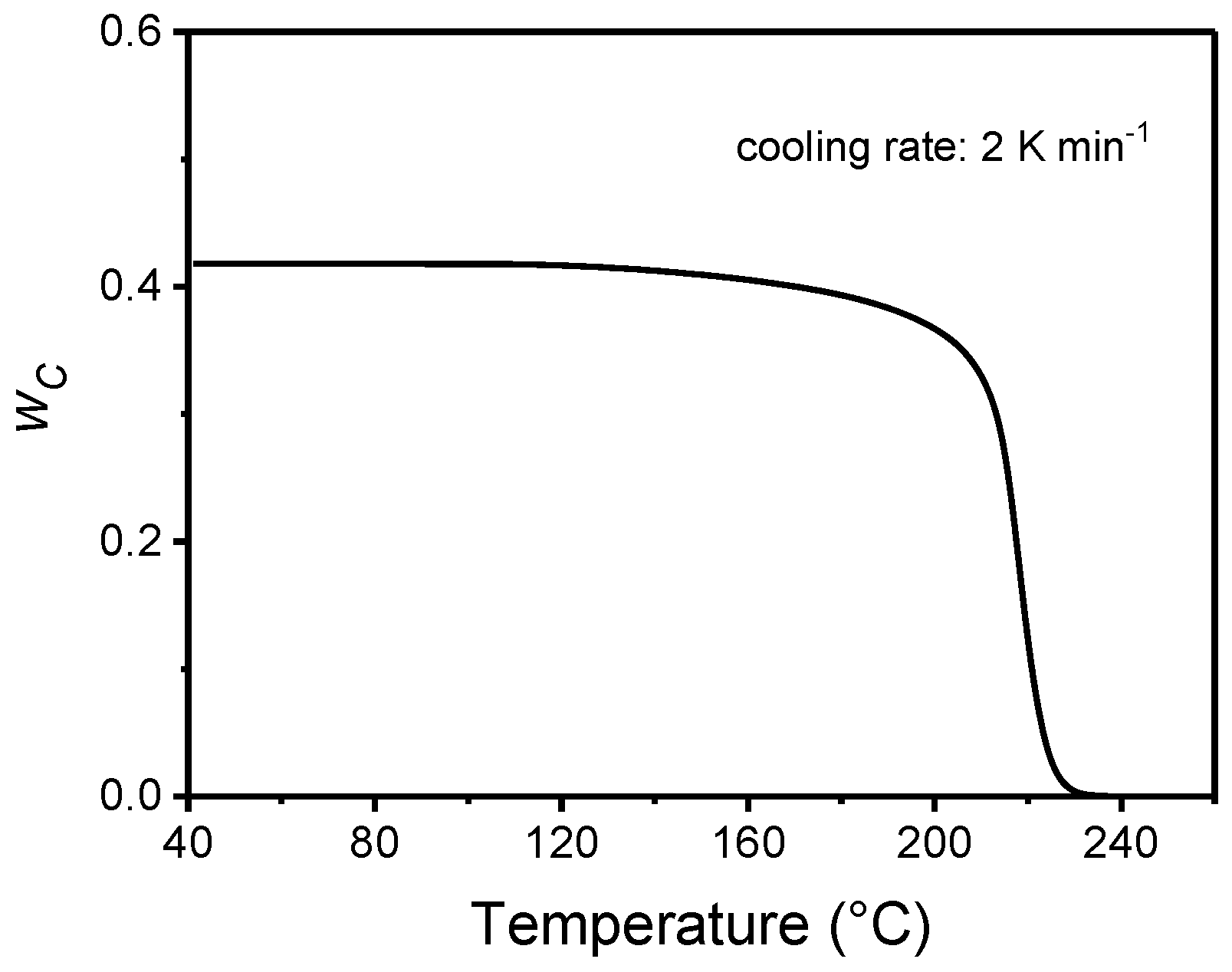
4. Non-Isothermal Crystallization and Stepwise Quasi-Isothermal Crystallization upon Cooling
5. Conclusions
Conflicts of Interest
References
- Schick, C. Temperature modulated differential scanning calorimetry (TMDSC—Basics and applications to polymers. In Handbook of Thermal Analysis and Calorimetry; Cheng, S.Z.D., Ed.; Elsevier Science B.V.: Amsterdam, The Netherlands, 2002; Volume 3, pp. 713–810. [Google Scholar]
- Wunderlich, B. Reversible crystallization and the rigid-amorphous phase in semicrystalline polymers. Prog. Polym. Sci. 2003, 28, 383–450. [Google Scholar] [CrossRef]
- Gill, P.S.; Sauerbrunn, S.R.; Reading, M. Modulated differential scanning calorimetry. J. Therm. Anal. 1993, 40, 931–939. [Google Scholar] [CrossRef]
- Okazaki, I.; Wunderlich, B. Reversible local melting in polymer crystals. Macromol. Rapid Commun. 1997, 18, 313–318. [Google Scholar] [CrossRef]
- Okazaki, I.; Wunderlich, B. Reversible melting in polymer crystals detected by temperature-modulated differential scanning calorimetry. Macromolecules 1997, 30, 1758–1764. [Google Scholar] [CrossRef]
- Androsch, R.; Wunderlich, B. Reversible crystallization and melting at the lateral surface of isotactic polypropylene crystals. Macromolecules 2001, 34, 5950–5960. [Google Scholar] [CrossRef]
- Schick, C.; Merzlyakov, M.; Minakov, A.; Wurm, A. Crystallization of polymers studied by temperature modulated calorimetric measurements at different frequencies. J. Therm. Anal. Calorim. 2000, 59, 279–288. [Google Scholar] [CrossRef]
- Goderis, B.; Reynaers, H.; Scherrnberg, R.; Mathot, V.B.F.; Koch, M.H.J. Temperature reversible transitions in linear polyethylene studied by TMDSC and time-resolved, temperature-modulated WAXS/SAXS. Macromolecules 2001, 34, 1779–1787. [Google Scholar] [CrossRef]
- Albrecht, T.; Armbruster, S.; Keller, S.; Strobl, G. Dynamics of surface crystallization and melting in polyethylene and poly(ethylene oxide) studied by temperature-modulated DSC and heat wave spectroscopy. Macromolecules 2001, 34, 8456–8467. [Google Scholar] [CrossRef]
- Wunderlich, B.; Jin, Y.; Boller, A. Mathematical description of differential scanning calorimetry based on periodic temperature modulation. Thermochim. Acta 1994, 238, 277–293. [Google Scholar] [CrossRef]
- Wunderlich, B. Modeling the heat flow and heat capacity of modulated differential scanning calorimetry. J. Therm. Anal. 1997, 48, 207–224. [Google Scholar] [CrossRef]
- Wurm, A.; Merzlyakov, M.; Schick, C. Reversible melting probed by temperature modulated dynamic mechanical and calorimetric measurements. Colloid Polym. Sci. 1998, 276, 289–296. [Google Scholar] [CrossRef]
- Androsch, R.; Moon, I.; Kreitmeier, S.; Wunderlich, B. Determination of heat capacity with a sawtooth-type, power-compensated temperature-modulated DSC. Thermochim. Acta 2000, 357–358, 267–278. [Google Scholar] [CrossRef]
- Merzlyakov, M.; Schick, C. Complex heat capacity measurements by TMDSC. Part 1. Influence of non-linear thermal response. Thermochim. Acta 1999, 330, 55–64. [Google Scholar] [CrossRef]
- Merzlyakov, M.; Schick, C. Optimization of experimental parameters in TMDSC. J. Therm. Anal. Calorim. 2000, 61, 649–659. [Google Scholar] [CrossRef]
- Merzlyakov, M.; Schick, C. Step response analysis in DSC—A fast way to generate heat capacity spectra. Thermochim. Acta 2001, 380, 5–12. [Google Scholar] [CrossRef]
- Androsch, R.; Wunderlich, B. Temperature modulated DSC using higher harmonics of the Fourier transform. Thermochim. Acta 1999, 333, 27–32. [Google Scholar] [CrossRef]
- Merzlyakov, M.; Schick, C. Simultaneous multi-frequency TMDSC measurements. Thermochim. Acta 2001, 377, 193–204. [Google Scholar] [CrossRef]
- Schick, C. Differential scanning calorimetry (DSC) of semicrystalline polymer. Anal. Bioanal. Chem. 2009, 395, 1589–1611. [Google Scholar] [CrossRef] [PubMed]
- Schick, C.; Mathot, V. (Eds.) Fast Scanning Calorimetry; Springer International Publishing: Switzerland, 2016. [Google Scholar]
- Shoifet, E.; Schulz, G.; Schick, C. Temperature modulated differential scanning calorimetry—Extension to high and low frequencies. Thermochim. Acta 2015, 603, 227–236. [Google Scholar] [CrossRef]
- Schick, C.; Wurm, A.; Mohamed, A. Vitrification and devitrification of the rigid amorphous fraction of semicrystalline polymers revealed from frequency-dependent heat capacity. Colloid Polym. Sci. 2001, 279, 800–806. [Google Scholar] [CrossRef]
- Androsch, R.; Wunderlich, B. Specific reversible melting of polymers. J. Polym. Sci. Polym. Phys. 2003, 41, 2039–2051. [Google Scholar] [CrossRef]
- Wurm, A.; Merzlyakov, M.; Schick, C. Crystallization of polymers studied by temperature-modulated techniques (TMDSC, TMDMA). J. Macromol. Sci. Phys. 1999, 38, 693–708. [Google Scholar] [CrossRef]
- Merzlyakov, M.; Wurm, A.; Zorzut, M.; Schick, C. Frequency and temperature amplitude dependence of complex heat capacity in the melting region of polymers. J. Macromol. Sci. Phys. 1999, 38, 1045–1054. [Google Scholar] [CrossRef]
- Scherrenberg, R.; Mathot, V.; Van Hemelrijck, A. The practical applicability of TMDSC to polymeric systems. Thermochim. Acta 1999, 330, 3–19. [Google Scholar] [CrossRef]
- Wurm, A.; Merzlyakov, M.; Schick, C. Isothermal crystallization of PCL studied by temperature modulated dynamic mechanical and TMDSC analysis. J. Therm. Anal. Calorim. 1999, 56, 1155–1161. [Google Scholar] [CrossRef]
- Wurm, A.; Merzlyakov, M.; Schick, C. Reversible melting during crystallization of polymers studied by temperature modulated techniques (TMDSC, TMDMA). J. Therm. Anal. Calorim. 2000, 60, 807–820. [Google Scholar] [CrossRef]
- Schick, C.; Wurm, A.; Merzlyakov, M.; Minakov, A.; Marand, H. Crystallization and melting of polycarbonate studied by temperature-modulated (TMDSC). J. Therm. Anal. Calorim. 2001, 64, 549–555. [Google Scholar] [CrossRef]
- Pak, J.; Wunderlich, B. Melting and crystallization of polyethylene of different molar mass by calorimetry. Macromolecules 2001, 34, 4492–4503. [Google Scholar] [CrossRef]
- Schick, C.; Wurm, A.; Mohammed, A. Formation and disappearance of the rigid amorphous fraction in semicrystalline polymers revealed from frequency dependent heat capacity. Thermochim. Acta 2003, 396, 119–132. [Google Scholar] [CrossRef]
- Xu, H.; Cebe, P. Heat capacity of isotactic polystyrene: Dual reversible crystal melting and relaxation of rigid amorphous fraction. Macromolecules 2004, 37, 2797–2806. [Google Scholar] [CrossRef]
- Righetti, M.C.; Tombari, E.; Di Lorenzo, M.L. Crystalline, mobile amorphous and rigid amorphous fractions in isotactic polystyrene. Eur. Polym. J. 2008, 44, 2659–2667. [Google Scholar] [CrossRef]
- Righetti, M.C.; Tombari, E. Crystalline, mobile amorphous and rigid amorphous fractions in poly(l-lactic acid) by TMDSC. Thermochim. Acta 2011, 522, 118–127. [Google Scholar] [CrossRef]
- Di Lorenzo, M.L.; Gazzano, M.; Righetti, M.C. The role of the rigid amorphous fraction on cold crystallization of poly(3-hydroxybutyrate). Macromolecules 2012, 45, 5684–5691. [Google Scholar] [CrossRef]
- Righetti, M.C.; Tombari, E.; Di Lorenzo, M.L. The role of the crystallization temperature on the nanophase structure evolution of poly[(R)-3-hydroxybutyrate]. J. Phys. Chem. B 2013, 117, 12303–12311. [Google Scholar] [CrossRef] [PubMed]
- Righetti, M.C.; Laus, M.; Di Lorenzo, M.L. Temperature dependence of the rigid amorphous fraction in poly(ethylene terephthalate). Eur. Polym. J. 2014, 58, 60–68. [Google Scholar] [CrossRef]
- Righetti, M.C.; Prevosto, D.; Tombari, E. Time and temperature evolution of the rigid amorphous fraction and differently constrained amorphous fractions in PLLA. Macromol. Chem. Phys. 2016, 217, 2013–2026. [Google Scholar] [CrossRef]
- Wunderlich, B. Macromolecular Physics; Volume 2 Crystal Nucleation, Growth, Annealing; Academic Press: New York, NY, USA, 1976; pp. 178–181. [Google Scholar]
- ATHAS Data Bank Available from Springer Materials. Available online: www.materials.springer.com Polymer Thermodynamics (accessed on 21 April 2017).
- Mathot, V.B.F. Thermal Characterization of states of matter. In Calorimetry and Thermal Analysis of Polymers; Mathot, V.B.F., Ed.; Hanser/Gardner: Cincinnati, OH, USA, 1994; pp. 105–167. [Google Scholar]
- Pan, P.; Inoue, Y. Polymorphism and isomorphism in biodegradable polyesters. Prog. Polym. Sci. 2009, 34, 605–640. [Google Scholar] [CrossRef]
- Pan, P.; Kai, W.; Zhu, B.; Dong, T.; Inoue, Y. Polymorphous crystallization and multiple melting behavior of poly(l-lactide): Molecular weight dependence. Macromolecules 2007, 40, 6898–6905. [Google Scholar] [CrossRef]
- Kawai, T.; Rahman, N.; Matsuba, G.; Nishida, K.; Kanaya, T.; Nakano, M.; Okamoto, H.; Kawada, J.; Usuki, A.; Honma, N.; et al. Crystallization and melting behavior of poly(l-lactic acid). Macromolecules 2007, 40, 9463–9469. [Google Scholar] [CrossRef]
- Pyda, M.; Bopp, R.C.; Wunderlich, B. Heat capacity of poly(lactic acid). J. Chem. Thermodyn. 2004, 36, 731–742. [Google Scholar] [CrossRef]
- Righetti, M.C.; Gazzano, M.; Di Lorenzo, M.L.; Androsch, R. Enthalpy of melting of α′- and α-crystals of poly(l-lactic acid). Eur. Polym. J. 2015, 70, 215–220. [Google Scholar] [CrossRef]
- Xu, H.; Ince, S.; Cebe, P. Development of the crystallinity and rigid amorphous fraction in cold-crystallized isotactic polystyrene. J. Polym. Sci. Polym. Phys. 2003, 41, 3026–3036. [Google Scholar] [CrossRef]
- Righetti, M.C.; Laus, M.; Di Lorenzo, M.L. Rigid amorphous fraction and melting behaviour of poly(ethylene terephthalate). Colloid Polym. Sci. 2014, 292, 1365–1374. [Google Scholar] [CrossRef]
- Righetti, M.C.; Di Lorenzo, M.L. Rigid amorphous fraction and multiple melting behavior in poly(butylene terephthalate) and isotactic polystyrene. J. Therm. Anal. Calorim. 2016, 126, 521–530. [Google Scholar] [CrossRef]
- Iannace, S.; Nicolais, L. Isothermal crystallization and chain mobility of poly(l-lactide). J. Appl. Polym. Sci. 1997, 64, 911–919. [Google Scholar] [CrossRef]
- Wang, Y.; Funari, S.S.; Mano, F.J. Influence of semicrystalline morphology on the glass transition of poly(l-lactic acid). Macromol. Chem. Phys. 2006, 207, 1262–1271. [Google Scholar] [CrossRef]
- Delpouve, N.; Saiter, A.; Mano, J.F.; Dargent, E. Cooperative rearranging region size in semi-crystalline poly(l-lactic acid). Polymer 2008, 49, 3130–3135. [Google Scholar] [CrossRef]
- Delpouve, N.; Saiter, A.; Dargent, E. Cooperatively length evolution during crystallization of poly(lactic acid). Eur. Polym. J. 2011, 47, 2414–2423. [Google Scholar] [CrossRef]
- Santa Cruz, C.; Strinbeck, N.; Zachman, H.G. Novel aspects in the structure of poly(ethylene terephthalate) as revealed by means of small angle X-ray scattering. Macromolecules 1991, 24, 5980–5990. [Google Scholar] [CrossRef]
- Alvarez, C.; Sics, I.; Nogales, A.; Denchev, Z.; Funari, S.S.; Ezquerra, T.A. Structure-dynamics relationship in crystallizing poly(ethylene terephthalate) as revealed by time resolved X-ray and dielectric methods. Polymer 2004, 45, 3953–3959. [Google Scholar] [CrossRef]
- Nogales, A.; Ezquerra, T.A.; Denchev, Z.; Šics, I.; Baltá Calleja, F.J.; Hsiao, B.S. Molecular dynamics and microstructure development during cold crystallization in poly(ether-ether-ketone) as revealed by real time dielectric and X-ray methods. J. Chem. Phys. 2001, 115, 3804–3813. [Google Scholar] [CrossRef]
- Toda, A.; Tomita, C.; Hikosaka, M. Temperature modulated DSC of irreversible melting of nylon 6 crystals. J. Therm. Anal. 1998, 54, 623–635. [Google Scholar] [CrossRef]
- Androsch, R.; Wunderlich, B. A study of the annealing of poly(ethylene-co-octene)s by temperature modulated and standard differential scanning calorimetry. Macromolecules 1999, 32, 7238–7247. [Google Scholar] [CrossRef]
- Righetti, M.C.; Di Lorenzo, M.L. Melting process of poly(butylene terephthalate) analyzed by temperature-modulated differential scanning calorimetry. J. Polym. Sci. Polym. Phys. 2004, 42, 2191–2201. [Google Scholar] [CrossRef]
- Huang, Z.; Marand, H.; Cheung, W.Y.; Guest, M. Study of crystallization processes in ethylene-styrene copolymers by conventional DSC and temperature-modulated calorimetry: Linear polyethylene and low styrene content copolymers. Macromolecules 2004, 37, 9922–9932. [Google Scholar] [CrossRef]
- Toda, A.; Oda, T.; Hikosaka, M.; Saruyama, Y. A new analyzing method of temperature modulated DSC of exo- or endo-thermic process: Application to polyethylene crystallization. Thermochim. Acta 1997, 293, 47–63. [Google Scholar] [CrossRef]
- Toda, A.; Oda, T.; Hikosaka, M.; Saruyama, Y. A new method of analysis transformation kinetics with temperature modulated differential scanning calorimetry: Application to polymer crystal growth. Polymer 1997, 38, 231–233. [Google Scholar] [CrossRef]
- Mathot, V.B.F. Temperature dependence of some thermodynamic functions for amorphous and semi-crystalline polymers. Polymer 1984, 25, 579–599. [Google Scholar] [CrossRef]
- Marand, H.; Huang, Z. Isothermal lamellar thickening in linear polyethylene: Correlation between the evolution of the degree of crystallinity and the melting temperature. Macromolecules 2004, 37, 6492–6497. [Google Scholar] [CrossRef]
- Boyd, R. Relaxation processes in crystalline polymers: Molecular interpretation—A review. Polymer 1985, 26, 1123–1133. [Google Scholar] [CrossRef]
- Androsch, R.; Wunderlich, B. Specific reversible melting of polyethylene. J. Polym. Sci. Polym. Phys. 2003, 41, 2157–2173. [Google Scholar] [CrossRef]
- Androsch, R. Reversible and irreversible crystallization in high-density polyethylene at low temperature. J. Therm. Anal. Calorim. 2004, 77, 1037–1043. [Google Scholar] [CrossRef]
- Schick, C.; Merzlyakov, M.; Wunderlich, B. Analysis of the reorganization of poly(ethylene terephthalate) in the melting range by temperature-modulated calorimetry. Polym. Bull. 1998, 40, 297–303. [Google Scholar] [CrossRef]
- Pyda, M.; Wunderlich, B. Reversible and irreversible heat capacity of poly(trimethylene terephthalate) analyzed by temperature-modulated differential scanning calorimtry. J. Polym. Sci. Polym. Phys. 2000, 38, 622–631. [Google Scholar] [CrossRef]
- Androsch, R.; Wunderlich, B. Analysis of the degree of reversibility of crystallization and melting in poly(ethylene-co-1-octene). Macromolecules 2000, 33, 9076–9089. [Google Scholar] [CrossRef]
- Pyda, M.; Di Lorenzo, M.L.; Pak, J.; Kamasa, P.; Buzin, A.; Grebowicz, J.; Wunderlich, B. Reversible and irreversible heat capacity of poly[carbonyl(ethylene-co-propylene)] by temperature-modulated differential scanning calorimetry. J. Polym. Sci. Polym. Phys. 2001, 39, 1565–1577. [Google Scholar] [CrossRef]
- Di Lorenzo, M.L.; Pyda, M.; Wunderlich, B. Reversible melting in nanophase-separated poly(oligoamide-alt-oligoether)s and its dependence on sequence length, crystal perfection, and molecular mobility. J. Polym. Sci. Polym. Phys. 2001, 39, 2969–2981. [Google Scholar] [CrossRef]
- Righetti, M.C.; Di Lorenzo, M.L.; Angiuli, M.; Tombari, E. Structural reorganization in poly(butylene terephthalate) during fusion. Macromolecules 2004, 37, 9027–9033. [Google Scholar] [CrossRef]
- Xu, H.; Cebe, P. Evaluation of the reversible contribution to the reversing heat capacity in isotactic polystyrene. Thermochim. Acta 2006, 442, 42–47. [Google Scholar] [CrossRef]
- Di Lorenzo, M.L. The melting process and the rigid amorphous fraction of cis-1,4-polybutadiene. Polymer 2009, 50, 578–584. [Google Scholar] [CrossRef]
- Di Lorenzo, M.L.; Wunderlich, B. Temperature-modulated calorimetry of the crystallization of polymers analyzed by measurements and model calculations. J. Therm. Anal. Calorim. 1999, 57, 459–472. [Google Scholar] [CrossRef]
- Coburn, J.C.; Boyd, R.H. Dielectric relaxation in poly(ethylene terephthalate). Macromolecules 1986, 19, 2238–2245. [Google Scholar] [CrossRef]
- Androsch, R.; Wunderlich, B. The link between rigid amorphous fraction and crystal perfection in cold-crystallized poly(ethylene terephthalate). Polymer 2005, 46, 12556–12566. [Google Scholar] [CrossRef]
- Righetti, M.C. The amorphous fractions of poly(lactic acid). Adv. Poly. Sci 2017, in press. [Google Scholar]
- Di Lorenzo, M.L. Crystallization behavior of poly(l-lactic acid). Eur. Polym. J. 2005, 41, 569–575. [Google Scholar]
- Ma, Q.; Georgiev, G.; Cebe, P. Constraints in semicrystalline polymers: Using quasi-isothermal analysis to investigate the mechanism of formation and loss of the rigid amorphous fraction. Polymer 2011, 52, 4562–4570. [Google Scholar] [CrossRef]
- Aharoni, S.M. Increased glass transition temperature in motionally constrained semicrystallline polymers. Polym. Advan. Technol. 1998, 9, 169–201. [Google Scholar] [CrossRef]
- Righetti, M.C.; Boggioni, A.; Laus, M.; Antonioli, D.; Sparnacci, K.; Boarino, L. Thermal and mechanical properties of PES/PTFE composites and nanocomposites. J. Appl. Polym. Sci. 2013, 130, 3624–3633. [Google Scholar] [CrossRef]
- Di Lorenzo, M.L.; Righetti, M.C. The three-phase structure of isotactic poly(1-butene). Polymer 2008, 49, 1323–1331. [Google Scholar] [CrossRef]
- Kolesov, I.; Androsch, R. The rigid amorphous fraction of cold-crystallized polyamide 6. Polymer 2012, 53, 4770–4777. [Google Scholar] [CrossRef]
- Martin, S.; Exposito, M.T.; Vega, J.F.; Martinez-Salazar, J. Microstructure and properties of branched polyethylene: Application of a three-phase structural model. J. Appl. Polym. Sci. 2013, 128, 1871–1878. [Google Scholar] [CrossRef]
- Nguyen, T.L.; Bédoui, F.; Mazeran, P.-E.; Guigon, M. Mechanical investigation of confined amorphous phase in semicrystalline polymers: Case of PET and PLA. Polym. Eng. Sci. 2015, 55, 397–405. [Google Scholar]
- Lin, J.; Shenogin, S.; Nazarenko, S. Oxygen solubility and specific volume of rigid amorphous fraction in semicrystalline poly(ethylene terephthalate). Polymer 2002, 43, 4733–4743. [Google Scholar] [CrossRef]
- Olson, B.G.; Lin, J.; Nazarenko, S.; Jamieson, A.M. Positron annihilation lifetime spectroscopy of poly(ethylene terephthalate): Contributions from rigid and mobile amorphous fractions. Macromolecules 2003, 36, 7618–7623. [Google Scholar] [CrossRef]
- Drieskens, M.; Peeters, R.; Mullen, J.; Franco, D.; Lemstra, P.J.; Hristova-Bogaerds, D.G. Structure versus properties relationship of poly(lactic acid). I. Effect of crystallinity on barrier properties. J. Polym. Sci. Pol. Phys. 2009, 47, 2247–2258. [Google Scholar] [CrossRef]
- Delpouve, N.; Stoclet, G.; Saiter, A.; Dargent, E.; Marais, S. Water barrier properties in biaxially drawn poly(lactic acid) Films. J. Phys. Chem. B 2012, 116, 4615–4625. [Google Scholar] [CrossRef] [PubMed]




© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Righetti, M.C. Crystallization of Polymers Investigated by Temperature-Modulated DSC. Materials 2017, 10, 442. https://doi.org/10.3390/ma10040442
Righetti MC. Crystallization of Polymers Investigated by Temperature-Modulated DSC. Materials. 2017; 10(4):442. https://doi.org/10.3390/ma10040442
Chicago/Turabian StyleRighetti, Maria Cristina. 2017. "Crystallization of Polymers Investigated by Temperature-Modulated DSC" Materials 10, no. 4: 442. https://doi.org/10.3390/ma10040442
APA StyleRighetti, M. C. (2017). Crystallization of Polymers Investigated by Temperature-Modulated DSC. Materials, 10(4), 442. https://doi.org/10.3390/ma10040442