
Understanding Drug Release Data through Thermodynamic Analysis

 ,
,
Abstract
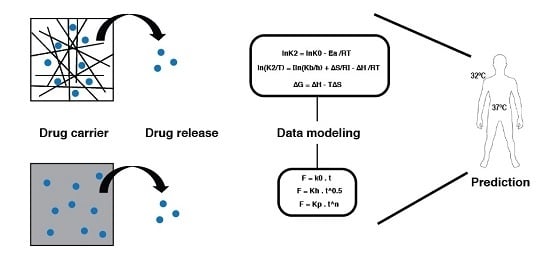
:
1. Introduction
2. Results and Discussion
2.1. Production of Systems
2.2. Scanning Electron Microscopy
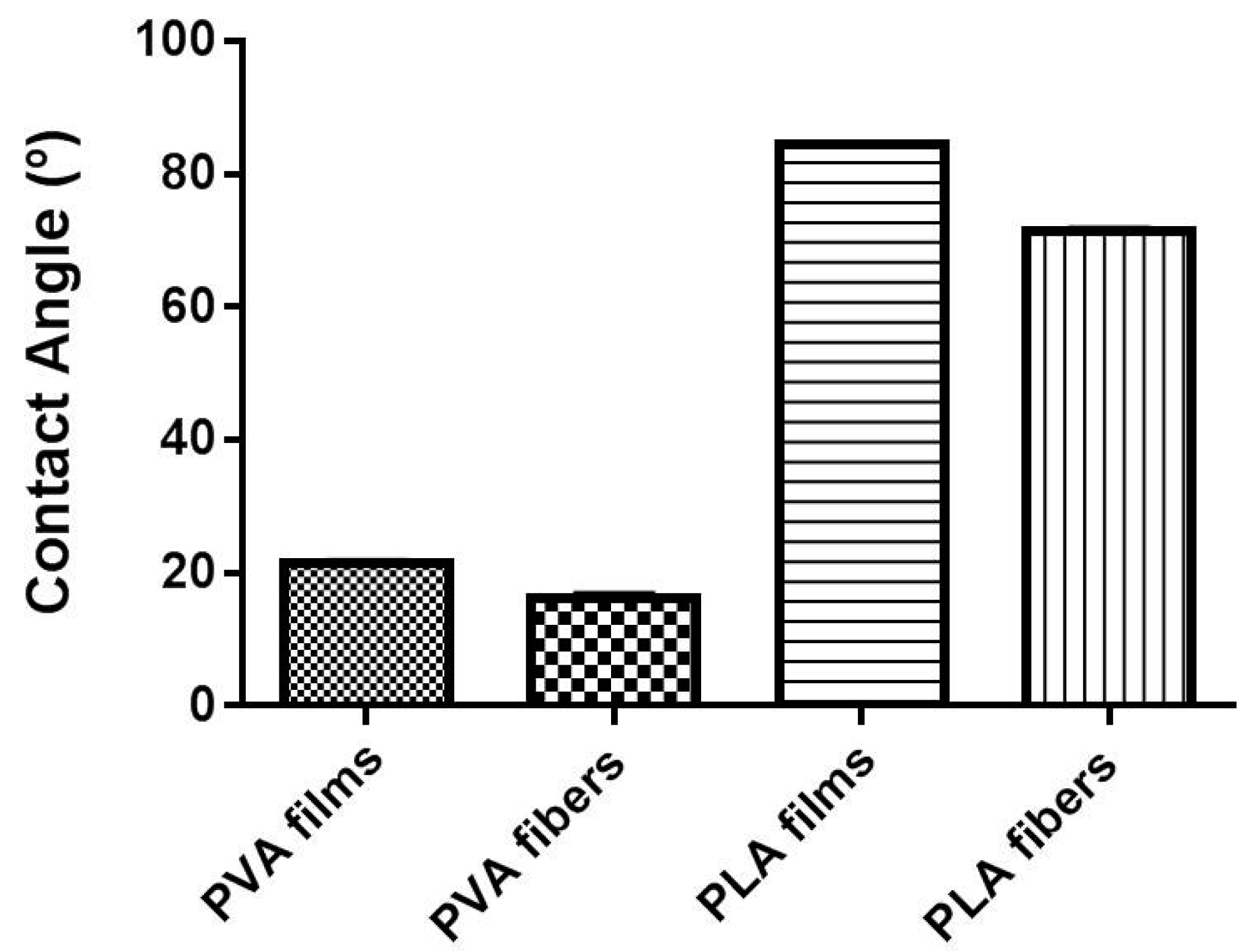
2.3. Superficial Wettability
2.4. Evaluation of the AmB Content in the Systems
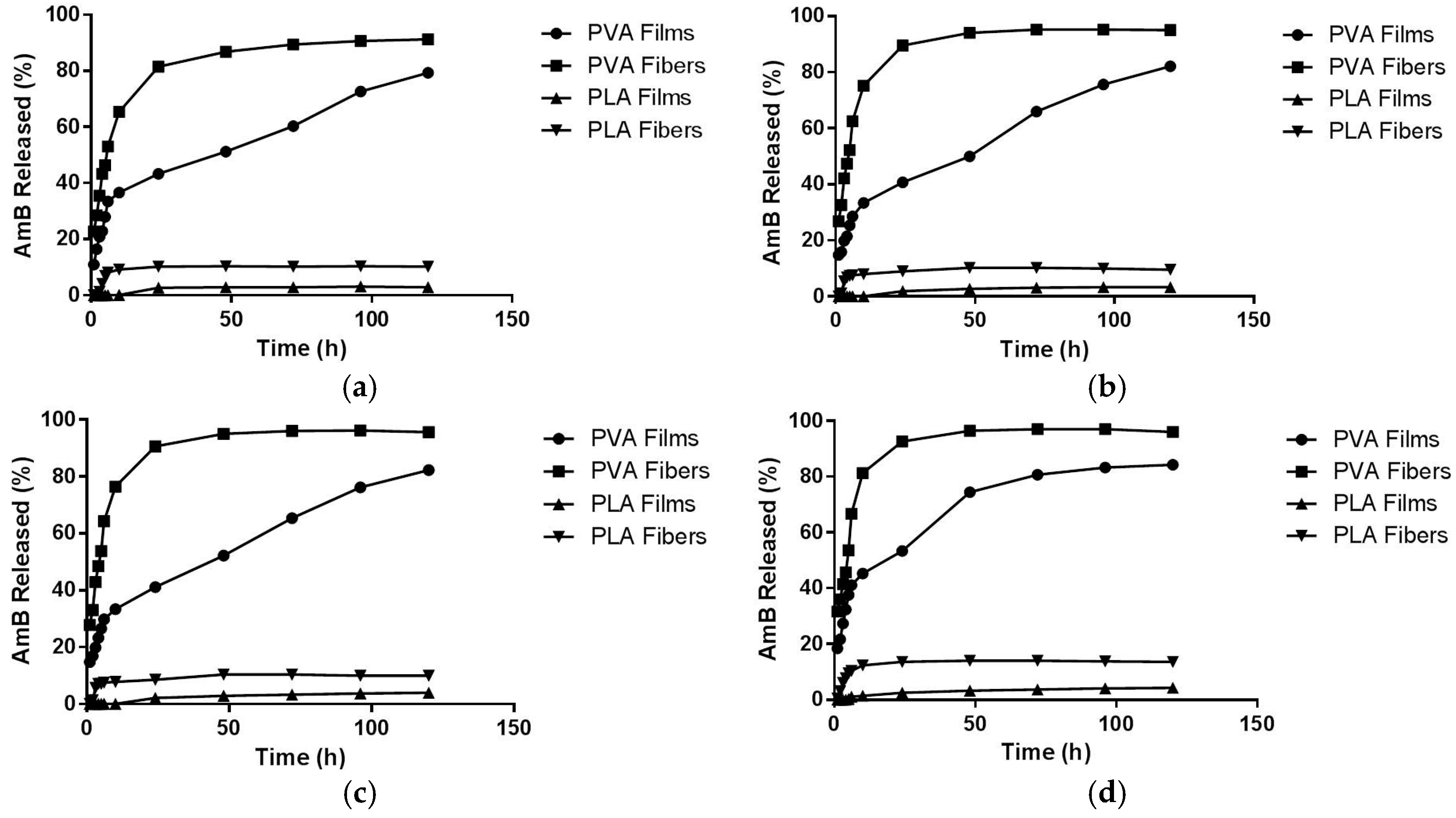
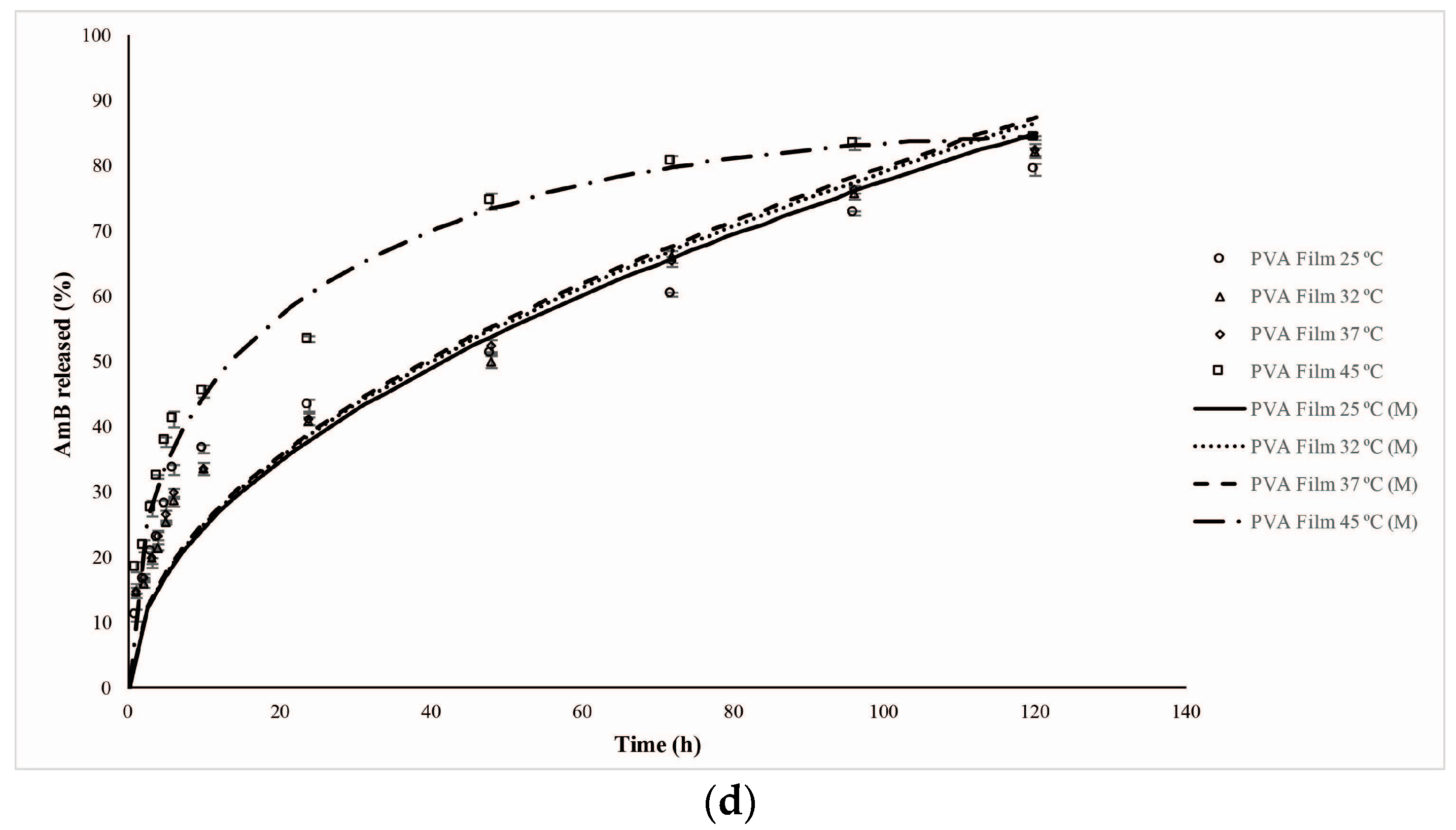
2.5. AmB Kinetics Release Profile
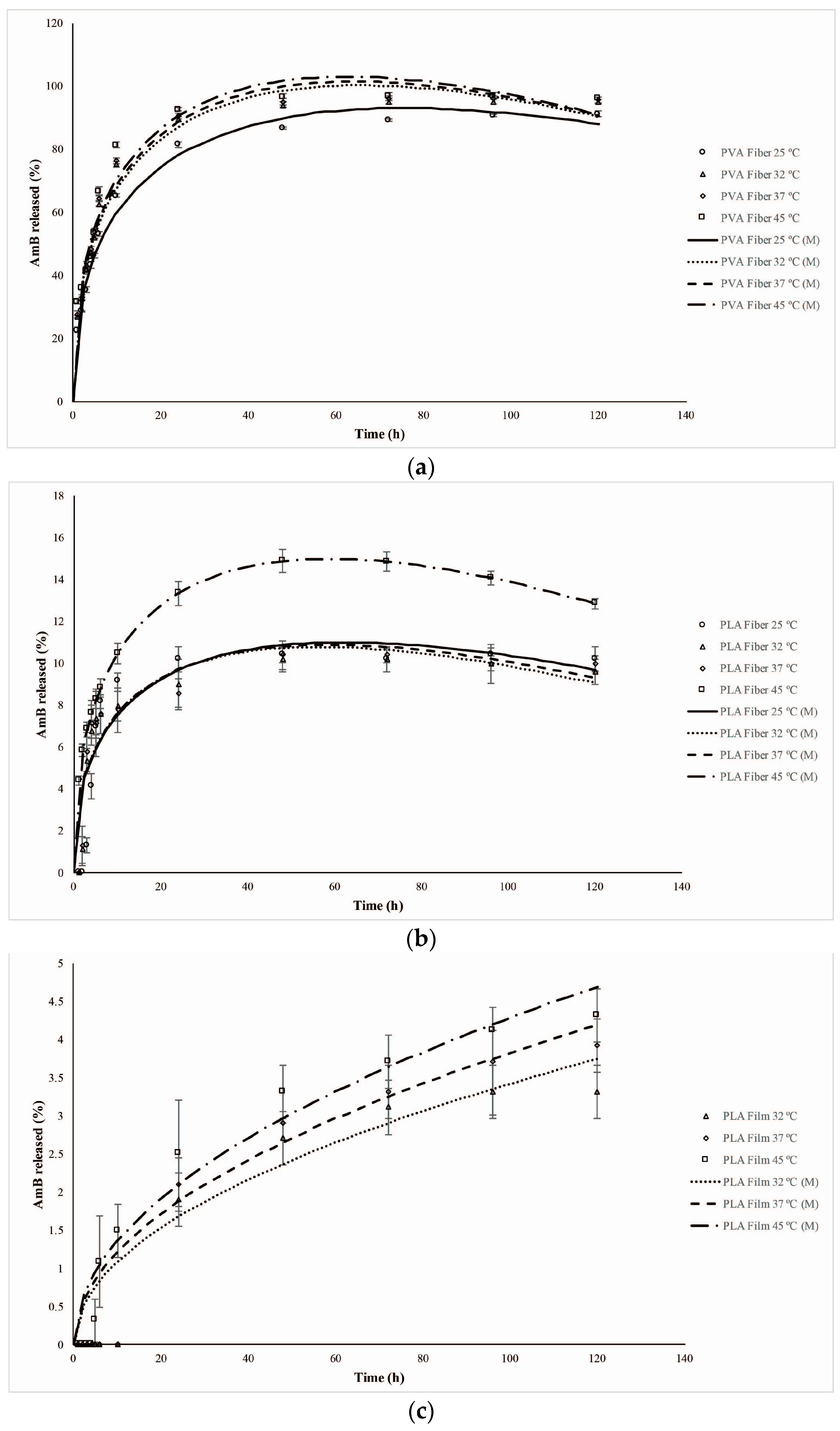
2.6. Mathematical Models of Releasing Kinetics
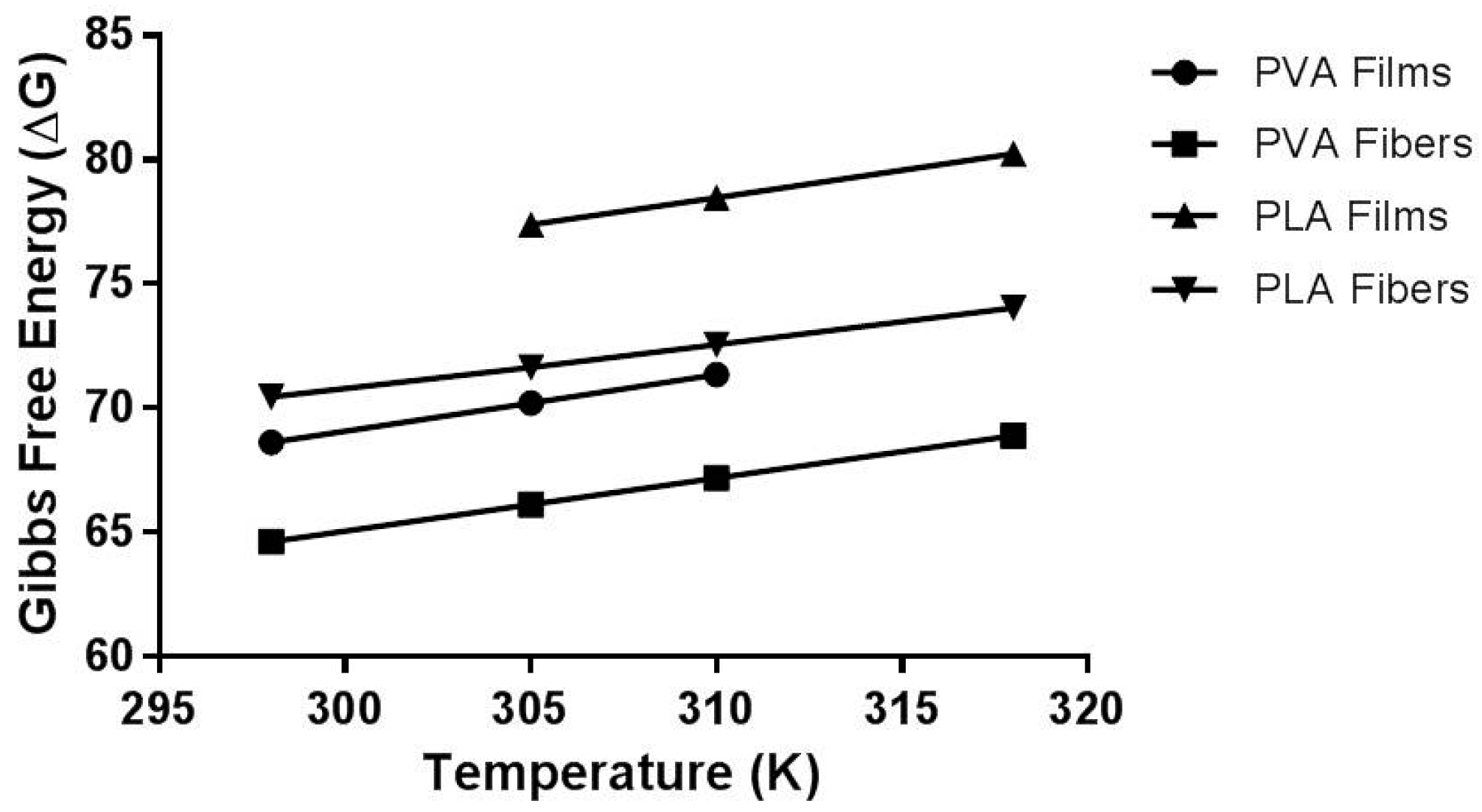
2.7. Thermodynamic Parameters
3. Materials and Methods
3.1. Materials
3.2. Methods
3.2.1. Development of Polymeric Systems
PVA Films
PVA Fibers
PLA Films
PLA Fibers
3.2.2. Factorial Design and Statistical Analysis of the Systems
3.2.3. Scanning Electron Microscopy
3.2.4. Superficial Wettability
3.2.5. Drug Entrapment Efficiency
3.2.6. Kinetic Release Assay
3.2.7. Mathematical Modeling of the Kinetics Release
3.2.8. Thermodynamic Parameters
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A
Equation List
References
- Narasimhan, B. Mathematical models describing polymer dissolution: Consequences for drug delivery. Adv. Drug Deliv. Rev. 2001, 48, 195–210. [Google Scholar] [CrossRef]
- Siepmann, J.; Siepmann, F. Mathematical modeling of drug dissolution. Int. J. Pharm. 2013, 453, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Lennernas, H.; Abrahamsson, B. The use of biopharmaceutic classification of drugs in drug discovery and development: Current status and future extension. J. Pharm. Pharmacol. 2005, 57, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Avdeef, A. Physicochemical profiling (solubility, permeability and charge state). Curr. Top. Med. Chem. 2001, 1, 277–351. [Google Scholar] [CrossRef] [PubMed]
- Beg, S.; Swain, S.; Rizwan, M.; Irfanuddin, M.; Malini, D.S. Bioavailability enhancement strategies: Basics, formulation approaches and regulatory considerations. Curr. Drug Deliv. 2011, 8, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Stella, V.J.; Nti-Addae, K.W. Prodrug strategies to overcome poor water solubility. Adv. Drug Deliv. Rev. 2007, 59, 677–694. [Google Scholar] [CrossRef] [PubMed]
- Liechty, W.B.; Kryscio, D.R.; Slaughter, B.V.; Peppas, N.A. Polymers for drug delivery systems. Annu. Rev. Chem. Biomol. Eng. 2010, 1, 149–173. [Google Scholar] [CrossRef] [PubMed]
- Kiryukhin, M.V. Active drug release systems: Current status, applications and perspectives. Curr. Opin. Pharmacol. 2014, 18, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Grund, S.; Bauer, M.; Fischer, D. Polymers in drug delivery—State of the art and future trends. Adv. Eng. Mater. 2011, 13, B61–B87. [Google Scholar] [CrossRef]
- Kaunisto, E.; Marucci, M.; Borgquist, P.; Axelsson, A. Mechanistic modelling of drug release from polymer-coated and swelling and dissolving polymer matrix systems. Int. J. Pharm. 2011, 418, 54–77. [Google Scholar] [CrossRef] [PubMed]
- Lao, L.L.; Peppas, N.A.; Boey, F.Y.; Venkatraman, S.S. Modeling of drug release from bulk-degrading polymers. Int. J. Pharm. 2011, 418, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Anseth, K.S.; Bowman, C.N.; Brannon-Peppas, L. Mechanical properties of hydrogels and their experimental determination. Biomaterials 1996, 17, 1647–1657. [Google Scholar] [CrossRef]
- Natarajan, J.V.; Nugraha, C.; Ng, X.W.; Venkatraman, S. Sustained-release from nanocarriers: A review. J. Control. Release 2014, 193, 122–138. [Google Scholar] [CrossRef] [PubMed]
- Barbanti, S.H.; Zavaglia, C.A.C.; Duek, E.A.R. Polímeros bioreabsorvíveis na engenharia de tecidos. Polímeros 2005, 15, 13–21. [Google Scholar] [CrossRef]
- De Souza Costa, E., Jr.; Mansur, H.S. Preparação e caracterização de blendas de quitosana/poli (álcool vinílico) reticuladas quimicamente com glutaraldeído para aplicação em engenharia de tecido. Quím. Nova 2008, 31, 1460–1466. [Google Scholar] [CrossRef]
- Hassan, C.M.; Peppas, N.A. Structure and applications of poly(vinyl alcohol) hydrogels produced by conventional crosslinking or by freezing/thawing methods. In Biopolymers PVA Hydrogels, Anionic Polymerisation Nanocomposites; Springer: Berlin/Heidelberg, Germany, 2000; pp. 37–65. [Google Scholar]
- Ramot, Y.; Haim-Zada, M.; Domb, A.J.; Nyska, A. Biocompatibility and safety of pla and its copolymers. Adv. Drug Deliv. Rev. 2016, 107, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Rowe, R.C.; Sheskey, P.J.; Quinn, M.E.; Press, P. Handbook of Pharmaceutical Excipients; Pharmaceutical Press: London, UK, 2009; Volume 6. [Google Scholar]
- Zhang, Y.; Huo, M.; Zhou, J.; Zou, A.; Li, W.; Yao, C.; Xie, S. DDsolver: An add-in program for modeling and comparison of drug dissolution profiles. AAPS J. 2010, 12, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Costa, P.; Lobo, J.M.S. Modeling and comparison of dissolution profiles. Eur. J. Pharm. Sci. 2001, 13, 123–133. [Google Scholar] [CrossRef]
- Higuchi, W.I. Diffusional models useful in biopharmaceutics. Drug release rate processes. J. Pharm. Sci. 1967, 56, 315–324. [Google Scholar] [CrossRef]
- Korsmeyer, R.W.; Gurny, R.; Doelker, E.; Buri, P.; Peppas, N.A. Mechanisms of solute release from porous hydrophilic polymers. Int. J. Pharm. 1983, 15, 25–35. [Google Scholar] [CrossRef]
- Peppas, N.A.; Sahlin, J.J. A simple equation for the description of solute release. III. Coupling of diffusion and relaxation. Int. J. Pharm. 1989, 57, 169–172. [Google Scholar] [CrossRef]
- Hopfenberg, H. Controlled Release Polymeric Formulations; American Chemical Society: Washington, DC, USA, 1976. [Google Scholar]
- Dash, S.; Murthy, P.N.; Nath, L.; Chowdhury, P. Kinetic modeling on drug release from controlled drug delivery systems. Acta Pol. Pharm. 2010, 67, 217–223. [Google Scholar] [PubMed]
- Maria, G.; Luta, I. Precautions in using global kinetic and thermodynamic models for characterization of drug release from multivalent supports. Chem. Pap. 2011, 65, 542–552. [Google Scholar] [CrossRef]
- Aragon, D.M.; Rosas, J.E.; Martinez, F. Relationship between the solution thermodynamic properties of naproxen in organic solvents and its release profiles from PLGA microspheres. J. Microencapsul. 2013, 30, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Hartsel, S.; Bolard, J. Amphotericin B: New life for an old drug. Trends Pharmacol. Sci. 1996, 17, 445–449. [Google Scholar] [CrossRef]
- Kayser, O.; Olbrich, C.; Yardley, V.; Kiderlen, A.F.; Croft, S.L. Formulation of Amphotericin B as nanosuspension for oral administration. Int. J. Pharm. 2003, 254, 73–75. [Google Scholar] [CrossRef]
- Hamill, R.J. Amphotericin b formulations: A comparative review of efficacy and toxicity. Drugs 2013, 73, 919–934. [Google Scholar] [CrossRef] [PubMed]
- Torrado, J.J.; Espada, R.; Ballesteros, M.P.; Torrado-Santiago, S. Amphotericin b formulations and drug targeting. J. Pharm. Sci. 2008, 97, 2405–2425. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Lee, T.R. Contact angle and wetting properties. In Surface Science Techniques; Bracco, G., Hols, B., Eds.; Springer Series: Berlin, Germany, 2013; p. 663. [Google Scholar]
- Buckton, G. The role of compensation analysis in the study of wettability, solubility, disintegration and dissolution. Int. J. Pharm. 1990, 66, 175–182. [Google Scholar] [CrossRef]
- Vasiliu, S.; Bunia, I.; Racovita, S.; Neagu, V. Adsorption of cefotaxime sodium salt on polymer coated ion exchange resin microparticles: Kinetics, equilibrium and thermodynamic studies. Carbohydr. Polym. 2011, 85, 376–387. [Google Scholar] [CrossRef]
- Xu, W.; Yang, Y. Drug release and its relationship with kinetic and thermodynamic parameters of drug sorption onto starch acetate fibers. Biotechnol. Bioeng. 2010, 105, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, Z.; Wang, X. Investigation on surface molecular conformations and pervaporation performance of the poly(vinyl alcohol) (PVA) membrane. J. Colloid Interface Sci. 2009, 333, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Ghorani, B.; Tucker, N. Fundamentals of electrospinning as a novel delivery vehicle for bioactive compounds in food nanotechnology. Food Hydrocoll. 2015, 51, 227–240. [Google Scholar] [CrossRef]
- Laidler, K.J.; King, M.C. Development of transition-state theory. J. Phys.Chem. 1983, 87, 2657–2664. [Google Scholar] [CrossRef]
- Efentakis, M.; Buckton, G. Modelling drug release from hydrophobic matrices by use of thermodynamic activation parameters. Int. J. Pharm. 1990, 60, 229–234. [Google Scholar] [CrossRef]
- Tapia, C.; Buckton, G.; Newton, J.M. Factors influencing the mechanism of release from sustained release matrix pellets, produced by extrusion/spheronisation. Int. J. Pharm. 1993, 92, 211–218. [Google Scholar] [CrossRef]
- Ferrero, C.; Massuelle, D.; Doelker, E. Towards elucidation of the drug release mechanism from compressed hydrophilic matrices made of cellulose ethers. II. Evaluation of a possible swelling-controlled drug release mechanism using dimensionless analysis. J. Control. Release 2010, 141, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Cano, A.I.; Cháfer, M.; Chiralt, A.; González-Martínez, C. Physical and microstructural properties of biodegradable films based on pea starch and PVA. J. Food Eng. 2015, 167, 59–64. [Google Scholar] [CrossRef]
- Fil, B.A.; Yilmaz, M.T.; Bayar, S.; Elkoca, M.T. Investigation of adsorption of the dyestuff astrazon red violet 3rn (basic violet 16) on montmorillonite clay. Braz J. Chem. Eng. 2014, 31, 171–182. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Concentration (µg of AmB/ mg of system) |
|---|---|
| PVA Films | 33.4 ± 0.1 |
| PVA Fibers | 11.6 ± 0.2 |
| PLA Films | 8.6 ± 0.2 |
| PLA Fibers | 10.8 ± 0.2 |
| System | Temperatures (°C/K) | % Release | Time (h) |
|---|---|---|---|
| PVA Films | 25/298 | 79.27 ± 1.60 | 120 |
| 32/305 | 82.10 ± 0.92 | 120 | |
| 37/310 | 82.30 ± 1.21 | 120 | |
| 45/318 | 84.32 ± 0.70 | 120 | |
| PVA Fibers | 25/298 | 91.19 ± 0.92 | 96 |
| 32/305 | 95.23 ± 0.61 | 72 | |
| 37/310 | 96.24 ± 1.05 | 72 | |
| 45/318 | 97.05 ± 0.35 | 72 | |
| PLA Films | 25/298 | 3.11 ± 0.61 | 96 |
| 32/305 | 3.31 ± 0.35 | 96 | |
| 37/310 | 3.52 ± 0.35 | 96 | |
| 45/318 | 4.12 ± 0.35 | 72 | |
| PLA Fibers | 25/298 | 10.18 ± 0.61 | 48 |
| 32/305 | 10.38 ± 0.35 | 48 | |
| 37/310 | 10.38 ± 0.31 | 48 | |
| 45/318 | 14.02 ± 0.12 | 48 |
| SYSTEM | Temperature (°C/K) | Mathematical Model | Equation | R2 Adjusted | RMSE | Constants |
|---|---|---|---|---|---|---|
| PVA Films | 25/298 | Higuchi | 0.91 | 6.34 | = 7.19 (min) −0.5 | |
| 32/305 | 0.93 | 6.10 | = 7.38 (min) −0.5 | |||
| 37/310 | 0.92 | 6.53 | = 7.52 (min) −0.5 | |||
| 45/318 | Peppas–Sahlin | 0.98 | 3.13 | =18.85 = −1.05 | ||
| PVA Fibers | 25/298 | Peppas–Sahlin | 0.98 | 3.65 | =29.21 = −1.86 | |
| 32/305 | 0.97 | 4.70 | = 30.13 = −2.33 | |||
| 37/310 | 0.97 | 4.91 | = 31.01 = −2.40 | |||
| 45/318 | 0.95 | 6.05 | = 32.08 = −2.50 | |||
| PLA Films | 25/298 | – | – | – | – | – |
| 32/305 | Higuchi | 0.87 | 0.56 | = 0.34 (min) −0.5 | ||
| 37/310 | 0.85 | 0.67 | = 0.38 (min) −0.5 | |||
| 45/318 | 0.90 | 0.55 | = 0.43 (min) −0.5 | |||
| PLA Fibers | 25/298 | Peppas–Sahlin | 0.72 | 2.40 | = 3.05 = −0.267 | |
| 32/305 | 0.73 | 1.77 | = 3.58 = −0.279 | |||
| 37/310 | 0.75 | 1.78 | = 4.03 = −0.28 | |||
| 45/318 | 0.82 | 1.95 | = 4.79 = −0.38 |
| System | Activation Energy (Ea, kJ·mol−1) | Enthalpy (ΔH, kJ·mol−1) | Entropy (ΔS, J/(kg·K)) | Gibbs Free Energy (ΔG) | |
|---|---|---|---|---|---|
| PVA Films | 2.032 ± 0.066 | 0.600 ± 0.015 | −0.2282 ± 0.001 | Temperature (K) | ΔG (kJ·mol−1) |
| 298 | 68.609 ± 0.455 | ||||
| 305 | 70.207 ± 0.467 | ||||
| 310 | 71.349 ± 0.474 | ||||
| 318 | - | ||||
| PVA Fibers | 3.677 ± 0.074 | 1.162 ± 0.036 | −0.2130 ± 0.001 | Temperature (K) | ΔG (kJ·mol−1) |
| 298 | 64.612 ± 0.027 | ||||
| 305 | 66.106 ± 0.028 | ||||
| 310 | 67.171 ± 0.028 | ||||
| 318 | 68.877 ± 0.028 | ||||
| PLA Films | 12.908 ± 1.475 | 10.961 ± 0.170 | −0.2177 ± 0.001 | Temperature (K) | ΔG (kJ·mol−1) |
| 298 | - | ||||
| 305 | 77.344 ± 0.344 | ||||
| 310 | 78.466 ± 0.350 | ||||
| 318 | 80.241 ± 0.332 | ||||
| PLA Fibers | 1.763 ± 0.017 | 15.214 ± 0.284 | −0.1849 ± 0.001 | Temperature (K) | ΔG (kJ·mol−1) |
| 298 | 70.440 ± 0.547 | ||||
| 305 | 71.635 ± 0.412 | ||||
| 310 | 72.560 ± 0.414 | ||||
| 318 | 74. 040 ± 0.418 | ||||
| Factor | Level (−1) | Level (+1) |
|---|---|---|
| Geometry | Film | Fiber |
| Composition | PLA | PVA |
| Dissolution Mathematical Model | Equation | Parameter (s) |
|---|---|---|
| Zero Order | (1) | |
| First Order | (2) | |
| Higuchi | (3) | |
| Korsmeyer–Peppas | (4), (5) | |
| Hopfenberg | . | (6), (7) |
| Baker–Lonsdale | (8) | |
| Peppas–Sahlin | (9), (10), (11) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Freire, M.C.L.C.; Alexandrino, F.; Marcelino, H.R.; Picciani, P.H.d.S.; Silva, K.G.d.H.e.; Genre, J.; Oliveira, A.G.d.; Egito, E.S.T.d. Understanding Drug Release Data through Thermodynamic Analysis. Materials 2017, 10, 651. https://doi.org/10.3390/ma10060651
Freire MCLC, Alexandrino F, Marcelino HR, Picciani PHdS, Silva KGdHe, Genre J, Oliveira AGd, Egito ESTd. Understanding Drug Release Data through Thermodynamic Analysis. Materials. 2017; 10(6):651. https://doi.org/10.3390/ma10060651
Chicago/Turabian StyleFreire, Marjorie Caroline Liberato Cavalcanti, Francisco Alexandrino, Henrique Rodrigues Marcelino, Paulo Henrique de Souza Picciani, Kattya Gyselle de Holanda e Silva, Julieta Genre, Anselmo Gomes de Oliveira, and Eryvaldo Sócrates Tabosa do Egito. 2017. "Understanding Drug Release Data through Thermodynamic Analysis" Materials 10, no. 6: 651. https://doi.org/10.3390/ma10060651